
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward robust lithium–sulfur batteries via advancing Li2S deposition†
Xun
Jiao
a,
Xiaoxia
Tang
a,
Jinrui
Li
a,
Yujiao
Xiang
a,
Cunpu
Li
 *a,
Cheng
Tong
*a,
Minhua
Shao
b and
Zidong
Wei
*a
*a,
Cheng
Tong
*a,
Minhua
Shao
b and
Zidong
Wei
*a
aState Key Laboratory of Advanced Chemical Power Sources, School of Chemistry and Chemical Engineering, China. E-mail: lcp@cqu.edu.cn; tongcheng@cqu.edu.cn; zdwei@cqu.edu.cn
bDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
First published on 1st May 2024
Abstract
Lithium–sulfur batteries (LSBs) with two typical platforms during discharge are prone to the formation of soluble lithium polysulfides (LiPS), leading to a decrease in the cycling life of the battery. Under practical working conditions, the transformation of S8 into Li2S is cross-executed rather than a stepwise reaction, where the liquid LiPS to solid Li2S conversion can occur at a high state of charge (SOC) to maintain the current requirement. Therefore, advancing Li2S deposition can effectively reduce the accumulation of LiPSs and ultimately improve the reaction kinetics. Herein, a “butterfly material” GeS2-MoS2/rGO is used as a sulfur host. Rich catalytic heterointerfaces can be obtained via the abundant S–S bonds formed between GeS2 and MoS2. MoS2 (left wing) can enhance LiPS adsorption, while the lattice-matching nature of Fdd2 GeS2 (right wing) and Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m Li2S can induce multiple nucleation and regulate the 3D growth of Li2S. Li2S deposition can be advanced to occur at 80% SOC, thereby effectively inhibiting the accumulation of soluble LiPSs. Attributed to the synergistic effect of catalytic and lattice-matching properties, robust coin and pouch LSBs can be achieved.
m Li2S can induce multiple nucleation and regulate the 3D growth of Li2S. Li2S deposition can be advanced to occur at 80% SOC, thereby effectively inhibiting the accumulation of soluble LiPSs. Attributed to the synergistic effect of catalytic and lattice-matching properties, robust coin and pouch LSBs can be achieved.
1 Introduction
As a promising alternative to lithium-ion batteries (LIBs), lithium–sulfur batteries (LSBs) have attracted widespread attention with their theoretical energy density of more than 2600 W h kg−1, as well as the eco-friendliness and low cost of sulfur.1–5 According to conventional understanding, the discharge of sulfur species is a stepwise reaction (Scheme 1(a)).6,7 During the discharge process, the reaction of sulfur species first undergoes a phase transition from solid S8 to liquid long-chain lithium polysulfides (LiPSs, Li2Sx, 6 ≤ x ≤ 8) (red line and rectangle in Scheme 1(a)), then to short-chain Li2S4 (blue line and rectangle in Scheme 1(a)), and finally to solid Li2S2/Li2S (green line and rectangle in Scheme 1(a)), which is called the “solid–liquid–solid” conversion mechanism.8–10 Throughout the discharge process, the accumulated soluble LiPSs will dissolve in the electrolyte, causing the loss of active material, which is known as the “shuttle effect”.11–13 However, in fact, under practical discharge working conditions, the conversion reactions of different sulfur species are cross-executed rather than stepwise (Scheme 1(b)).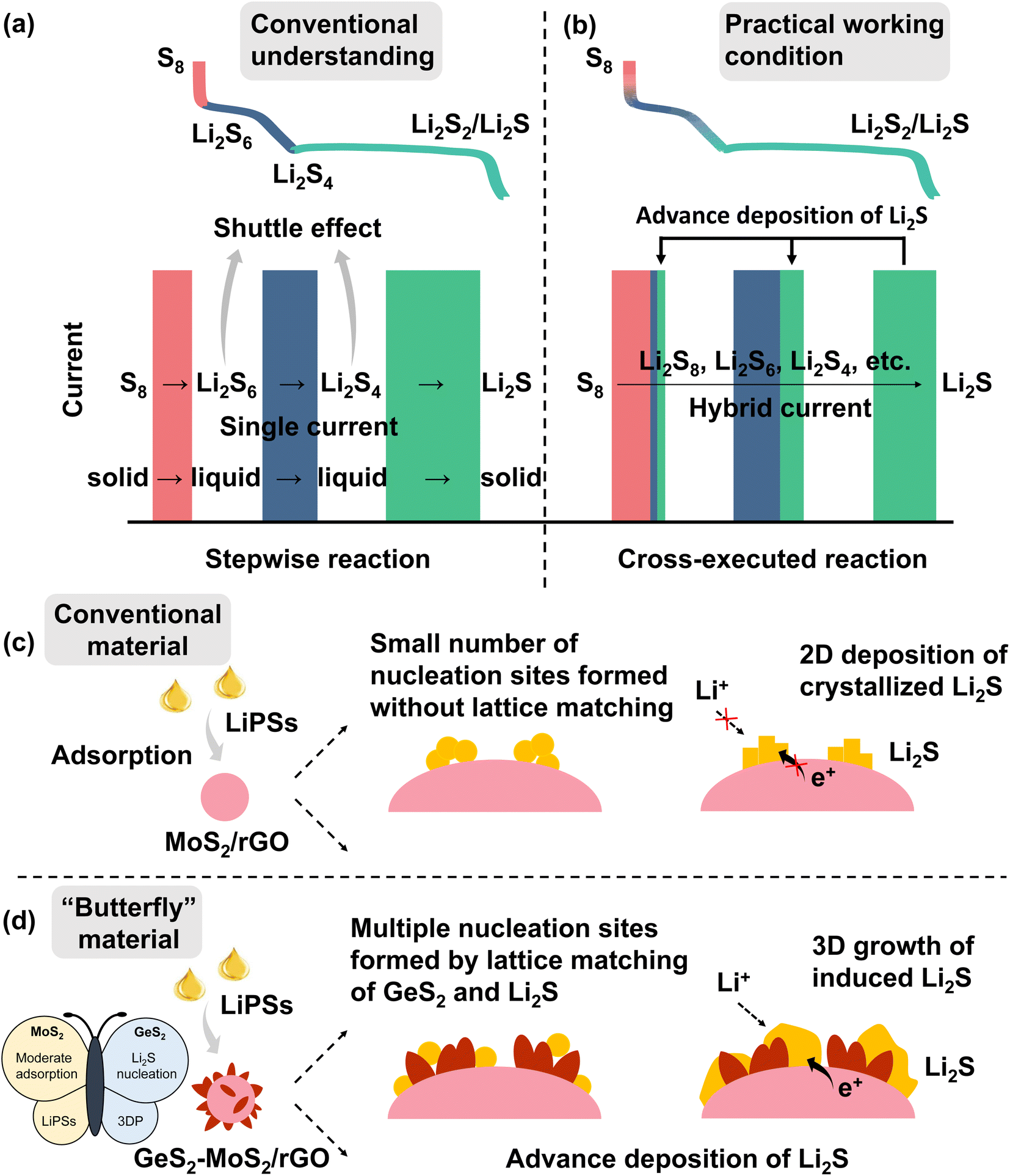 | ||
| Scheme 1 Conventional understanding (a) and practical working conditions (b) of discharge of LSBs (the red line and rectangle are regarded as solid–liquid reactions from S8 to Li2S6, the blue line and rectangle are regarded as liquid–liquid reactions from Li2S6 to Li2S4, and the green line and rectangle are regarded as liquid–solid reactions from Li2S4 to Li2S). In contrast to the conventional understanding of a stepwise reaction (a), the practice cycling condition is in fact a cross-executed reaction (b). Strengthening the hybrid current during cycling of LSBs can promote the adsorption and conversion of sulfur species and ultimately enhance the redox kinetics of the batteries; (c and d) schematic illustration of LiPS conversion and Li2S growth on the MoS2/rGO surface (c) and GeS2-MoS2/rGO surface (d). (d) The designed “butterfly material”: MoS2 (left wing) can enhance LiPS adsorption, while the lattice-matching nature of Fdd2 GeS2 (right wing) and Fmm Li2S can induce multiple nucleation and regulate the 3D growth of Li2S. The wings of the butterfly enable advanced deposition of Li2S. | ||
During the discharge process, when the preceding reactions cannot meet the current demand, the subsequent reactions will participate in the electrochemical reaction, thus forming a “hybrid current”.14 For example at 0.1 A, the number of electrons transferred on the electrode per second is determined (0.1/Faraday constant). During the discharge process of LSBs, the conversion of Li2S6 to Li2S4 cannot provide enough electrons in the electrode reaction, and the subsequent reaction of Li2S4 to Li2S2/Li2S will be involved simultaneously. So, the practical discharge/charge process produces a hybrid current, not a single current. The existence of hybrid current provides an opportunity to address the shuttle effect of LiPSs. If the adsorption of LiPSs is enhanced and then rapidly converted to Li2S2/Li2S, the subsequent liquid–solid reaction can be involved in the whole discharge process, which effectively reduces the accumulation of soluble LiPSs and greatly improves the redox kinetics of the battery. And the conversion of LiPSs to Li2S is the rate-determining step in the sulfur reduction reaction.15 Therefore, it is desirable to find a method to enhance the adoption of LiPSs and promote Li2S growth, and to advance the deposition of Li2S at a high state of charge (SOC). The advanced deposition of Li2S will effectively reduce the accumulation of liquid LiPSs, as well as facilitate the conversion of sulfur species.
