
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,3-Dipolar cyclisation reactions of nitriles with sterically encumbered cyclic triphosphanes: synthesis and electronic structure of phosphorus-rich heterocycles with tunable colour†
Mitchell A.
Nascimento
 a,
Etienne A.
LaPierre
*a,
Brian O.
Patrick
b,
Jade E. T.
Watson
a,
Lara
Watanabe
c,
Jeremy
Rawson
c,
Christian
Hering-Junghans
*de and
Ian
Manners‡
a
a,
Etienne A.
LaPierre
*a,
Brian O.
Patrick
b,
Jade E. T.
Watson
a,
Lara
Watanabe
c,
Jeremy
Rawson
c,
Christian
Hering-Junghans
*de and
Ian
Manners‡
a
aDepartment of Chemistry, University of Victoria, 3800 Finnerty Rd, Victoria, British Columbia V8P 5C2, Canada. E-mail: elapierre@uvic.ca
bDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada
cDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Avenue, Windsor, Ontario N9B 3P4, Canada
dLeibniz Institut für Katalyse e.V. (LIKAT), A.-Einstein-Str.3a, 18059 Rostock, Germany. E-mail: christian.hering-junghans@catalysis.de
eInstitut für Chemie, Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany
First published on 19th June 2024
Abstract
We describe the synthesis, solid state and electronic structures of a series of tunable five-membered cationic and charge-neutral inorganic heterocycles featuring a P3CN core. 1-Aza-2,3,4-triphospholenium cations [(PR)3N(H)CR′]+, [1R]+ (R′ = Me, Ph, 4-MeOC6H4, 4-CF3C6H4) were formed as triflate salts by the formal [3 + 2]-cyclisation reactions of strained cyclic triphosphanes (PR)3 (R = tBu, 2,4,6-Me3C6H2 (Mes), 2,6-iPr2C6H3 (Dipp), 2,4,6-iPr3C6H2 (Tipp)) with nitriles R′CN in the presence of triflic acid. The corresponding neutral free bases (PR)3NCR′ (2R) were readily obtained by subsequent deprotonation with NEt3. The P3CN cores in 2R show an envelope conformation typical for cyclopentenes and present as yellow to orange compounds in the solid state as well as in solution depending on both substituents R and R′ in (PR)3NCR′. The P3CN cores in [1R]+ show a significant deviation from planarity with increasing steric bulk of the R groups at phosphorus, which results in a decrease in the HOMO–LUMO gap and distinct low-energy UV-Visible absorption bands. This allows access to colours spanning red, blue, indigo, and magenta. TD-DFT calculations provide valuable insight into this phenomenon and indicate an intramolecular charge-transfer from the HOMO located on the P3 framework to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–R′-based LUMO in the cationic species. The cations [1R]+ represent rare examples of phosphorus-rich heterocycles with tunable colour, which can be incorporated into polymers by post-polymerization modification to afford coloured polymers, which demonstrate utility as both proton and ammonia sensors.
C–R′-based LUMO in the cationic species. The cations [1R]+ represent rare examples of phosphorus-rich heterocycles with tunable colour, which can be incorporated into polymers by post-polymerization modification to afford coloured polymers, which demonstrate utility as both proton and ammonia sensors.
Introduction
Decades of intensive research into catalytic and stoichiometric organic transformations have resulted in myriad routes for C–C and C–X bond formation.1–5 In contrast, analogous routes to bonds between main group elements are poorly developed.6–10 In main group chemistry reductive coupling reactions or salt metatheses continue to represent the state of the art, but often involve aggressive reaction conditions that restrict the introduction of functional groups and lead to the formation of unwanted byproducts. The adaptation of well-established organic methodologies to inorganic substrates offers the possibility of new or improved routes to molecular and polymeric inorganic species with a wide range of potential applications.11–15A common atom-economic route to organic heterocycles is the Huisgen cycloaddition of 1,3-dipoles with dipolarophiles. Huisgen first demonstrated the thermal [3 + 2]-cycloaddition of organic azides with alkynes, which proceeds without regioselectivity.16 Through the use of Cu-catalysts, Meldal,17 Sharpless and Fokin,18 concurrently and independently developed a regioselective process. Accordingly, a wide range of 4-triazoles can be easily accessed at room temperature (Scheme 1, i). In terms of using main-group multiply bonded systems as dipolarophiles, the formal [3 + 2]-cycloadditions with organic azides have been extended to phosphaalkynes (Scheme 1, iii),19–22 cyaphides,23,24 arsadiazoniums and arsaalkynes25 to give 5-membered heterocycles selectively with 100% atom-economy. Vicinal donor–acceptor cyclopropanes (DACs) represent a class of “disguised” or “masked” 1,3-dipoles. Electron donating and withdrawing groups in vicinal position polarise the C–C bond and stabilise partial positive and negative charges (Scheme 1, ii), respectively, thereby introducing significant 1,3-dipolar character.26,27 These DACs react with dipolarophiles to yield a plethora of organic heterocycles akin to the Huisgen–Sharpless cycloaddition. Given their utility, cyclisation reactions are an emergent methodology in phosphorus chemistry for the synthesis of inorganic ring systems.28–34
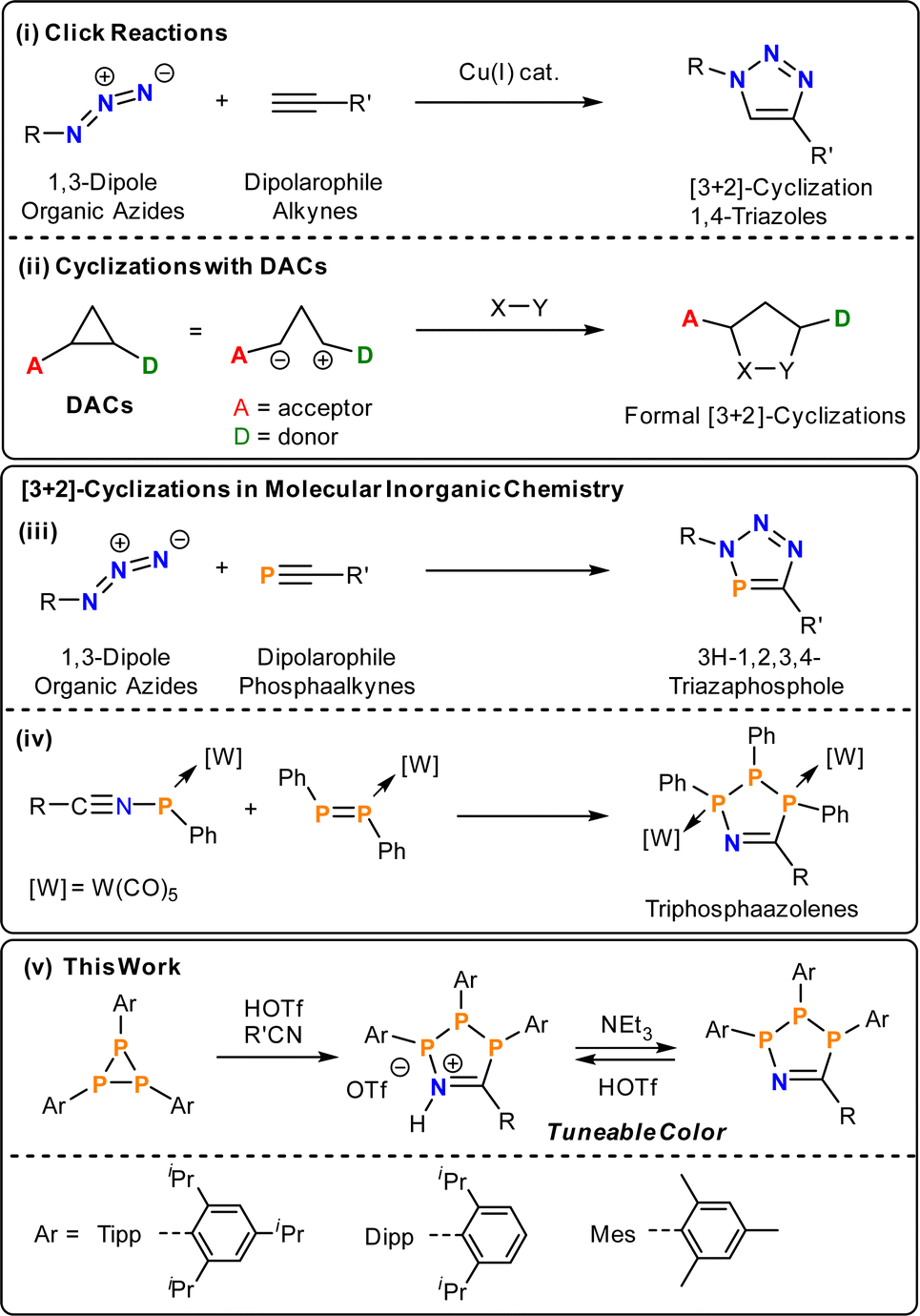 | ||
| Scheme 1 (i) Prototypical Huisgen–Sharpless cycloadditions; (ii) generalized donor–acceptor cyclopropane cycloaddition; (iii) [3 + 2]-cycloaddition between phosphaalkynes and azides; (iv) formation of P3CN-heterocycles in the coordination sphere of W(CO5); (v) summary of this study. | ||
Triphosphiranes, cyclic phosphanes of the general formula (PR)3, can be considered as heavier cyclopropane analogs by isolobal replacement of CR2 for PR.35 Accordingly, triphosphiranes have a long history as starting materials for the synthesis of inorganic ring systems and the field was recently reviewed.36 Recent examples include phosphenium ion insertion and Lewis acid activation reactions.37,38 In general, the insertion of organic molecules into P–P bonds is a valuable tool for the construction of new phosphorus compounds.39
By analogy to DACs, the triphosphirane (tBuP)3 can be activated by either Brønsted (HOTf, OTf = [SO3CF3]−) or Lewis acids such as (Ph3Sb(Cl)OTf), through polarization of one P–P bond and subsequently react with nitriles R′CN to give five-membered P3CN-species.40 In the absence of HOTf or MeOTf no reaction with nitriles is observed. This methodology provides an operationally simple and rapid route to 1-aza-2,3,4-triphospholenium salts [P3tBu3N(H)CR′]OTf and their neutral 1-aza-2,3,4-triphospholene P3tBu3NCR′ congeners, which can be cycled between neutral and cationic states by addition of acid or base, respectively. The neutral rings were previously only accessible through formal [3 +2]-cyclisations of W(CO)5-coordinated diphosphenes and nitrilium phosphanylides, respectively (Scheme 1, iv).41 However, both the cationic [P3tBu3N(H)CR′]+ as well as the neutral P3tBu3NCR′ rings are either colourless or pale yellow and show no evidence for low-energy electronic transitions.
