
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective phosphine-catalyzed [6 + 1] annulations of α-allyl allenoates with 1,1-bisnucleophiles†
Jingxiong
Lai
,
Wei
Cai
* and
You
Huang
 *
*
State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: wcai@nankai.edu.cn; hyou@nankai.edu.cn
First published on 21st June 2024
Abstract
Organocatalytic annulations between allenes and bisnucleophiles represent one of the most convenient routes to various carbocycles and heterocycles. However, most examples are limited to the formation of five- and six-membered rings, probably owing to relatively easy handling of short-chained biselectrophiles. Here we report long-chained α-allyl allenoate-derived 1,6-biselectrophiles for the first time, enabling a phosphine catalyzed [6 + 1] annulation with readily available 1,1-bisnucleophilic reagents. The reaction proceeds via a tandem γ-umpolung addition and δ′-addition process, smoothly constructing both seven-membered N-heterocycles and carbocycles with a broad scope of substrates, high atom economy and excellent enantioselectivity (up to 99% yield and up to 96% ee). Mechanistic experiments revealed a conversion of the 1,6-dipole into a 1,6-biselectrophilic intermediate through proton abstraction.
Introduction
Phosphine-catalyzed annulation of functionalized allenes and bisnucleophiles represents a straightforward and efficient strategy for the construction of cyclic compounds and has garnered considerable and enduring interest since Lu's groundbreaking research in 2002.1,2 The reaction proceeds via two tandem nucleophilic additions of bisnucleophiles to allenoates. With various allene-derived biselectrophiles serving as C1,3 C2 (ref. 4) or C3 (ref. 4d and 5) synthons, a diverse range of annulations have been realized through classical β-Michael addition and α,γ-umpolung addition, mainly providing a variety of five- and six-membered rings in the presence of 1,n-biseletrophiles (n = 1–3), albeit with sporadic racemic examples on [2 + 5] annulations.2 To extend the chain-length in annulations, the introduction of novel reaction sites into allenes has emerged as a prevailing trend over the last two decades, and the scatter plots mathematically show the annulation pattern of such reactions (Scheme 1A).1c,6 Compared with those short-chained allene-derived synthons, longer-chained synthons are more difficult to handle in annulation reactions, because more flexible allene-derived biselectrophiles may lead to more difficult control of reactivity and selectivity.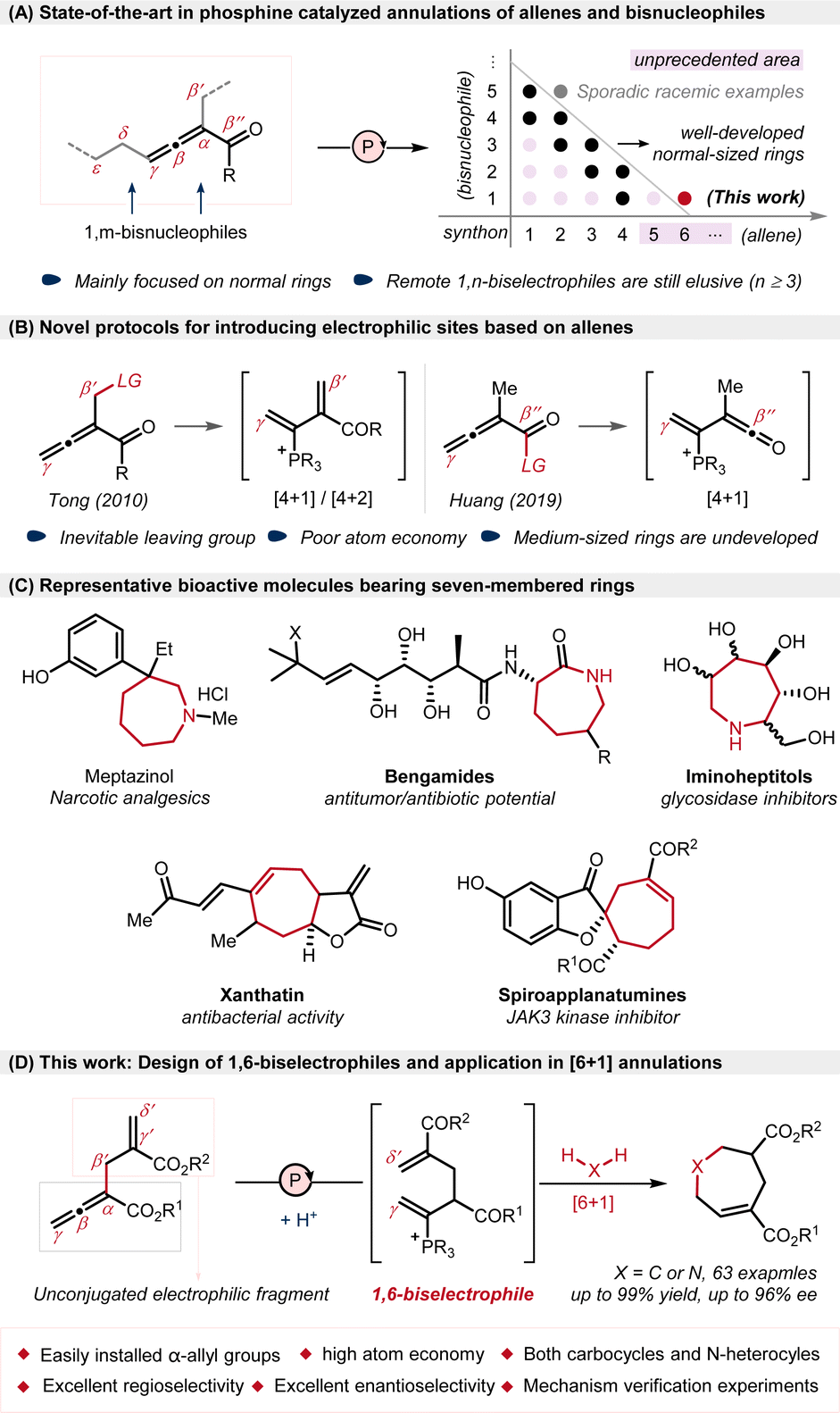 | ||
| Scheme 1 Context of the work. | ||
Thus far, only two categories of allenes have been effectively established as longer-chained biseletrophiles for C4 synthons under phosphine-catalyzed conditions (Scheme 1B). In 2010, Tong exquisitely introduced an acetate group at β′-position as leaving group and generated phosphonium diene intermediates as 1,4-biseletrophiles.7 Racemic and enantiomerically enriched [4 + 1] and [4 + 2] annulations were achieved via sequential γ-umpolung addition and β′-addition with 1,1- and 1,2-bisnucleophiles.8,9 In addition, another allene-derived 1,4-biseletrophile is also reported by Huang et al., who used β′′-oxazolidinyl-substituted allenyl imide as a C4 synthon, achieving [4 + 1] annulations via tandem β′′-addition and γ-addition.10 On the basis of these two types of leaving group-containing allenes, various other types of annulations have been developed with different bisnucleophiles, generating 5- or 6-membered rings.11 Furthermore, although both the δ-acetoxy-substituted allenoate developed by Tong and the γ-alkenyl allenoate developed by our group exhibited δ-addition and remote 1,7-addition reactivity,12 the construction of medium-sized rings through the annulations of long-chained allene-derived remote biselectrophiles still remains an elusive challenge,13 probably owing to unfavourable enthalpic and entropic effects in the synthesis of medium-sized rings,14 as well as the limited availability of long-chained allene-derived biselectrophiles.