According to many research studies, well-designed sulfur hosts with moderate adsorption ability and catalytic activity can alleviate the shuttle effect of LiPSs and improve reaction kinetics of LSBs.16–18 Whereas, it is known that the discharging product Li2S is an electronic–insulating ionic compound, which is difficult to deposit and grow rapidly on the substrate interface.19–21 In general, Li2S deposition begins with nucleation on the conductive substrate, and then increases at the interface of nucleation, substrate, and electrolyte.22 With the deposition and accumulation of Li2S, an insulating Li2S crystal structure is gradually formed on the conductive interface, leading to a gradual slowdown of Li2S growth, which ultimately limits the efficiency of electrochemical conversion in LSBs.23,24 Moreover, the first step, solid–solid decomposition of crystalline Li2S, produces an ultra-high overpotential.25 Therefore, developing the catalytic interconversion between LiPSs and Li2S, as well as improving the deposition and decomposition efficiency of Li2S, are particularly important towards the high performance LSBs.
In this regard, we designed and fabricated a “butterfly material” GeS2-MoS2/rGO. The nanosheet hierarchical petal-spherical GeS2-MoS2 heterostructure can enhance the reaction kinetics of LiPSs and advance the Li2S deposition. MoS2 (left wing) can enhance the LiPS adsorption, and GeS2 (right wing) can induce 3D Li2S deposition. And the introduction of reduced graphene oxide (rGO), a conductive carbon material, into the heterostructure can not only further enhance the electrical conductivity, but relieve the mechanical stress caused by the volume change of the electrode material during the cycling. The lattice-matching nature between orthorhombic GeS2 (Fdd2) and cubic Li2S (Fmm) can guide Li2S growth in a 3D model, which reduces the Li2S transverse diffusion and avoids the catalyst surface passivation. What's more, the three-dimensional (3D) model deposited Li2S also ensures that the interface always provides channels for ionic and electronic conduction, exposing sufficient catalytically active sites for the conversion of Li2S.26 The wings of the butterfly promote Li2S growth and regulate the Li2S deposition behavior, and finally advance the Li2S formation at a high SOC (Scheme 1(c and d)). Therefore, robust LSBs with long-term cycling stability and potential for practical applications can be achieved.
2 Results and discussion
2.1 Material design
The heterostructure design combines the advantages of different components and provides a manipulable electronic structure, which is a good choice for promoting redox kinetics in LSBs.27,28 In the selection of catalysts, two-dimensional materials have been extensively studied in electrochemistry due to their unique physical and chemical properties.29 And molybdenum-based materials have been widely used in LIBs and LSBs due to their chemical stability and environmental friendliness.30 Among them, MoS2 is one of the most typical representatives of a tri-atomic layer (S–Mo–S) accumulated by weak van der Waals forces. MoS2 possesses abundant active sites, moderate adsorption ability toward LiPSs, and good electronic conductivity, which enables rapid liquid–liquid conversion and provides a high-speed electrochemical conversion pathway.31More importantly, to improve sulfur utilization and redox kinetics, it is critical to introduce substrates capable of coordinating the Li2S deposition process: Fdd2 GeS2, and the b-axis of the lattice is equal to 22.67 Å, which is four times that of the typical reduction product of LSBs, Fmm Li2S (b = 5.67 Å). The lattice mismatch (f) between Fdd2 GeS2 and Fmm Li2S can be calculated from eqn (1):32
| f = (αs − αg)/αs | (1) |
m Li2S can be readily grown on Fdd2 GeS2. That is, in the beginning, Fmm Li2S selectively nucleates on one facet of the Fdd2 GeS2 substrate, and subsequently nucleates and grows on the other facets of Fdd2 GeS2, which ultimately produces multi-site deposition and 3D growth of Li2S. Therefore, the interface of the GeS2-MoS2 heterostructure always maintains a conductive network and Li+ transport channels as Li2S growth increases.
From the density functional theory (DFT) calculations, we can find that rich GeS2-MoS2 heterointerfaces can be constructed by the easily formed S–S bonds between GeS2 and MoS2. As displayed in Fig. 1(a), GeS2 (311) and MoS2 (002) planes are selected to construct the GeS2-MoS2 heterostructure. Abundant S–S bonds are formed between heterointerfaces, and the differential charge density diagram shows a significant charge accumulation and depletion at the interface of GeS2 and MoS2 in the heterostructure. And the interaction of rich catalytic heterointerfaces can favor intensive charge transfer. The charge transfer analysis is shown to allow charge redistribution at the GeS2-MoS2 interface, and it can be reasonably inferred that the GeS2-MoS2 heterostructure facilitates the interfacial charge transfer.