Phosphorus containing acyclic and cyclic species are of general interest as OLED emitters42–44 and fluorophores45–47 – with examples ranging from small molecule photoswitches48 to phosphorus analogues of common dyes.49–51 Phosphamethine cyanines were first reported as red crystalline solids by Dimroth in 1964, and subsequent examples exhibit colours spanning yellow, orange, and red as a function of the P-substituents and thus these species have been used as phosphorus-based dyes.52–59 The colour in the base-stabilised phosphasilene carbene-SiCl2P-Tipp (Tipp = 2,4,6-iPr3C6H2) ranges from deep blue, when cyclic alkyl amino carbenes (cAACs) are employed (Fig. 1a, A) or red when using the NHC IPr (IPr = (HCNDipp)2C, Dipp = 2,6-iPr2C6H3; Fig. 1a, B).60 Phosphonium species47,61,62 and phospholes48,61–68 have also shown colours that include red, orange, green, blue, and purple, although these species achieve their colour on account of large polycyclic aromatic systems with appended or incorporated phosphorus centres.69 Phosphorus radicals, as expected, form intensely-coloured compounds across the visible spectrum as well.70–73 However, there are few instances of colour in closed-shell phosphorus-rich or catenated phosphorus species. Among those are deep red cyclic 1-aza-2,3,4-triphospholide anions (Fig. 1a, C),74 or a unique example of a five-membered GaP2CO species with a 1,2-diphospha-1,3-butadiene recently reported by Schulz (Fig. 1a, D).75
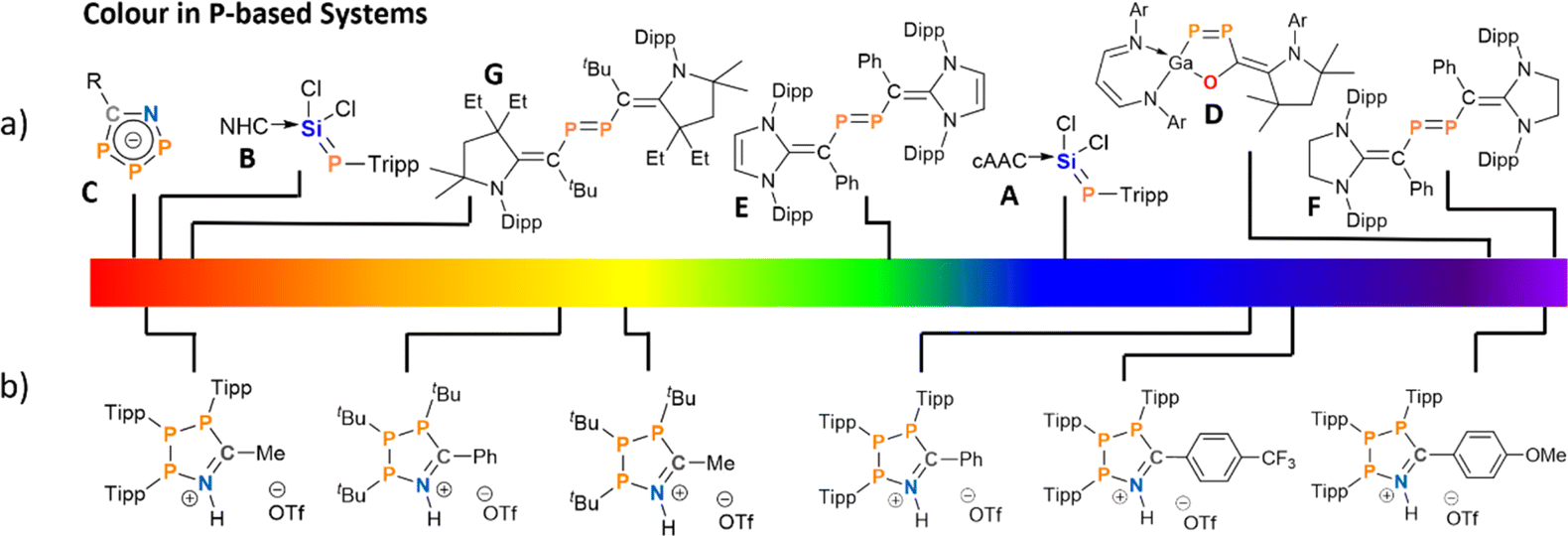 | ||
| Fig. 1 Previously reported coloured (poly-)phosphorus species (a) and coloured phosphorus species reported in this work (b), shown in line with their visible colours. | ||
Another class of P-rich compounds are diphosphenes, which commonly present as yellow to orange solids.76 Introduction of N-heterocyclic vinyl (NHV) substituents at phosphorus, offer a tool to effectively tune the colour of such diphosphenes of the type (NHCCR)2P2 through extended conjugation. Colours include green when (IPr)CPh-substituents are utilized (Fig. 1a, E), or magenta when (SIPr)CPh-substituents (SIPr = (H2CNDipp)2C; Fig. 1a, F) are used.77 When (EtcAAC)CtBu-substituents are introduced, deep red crystalline solids are obtained (Fig. 1a, G).78 Notably, the colour in these examples predominantly arises from π–π* transitions, and tuning the energy of absorption through distal or late-stage modification is often non-trivial.
In this contribution we show that the colour of 1-aza-2,3,4-triphospholenium salts can be effectively tuned through modification of the steric demand of the P-substituents, and through the electronic properties of the nitrile coupling partner (Fig. 1b). This is achieved by using aryl-substituted triphosphiranes (PAr)3 (Ar = Mes, 2,4,6-Me3C6H2; Dipp; Tipp, 2,4,6-iPr3C6H2) as starting materials.79 The colouration can be traced to intramolecular charge transfer processes, and we also show switchable colour when going from the cationic to the neutral state through addition of external base. This feature was used to prepare a Brønsted acid–base responsive polymer.
Results
Synthesis and NMR spectroscopic characterisation of cationic and neutral P3CN heterocycles [1R]+ and 2R
Our investigation into the effect of steric hindrance on the Brønsted acid-mediated [3 + 2]-cyclisation of cyclic triphosphanes began with the reaction of (PTipp)3 with an excess of acetonitrile (MeCN) in the presence of HOTf (Scheme 2). (PTipp)3 was dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of toluene/MeCN, which, after addition of one equivalent of neat HOTf at 20 °C, resulted in a deep red homogeneous solution. This colouration was unexpected, given that analogous reactions with (PtBu)3 yield yellow solutions and pale-yellow solid products (Fig. 2). An aliquot of the red reaction mixture analysed by 31P{1H} NMR spectroscopy showed conversion to a single phosphorus containing species with an AMX spin system consistent with formation of the 1-aza-2,3,4-triphospholenium ring [1Tipp]+ (R′ = Me) (Fig. 2, top right).
:1 v/v mixture of toluene/MeCN, which, after addition of one equivalent of neat HOTf at 20 °C, resulted in a deep red homogeneous solution. This colouration was unexpected, given that analogous reactions with (PtBu)3 yield yellow solutions and pale-yellow solid products (Fig. 2). An aliquot of the red reaction mixture analysed by 31P{1H} NMR spectroscopy showed conversion to a single phosphorus containing species with an AMX spin system consistent with formation of the 1-aza-2,3,4-triphospholenium ring [1Tipp]+ (R′ = Me) (Fig. 2, top right).