In consideration of widely-existing of chiral medium-sized rings in natural products and bioactive molecules15–17 (Scheme 1C), the development of long-chained allene-derived biselectrophiles to construct medium-sized rings, especially enantioselectively, would be in high demand. Herein, we report a phosphine-catalyzed [6 + 1] annulation of α-allyl allenoates with 1,1-bisnucleophiles, achieving a general and economical approach for the synthesis of seven-membered rings (Scheme 1D) which is different with the previous works through [4 + 3] annulation.18 Notable features of this strategy include: (a) the longest-chained allene-derived biselectrophile, acting as C6 synthon to enable the rarely-reported [6 + 1] annulation; (b) both N-heterocycles and carbocycles produced with broad scope and great efficiency (up to 99% yield and 96% ee); (c) high atom economy without the need of leaving groups; (d) excellent regioselectivity without observing potential α- and β-additions, as well as competing self-cyclization.19
Results and discussion
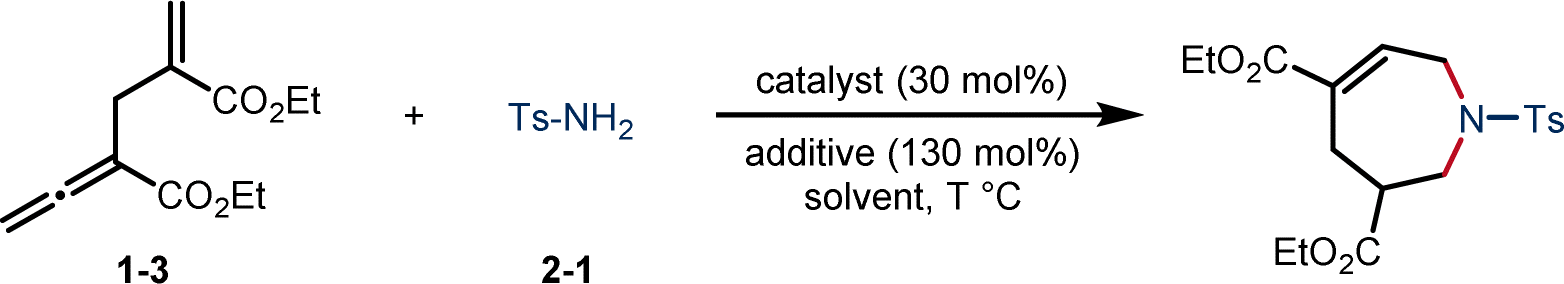
Recently, a phosphine-catalyzed cycloaddition of α-allyl allenoates with 3-formylchromones was developed by our group.20 Inspired by this success, we envisioned that the conversion of α-allyl allenoate-derived 1,6-dipole into 1,6-biselectrophile through proton abstraction may be feasible, thus facilitating a distinct [6 + 1] annulation with readily accessible and cost-effective 1,1-bisnucleophilic reagents. Following this hypothesis, we commenced the study with the screening of α-allyl allenoate 1-3 and p-toluenesulfonamide 2-1 in the presence of tertiary phosphine catalysts (Table 1). To our delight, the use of PBu3 in CHCl3 at 25 °C for 24 h provided the desired azepine 3 in 61% yield (entry 1), while the base additives such as K2CO3 or Cs2CO3 did not promote the reaction (entries 2 and 3), probably because the addition of base inhibited the proton transfer process, leading to more difficult formation of 1,6-bielectrophilic intermediate.13 Notably, 4 Å MS reduced the yield while H2O slightly improved the yield (entries 4 and 5), suggesting a water–tolerant reaction. When the ratio of 1-3 to 2-1 was changed from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 to 2:1, the yield of 3 was improved to 80% with a slightly prolonged reaction time (entry 6). Other screened solvents provided unsatisfactory performances (entries 7–9), and elevating the temperature also decreased the yield. On the other hand, reducing the temperature to 0 °C led to slightly higher yields, yet requiring a longer reaction time (entries 10 and 11). Importantly, the structure of catalyst had strong influence on the reactivity (entries 12–14), and other phosphines or an amine catalyst were in general less effective. In addition, control experiment showed that the absence of PBu3 resulted in no reactivity, further confirming a vital role of phosphine catalyst in the [6 + 1] reaction (entry 15).