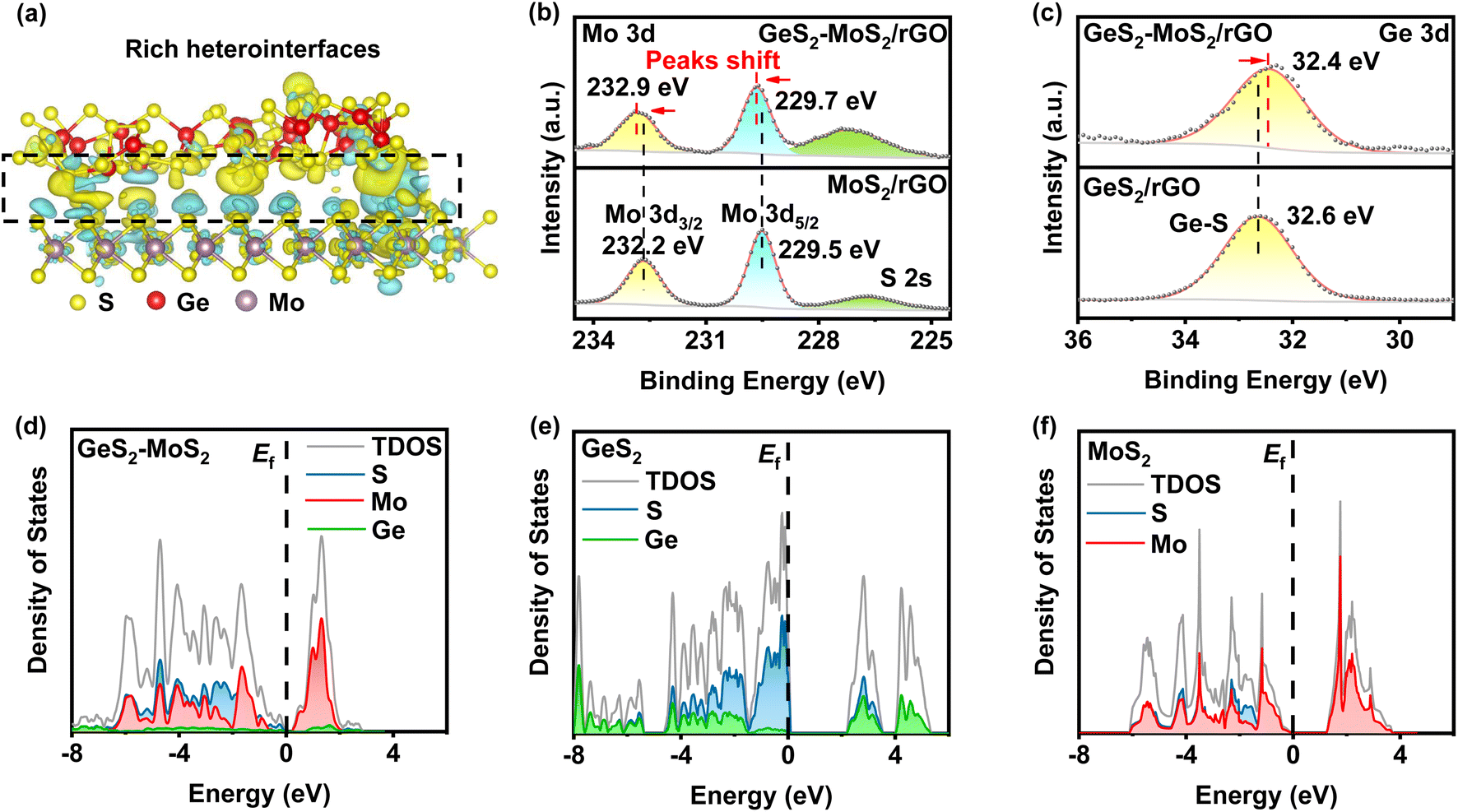 | ||
| Fig. 1 (a) Interfacial charge density difference of GeS2-MoS2 (yellow: electron accumulation; cyan: electron depletion). The strong bonding interactions of the heterostructure form rich catalytic heterointerfaces and multiple nucleation sites of Li2S. (b) High-resolution Mo 3d XPS spectra of GeS2-MoS2/rGO and MoS2/rGO. The Mo 3d peaks of GeS2-MoS2/rGO shift towards the higher binding energy region, compared with those for MoS2/rGO. (c) High-resolution Ge 3d XPS spectra of GeS2-MoS2/rGO and GeS2/rGO. Electron transfer from MoS2 to GeS2 in the GeS2-MoS2 heterostructure. (d–f) Calculated pDOS near the Fermi level of GeS2-MoS2, GeS2 and MoS2. GeS2-MoS2 possesses the smallest band gap, enhancing the conversion of LiPSs. | ||
X-ray photoelectron spectroscopy (XPS) analysis was performed to experimentally confirm the interactions between GeS2 and MoS2 and study the chemical state of the different elements (Fig. 1(b, c) and S1†). The S 2p spectrum in Fig. S1(a)† shows two peaks at 163.8 and 162.7 eV, which correspond to S 2p1/2 and S 2p3/2 of S2− species in GeS2-MoS2/rGO.34 In terms of the Mo 3d spectrum (Fig. 1(b)), the two major peaks at 232.9 eV (Mo 3d3/2) and 229.7 eV (Mo 3d5/2) of GeS2-MoS2/rGO are assigned to Mo4+ ions in MoS2.35 In the Ge 3d spectrum (Fig. 1(c)), the peak at 32.4 eV is a typical bonding of the Ge4+ ion in GeS2.36 More importantly, the Mo 3d spectrum of MoS2/rGO displays an obvious positive shift with the addition of GeS2, which is associated with electron transfer and strong interaction in the heterointerfaces, consistent with the interfacial charge arrangement predicted by DFT calculations.
Conductivity tests experimentally demonstrated the higher conductivity of GeS2-MoS2/rGO compared to GeS2/rGO and MoS2/rGO. Fig. S2† displays the current variations over 3000 s of testing at a constant voltage of 1.0 V to compare the conductivity of different catalysts. The electronic conductivity (σ) is calculated according to eqn (2) and (3):37
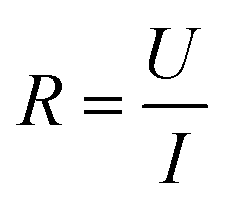 | (2) |
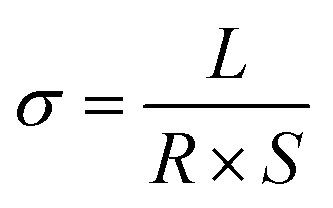 | (3) |
In eqn (2), U is the constant voltage (1.0 V), I is the average current (A) from 100 to 3000 s, and R is the calculated resistance (1/S). In eqn (3), L is the thickness of the sample (mm) and S is the area of the sample (132.665 mm2). The σ values of different catalysts are listed in Table S1.† And the result shows that GeS2-MoS2/rGO has the largest σ value, demonstrating the enhanced electrical conductivity of the heterostructure.
The projected densities of states (pDOSs) are then shown in Fig. 1(d–f) to assess the electronic structure differences of the different catalysts. All three catalysts have semiconductor properties, with the GeS2-MoS2 heterostructure showing the smallest band gap (≈ 0.19 eV), much smaller than that of GeS2 (≈ 2.14 eV) and MoS2 (≈ 1.21 eV). These results demonstrate that the GeS2-MoS2 heterostructure has good electrical conductivity and enhanced adsorption energies with LiPSs, which is attributed to the rich catalytic heterointerfaces and strong interfacial synergistic effect.