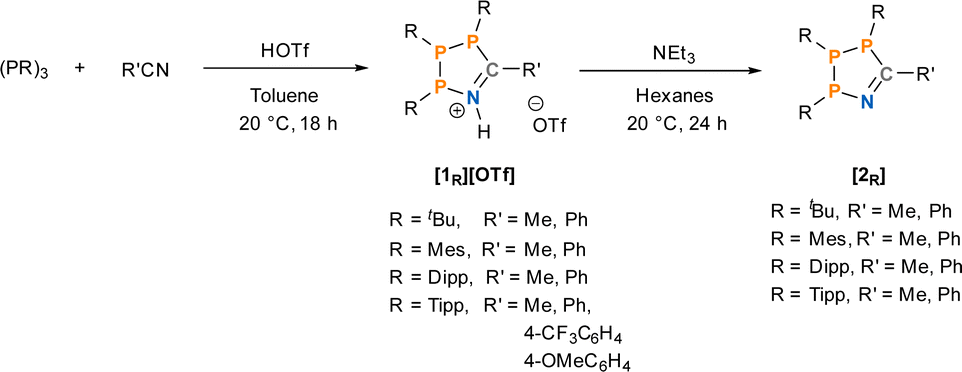 | ||
| Scheme 2 Synthesis of 1-aza-2,3,4-triphospholenium triflate salts and their corresponding free bases in this work with a labelling notation, where R describes the substitution at phosphorus and R′ describes the nitrile substituent. | ||
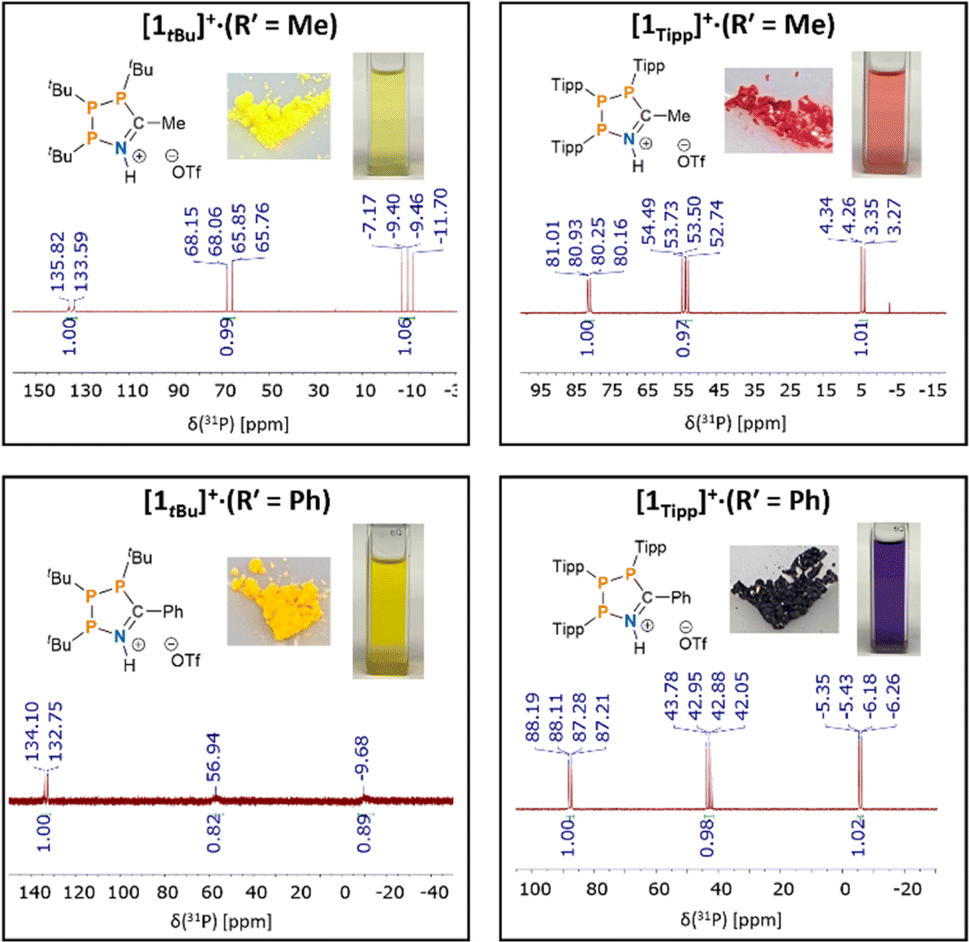 | ||
| Fig. 2 31P{1H} NMR spectra in CDCl3 for a comparative set of 1-aza-2,3,4-triphospholenium cations, and photographs of their [OTf]− salts in the solid state and in solution (CH2Cl2). | ||
Compared to previously synthesized [1tBu]+ (R′ = Me) the most downfield signal in species [1Tipp]+ (R′ = Me) is shielded by approximately 52 ppm, while both JPP coupling constants are smaller than in [1tBu]+ (R′ = Me) by approximately 120 Hz and 70 Hz, respectively (Fig. 2, top left). Removal of the volatiles followed by washing of the tacky residue with n-hexane yielded a red powder. 1H NMR spectra in CDCl3 (ESI Fig. S-1†) revealed inequivalent methine environments for the Tipp substituents, as well as a characteristic doublet of doublets (JHP = 10.2 Hz, JHH = 4.6 Hz) corresponding to the methyl resonance of the incorporated acetonitrile-derived moiety. A broad singlet at 12.61 ppm is assigned to the H–N resonance of the protonated skeletal nitrogen atom. In the 19F{1H} NMR spectrum in CDCl3 (ESI Fig. S-2†) a singlet at δF = −78.7 ppm, indicated a non-interacting triflate anion (cf. [Me3SiN(C6H10)P(C6H10)][O3SCF3] δF = −78.8 ppm).80
Given that (PTipp)3 demonstrated facile MeCN insertion chemistry, we sought to extend the synthetic protocol to (PDipp)3 and (PMes)3. [1Dipp]+ (R′ = Me) was prepared similarly and was found to be virtually identical to [1Tipp]+ (R′ = Me) in colour, with similar 31P, 1H and 19F NMR spectroscopic characteristics (ESI Fig. S-41 to S-43†); in contrast, the attempted synthesis of [1Mes]+ (R′ = Me) was less straightforward. After conducting a similar work-up to that used for the isolation of [1Tipp]+ (R′ = Me) and [1Dipp]+ (R′ = Me), a yellow powder was obtained. Recrystallisation of the powder from PhF/n-pentane afforded crystals which, when dissolved in CDCl3 and subsequently analysed by 31P{1H} NMR spectroscopy, showed the anticipated AMX spin system, alongside impurities characterized by resonances at −59.5 ppm (singlet, ca. 7%) and −18.9 (two peaks, ca. 2%) (ESI Fig. S-70†).
A second recrystallisation of [1Mes]+ (R′ = Me) by allowing a PhF/Et2O solution layered with n-pentane to stand for a month at −30 °C resulted in a mixture of yellow prisms of [1Mes]+ (R′ = Me) and a small amount of yellow plates that were crystallographically identified as the Et2O solvate of (PMes)6 (ESI Section 5.18†). (PMes)6 has been previously observed as a side-product of the reaction between Na2[P4Mes4] with [Rh(COD)Cl]2 by Hey-Hawkins and coworkers by means of single-crystal X-ray crystallography.81 The mechanism of the formation of (PMes)6 is currently unknown.
Next, we synthesized the benzonitrile-derived series [1Tipp]+ (R′ = Ph), [1Dipp]+ (R′ = Ph), [1Mes]+ (R′ = Ph) and [1tBu]+ (R′ = Ph) to provide a comparison in their properties with those of [1R]+ (R′ = Me). In this case (PTipp)3 was dissolved in a 2:1 v/v solution of toluene/PhCN, giving a colourless solution. Addition of one equivalent of neat triflic acid at 20 °C resulted in a deep blue solution. After removal of the volatiles, the dark blue residue of [1Tipp]+ (R′ = Ph) was washed with n-hexane to yield a blue powder, whose 31P{1H} NMR spectrum in CDCl3 consists of an AMX spin system, which is virtually identical to that of [1Tipp]+ (R′ = Me) (Fig. 2, bottom right). The analogous reaction was repeated with (PDipp)3, again yielding a blue powder of [1Dipp]+ (R′ = Ph) possessing the characteristic AMX spin system in the 31P{1H} NMR spectrum (ESI Fig. S-56†), likewise virtually identical to [1Dipp]+ (R′ = Me). In the case of [1Mes]+ (R′ = Ph), we obtained an orange powder, as opposed to the yellow [1Mes]+ (R′ = Me), that gave rise to an AXY spin system with second order effects in the 31P{1H} NMR spectrum in CDCl3, similar to that observed in [1Mes]+ (R′ = Me). We note that [1Mes]+ (R′ = Ph) has greater stability in solution than [1Mes]+ (R′ = Me) and that the only impurities that form upon dissolution correlate to (PMes)4 as evidenced by 31P NMR spectroscopy. Finally, [1tBu]+ (R′ = Ph), was synthesized and isolated as a yellow powder with an AMX 31P{1H} NMR spectrum featuring broad signals (Fig. 2, bottom left).
To probe the effect of electron-donating and electron withdrawing substituents on [1Tipp]+ (R′ = Ph), p-MeOC6H4CN and p-CF3C6H4CN were used in place of PhCN to synthesize [1Tipp]+ (R′ = p-MeOC6H4) and [1Tipp]+ (R′ = p-CF3C6H4), respectively. [1Tipp]+ (R′ = p-MeOC6H4) formed a magenta solution with 31P{1H} NMR spectra bearing an AMX spin system in which the M and X nuclei are more shielded relative to [1Tipp]+ (R′ = Ph). [1Tipp]+ (R′ = p-CF3C6H4), contrarily, afforded royal blue solutions, with a similar AMX spin system in the 31P{1H} NMR spectrum, in which the M and X nuclei are shifted downfield relative to [1Tipp]+ (R′ = Ph). As these compounds could be potentially used as tunable P-based dyes, the photo-stability of a prototypical species was investigated. Irradiation of [1Tipp]+ (R′ = Ph) with a broad band solar irradiation LED (480 mW, see ESI Fig. S-115 and S-116† for lamp and 31P NMR spectra) for 72 h revealed no discernible decomposition as assayed by 31P{1H} NMR spectrometry in CDCl3, demonstrating the photostability of these 1-aza-2,3,4-triphospholenium salts.
Subsequently, the formation and properties of the corresponding free bases, 1-aza-2,3,4-triphospholenes 2R (R′ = Me and Ph) were explored, to determine if this unexpected and intriguing colouration was unique to the protonated heterocycles. The free base 2Tipp (R′ = Me) was synthesized through the addition of NEt3 to a slurry of [1Tipp]+ (R′ = Me) in n-hexane, and was isolated as a yellow solid after removal of [Et3NH]OTf by filtration and of volatiles in vacuo. The 1H NMR spectrum lacks the N–H signal, consistent with quantitative deprotonation. Loss of triflate, and thus formation of [HNEt3]OTf was indicated by the absence of detectable 19F NMR signals in isolated 2Tipp (R′ = Me). The 31P{1H} NMR spectrum of 2Tipp (R′ = Me) also consists of an AMX spin system, however, with notably shielded signals for the A and M nuclei compared to previously reported 2tBu (R′ = Me).402Dipp (R′ = Me) had similar colouration to 2Tipp (R′ = Me), while the 31P{1H} NMR spectrum exhibits a second order AXY spin system with the A nucleus shifting downfield to 98.6 ppm compared to the chemical shift of 82.3 ppm observed for the A nucleus of 2Tipp (R′ = Me). 2Mes (R′ = Me) is colourless in solution and exhibits an AMX spin system distinct from that of 2Dipp (R′ = Me) and 2Tipp (R′ = Me). 2Mes (R′ = Me) and also proved to be stable in solution, in contrast to its protonated analogue [1Mes]+ (R′ = Me), as NMR spectra collected on the solution after several weeks showed no significant change. In the case of the 2R (R′ = Ph) series, no significant changes were observed in the 31P{1H} NMR chemical shifts compared to the 2R (R′ = Me) series. When single crystals of 2Mes (R′ = Ph) were dissolved in CDCl3, partial decomposition was noted, as evidenced by formation of an unknown impurity which appears as a singlet in the 31P{1H} NMR spectrum at 62.4 ppm in CDCl3.
Not only are the neutral 1-aza-2,3,4-triphospholenes readily synthesized through deprotonation with NEt3, these free bases are near quantitatively re-protonated with triflic acid to again form the 1-aza-2,3,4-triphospholenium salts [1R]OTf, which may again be reversibly deprotonated. This ability to reversibly convert between cationic and free base rings provides a second route to forming these heterocycles in high purity, and also provides a useful colorimetric switch (vide infra).