:1.2 to 2:1, the yield of 3 was improved to 80% with a slightly prolonged reaction time (entry 6). Other screened solvents provided unsatisfactory performances (entries 7–9), and elevating the temperature also decreased the yield. On the other hand, reducing the temperature to 0 °C led to slightly higher yields, yet requiring a longer reaction time (entries 10 and 11). Importantly, the structure of catalyst had strong influence on the reactivity (entries 12–14), and other phosphines or an amine catalyst were in general less effective. In addition, control experiment showed that the absence of PBu3 resulted in no reactivity, further confirming a vital role of phosphine catalyst in the [6 + 1] reaction (entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive | Solvent | yieldb (%) |
| a Reaction conditions: 1-3 (0.2 mmol), 2-1 (0.24 mmol), catalyst (30 mol%) and additives (130 mol%) in solvent (2.0 mL) at 25 °C for 24 hours. b Isolated yields. c 1-3 (0.4 mmol), 2-1 (0.2 mmol). d 60 °C. e 0 °C, 48 hours. | ||||
| 1 | PBu3 | — | CHCl3 | 61 |
| 2 | PBu3 | K2CO3 | CHCl3 | 43 |
| 3 | PBu3 | Cs2CO3 | CHCl3 | 24 |
| 4 | PBu3 | 4 Å MS | CHCl3 | 56 |
| 5 | PBu3 | H2O | CHCl3 | 62 |
| 6c | PBu3 | — | CHCl3 | 80 |
| 7c | PBu3 | — | DCE | 77 |
| 8c | PBu3 | — | Toluene | 64 |
| 9c | PBu3 | — | Et2O | 56 |
| 10d | PBu3 | — | CHCl3 | 70 |
| 11e | PBu3 | — | CHCl3 | 81 |
| 12c | DMAP | — | CHCl3 | NR |
| 13c | (4-MeOC6H4)3P | — | CHCl3 | Trace |
| 14c | PCy3 | — | CHCl3 | 47 |
| 15c | — | — | CHCl3 | NR |
With the optimal reaction conditions in hand, we set out to explore the generality of this [6 + 1] annulation. The scope was first explored with various sulfonamides (Scheme 2). Various sulfonamides with aryl rings bearing both electron-withdrawing and electron-donating para-substituents were well tolerated, affording 7-membered N-heterocycles with 61–81% yield (3–8). The o-tolyl and phenyl-substituted sulfonamides also participated in the reaction smoothly (9 in 64% yield and 10 in 84% yield). In addition, alkyl and heteroaryl sulfonamides provided satisfactory yields (11 in 82% yield, 12 in 80% yield and 13 in 66% yield). Different allenoates were tolerated well in this reaction, and the steric hindrance of the ester groups at both α- and γ′-positions had a small influence on the yield (14–19, 48–79% yield). To our delight, valdecoxib and celecoxib were successfully transformed into the corresponding cycloadducts (20, 66% yield, and 21, 73% yield).
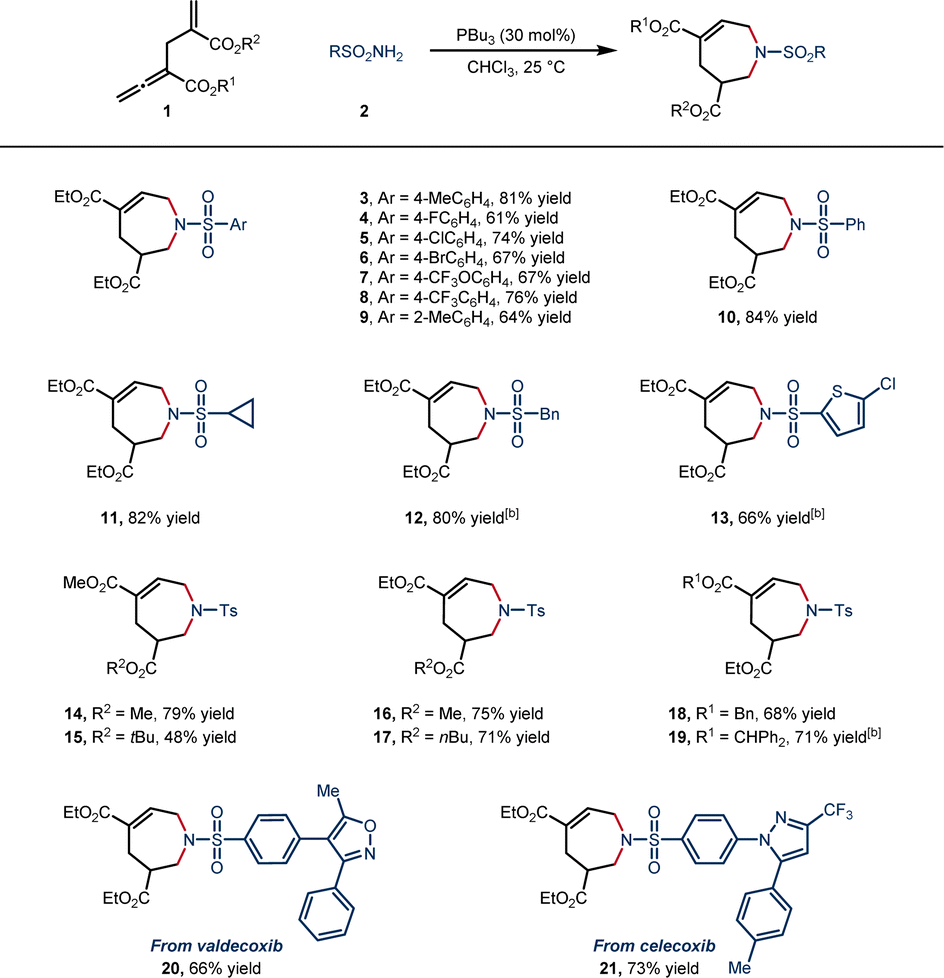 | ||
| Scheme 2 Scope of 7-membered N-heterocycles. aReactions of 1 (0.20 mmol) and 2 (0.40 mmol) were carried out in the presence of PBu3 (0.06 mmol) in 2.0 mL of CHCl3 at 25 °C for 24 h. Isolated yields. b20 eq. H2O was added. | ||
Given the wide availability of 1C,1C-bisnucleophilic reagents, we then investigated the feasibility of the [6 + 1] annulation in the synthesis of 7-membered carbocycles (Scheme 3). We were pleased to find that this transformation was indeed successful with readily available malononitrile and malonate diesters, providing the corresponding cycloheptenes with good efficiency under the standard conditions (22–25, 71–88% yields). The steric hindrance of the ester groups at both α- and γ′-positions did not affect the yields (26–31, 83–90% yield). In addition, the citronellol-derived malonate proceeded smoothly to afford 32 in 71% 1H NMR yield. Non-symmetrical 1C,1C-bisnucleophilic substrates also provided the desired products (33–38, 61–90% yield, 1:1–3.8:1 dr), and the structure of 34 was confirmed by single-crystal X-ray diffraction.21 Heterocyclic spiroheptenes were generated when cyclic 1C,1C-bisnucleophilic reagents were employed. An array of benzofuranones bearing electron-withdrawing or electron-donating substituents on the aryl rings were tolerated and delivered the spiro products with high efficiency (39–48, 59–99% yield, 1:1–1.3:1 dr). Notably, compound 48 is a spiroapplanatumine analogue.17f Other cyclic skeleton such as benzothiophene, benzolactone, and indolone were also tolerated, providing the desired products (49–52) in up to 95% yield, albeit with low diastereoselectivity (1:1–3.2:1 dr).