To determine the valence and chemical coordination environment changes of the GeS2-MoS2 heterostructure, Mo and Ge K-edge X-ray absorption fine structure (XAFS) spectra were further measured. The X-ray absorption near-edge structure (XANES) spectra of the Mo K-edge in Mo foil, GeS2-MoS2/rGO and MoS2/rGO are displayed in Fig. 2(a). In the enlarged illustration, the pre-edge feature of GeS2-MoS2/rGO and MoS2/rGO show a shift to higher energy compared to that of Mo foil. Because Mo is oxidized to a higher state, the valence state of Mo in GeS2-MoS2/rGO is slightly higher than that in MoS2/rGO, which is consistent with the XPS results of Mo 3d (Fig. 1(b)). In addition, the Ge valence state in GeS2-MoS2/rGO is lower than that in GeS2/rGO (Fig. 2(b)), demonstrating the electron transfer from MoS2 to GeS2 in the heterointerfaces.38 According to the R-space of the extended X-ray absorption fine structure (EXAFS) in Fig. 2(c), the EXAFS spectra of GeS2-MoS2/rGO and MoS2/rGO exhibit two main peaks at around 1.9 and 2.6 Å, corresponding to Mo–S and Mo–Mo bonds, respectively.39 And the EXAFS spectra in Fig. 2(d) show that the GeS2-MoS2/rGO and GeS2/rGO peaks are similar to those of Ge foil, but the positions of the peaks are slightly lower than those of Ge foil, indicating that Ge is bonded with other elements. Thus, the EXAFS spectra of GeS2-MoS2/rGO and GeS2/rGO are fitted, corresponding to Ge–Ge and Ge–S bonds, respectively (Fig. S3†). Fig. 2(e and f) display the oscillation curves of the Mo and Ge K-edge for different samples in the 0–12 Å−1 K range. The decrease in the oscillation intensity suggests a periodic decrease, which is due to the formation of rich heterointerfaces between the two crystal phases (MoS2 and GeS2) in GeS2-MoS2/rGO by charge transfer. The wavelet transform (WT) is considered to be a good complement to the Fourier-transform (FT) for separating backscattered atoms in both R-space and K-space resolution and displaying atomic dispersion.40Fig. 2(g and h) exhibit the Mo K-edge and Ge K-edge WT results of different samples, respectively. For GeS2-MoS2/rGO, MoS2/rGO, Mo foil, GeS2/rGO, and Ge foil, the fitted parameters of the R-space are shown in Tables S2 and S3,† which further suggests that the lattice distortion and interfacial charge redistribution are expected to contribute to the electrochemical performance of LSBs. Furthermore, the absence of Ge–Mo bonds in the GeS2-MoS2 heterostructure indicates that the heterointerfaces are mediated by S for charge transfer, which is consistent with the DFT results.
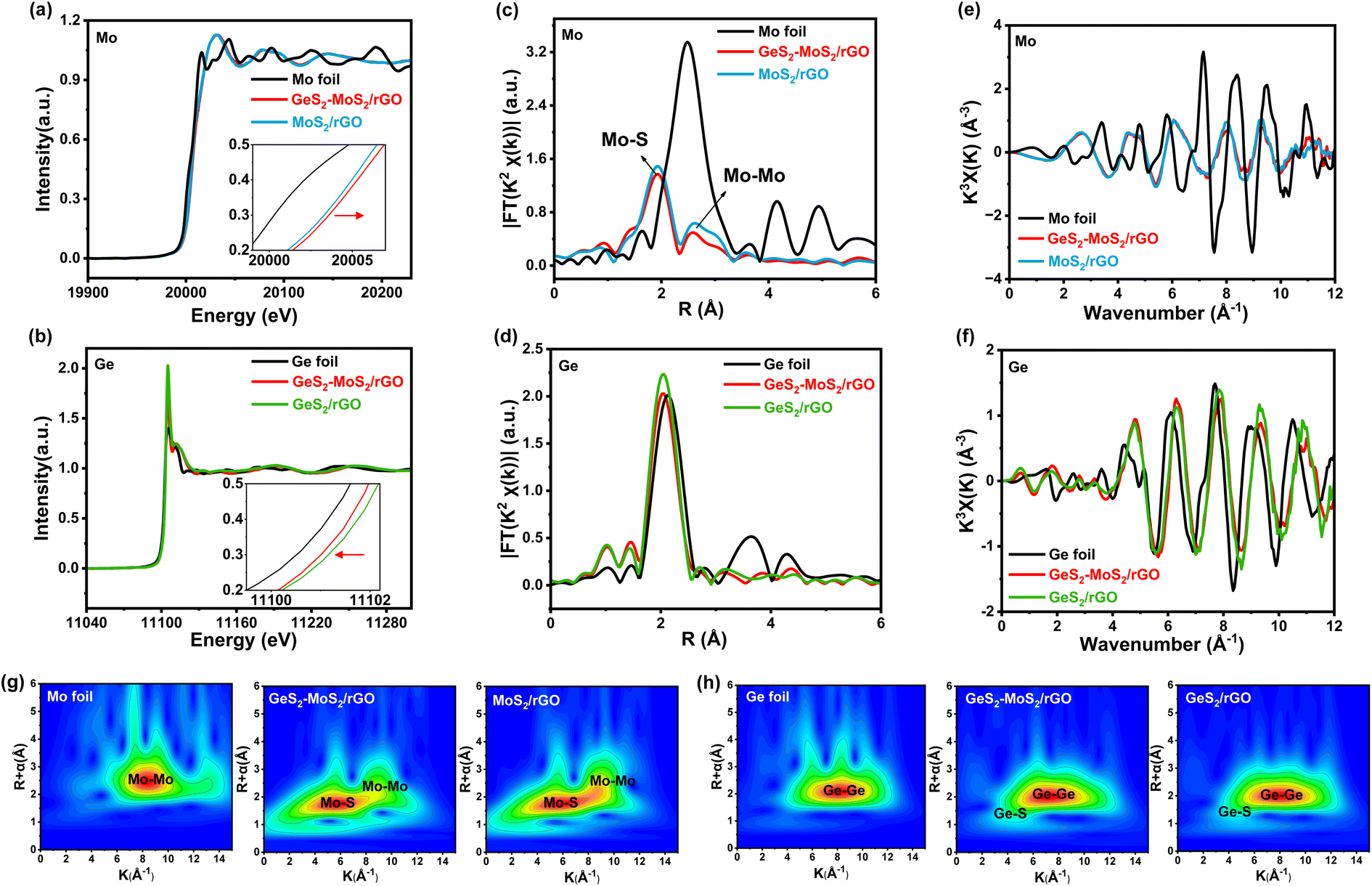 | ||
| Fig. 2 (a) Mo K-edge XANES spectra of Mo foil, GeS2-MoS2/rGO and MoS2/rGO. (b) Ge K-edge XANES spectra of Ge foil, GeS2-MoS2/rGO and GeS2/rGO. Electron transfer from MoS2 to GeS2 in the heterointerfaces. (c) R-space of EXAFS analysis of Mo in Mo foil, GeS2-MoS2/rGO and MoS2/rGO. (d) R-space of EXAFS analysis of Ge in Ge foil, GeS2-MoS2/rGO and GeS2/rGO. (e) Mo K-edge EXAFS oscillations of Mo foil, GeS2-MoS2/rGO andMoS2/rGO. (f) Ge K-edge EXAFS oscillations of Ge foil, GeS2-MoS2/rGO and GeS2/rGO. (g) WT contour plots at the Mo K-edge of Mo foil, GeS2-MoS2/rGO and MoS2/rGO. (h) WT contour plots at the Ge K-edge of Ge foil, GeS2-MoS2/rGO and GeS2/rGO. No Ge–Mo bond can be observed, suggesting that the heterointerfaces undergoes charge transfer mediated by S, which is consistent with the above DFT results. | ||
2.2 Lattice-matching nature between GeS2 and Li2S
The material ratios are optimized based on the morphology and cycling capacity of the GeS2-MoS2/rGO heterostructure, and the element content (Table S4†) was measured by inductively coupled plasma-optical emission spectrometry (ICP-OES). Heterostructures with three different ratios (MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GeS2 = 0.7, 0.9 and 1.1) were prepared for morphology and capacity characterization (Fig. S4†). When MoS2:GeS2 = 0.7, a large amount of GeS2 accumulates on the surface of MoS2. This hinders the contact between MoS2 and LiPSs and weakens the adsorption effect of the heterostructure on LiPSs, leading to a rapid capacity decay (capacity retention of 77.29% after 300 cycles at 0.5C). When MoS2:GeS2 = 1.1, MoS2 agglomerates heavily in the heterostructure, and only a small number of GeS2 layers are attached to the MoS2 surface. The reduced pores lead to a decrease in contact