X-ray structural characterisation of the P3NC heterocycles [1R]+ and 2
Characterisation of each of the aforementioned heterocycles [1R]+ and 2R in the solid state was performed by single crystal X-ray diffraction (SC-XRD) experiments to investigate a potential structural rationale for the unexpected colouration. Structures, key bond lengths and angles for all species can be found in Section 5 of the ESI,† while representative structures of [1R]+ (R′ = Ph) and of [2R] (R′ = Ph) for R = Tipp, Dipp, and Mes are shown in Fig. 3. For this discussion, we will refer to the phosphorus atoms of the central P3CN framework as PC (phosphorus bound to the nitrile-derived carbon atom), PN (phosphorus bound to the nitrile nitrogen atom), and PP for the phosphorus atom between PC and PN. First, the down, down, up arrangement of the aryl substituents with respect to the P3 unit is maintained in all species when compared to (PAr)3. Additionally, there is a close O1–H1 contact (d(O–H) ca. 1.8 Å) between the nitrilium unit in [1R]+ and the triflate counter anion, indicative of H-bonding in the solid state, while the 19F NMR data clearly indicates a weakly-interacting OTf ion in solution. One feature common to the three cationic rings are minimally longer PN–PP (R′ = Ph, [1Tipp]+ 2.2458(6), [1Dipp]+ 2.243(1), [1Mes]+ 2.238(1) Å) compared to the PP–PC distances (R′ = Ph, [1Tipp]+ 2.2262(4), [1Dipp]+ 2.212(1), [1Mes]+ 2.206(1) Å), though all of these are within the range of typical P–P single bonds (cf. Σrcov(P–P) = 2.22 Å),82 also when compared to triphosphirane precursor (PDipp)3 (cf. d(P–P) 2.1991(4), 2.2440(4), 2.2124(3) Å).79 The C–N distances are ca. 1.31 Å, minimally longer than an ideal double bond (cf. Σrcov(CN) = 1.27 Å),82 while the PN–N and PC–C distances are best described as minimally shortened single bonds. All P atoms are in a trigonal pyramidal coordination environment with the smallest sum of angles at the central PP atoms (Σ < (P); R′ = Ph, [1Tipp]+ 296.46, [1Dipp]+ 299.69, [1Mes]+ 301.49°), with similar values for PN, while Σ < (P) at the PC atom is considerably larger by ca. 10 °C. In the corresponding neutral species 2R the PN–PP (R′ = Ph; 2Tipp 2.256(6); 2Dipp 2.2725(6); 2Mes 2.2496(8), 2.256(1) Å) bonds are again longer than the PP–PC (R′ = Ph; 2Tipp 2.201(1); 2Dipp 2.1972(5); 2Mes 2.1907(8), 2.1862(8) Å) bonds, with a greater difference when compared to the cationic rings. Upon proton abstraction the C–N distance decreases to ca. 1.28 Å, in line with a double bond. The PN–N distances shorten considerably (davg.(PN–N) = 1.70 Å), while the PC–C distances increase to an average value of ca. 1.86 Å. The sum of angles at the P atoms are similar to those in the cationic derivatives.
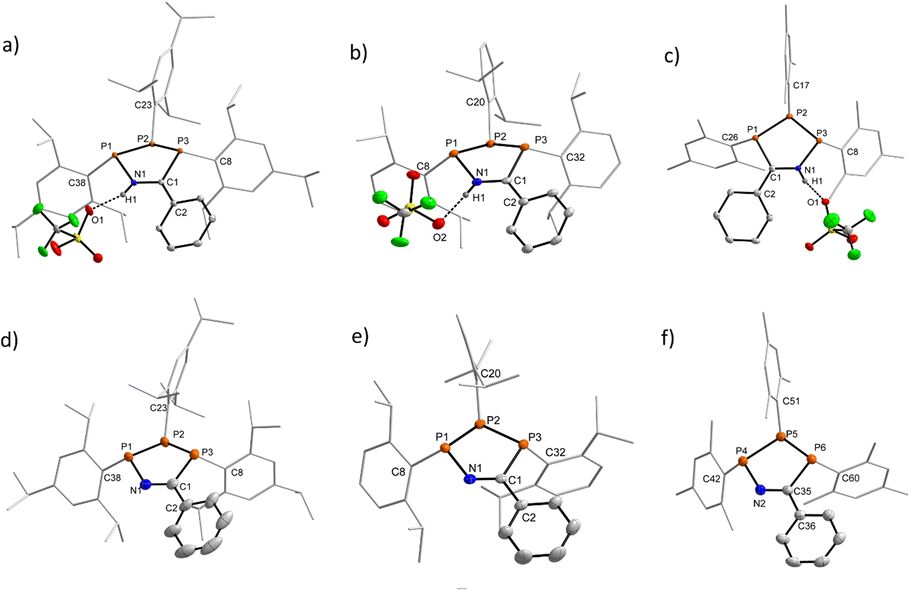 | ||
| Fig. 3 Solid-state structures of [1Tipp]+ (R′ = Ph) (a), [1Dipp]+ (R′ = Ph) (b), [1Mes]+ (R′ = Ph) (c), 2Tipp (R′ = Ph) (d), 2Dipp (R′ = Ph) (e), and 2Mes (R′ = Ph) (f). Thermal ellipsoids are drawn at the 50% probability level. Tipp, Dipp, and Mes substituents depicted as wireframe for clarity, and all hydrogen atoms except H1 in [1R]+ have been omitted for clarity. For 2Mes (R′ = Ph) only one of two independent molecules in the asymmetric unit is shown. | ||
When analysing the molecular structures of all synthesized species, two distinct structural features are apparent in the Dipp and Tipp series when compared to the Mes and tBu series. Firstly, a clear shortening in the distance between PN and PC atoms in [1Tipp]+ and [1Dipp]+ (R′ = Me and Ph) species is observed, resulting in 4.5–5.5% shorter distances compared to the analogous [1tBu]+ (R′ = Me and Ph) species (e.g. cf.[1Tipp]+ (R′ = Ph) 3.1149(7) Å; [1tBu]+ (R′ = Ph) 3.2549(6) Å, see ESI Section 5.19† for full details). Conversely, the free bases show no appreciable trend in lengthening or shortening of the PN–PC distance, indicating that this phenomenon is specific to the cationic rings bearing sterically encumbering aryl substituents.
Secondly, we observed a substantially increased bending in both [1Tipp]+ and [1Dipp]+ (R′ = Me and Ph) heterocycles compared to their [1tBu]+ and [1Mes]+ congeners (Fig. 4). The angle of bending, θ, is measured from the horizontal PC–CN–PN plane downward to the plane formed by the three catenated phosphorus atoms (see top of Fig. 4).
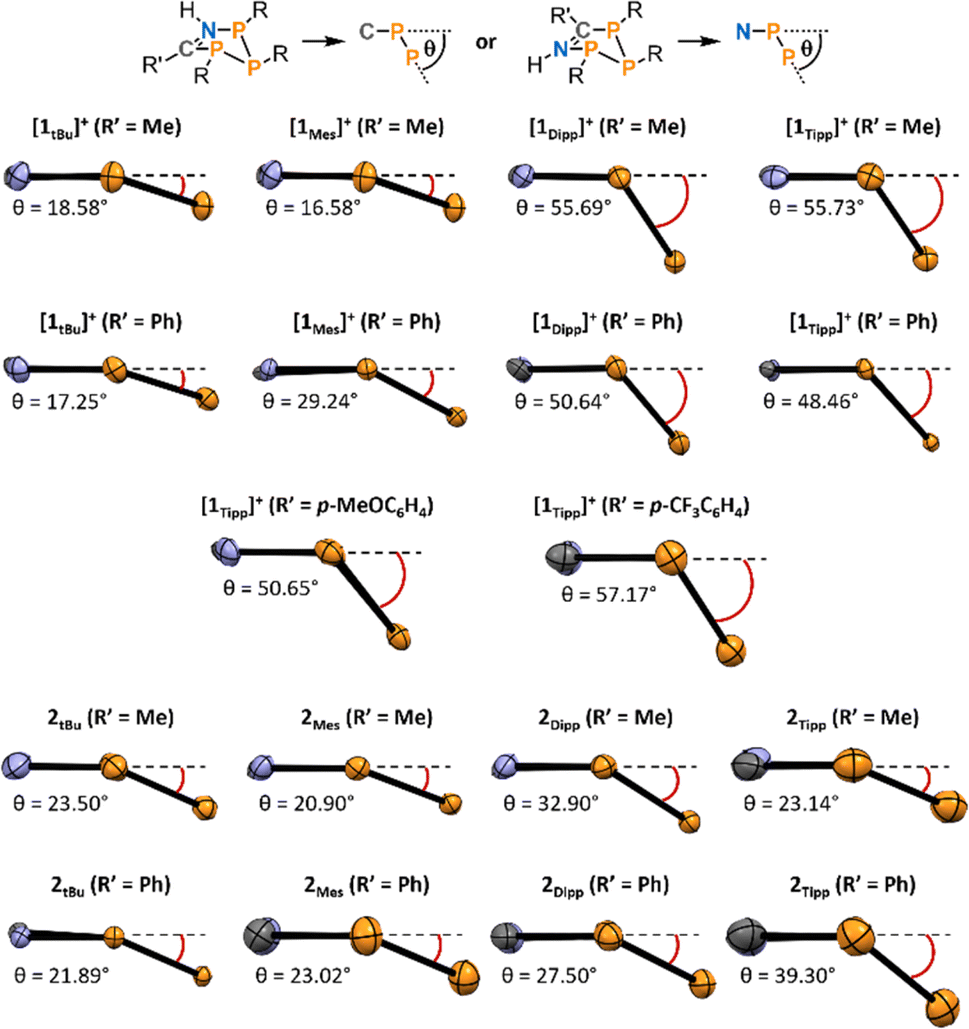 | ||
| Fig. 4 Side-on view of the central P3CN rings in the molecular structures as determined by SC-XRD experiments of [1R]+ and 2R demonstrating the angle of bending of each species (P = orange, C = grey, N = blue). View was chosen such that the bent-out-of-plane PP atom sits to the right, and all θ values are measured from the horizontal PC–C–N–PN plane (represented as a dotted line parallel to the bottom of the page) downward to the plane formed by the three phosphorus atoms. Thermal ellipsoids are drawn at the 50% probability level. | ||
Using this parameter, [1tBu]+ (R′ = Me and Ph) are minimally bent, with the angles between the PN–PP–PC plane and the PN–NC–PC plane being 18.58° and 17.25°, typical of cyclopentanes, while these angles are substantially increased to 55.73° and 48.46° in [1Tipp]+ (R′ = Me and Ph), respectively. Comparing the complete set of metrical parameters for [1R]+ (R′ = Ph) and 2R (R′ = Ph), it can be seen that the change in bending angle as steric bulk increases is greater for the [1R]+·(R′ = Ph) series than in their neutral analogues. Similar structural distortions are also observed in the electronically modified compounds [1Tipp]+ (R′ = p-MeOC6H4, p-CF3C6H4), which display bending angles of 50.65° and 57.17°, respectively, the latter being the most bent of the series. This substantial bending is also linked to changes in the PN–PC distance in these species, with [1Tipp]+ (R′ = p-MeOC6H4CN) displaying a 4.86% shortening and [1Tipp]+ (R′ = p-CF3C6H4CN) displaying a 6.76% shortening (ESI Section 5.19†).