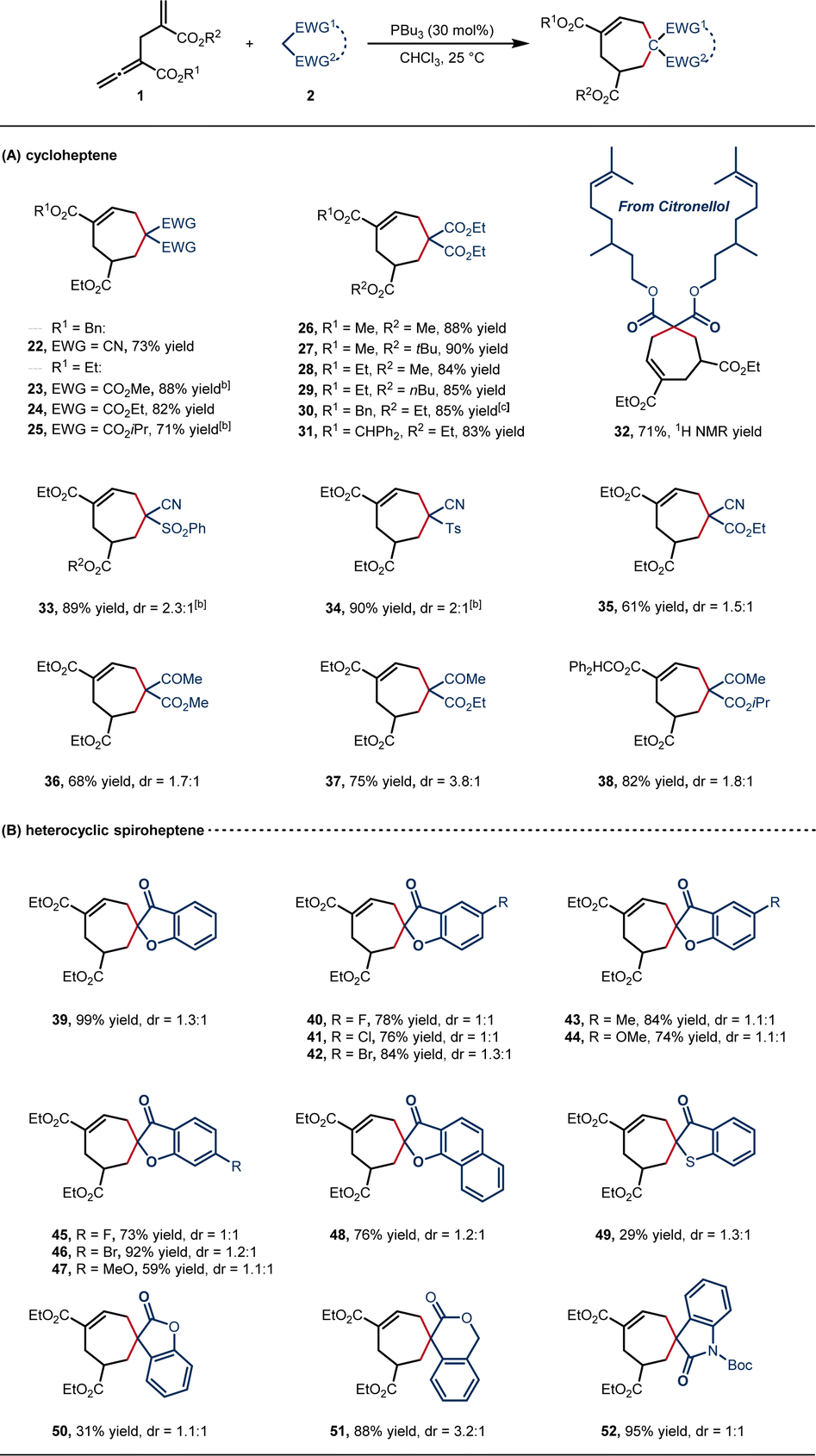 | ||
| Scheme 3 Scope of seven-membered carbocycles. aReactions of 1 (0.20 mmol) and 2 (0.40 mmol) were carried out in the presence of PBu3 (0.06 mmol) in 2.0 mL of CHCl3 at 25 °C for 24 h. Isolated yields. dr value determined by 1H NMR. b20 eq. H2O was added. c82% yield, 23% ee under (R)-SITCP catalysis. | ||
We then turned to explore the asymmetric version of [6 + 1] annulation reaction (Scheme 4). Catalyst screening indicated that (R)-SITCP was the most efficient catalyst among many privileged chiral phosphines (see the ESI†), providing 53–55 in 55–71% yield with good enantioselectivities (81–86% ee).