between the electrolyte and the material, which affects ionic conduction and ultimately electrochemical performance (capacity retention of 79.51% after 300 cycles at 0.5C). When MoS2:GeS2 = 0.9, the hierarchical heterostructure can significantly expand the contact area between the electrode and electrolyte, and therefore increase the active reaction and storage sites for LiPSs and Li2S. The nanosheets in the heterostructure can greatly shorten the ion transport path, which increases the reversible capacity of the battery (capacity retention of 89.17% after 300 cycles at 0.5C). As a result, in this work, we chose a heterostructure with a MoS2 to GeS2 ratio of 0.9 as the sulfur host for the study, obtaining excellent electrochemical cycling preformance and enhanced redox kinetics.
:GeS2 = 0.7, 0.9 and 1.1) were prepared for morphology and capacity characterization (Fig. S4†). When MoS2:GeS2 = 0.7, a large amount of GeS2 accumulates on the surface of MoS2. This hinders the contact between MoS2 and LiPSs and weakens the adsorption effect of the heterostructure on LiPSs, leading to a rapid capacity decay (capacity retention of 77.29% after 300 cycles at 0.5C). When MoS2:GeS2 = 1.1, MoS2 agglomerates heavily in the heterostructure, and only a small number of GeS2 layers are attached to the MoS2 surface. The reduced pores lead to a decrease in contact between the electrolyte and the material, which affects ionic conduction and ultimately electrochemical performance (capacity retention of 79.51% after 300 cycles at 0.5C). When MoS2:GeS2 = 0.9, the hierarchical heterostructure can significantly expand the contact area between the electrode and electrolyte, and therefore increase the active reaction and storage sites for LiPSs and Li2S. The nanosheets in the heterostructure can greatly shorten the ion transport path, which increases the reversible capacity of the battery (capacity retention of 89.17% after 300 cycles at 0.5C). As a result, in this work, we chose a heterostructure with a MoS2 to GeS2 ratio of 0.9 as the sulfur host for the study, obtaining excellent electrochemical cycling preformance and enhanced redox kinetics.
The morphology of the prepared GeS2-MoS2/rGO was characterized, which confirmed the formation of the heterostructure and heterointerfaces. As shown by the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) results (Fig. 3(a) and S5(a, b)),† the GeS2-MoS2/rGO heterostructure exhibits uniformly hierarchical petal-spherical particles. And the nanosheets in GeS2-MoS2/rGO clearly show hierarchical structures, indicating that the heterostructure effectively expands the contact area between the electrode and electrolyte as well as exposes abundant active sites. These advantages can significantly shorten the ion transport path and improve the kinetics of the reaction. The interface of the GeS2-MoS2/rGO heterostructure is shown in the high-resolution TEM (HRTEM) image (Fig. 3(b)). The lattice fringe spacing of 0.62 nm is assigned to the (002) plane of hexagonal MoS2, and 0.34 nm corresponds to the (311) plane of orthorhombic GeS2, further revealing the formation of the heterostructure. According to the energy dispersive X-ray (EDX) analysis (Fig. S5(c–f)),† the distribution of S, Mo, and Ge elements of GeS2-MoS2/rGO is clearly observed, where GeS2 nanosheets are dispersed on the MoS2 samples. Atomic force microscopy (AFM) measurement was carried out to determine the thickness of the nanosheets in GeS2-MoS2/rGO (Fig. S6†). The results show that the particle distribution of GeS2-MoS2/rGO is 100–500 nm with a thickness of about 6 nm. The ultra-thin nanosheets effectively shorten the ion and electron transport paths and accelerate the surface charge transfer rate, ultimately improving the redox kinetics of LSBs. In addition, the morphology analyses of MoS2/rGO and GeS2/rGO can also prove the successful synthesis of materials and the uniform distribution of elements (Fig. S7 and S8†). More interestingly, the EDX results of GeS2-MoS2/rGO show that GeS2 nanosheets are generated more on the outer petals in the hierarchical GeS2-MoS2/rGO, suggesting that GeS2 nanosheets in the heterostructure grow epitaxially along the conductive MoS2 core. These unique hierarchical petal-spherical GeS2-MoS2/rGO heterostructures provide an effectively shortened ion transport path, which facilitates redox kinetics of LSBs.