UV-Vis spectroscopic studies of [1R]+ and 2R
UV-Visible spectra were obtained in CH2Cl2 solution for each of the cyclic species to assess whether the observed data would be correlated to the steric profiles of the aryl substituents, or the characteristics of the nitrile used. Given that we were unable to successfully crystallise sufficiently pure 2Mes (R′ = Me), and that [1Mes]+ (R′ = Me) and 2Mes·(R′ = Ph) always undergo partial decomposition in solution, even when using single crystals, UV-Vis spectra were not acquired for these species.All compounds studied by UV-Vis spectroscopy show broad absorption bands in the 300–400 nm region typically associated with the aryl substituents at phosphorus (Table 1, λ2). However, the spectra of all cationic rings (except [1tBu]+ (R′ = Me)) also exhibit a comparatively low-energy absorption band (Table 1, λ1) which follows Beer's Law. The low-energy absorption band observed in [1R]+ (R′ = Ph) is, on average, bathochromically shifted by 59 nm compared to the absorption band of the analogous [1R]+ (R′ = Me) species (Fig. 5, top), which is attributed to increased electronic conjugation involving the π-system of the aromatic Ph substituent (vide infra). When the steric bulk is increased at phosphorus (where tBu < Mes < Dipp ≈ Tipp), the bending angle of the P3CN-rings increases significantly, resulting in a progressive red-shift in the value of the low-energy absorption bands.
| Species | Angle (°) | λ CT (nm) | Species | Angle (°) | λ CT (nm) |
|---|---|---|---|---|---|
| [1tBu]+ (R′ = Ph) | 17.25 | 419 | 2tBu (R′ = Ph) | 21.89 | 382 |
| [1Mes]+ (R′ = Ph) | 29.24 | 488 | 2Mes (R′ = Ph) | 23.02 | NA |
| [1Dipp]+ (R′ = Ph) | 50.64 | 550 | 2Dipp (R′ = Ph) | 27.5 | 461 |
| [1Tipp]+ (R′ = Ph) | 48.46 | 560 | 2Tipp (R′ = Ph) | 39.3 | 472 |
| [1Tipp]+ (R′ = p-MeOC6H4) | 50.65 | 555 | |||
| [1Tipp]+ (R′ = p-CF3C6H4) | 57.17 | 583 |
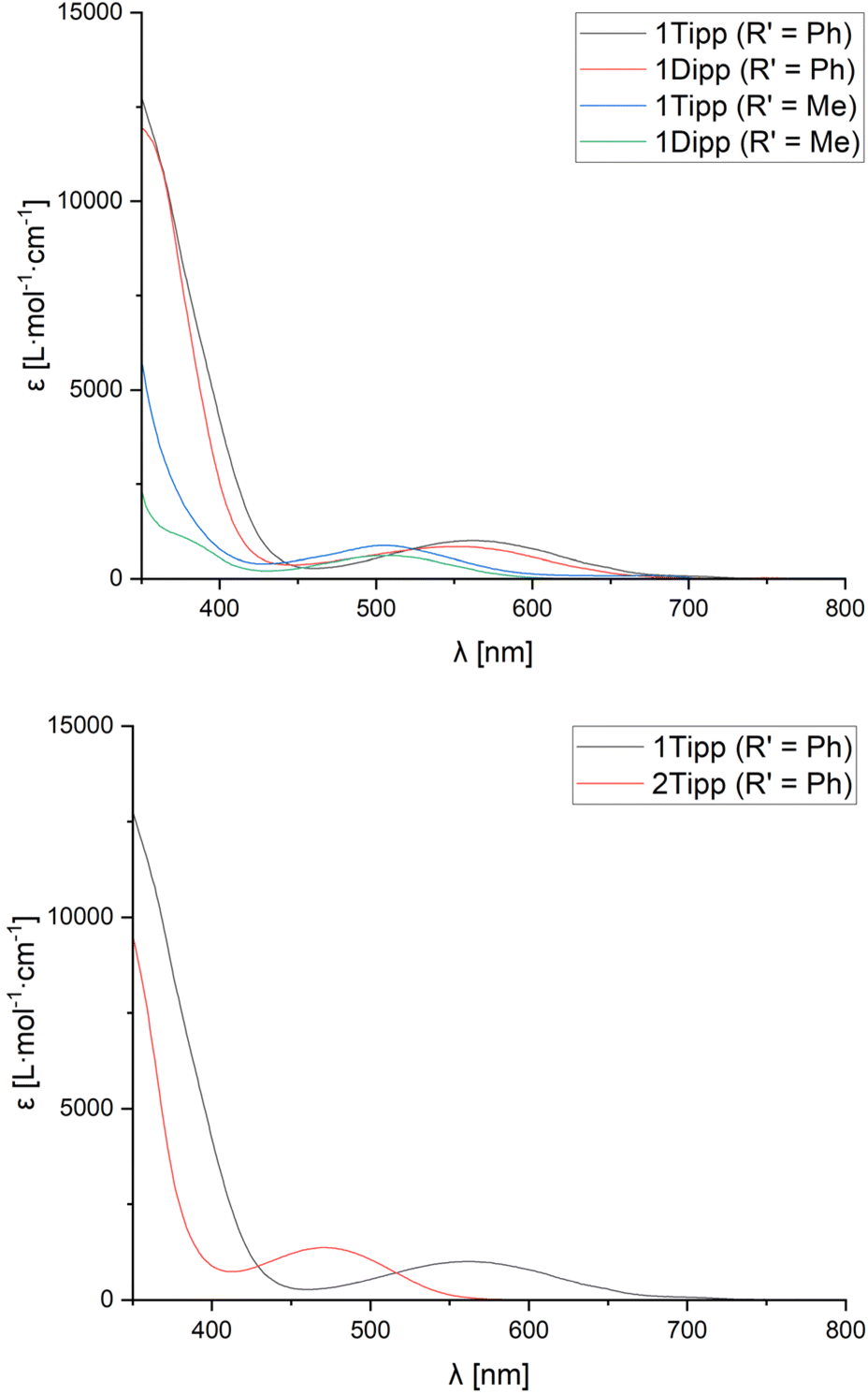 | ||
| Fig. 5 UV-visible spectra of [1Tipp]+ (R′ = Ph, Me) and [1Dipp]+ (R′ = Ph, Me) in CH2Cl2 solution at room temperature (top) and of 1Tipp (R′ = Ph) compared to 2Tipp (R′ = Ph) (bottom). | ||
Unlike [1R]+ (R′ = Me), the spectra of 2R (R′ = Me) showed no evidence of low-energy absorption bands between 300-800 nm, while spectra of 2R (R′ = Ph) species showed low-energy absorption bands that are hypsochromically shifted relative to those observed in [1R]+ (R′ = Ph) (Table 1) and manifest in a change of observed colouration to colourless, yellow, or orange in all species 2R (Fig. 5, bottom). In comparing [1Tipp]+ (R′ = Ph) to [1Tipp]+ (R′ = p-MeOC6H4 or p-CF3C6H4), we note that [1Tipp]+ (R′ = p-MeOC6H4) bears a hypsochromically shifted λ1 compared to [1Tipp]+ (R′ = Ph), while the λ1 associated with [1Tipp]+ (R′ = p-CF3C6H4) is bathochromically shifted by 20 nm compared to [1Tipp]+ (R′ = Ph). When considering the Hammett parameter of the para-substituent at R′ a correlation is found between an increasing Hammett parameter σp, and an increase in the wavelength of the low-energy absorption band λ1 (p-MeO-C6H4 < p-H-C6H4 < p-CF3-C6H4).
DFT studies of [1R]+ and 2R
To shed further light on the unusual nature of the UV-visible absorption bands and their seemingly strong correlation to the bending angle in cations [1R]+, the electronic structures of [1Tipp]+ and [1tBu]+ (R′ = Me), 2Tipp and 2tBu (R′ = Me), [1R]+ (R′ = Ph), and 2R (R′ = Ph) were investigated by DFT calculations on the BP86-D3/def2SVP level of theory, while taking the molecular structures from SC-XRD experiments as a structural basis. In all cationic species [1R]+ the triflate ion was explicitly retained. Time-dependent density functional theory (TD-DFT) calculations were performed at the B3LYP/cc-pVTZ (in both the gas phase and using the polarizable continuum solvation model (PCM) for CH2Cl2 (smd = DCM), for comparison) level of density functional theory using the respective BP86-D3/def2-SVP optimized gas-phase S0 geometries, which were found to be in excellent agreement with the experimentally determined solid state structures (for complete computational details, including a comparison of the structural parameters, please refer to the ESI p. S-150 ff.†). The UV-Vis spectra of all species were reproduced well by the calculations (cf. compare ESI p. S-155 ff.†) in the gas phase. There is no significant difference between the data calculated in the gas phase and those using a PCM for solvation. The calculated lowest energy transitions (λ1) of [1R]+ correspond to a HOMO–LUMO transition, with all other transitions associated with transitions from the HOMO−1 and HOMO−2 orbitals to the LUMO or higher unoccupied molecular orbitals (ESI Section 6†). To better understand the origin of colour in these species, it is useful to assess the differences in the HOMO and LUMO orbitals of [1tBu]+ (blue-shifted absorption), and those in [1Tipp]+ which absorbs at longer wavelengths (Fig. 6), and shows a considerably larger bending angle θ. In the case of [1Tipp]+ (R′ = Ph), the HOMO is best described mainly (ca. 74%) as linear combination of the n(P) orbitals (e.g. non-bonding combination, P lone pairs), with minimal delocalization into the CN (ca. 5%) and Tipp (ca. 7%) π-systems. The LUMO, by comparison, is mainly delocalized over the NCPh unit and has π* character, with small contributions at PC, PN and PP. The HOMO in [1tBu]+ (R′ = Ph) is similar to that of [1Tipp]+ (R′ = Ph), while the LUMO again has mainly NCPh π* character, however, it lacks the contribution from PP observed in [1Tipp]+ (R′ = Ph).