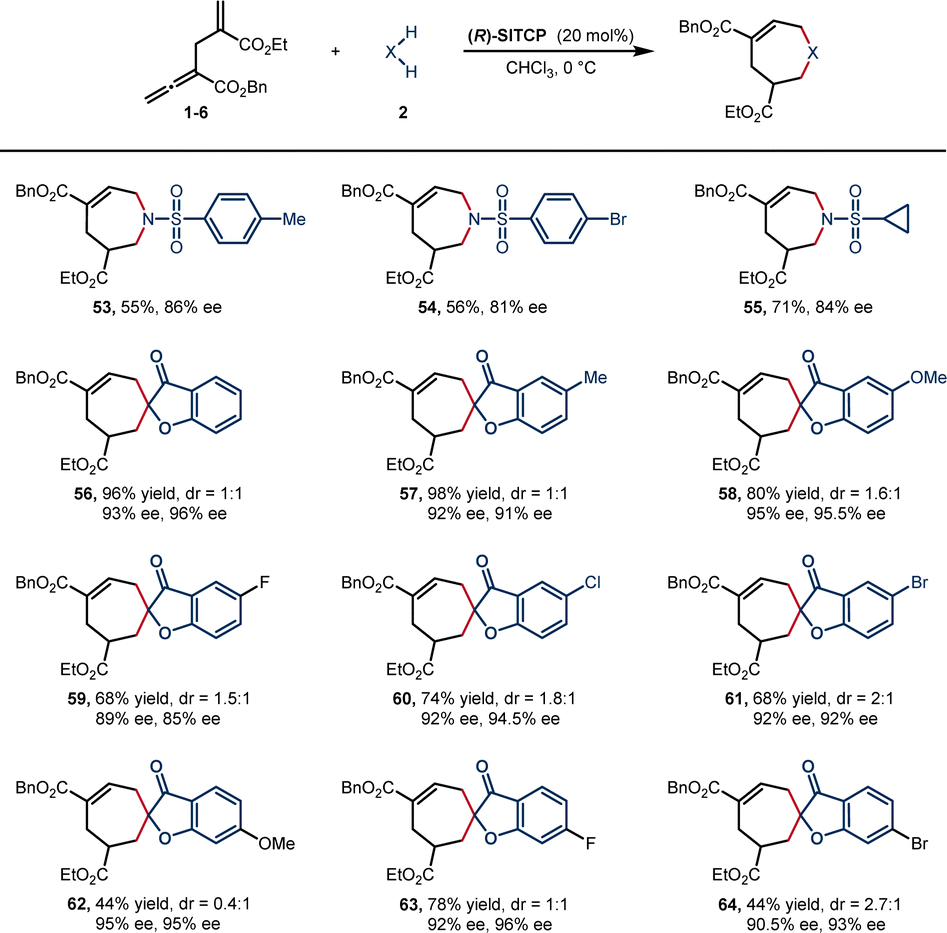 | ||
| Scheme 4 Asymmetric substrate scope. aReactions of 1 (0.05 mmol) and 2 (0.10 mmol) were carried out in the presence of (R)-SITCP (0.01 mmol) in 0.5 mL of CHCl3 at 0 °C for 24 h. Isolated yields. dr values were determined by 1H NMR. ee values were determined by HPLC. | ||
Benzofuranone spiroheptenes were also smoothly generated under the same reaction conditions, producing the desired products 56–64 in 44–98% yields with good enantioselectivities (mostly more than 90% ee). Asymmetric reaction of 1,3-dicarbonyl compound was also tested, albeit with low enantioselectivity (A-30, 23% ee, see Scheme 3 and ESI† for details).
The synthetic utility of this [6 + 1] annulation was then explored (Scheme 5). To our delight, the scale-up reactions gave higher yields, providing N-heterocycle 5 and carbocycle 24 in 79% and 87% yield, respectively. Consequently, these products can be easily converted into various 7-membered ring compounds. The Pd/C-catalyzed hydrogenation of internal alkenes afforded 65 in 61% and 68 in 89% yield. In addition, the internal alkenes were able to converted into bicyclo[5.3.0] skeleton 66 in 91% and 69 in 50% yield via TFA-mediated [3 + 2] annulation, and dihydroxylated 67 in 62% yield via RuCl3/NaIO4-catalyzed oxidation.
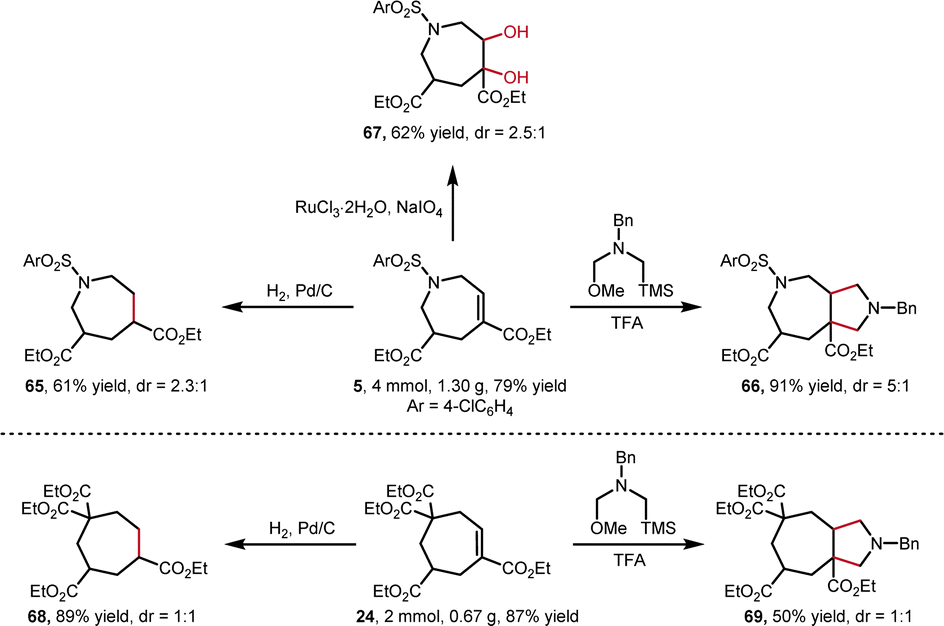 | ||
| Scheme 5 Synthetic applications. | ||
To gain mechanistic insight into this [6 + 1] reaction, a deuterium-labeling experiment was first performed (Scheme 6A). With the addition of 20 eq. D2O, the corresponding product 70 was obtained in 99% yield, with the β- and γ′-positions deuterated in ratios of 66% and 35%, respectively, suggesting a D2O-assisted proton transfer process.22 The efficiency of γ-umpolung addition reaction of TsNH2 (2-1) to α-methyl alleonate was tested under the standard conditions (Scheme 6B).