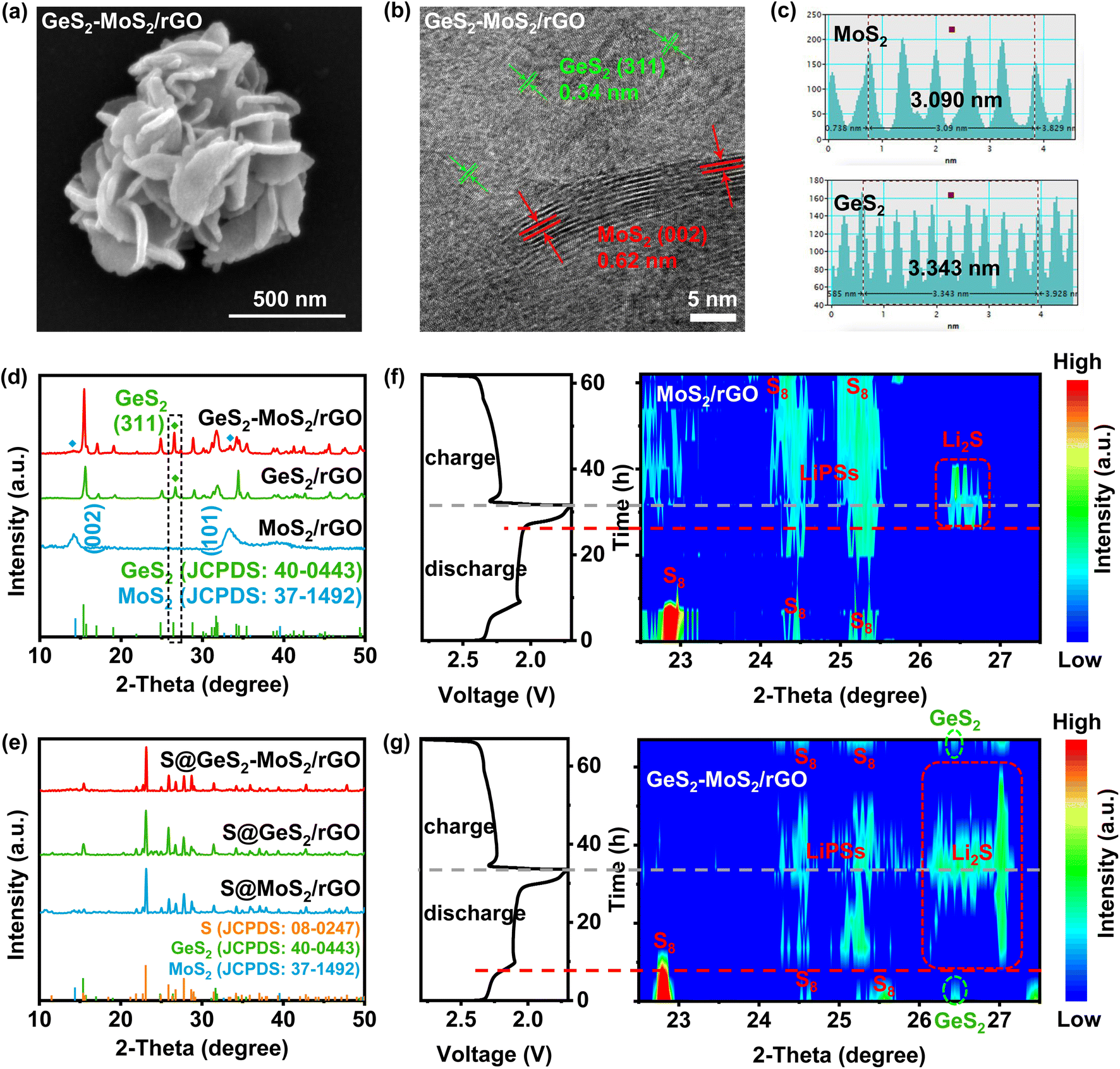 | ||
| Fig. 3 (a) SEM images of GeS2-MoS2/rGO. A petal-spherical morphology can be observed. (b) HRTEM image of GeS2-MoS2/rGO and (c) the corresponding reduced FFT patterns. The GeS2 (311) and MoS2 (002) lattice fringes can be determined. (d) XRD patterns of GeS2-MoS2/rGO, MoS2/rGO and GeS2/rGO. (e) XRD patterns of S@GeS2-MoS2/rGO, S@MoS2/rGO and S@GeS2/rGO. (f and g) The initial discharge/charge profiles and corresponding in situ XRD contour plots of MoS2/rGO and GeS2-MoS2/rGO batteries, respectively. The lattice-matching nature between Fdd2 GeS2 and Fmm Li2S significantly advances the Li2S deposition at about 60% SOC for the GeS2-MoS2/rGO battery. Also, the GeS2-MoS2/rGO battery exhibits a much weaker and reversible LiPS peak compared with the MoS2/rGO battery, implying that the LiPS shuttling is significantly restrained by the GeS2-MoS2/rGO “butterfly material”. | ||
The X-ray diffraction (XRD) pattern of GeS2-MoS2/rGO in Fig. 3(d) displays the characteristic diffraction peaks of orthorhombic GeS2 (JCPDS no. 40-0443) and two diffraction peaks at 14.38° and 32.68° which correspond to hexagonal MoS2 (JCPDS no. 37-1492). The XRD result of GeS2-MoS2/rGO shows mixed peaks of GeS2 and MoS2 phases, implying the coexistence of GeS2 and MoS2 to construct the heterostructure. And the XRD patterns (Fig. 3(e)) of different catalysts after sulfur loading exhibit the presence of a cubic sulfur crystal structure (JCPDS no. 08-0247). In addition, the sulfur content was determined with a thermogravimetric (TG) analyzer (Fig. S9†), and the sulfur contents of GeS2-MoS2/rGO, GeS2/rGO and MoS2/rGO are approximately 71.7, 71.9 and 71.6 wt%, respectively. Fig. S10† exhibits the Brunauer–Emmett–Teller (BET) results of various catalysts. And the specific surface area (SSA) of the GeS2-MoS2/rGO heterostructure is 141.232 m2 g−1, which is well above that of MoS2/rGO (57.085 m2 g−1) and GeS2/rGO (2.434 m2 g−1). This result further demonstrates that the hierarchical heterostructure exposes more active sites and increases the contact area between the electrode and electrolyte.
As discussed previously, the lattice-matching nature between Fdd2 GeS2 and Fmm Li2S can induce Li2S multi-site nucleation and 3D growth. The evolution of sulfur species during the electrochemical process was monitored by in situ characterization (Fig. 3(f and g)). Throughout the electrochemical reaction, in situ XRD shows the conversion from S8 to LiPSs and finally to Li2S. For the MoS2/rGO battery (Fig. 3(f)), at the beginning of the discharge process, the XRD diffraction peaks of S8 can be clearly seen. The broad peak at 24–25.5° corresponds to long-chain LiPSs.41,42 The Li2S peak appeared at about 20% SOC, and corresponds to the posterior liquid-to-solid or solid-to-solid discharge intervals for LSBs. Also, we can find that the LiPS peak for the MoS2/rGO battery is broad and strong, and corresponds to more accumulated liquid LiPSs and a severe shuttle effect. However, in comparison, for the GeS2-MoS2/rGO battery (Fig. 3(g)), a characteristic peak of cubic Li2S (111) appears at 26.3–27° at high SOC (80% SOC), which is superior to that of the MoS2/rGO battery (60% SOC). The Li2S peak is in accordance with the orthorhombic GeS2 (311) plane (26.4°),43 suggesting that the lattice-matching nature between Fdd2 GeS2 and Fmm Li2S significantly advances the Li2S deposition. Also, we can observe that the LiPS peak for the GeS2-MoS2/rGO battery was much weaker, and it disappeared after the battery was fully charged. By comparing these results with the Li2S peak at 20% SOC which appeared late and the LiPS peak at 100% SOC which did not disappear, of the MoS2/rGO battery, we can claim that the accumulation of LiPSs is significantly restrained in the GeS2-MoS2/rGO battery.