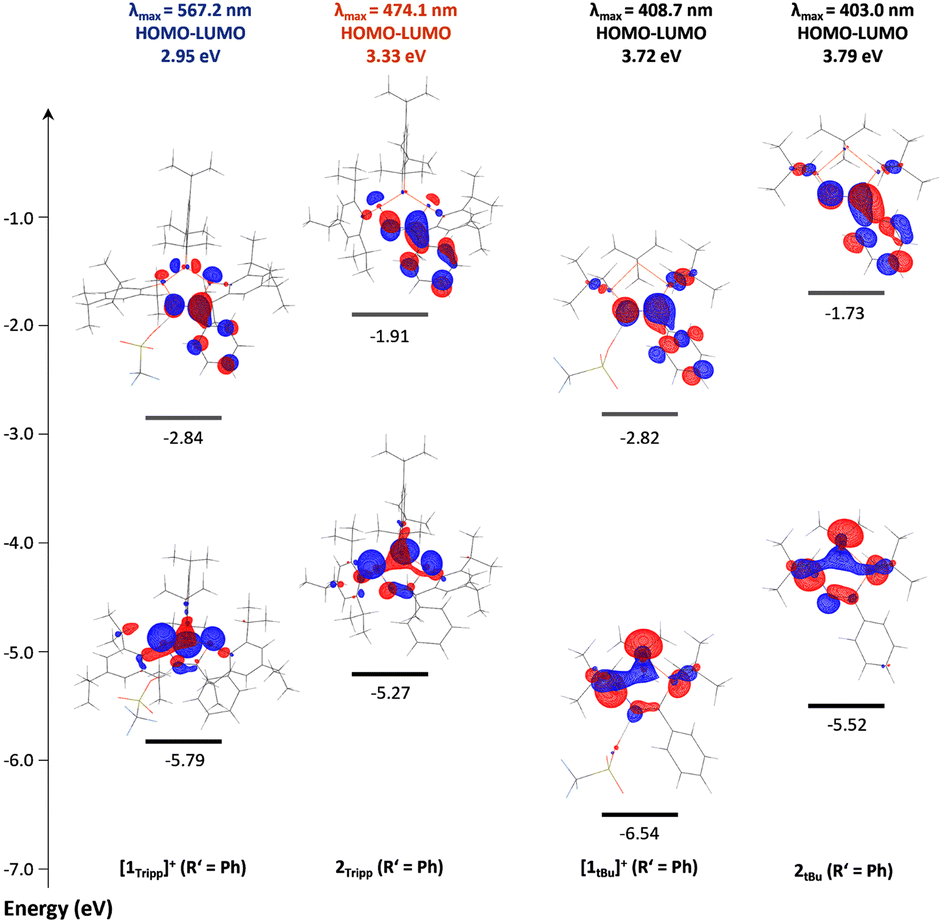 | ||
| Fig. 6 HOMO and LUMO frontier orbitals of [1Tipp]+ (R′ = Ph), 2Tipp (R′ = Ph), [1tBu]+ (R′ = Ph), and 2tBu (R′ = Ph), with lowest-energy charge transfer energies and corresponding absorption maxima. | ||
While the electronic differences in [1tBu]+ and [1Tipp]+ (R′ = Ph) are seemingly small, the structural effects imposed by the bulky Tipp substituents on the molecule have a large influence on the HOMO–LUMO gap in these two species. For example, the ring bending in [1Tipp]+ (R′ = Ph) substantially destabilises the HOMO compared to that in [1tBu]+ (R′ = Ph) while leaving the energy level of the LUMO virtually identical, resulting in a smaller HOMO–LUMO gap that allows a lower energy absorption in [1Tipp]+ (R′ = Ph). In the case of 2R, it should first be noted that deprotonation of both [1Tipp]+ and [1tBu]+ (R′ = Ph) results in destabilisation of both the HOMO and LUMO, resulting in a greater delocalisation of the HOMO across the entire PN–PP–PC framework and onto the PhCN moiety. However, Tipp substitution in 2Tipp (R′ = Ph) results in destabilisation of the HOMO but stabilisation of the LUMO relative to 2tBu (R′ = Ph), again resulting in a smaller HOMO–LUMO gap and allowing for orange colouration to occur compared to the pale yellow 2tBu (R′ = Ph). To shed light on the nature of the HOMO–LUMO transition and the resultant unexpected range of colouration, we calculated the D indices of each species.
The D index is used to qualitatively assess the distance of donor–acceptor electron regions within a molecule by calculating the positive and negative barycenters of the orbitals involved in an electronic transition – in this case, the HOMO and LUMO – and then subsequently calculating the distance between these two barycenters. For values greater than 1.6 Å it can be determined that charge is flowing between the two barycenters, especially in cases molecules are not symmetric and where orbitals involved in the electronic transition have minimal spatial overlap, as is the case in [1R]+. In this way, the D index qualitatively supports the existence of charge transfer across a molecule.83,84 A comparison across the series of [1R]+ (R′ = Ph), reveals D indices of ≥2.3 Å, in line with a HOMO–LUMO charge transfer event from the non-bonding linear combination of n(P) orbitals to the N(H)CPh moiety in these species. This intramolecular charge transfer is nicely illustrated by plotting the charge density difference between S0 and S1 state (Fig. 7). We also observe large D values for the free bases 2Tipp (R′ = Ph) and 2tBu (R′ = Ph), while [1Tipp]+ (R′ = Me), 2Tipp (R′ = Me), and 2tBu (R′ = Me) all showed D values of less than 1.4 Å. This is in line with the colouration of benzonitrile-derived free bases, while acetonitrile-derived free bases appear weakly coloured, and computationally bear no significant HOMO–LUMO charge-transfer phenomena. However, there is no clear relation between the D indices and the bending angles of the P3CN-rings (Fig. S-189†).
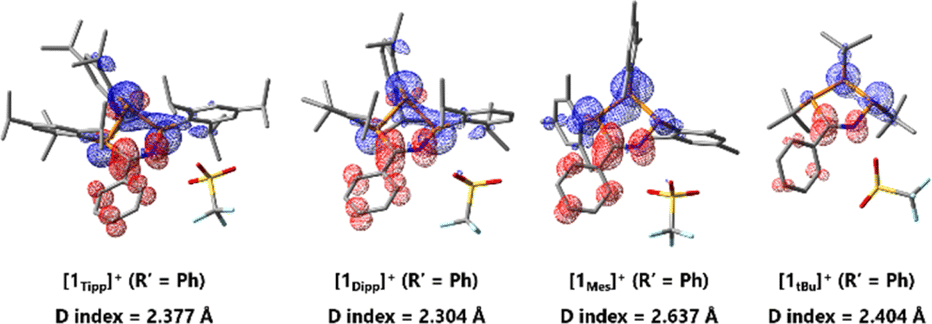 | ||
| Fig. 7 Charge density difference diagrams and D indices of the first excited state (HOMO–LUMO) illustrating charge transfers during excitation of [1Tipp]+, [1Dipp]+, [1Mes]+, and [1tBu]+ (R′ = Ph); blue regions correspond to electron donor regions and red regions correspond to electron acceptor regions. | ||
Preparation of a polymer-based Brønsted acid–base sensor
Having shown reversible H+- and base-induced switching between the neutral triphosphaazolene and cationic triphosphaazolenium states, we were intrigued by the idea of incorporating these P3CN-species into a polymer through post-polymerization functionalization. Immobilization of such polymer should then enable to manufacture a chemical H+ sensor. First, low-dispersity poly(4-cyanostyrene) (P4CS) (Mn = 109.4 kDa, Đ = 1.14) was synthesised through an established reversible addition fragmentation chain-transfer polymerization (RAFT) protocol using 4-cyano-4-(phenylcarbothioylthio)pentanoic acid (CPADB) as RAFT-agent and azobisiobutyronitrile (AIBN) as initiator.85 Post-polymerization functionalization to synthesise a co-polymer was effected by suspending the as-synthesised poly(4-cyanostyrene) in CH2Cl2 and stepwise addition of 0.5 eq. of both (PTipp)3 and HOTf, which immediately afforded a purple suspension. After stirring for 12 h and precipitation with n-hexane P4CS-co-([1TippH][OTf] R′ = Ph) was afforded in 74% yield as purple beads. This insoluble purple polymer was suspended in toluene and an excess of neat NEt3 was added, resulting in the purple beads developing an orange colouration; continued stirring for 24 h gave an orange solution. The neutral, soluble polymer P4CS-co-(2Tipp R′ = Ph) was afforded in good yields as an orange powder after workup (Scheme 3, top), which according to 31P NMR spectroscopy contained, in addition to the expected 1:1:1 resonances of the 1-aza-2,3,4-triphospholene, minimal amounts of an unidentified impurity (δ(31P) = 59.6 ppm), which could not be removed by further purification and therefore seems likely to be trapped in the polymer matrix (cf. ESI p. S-15 ff.†).