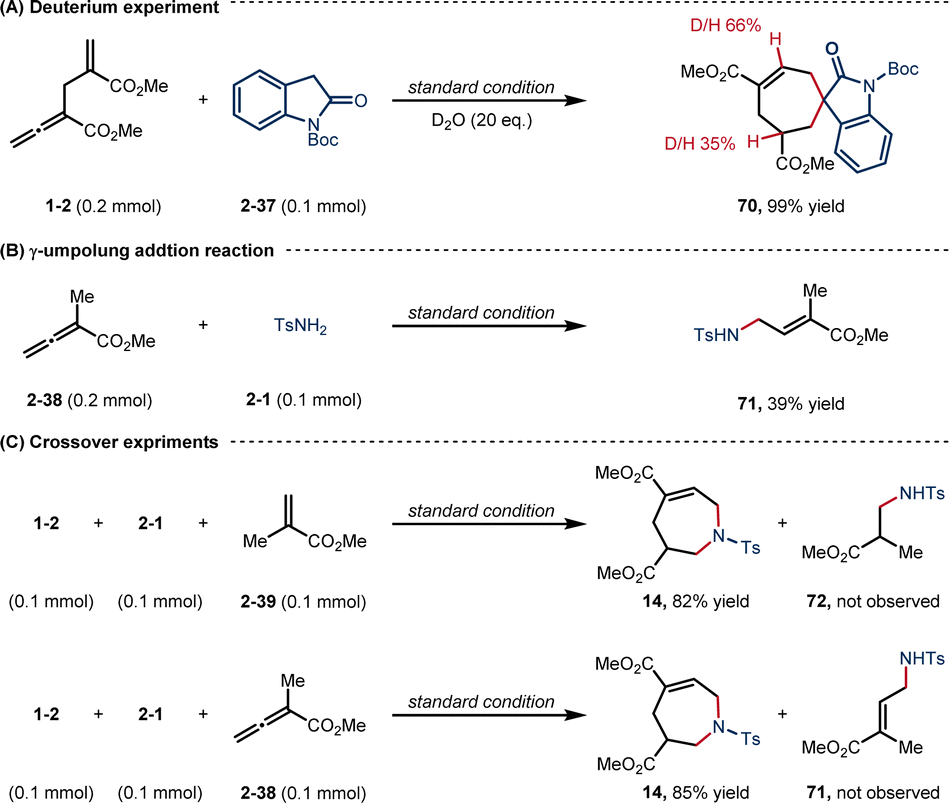 | ||
| Scheme 6 Mechanistic investigations. | ||
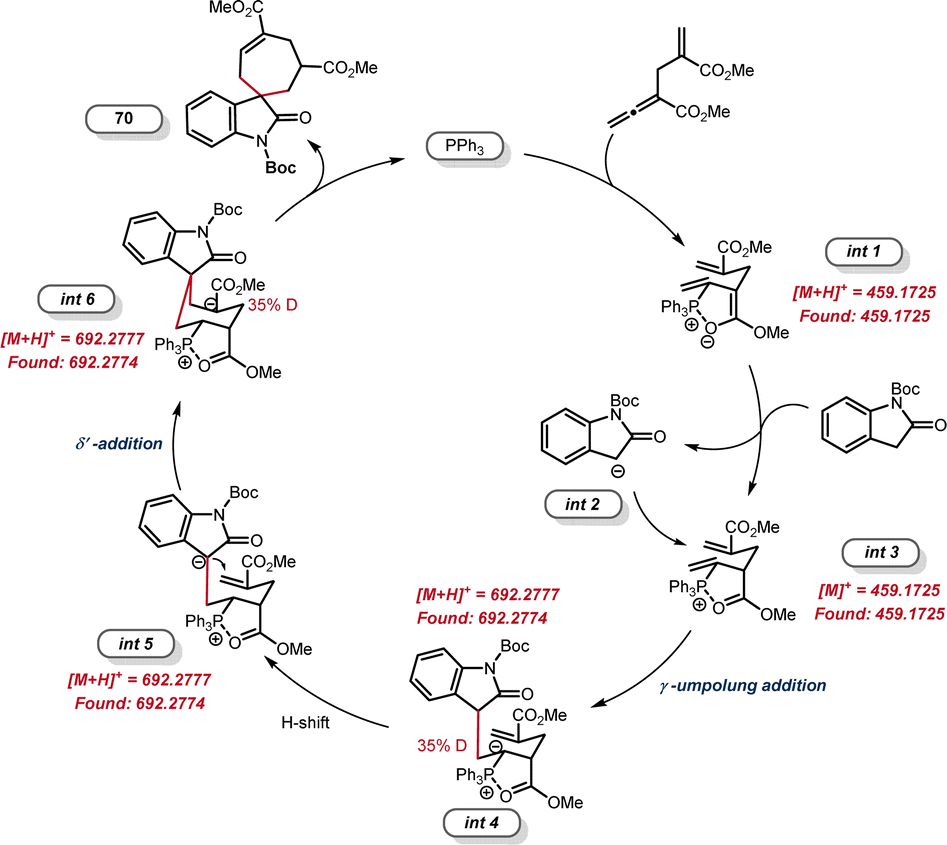
The yield of product 71 (39%) was much lower than that of [6 + 1] annulation (3, 81% yield), indicating that the γ-umpolung addition alone was not as smooth as the two tandem additions towards annulation. Both crossover experiments with substrates 2-39 or 2-38 furnished only the non-crossover cycloadduct 14 (Scheme 6C). TsNH2 did not undergo Michael addition with the α-allyl-like substrate 2-39, implying that the alkenyl phosphonium fragment is favoured over the acrylate fragment for the first nucleophilic addition. Interestingly, no products generated from only γ-umpolung addition were detected in the presence of 2-38, which may be attributed to the fact that the δ′-addition towards facilitating the current [6 + 1] reaction did not undergo in the reverse γ-addition. To comfirm the key intermediates, the reaction mixture of allenoate 1-2 and indolone 2-37 in the presence of PPh3 under the standard conditions (0.5 h) was analyzed by high-resolution mass spectrometry (ESI-HRMS). Two major ion signals at 459.1725 and 692.2774 suggested the formation of zwitterionic species int 1 and phosphonium int 3, and carbanionic int 4 to int 6 (Fig. 1).