2.3 Synergistic “butterfly” to realize Li2S 3D growth
The GeS2-MoS2 heterostructure can effectively catalyze liquid–solid reaction kinetics and regulate the Li2S growth process (Fig. 4(a)). To investigate the electrochemical stability and electrocatalytic activity of different catalysts toward polysulfide conversion, cyclic voltammetry (CV) tests were performed in symmetric batteries at a scan rate of 20 mV s−1 (Fig. 4(b)). GeS2-MoS2/rGO exhibits two reduction peaks at −0.25 and −0.71 V, which are related to the reduction of S8 to Li2S6 and then Li2S6 to Li2S2/Li2S. And the latter well-defined oxidation peaks at 0.25 and 0.71 V are associated with the oxidation of Li2S2/Li2S to Li2S6 and Li2S6 to S8.44,45 The CV curve of GeS2-MoS2/rGO without Li2S6 electrolyte presents a characteristic rectangle of pure capacitive behavior, indicating that Li2S6 is the only electrochemically active species. Moreover, the stronger peak current densities and smaller overpotential (ΔE) of GeS2-MoS2/rGO compared to GeS2/rGO and MoS2/rGO indicate more robust interfacial stability and electrocatalytic performance.46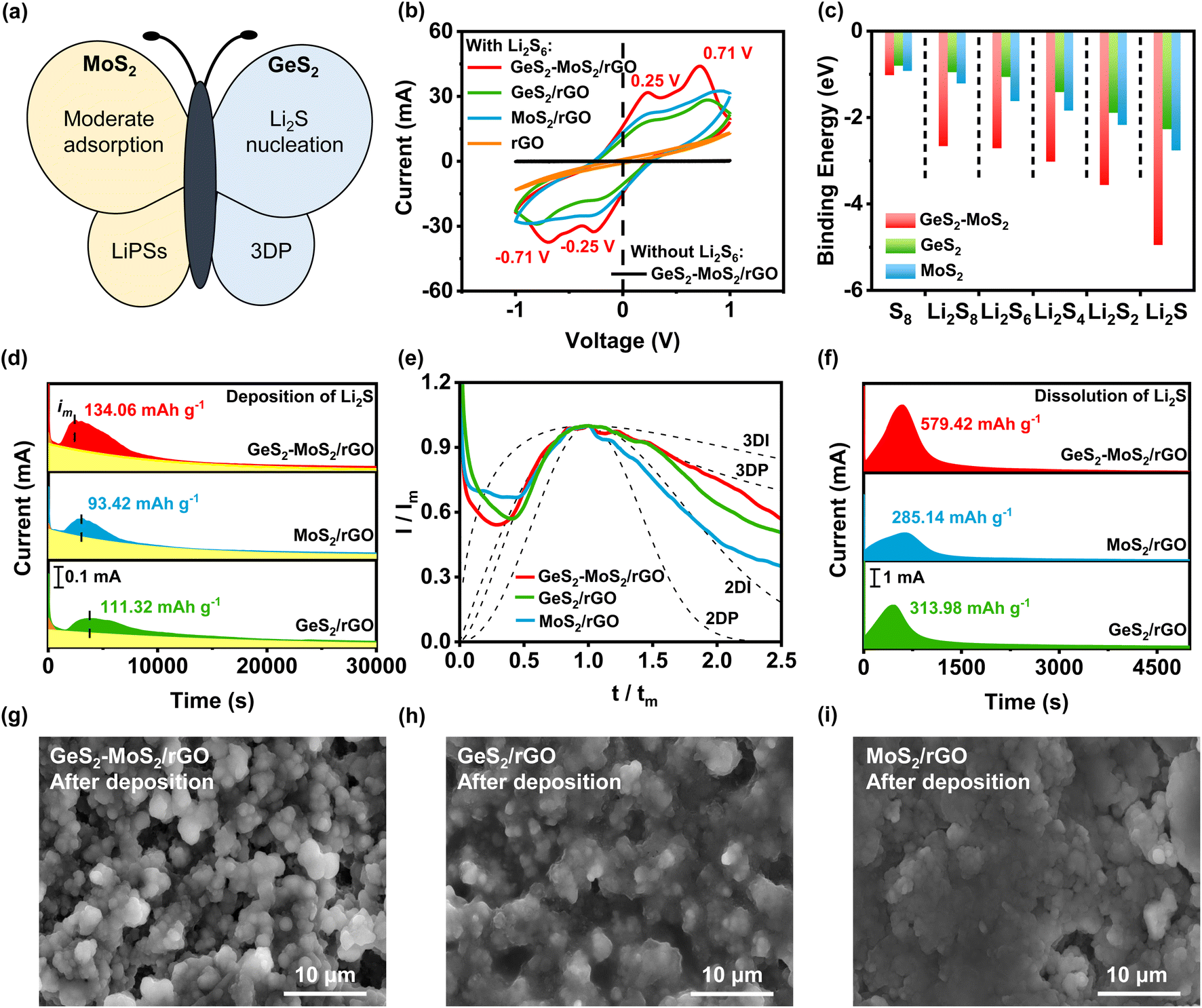 | ||
| Fig. 4 (a) Schematic illustration of the GeS2-MoS2 heterostructure. The butterfly heterostructure is designed as a sulfur host to facilitate the conversion of LiPSs and promote the growth of Li2S synergistically. (b) CV curves of different catalysts in symmetric batteries at a scan rate of 20 mV s−1. GeS2-MoS2/rGO has the strongest peak current density and the smallest overpotential; (c) binding energies of S8 and Li2Sx (x = 1, 2, 4, 6, and 8) species on different catalysts. The GeS2-MoS2 heterostructure has the strongest binding energy with LiPSs, effectively promoting the catalytic effect. (d) Potentiostatic discharge profiles at 2.05 V with Li2S8 catholyte on different samples. The GeS2-MoS2/rGO heterostructure with rich catalytic heterointerfaces can achieve rapid conversion of LiPSs and advanced deposition of Li2S. (e) Dimensionless transient (symbols) of different samples in comparison with theoretical 2D and 3D models (Im: peak current; tm: time needed to achieve the peak current). Li2S growth follows a typical 3DP mode in the GeS2-MoS2/rGO heterostructure. (f) Potentiostatic charge profiles of different samples at 2.40 V for Li2S dissolution. (g–i) SEM images of GeS2-MoS2/rGO, GeS2/rGO and MoS2/rGO after Li2S deposition. The uniform and radial deposition of Li2S is achieved on the surface of the GeS2-MoS2/rGO heterostructure. | ||
The interactions between different catalysts and LiPSs were investigated by calculating the adsorption energies of various hosts on LiPSs (Fig. 4(c)). And the optimized adsorption configurations of LiPS (Li2S, Li2S2, Li2S4, Li2S6, Li2S8, and S8) species on the GeS2-MoS2 heterostructure, and MoS2 (002) and GeS2 (311) surfaces are displayed in Fig. S11–S13.†Fig. 4(c) shows the strongest binding energies (Eb) between the GeS2-MoS2 heterostructure and LiPSs, which indicates that their strong interactions can effectively balance the adsorption/dissociation and catalytic performance. Overall, the DFT results demonstrate that the heterostructure after the addition of GeS2 to MoS2 enhances the chemisorption ability of LiPSs with strong charge transfer and multiple adsorption sites.