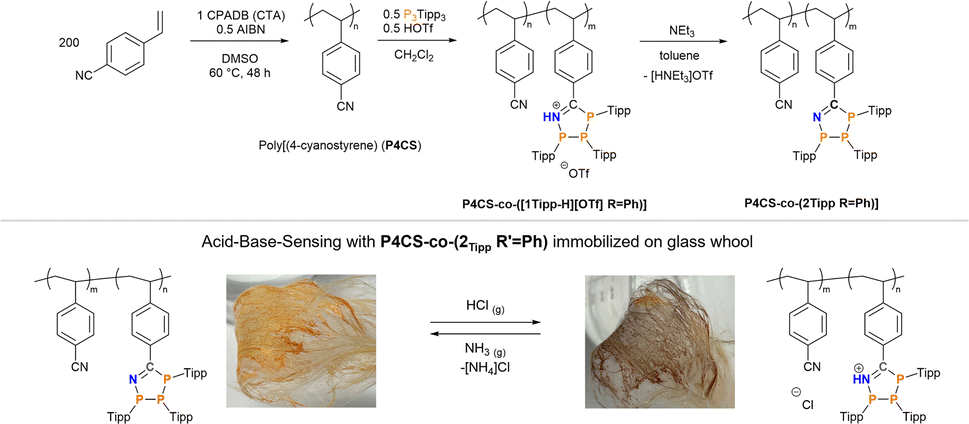 | ||
| Scheme 3 Top: Synthesis of the co-polymers P4CS-co-([1Tipp-H][OTf] R = Ph) and its neutral congener P4CS-co-(2Tipp R = Ph). Bottom: Acid–base sensing with P4CS-co-(2Tipp R = Ph) immobilized on glass wool. | ||
Compared to the starting poly(4-cyanostyrene) the dispersity (Đ = 3.8) of P4CS-co-(2Tipp R′ = Ph) is considerably higher, which may be a result of phosphorus lone pairs in the polymer interacting with the GPC column. To immobilise P4CS-co-(2Tipp R′ = Ph), oven-dried glass wool was dipped into a toluene solution of the neutral polymer and after drying for 24 h pale orange glass fibers were obtained. Sensing was tested by placing the bundled fibers in the headspace of a beaker containing concentrated aqueous (35%) HCl, which resulted in a noticeable colour change to purple (Scheme 3, bottom right), characteristic of P4CS-co-([1TippH][Cl] R′ = Ph). When these purple fibers were exposed to NH3 vapours by suspending the sample above saturated aqueous ammonium hydroxide, the purple colour vanished and the characteristic orange of P4CS-co-(2Tipp R′ = Ph) was again observed (Scheme 3, bottom left). This procedure could be repeated at least five times, without noticeable build-up of [NH4]Cl on the glass fibers. Future studies will focus on obtaining P3CN-containing polymers that can be more easily processed to harness the proton-responsiveness in simple sensors more effectively.
Discussion
A rare example of tunable colouration in phosphorus-rich main group species is described via intramolecular charge transfer, as demonstrated prototypically in compound [1Tipp]+. While colour in poly-phosphorus species is not uncommon and can even be tuned electronically, colour typically arises from π–π* transitions, and this, to the best of our knowledge, is the first report on sterically-induced tuning of the HOMO energy, to enable facile modification of the colour in P3CN ring systems. The low-energy charge transfer arises from perturbation of the HOMO–LUMO energy levels, which can be related to the structural bending (θ) upon N-protonation of the P3CN heterocycle and is further enhanced by increased steric demand of the phosphorus substituents. This is corroborated by a strong correlation between the wavelength of charge-transfer (λCT, previously labelled as λ1) as a function of the bending angle as observed in Table 1 and Fig. 8. Furthermore, solid-state UV-Vis spectra of a representative set of samples show near-identical λCT values to that of the analogous solution UV-Vis spectra (ESI Section 3, cf.[1Tipp]+ (R′ = Me) Fig. S-95 and S-96 ff.†), indicating that the bending angles are similar in solution and the solid-state. Additionally, modification of the R′ substituent affects the λCT as shown in Fig. 5, presumably by modulation of the LUMO energy level. In silico investigations into this charge transfer phenomenon reveal that the sterically-induced bending found in [1R]+ (R = Dipp, Tipp) raises the energy of the HOMO to allow for intramolecular charge transfer to occur in the visible region (ESI Section 6†). Further, these systems exhibit notable photostability in the solid state (vide supra), and further tuning of the HOMO–LUMO gap can be readily effected by varying the nature of the nitrile used in the synthesis.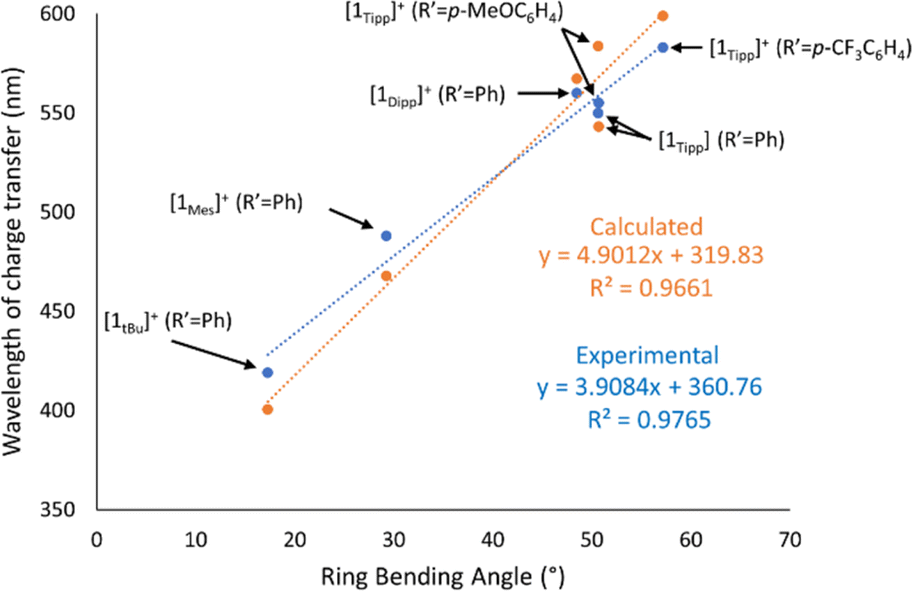 | ||
| Fig. 8 Graph depicting wavelength of charge transfer as a function of crystallographic ring-bending angle in [1R]+ species both experimentally (blue) and with calculated absorption maxima by TD-DFT (orange). | ||
Conclusions
This contribution presents a thorough investigation of the structural and photophysical properties of 1-aza-2,3,4-triphospholenes and related 1-aza-2,3,4-triphospholenium cations, some of which exhibit strong colouration due to a hitherto unrealized charge transfer phenomenon, resulting in an unprecedented potential for control over colouration in phosphorus-rich molecules. Complexes [1R]+ show colouration associated with low-energy absorption bands that become increasingly red-shifted as steric bulk at the cyclic phosphane substituent is increased. Use of benzonitrile as a substrate allows access to yellow, red, and blue colours as steric bulk at phosphorus is increased in [1R]+, and colouration may be further tuned by the variation of the aryl nitrile substrate, for example use of para-substituted phenyl nitriles, as was shown for selected examples. UV-Visible spectroscopy revealed that [1Tipp]+ and [1Dipp]+ have broad, red-shifted absorption bands characteristic of intramolecular charge transfers. Single crystal X-ray crystallography revealed increasing ring bending of [1R]+ as steric encumbrance at the phosphorus centres are increased, while 2R species conversely remain closer to planarity. A linear correlation was found between the angle of ring bending and the wavelength of the low-energy absorption bands across all benzonitrile-derived species (R′ = Ph), indicating a relationship between ground-state structure and the photophysical properties in this family of compounds. TD-DFT calculations support an intramolecular HOMO–LUMO (n(P) → π*) charge-transfer to which the observed colouration is ascribed. This unusual structure–property relationship provides insight for the rational design of pnictogen-rich charge transfer materials, while the facile and modular synthesis, along with photostability, tunable colour and proton responsiveness of 1-aza-2,3,4-triphospholenes, suggest these heterocycles may have considerable utility in numerous applications. As proof-of-concept, covalent incorporation of these moieties into a 4-cyanostyrene polymer backbone through post-polymerization modification allowed for expedient generation of an air-stable polymer capable of reversible Brønsted acid–base colorimetric chemosensing.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds [1Tipp]+ (R′ = Me) (CCDC No. 2293093), [1Tipp]+ (R′ = Ph) (2293094), [1Dipp]+ (R′ = Me) (2293086), [1Dipp]+ (R′ = Ph) (2293087), [1Mes]+ (R′ = Me) (2293088), [1Mes]+ (R′ = Ph) (2293089), [1tBu]+ (R′ = Ph) (2293090), 2Tipp (R′ = Me) (2293100), 2Tipp (R′ = Ph) (2293101), 2Dipp (R′ = Me) (2293095), 2Dipp (R′ = Ph) (2293096), 2Mes (R′ = Me) (2293097), 2Mes (R′ = Ph) (2293098), 2tBu (R′ = Me) (2293099), [1Tipp]+ (R′ = p-MeOC6H4) (2293092), [1Tipp]+ (R′ = p-CF3C6H4) (2293091) and P6Mes6 (2293102) have been deposited at the CCDC under 2293086–2293102 and can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/structures/. The geometries of all theoretically studied molecules have been uploaded as a separate xyz-file to provide a more intuitive view of the calculated 3D structures. A video of the acid–base-sensing has been uploaded as a separate file.Author contributions
M. A. N., E. A. L., J. E. T. W. and C. H.-J. carried out the experimental work. B. O. P. was responsible for SC-XRD experiments. L. W. and J. E. R. carried out the EPR measurements. The experimental work was designed by M. A. N., E. A. L., I. M. and C. H.-J., while supervision was shared between E. A. L., I. M. and C. H.-J. DFT studies were performed and evaluated by C. H.-J. The ESI was written through contributions from all authors. The manuscript was drafted by M. A. N. and finalized through contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. N. thanks NSERC for a CGS Doctoral Scholarship, and the University of Victoria for a Doctoral Fellowship and a President's Research Scholarship. E. A. L. thanks NSERC for a Post-doctoral Fellowship. C. H-J. thanks the German Scholar Organization for funding through a KT boost fund. J. M. R. thanks the NSERC DG and RTI programs for funding. I. M. thanks the Canadian government for a Canada 150 Research Chair and NSERC for a Discovery Grant, and the University of Victoria for start-up funds. Dr Marcus Klahn is acknowledged for help with MS measurements. Dr Emmanuel Hupf (University of Bremen) is acknowledged for helpful discussions regarding TD-DFT calculations.Notes and references
-
G. Natta, From the Stereospecific Polymerization to the Asymmetric Autocatalytic Synthesis of Macromolecules, in Nobel Lectures, Chemistry 1963-1970, Elsevier Publishing Company, Amsterdam, 1972 Search PubMed
.