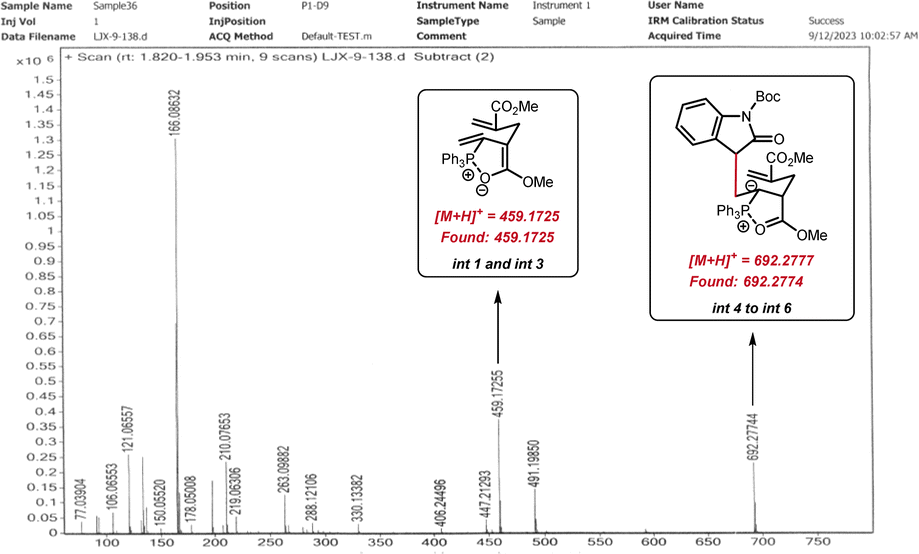 | ||
| Fig. 1 HRMS analysis of the reaction system. | ||
Based on these results, a plausible catalytic cycle was outlined in Fig. 2. Initially, the nucleophilic addition of the phosphine catalyst to allenoate 2-3 leads to the generation of 1,6-dipole with a chair conformation that was stabilized by a five-membered P–O ring (int 1).23 This species forms 1,6-biselectrophilic int 3 after hydrogen abstraction from the 1,1-bisnucleophilic reagent. Addition of the nucleophilic int 2 to the γ-position of int 3 then provides the carbanionic int 4, which is consistent with the deuterium labeling results. The subsequent 1,3-H shift generates int 5, which undergoes intramolecular δ′-addition to give the carbanionic int 6. Finally, the second 1,3-H shift and elimination of the phosphine catalyst afforded the [6 + 1] cycloadduct 70.
 | ||
| Fig. 2 Plausible reaction mechanism. | ||
Conclusions
In summary, we have developed an unprecedented phosphine-catalyzed [6 + 1] annulation using α-allyl allenoate as a long-chained C6 synthon with various readily available 1,1-bisnucleophiles, providing both 7-membered N-heterocycles and carbocycles with broad scope, high atom economy, excellent regioselectivity, and great efficiency. High enantioselectivity can be achieved by employing (R)-SITCP as the chiral catalyst. The synthetic utility including scale-up synthesis and facile transformation into bicyclo[5.3.0] products, has also been well demonstrated. Mechanistic experiments suggested the formation of key remote 1,6-biselectrophilic intermediate and the plausible catalytic cycle. Our investigations on the formation of other medium-sized rings through the newly designed C6-biselectrophiles are under the way in the laboratory.Data availability
General information, detailed experimental procedures, characterization data for compounds, and NMR, HPLC spectra are available in the ESI.†Author contributions
J. X. L., W. C. and Y. H. proposed the project, designed the experiments, and wrote the manuscript. J. X. performed the whole experiment. J. X. L., W. C. and Y. H. performed the analysis of experimental data. Y. H. supervised the whole project. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22171147 and 21871148) for generous financial support for our programs. We thank Prof. Mengchun Ye of Nankai University for helpful discussions.Notes and references
- Selected reviews: (a) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140–1152 RSC; (b) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (c) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- C. Lu and X. Lu, Org. Lett., 2002, 4, 4677–4679 CrossRef CAS PubMed.
- J. Szeto, V. Sriramurthy and O. Kwon, Org. Lett., 2011, 13, 5420–5423 CrossRef CAS PubMed.
- (a) B. Liu, R. Davis, B. Joshi and D. W. Reynolds, J. Org. Chem., 2002, 67, 4595–4598 CrossRef CAS PubMed; (b) Z. Lu, S. Zheng, X. Zhang and X. Lu, Org. Lett., 2008, 10, 3267–3270 CrossRef CAS PubMed; (c) P. Xie, W. Lai, Z. Geng, Y. Huang and R. Chen, Chem.–Asian J., 2012, 7, 1533–1537 CrossRef CAS PubMed; (d) J. Hu, W. Dong, X.-Y. Wu and X. Tong, Org. Lett., 2012, 14, 5530–5533 CrossRef CAS PubMed.
- D. Wang, W. Liu, Y. Hong and X. Tong, Org. Lett., 2018, 20, 5002–5005 CrossRef CAS PubMed.
- Selected examples: (a) X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716–4717 CrossRef CAS PubMed; (b) E. Li, Y. Huang, L. Liang and P. Xie, Org. Lett., 2013, 15, 3138–3141 CrossRef CAS PubMed; (c) N. Li, P. Jia and Y. Huang, Chem. Commun., 2019, 55, 10976–10979 RSC.
- Q. Zhang, L. Yang and X. Tong, J. Am. Chem. Soc., 2010, 132, 2550–2551 CrossRef CAS PubMed.
- (a) X. Han, W. Yao, T. Wang, Y. R. Tan, Z. Yan, J. Kwiatkowski and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 5643–5647 CrossRef CAS PubMed; (b) D. T. Ziegler, L. Riesgo, T. Ikeda, Y. Fujiwara and G. C. Fu, Angew. Chem., Int. Ed., 2014, 53, 13183–13187 CrossRef CAS PubMed; (c) S. Kramer and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 3803–3806 CrossRef CAS PubMed; (d) X. Tang, H. Ni and Y. Lu, Org. Chem. Front., 2021, 8, 4485–4489 RSC; (e) Y. Zhang, D. Wang and X. Tong, Chem. Commun., 2021, 57, 3488–3491 RSC.
- (a) H. Jang, W. Liu, X. a. Zhang Sean and W. Liao, Chem. Res. Chin. Univ., 2016, 32, 385–389 CrossRef CAS; (b) X. Tang, C. X. A. Tan, W.-L. Chan, F. Zhang, W. Zheng and Y. Lu, ACS Catal., 2021, 11, 1361–1367 CrossRef CAS.