-
K. Ziegler, Consequences and Development of an Invention, in Nobel Lectures, Chemistry 1963-1970, Elsevier Publishing Company, Amsterdam, 1972 Search PubMed
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed
-
S. Harder, Early Main Group Metal Catalysis, 2020, pp. 151–173 Search PubMed
- B. T. Novas and R. Waterman, ChemCatChem, 2022, 14, e202200988 CrossRef CAS
- S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482 RSC
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed
- R. J. Less, R. L. Melen, V. Naseri and D. S. Wright, Chem. Commun., 2009, 4929–4937 RSC
- R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC
- R. L. Melen, Chem. Soc. Rev., 2016, 45, 775–788 RSC
- A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2016, 45, 922–953 RSC
- F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS PubMed
- I. Manners, Angew. Chem., Int. Ed., 1996, 35, 1602–1621 CrossRef
- A. W. Knights, M. A. Nascimento and I. Manners, Polymer, 2022, 247, 124795 CrossRef CAS
- I. Manners, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 179–191 CrossRef CAS
-
(a) R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed
-
(a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS
- W. Rösch, T. Facklam and M. Regitz, Tetrahedron, 1987, 43, 3247–3256 CrossRef
- J. A. W. Sklorz, S. Hoof, M. G. Sommer, F. Weißer, M. Weber, J. Wiecko, B. Sarkar and C. Müller, Organometallics, 2014, 33, 511–516 CrossRef CAS
- J. A. W. Sklorz and C. Müller, Eur. J. Inorg. Chem., 2016, 595–606 CrossRef CAS
- M. Papke, L. Dettling, J. A. W. Sklorz, D. Szieberth, L. Nyulászi and C. Müller, Angew. Chem., Int. Ed., 2017, 56, 16484–16489 CrossRef CAS PubMed
- T. Görlich, D. S. Frost, N. Boback, N. T. Coles, B. Dittrich, P. Müller, W. D. Jones and C. Müller, J. Am. Chem. Soc., 2021, 143, 19365–19373 CrossRef PubMed
-
(a) E. S. Yang, A. Mapp, A. Taylor, P. D. Beer and J. M. Goicoechea, Chem.–Eur. J., 2023, 29, e202301648 CrossRef CAS PubMed
- G. Pfeifer, M. Papke, D. Frost, J. A. W. Sklorz, M. Habicht and C. Müller, Angew. Chem., Int. Ed., 2016, 55, 11760–11764 CrossRef CAS PubMed
- N. R. O'Connor, J. L. Wood and B. M. Stoltz, Isr. J. Chem., 2016, 56, 431–444 CrossRef PubMed
- P. Singh, R. K. Varshnaya, R. Dey and P. Banerjee, Adv. Synth. Catal., 2020, 362, 1447–1484 CrossRef CAS
- A. Villinger, P. Mayer and A. Schulz, Chem. Commun., 2006, 1236–1238 RSC
- A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2008, 47, 603–606 CrossRef CAS PubMed
- A. Velian and C. C. Cummins, Science, 2015, 348, 1001–1004 CrossRef CAS PubMed
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed
- X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed
- A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 8536–8541 CrossRef CAS PubMed
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef
-
(a) T. Wellnitz and C. Hering-Junghans, Eur. J. Inorg. Chem., 2021, 2021, 8–21 CrossRef CAS
- C. A. Dyker, N. Burford, G. Menard, M. D. Lumsden and A. Decken, Inorg. Chem., 2007, 46, 4277–4285 CrossRef CAS PubMed
- M. H. Holthausen, D. Knackstedt, N. Burford and J. J. Weigand, Aust. J. Chem., 2013, 66, 1155–1162 CrossRef CAS
- E. Gorbachuk, T. Grell, E. Hey-Hawkins and D. Yakhvarov, Eur. J. Inorg. Chem., 2024, 27, e202300751 CrossRef CAS
- S. S. Chitnis, H. A. Sparkes, V. T. Annibale, N. E. Pridmore, A. M. Oliver and I. Manners, Angew. Chem., Int. Ed., 2017, 56, 9536–9540 CrossRef CAS PubMed
- N. Hoffmann, C. Wismach, P. G. Jones, R. Streubel, N. H. T. Huy and F. O. Mathey, Chem. Commun., 2002, 454–455 RSC
- L. S. Sapochak, A. B. Padmaperuma, P. A. Vecchi, X. Cai and P. E. Burrows, Proc. SPIE, 2007, 6655, 665506–665511 CrossRef
- S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 4233–4243 RSC
- D. Joly, D. Tondelier, V. Deborde, W. Delaunay, A. Thomas, K. Bhanuprakash, B. Geffroy, M. Hissler and R. Réau, Adv. Funct. Mater., 2012, 22, 567–576 CrossRef CAS
-
M. Stolar and T. Baumgartner, P-Containing Heteroarenes: Synthesis, Properties, Applications, in Polycyclic Arenes and Heteroarenes, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 309–330 Search PubMed
- M. P. Duffy, W. Delaunay, P. A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC
- A. Belyaev, P. T. Chou and I. O. Koshevoy, Chem.–Eur. J., 2021, 27, 537–552 CrossRef CAS PubMed
- N. M.-W. Wu, M. Ng, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 15142–15150 CrossRef CAS PubMed
- X.-D. Jiang, J. Zhao, D. Xi, H. Yu, J. Guan, S. Li, C.-L. Sun and L.-J. Xiao, Chem.–Eur. J., 2015, 21, 6079–6082 CrossRef CAS PubMed
- A. M. Christianson and F. P. Gabbaï, Inorg. Chem., 2016, 55, 5828–5835 CrossRef CAS PubMed
- Y. Qiu, J. C. Worch, D. N. Chirdon, A. Kaur, A. B. Maurer, S. Amsterdam, C. R. Collins, T. Pintauer, D. Yaron, S. Bernhard and K. J. T. Noonan, Chem.–Eur. J., 2014, 20, 7746–7751 CrossRef CAS PubMed
- K. Dimroth and P. Hoffmann, Angew. Chem., Int. Ed. Engl., 1964, 3, 384 Search PubMed
- R. Allmann, Angew. Chem., Int. Ed., 1965, 4, 150–151 CrossRef
-
K. Dimroth, Delocalized phosphorus-carbon double bonds, in Phosphorus-Carbon Double Bonds, Springer-Verlag, 1964, vol. 38, pp. 1–147 Search PubMed
- P. Jutzi, Angew. Chem., Int. Ed. Engl., 1975, 14, 232–245 CrossRef
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, Chem. Commun., 2005, 1965–1967 RSC
- J. F. Binder, A. M. Corrente and C. L. B. Macdonald, Dalton Trans., 2016, 45, 2138–2147 RSC
- C. L. B. Macdonald, J. F. Binder, A. A. Swidan, J. H. Nguyen, S. C. Kosnik and B. D. Ellis, Inorg. Chem., 2016, 55, 7152–7166 CrossRef CAS PubMed
- A. Schmidpeter, S. Lochschmidt and A. Willhalm, Angew. Chem., Int. Ed., 1983, 22, 545–546 CrossRef
- S. Roy, P. Stollberg, R. Herbst-Irmer, D. Stalke, D. M. Andrada, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 150–153 CrossRef CAS PubMed
- N. M.-W. Wu, H.-L. Wong and V. W.-W. Yam, Chem. Sci., 2017, 8, 1309–1315 RSC
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, W.-T. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 11504–11508 CrossRef CAS PubMed
- X. He, J. Borau-Garcia, A. Y. Y. Woo, S. Trudel and T. Baumgartner, J. Am. Chem. Soc., 2013, 135, 1137–1147 CrossRef CAS PubMed
- F. Riobé, R. Szűcs, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio, J. Buendía, L. Sánchez, R. Réau, L. Nyulászi and M. Hissler, Chem.–Eur. J., 2015, 21, 6547–6556 CrossRef PubMed
- J. C.-H. Chan, H.-L. Wong, W.-T. Wong and V. W.-W. Yam, Chem.–Eur. J., 2015, 21, 6936–6948 CrossRef CAS PubMed
- T. Higashino, T. Yamada, T. Sakurai, S. Seki and H. Imahori, Angew. Chem., Int. Ed., 2016, 55, 12311–12315 CrossRef CAS PubMed
- W. Delaunay, R. Szűcs, S. Pascal, A. Mocanu, P. A. Bouit, L. Nyulászi and M. Hissler, Dalton Trans., 2016, 45, 1896–1903 RSC
- H. Omori, S. Hiroto, Y. Takeda, H. Fliegl, S. Minakata and H. Shinokubo, J. Am. Chem. Soc., 2019, 141, 4800–4805 CrossRef CAS PubMed
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed
- M. Stolar, J. Borau-Garcia, M. Toonen and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 3366–3371 CrossRef CAS PubMed
- C. R. Bridges, A. M. Borys, V. A. Béland, J. R. Gaffen and T. Baumgartner, Chem. Sci., 2020, 11, 10483–10487 RSC
- C. Reus, M. Stolar, J. Vanderkley, J. Nebauer and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 11710–11717 CrossRef CAS PubMed
- B. Das, A. Makol and S. Kundu, Dalton Trans., 2022, 51, 12404–12426 RSC
- A. Petrov, L. Conrad, N. T. Coles, M. Weber, D. Andrae, A. Zagidullin, V. Miluykov and C. Müller, Chem.–Eur. J., 2022, 28, e202203056 CrossRef CAS PubMed
- M. K. Sharma, H. M. Weinert, B. Li, C. Wölper, J. T. Henthorn, G. E. Cutsail Iii, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2023, 62, e202309466 CrossRef CAS PubMed
- L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS
- D. Rottschäfer, M. K. Sharma, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8127–8134 CrossRef PubMed
- L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed
- A. Schumann, F. Reiß, H. Jiao, J. Rabeah, J.-E. Siewert, I. Krummenacher, H. Braunschweig and C. Hering-Junghans, Chem. Sci., 2019, 10, 7859–7867 RSC
- C. Hering, A. Schulz and A. Villinger, Chem. Sci., 2014, 5, 1064–1073 RSC
- S. Gómez-Ruiz, R. Frank, B. Gallego, S. Zahn, B. Kirchner and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2011, 2011, 739–747 CrossRef
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed
- L. Huet, A. Perfetto, F. Muniz-Miranda, M. Campetella, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2020, 16, 4543–4553 CrossRef CAS PubMed
- J. Jin, Y. Li, D. Cao, S. Wang and X. Yan, Angew. Chem., Int. Ed., 2023, 62, e202306169 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, NMR spectra, UV-Vis, crystallographic, and computational details. CCDC 2293086–2293102. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02497d |
| ‡ Deceased December 3rd, 2023 (I. M.). |
| This journal is © The Royal Society of Chemistry 2024 |