- (a) Z.-H. Cao, Y.-H. Wang, S. J. Kalita, U. Schneider and Y.-Y. Huang, Angew. Chem., Int. Ed., 2019, 59, 1884–1890 CrossRef PubMed; (b) Y.-C. Li, Y.-H. Wang, S. J. Kalita and Y.-Y. Huang, Adv. Synth. Catal., 2023, 365, 2487–2510 CrossRef CAS; (c) Z.-Q. Zhang, Z.-K. Zhang, Y.-H. Wang, B.-T. Chen, F.-K. He, Y.-L. Wang, T. Shu and Y.-Y. Huang, J. Org. Chem., 2024, 89, 6607–6614 CrossRef CAS PubMed.
- (a) Y.-H. Wang, Z.-N. Zhao, S. J. Kalita and Y.-Y. Huang, Org. Lett., 2021, 23, 8147–8152 CrossRef CAS PubMed; (b) S. Debnath, A. S. Kumar, S. Chauhan and K. C. Kumara Swamy, J. Org. Chem., 2021, 86, 11583–11598 CrossRef CAS PubMed.
- J. Feng and Y. Huang, ACS Catal., 2020, 10, 3541–3547 CrossRef CAS.
- Selected examples: (a) C. Ni, J. Chen, Y. Zhang, Y. Hou, D. Wang, X. Tong, S.-F. Zhu and Q.-L. Zhou, Org. Lett., 2017, 19, 3668–3671 CrossRef CAS PubMed; (b) D. Wang and X. Tong, Org. Lett., 2017, 19, 6392–6395 CrossRef CAS PubMed; (c) J. Xing, Y. Lei, Y.-N. Gao and M. Shi, Org. Lett., 2017, 19, 2382–2385 CrossRef CAS PubMed; (d) J. Feng and Y. Huang, Chem. Commun., 2019, 55, 14011–14014 RSC; (e) J. Feng, Y. Chen, W. Qin and Y. Huang, Org. Lett., 2020, 22, 433–437 CrossRef CAS PubMed.
- (a) G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 CrossRef CAS; (b) G. A. Molander, Acc. Chem. Res., 1998, 31, 603–609 CrossRef CAS.
- J. Otevrel, M. Eugui, S. Ričko and K. A. Jørgensen, Nat. Synth., 2023, 2, 1142–1158 CrossRef.
- Selected reviews: (a) E. Rodriguez, G. H. N. Towers and J. C. Mitchell, Phytochemistry, 1976, 15, 1573–1580 CrossRef CAS; (b) W. He, M. Cik, G. Appendino, V. L. Puyvelde, E. J. Leysen and D. N. Kimpe, Mini-Rev. Med. Chem., 2002, 2, 185–200 CrossRef CAS PubMed; (c) H.-B. Wang, X.-Y. Wang, L.-P. Liu, G.-W. Qin and T.-G. Kang, Chem. Rev., 2015, 115, 2975–3011 CrossRef CAS PubMed.
- (a) B. Holmes and A. Ward, Drugs, 1985, 30, 285–312 CrossRef CAS PubMed; (b) I. Tietjen, D. E. Williams, S. Read, X. T. Kuang, P. Mwimanzi, E. Wilhelm, T. Markle, N. N. Kinloch, C. N. Na-phen and K. Tenney, Antiviral Res., 2018, 152, 94–103 CrossRef CAS PubMed; (c) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (d) I. Bruce, G. W. J. Fleet, B. Cenci di Bello and I. Winchester, Tetrahedron Lett., 1989, 30, 7257–7260 CrossRef CAS; (e) P. A. Vans, D. E. Negru and D. Shang, Angew. Chem., Int. Ed., 2015, 54, 4768–4772 CrossRef PubMed; (f) Q. Luo, X.-Y. Wei, J. Yang, J.-F. Luo, R. Liang, Z.-C. Tu and Y.-X. Cheng, J. Nat. Prod., 2017, 80, 61–70 CrossRef CAS PubMed.
- (a) K. Kumar, R. Kapoor, A. Kapur and M. P. S. Ishar, Org. Lett., 2000, 2, 2023–2025 CrossRef CAS PubMed; (b) S. Zheng and X.-Y. Lu, Org. Lett., 2009, 11, 3978–3981 CrossRef CAS PubMed; (c) R. Na, C. Jing, Q. Xu, H. Jiang, X. Wu, J. Shi, J. Zhong, M. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard, H. Guo and O. Kwon, J. Am. Chem. Soc., 2011, 133, 13337–13348 CrossRef CAS PubMed; (d) R. Zhou, J. Wang, C. Duan and Z. He, Org. Lett., 2012, 14, 6134–6137 CrossRef CAS PubMed; (e) J. Wu, Y. Tang, W. Wei, Y. Wu, Y. Li, J. Zhang, Y. Zheng and S. Xu, Angew. Chem., Int. Ed., 2018, 57, 6284–6288 CrossRef CAS PubMed; (f) Z. Dai, J. Zhu, J. Wang, W. Su, F. Yang and Q. Zhou, Adv. Synth. Catal., 2019, 362, 545–551 CrossRef; (g) K. Zhang, L. Cai, S. Hong and O. Kwon, Org. Lett., 2019, 21, 5143–5146 CrossRef CAS PubMed.
- (a) K. Gao, Y. G. Zhang, Z. Wang and H. Ding, Chem. Commun., 2019, 55, 1859–1878 RSC; (b) K. Selvaraj, S. Chauhan, K. Sandeep and K. C. K. Swamy, Chem. Asian. J., 2020, 15, 2380–2402 CrossRef CAS PubMed.
- J. Lai and Y. Huang, Chem. Commun., 2023, 59, 13215–13218 RSC.
- Deposition numbers 2211963 (34) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- Y.-Z. Xia, Y. Liang, Y.-Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y.-H. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470–3471 CrossRef CAS PubMed.
- (a) C.-W. Cho and M. J. Krische, Angew. Chem., Int. Ed., 2004, 43, 6689–6691 CrossRef CAS PubMed; (b) R. K. Thalji and W. R. Roush, J. Am. Chem. Soc., 2005, 127, 16778–16779 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2211963. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02487g |
| This journal is © The Royal Society of Chemistry 2024 |