To reveal the LiPS trapping ability of the as-prepared catalysts, the adsorption experiments were conducted by immersing the samples with the same content in Li2S6 solution (Fig. S14†). In the adsorption optical images in Fig. S14(a),† the original orange-brown solution containing GeS2-MoS2/rGO becomes slightly lighter after 1 h compared to the other two hosts, and all catalysts become colorless after 6 h. Furthermore, ex situ ultraviolet-visible (UV-vis) absorption spectra were examined to evaluate the concentration changes of the Li2S6 solution used. As shown in Fig. S14(b),† the Li2S6 absorption band in the 400–500 nm region almost disappears for the GeS2-MoS2/rGO host, which demonstrates that GeS2-MoS2/rGO can effectively anchor LiPSs. These results suggest a strong chemical interaction between GeS2-MoS2/rGO and LiPSs, which is attributed to the rich interfacial interaction of the heterostructure.
Li2S nucleation and dissolution experiments were conducted to study the liquid–solid reaction kinetics and Li2S deposition process of different catalysts.47 According to Faraday's law, the Li2S deposition capacities of GeS2-MoS2/rGO, GeS2/rGO, and MoS2/rGO were calculated to be 134.06, 111.32, and 93.42 mA h g−1, respectively (Fig. 4(d)). The growth of Li2S is closely related to the deposition kinetics, which determines the deposition capacity of Li2S and the reversibility of LSBs. Cui et al.48 preliminarily demonstrated that the polar sites can strongly adsorb LiPSs and significantly reduce the interfacial impedance of Li2S deposition. First, Li2S nucleates on the cathode substrate by overcoming the interfacial impedance between the electrolyte and the substrate. Subsequently, LiPSs will be converted to Li2S by adsorption and simultaneously precipitated as Li2S.49 As shown in Fig. 4(d), the depositional curve shows a clear hill-like shape, with a period of incubation ahead (when the current reaches im). And the incubation process is related to the reduction of long chain LiPSs (i.e., Li2S8 and Li2S6) to short chain Li2S4. Because of the better electrical conductivity of MoS2 than GeS2, MoS2 has a stronger adsorption effect on LiPSs, which can promote the conversion of LiPSs to Li2S (current reaches im faster). Compared with GeS2/rGO, the peak current of the Li2S deposition curve of MoS2/rGO appears earlier and the peak current is enhanced (peak current of MoS2/rGO is 0.19 mA at 3035 s and that of GeS2/rGO is 0.14 mA at 3747 s), indicating a faster response to Li2S nucleation. Moreover, the Li2S deposition process ends prematurely with MoS2/rGO, which is due to the lack of 3D nucleation leading to premature passivation of the cathode substrate. So, because of the lattice-matching nature between Fdd2 GeS2 and Fmm Li2S, GeS2 can induce multi-site nucleation and 3D deposition of Li2S, and the deposition capacity of GeS2/rGO is higher than that of MoS2/rGO. More importantly, due to the synergistic effect of MoS2 and GeS2, the GeS2-MoS2/rGO heterostructure with rich catalytic heterointerfaces can achieve rapid conversion of LiPSs and high Li2S precipitation. Thus, the GeS2-MoS2/rGO heterostructure has the earliest Li2S deposition current (0.23 mA at 2518 s) and the highest deposition capacity. These results suggest that hierarchical GeS2-MoS2/rGO with rich catalytic heterointerfaces provides more active sites for achieving rapid conversion of LiPSs and advanced deposition of Li2S.
To investigate the Li2S growth behavior of different catalysts, a dimensionless diagnostic analysis of the current–time curves obtained from Li2S nucleation tests was conducted according to the Scharifker–Hills model (Fig. 4(e) and eqn (S1)–(S4)†).42,50 Four classical electrochemical deposition models are used to fit the current–time responses obtained in chronoamperometric tests. Among them, two-dimensional progressive (2DP) and two-dimensional transient (2DI) nucleation are controlled by incorporating adatoms into the lattice interface. And three-dimensional progressive (3DP) and three-dimensional transient (3DI) nucleation are achieved by volume diffusion controlled growth.51 For GeS2/rGO, Li2S growth shows a mixed 2DI and 3DP mode, while a typical 2DI mode is presented in MoS2/rGO. In comparison, because of the lattice-matching nature between Fdd2 GeS2 and Fmm Li2S, GeS2 induces the deposition and growth of Li2S during the discharge process, thus showing a tendency of 3D model. However, the typical 3DP model is not presented in GeS2/rGO, probably due to the weak electrical conductivity and insufficient reactive sites of GeS2. In the GeS2-MoS2/rGO heterostructure, Li2S growth follows a typical 3DP mode. GeS2 is grown on the epitaxial petals of conductive-core MoS2, and lattice-matching between Fdd2 GeS2 and Fmm Li2S induces rapid and multi-site nucleation of Li2S on the surface of the heterostructure, which enables the multi-site deposition and 3D growth of Li2S.25,52
Furthermore, Li2S dissolution experiments verified the excellent kinetic properties of GeS2-MoS2/rGO (Fig. 4(f)). As a result, the GeS2-MoS2/rGO catalytic electrode exhibits higher current density and Li2S dissolution capacity (579.42 mA h g−1) compared to the GeS2/rGO (285.14 mA h g−1) and MoS2/rGO (313.98 mA h g−1) electrodes. The first step of solid–solid decomposition of Li2S is the slowest step in the charge process, resulting in ultra-high overpotential. These results indicate that the introduced GeS2-MoS2/rGO catalyst can effectively reduce the decomposition barriers and accelerate the charge process.
The phenomenon of Li2S growth can also be obtained from the SEM morphologies of the deposited electrodes (Fig. 4(g–i)). Fig. 4(g) shows the uniform and radial deposition of Li2S on the GeS2-MoS2/rGO heterostructure. And the deposition of Li2S on GeS2/rGO has a tendency of radial growth, which is in agreement with the deposition model and lattice-matching (Fig. 4(e)). However, the deposition of Li2S on MoS2/rGO forms a dense coating covering the catalyst, which hindered the subsequent Li2S deposition (Fig. 4(i)). The 3DP model of Li2S nucleation in GeS2-MoS2/rGO suggests that the sufficient active sites of heterointerfaces and the lattice-matching between Fdd2 GeS2 and Fmm Li2S can guide the radial Li2S growth, thus balancing surface transverse atomic diffusion and mass transport in the electrolyte.53 Therefore, the large accumulation of Li2S caused by the passivation of the electrode surface can be effectively avoided, as demonstrated in Fig. 1(d). Li2S 3D growth of GeS2-MoS2/rGO shortens the ion/electron diffusion path and exposes sufficient catalytically active sites for Li2S conversion. More importantly, the ionic and electronic conduction networks are always present on the surface of the heterostructure, thus consistently providing an efficient pathway for LiPS conversion as well as excellent redox kinetics.
2.4 Electrochemical performance of LSBs
CV tests were performed on S@GeS2-MoS2/rGO, S@MoS2/rGO and S@GeS2/rGO cathodes to investigate the redox kinetics of different catalysts (Fig. 5(a)). All cathodes show two representative cathodic and anodic peaks, respectively. The two cathodic peaks (peak I and peak II) are attributed to the reduction of S8 molecules to long-chain LiPSs (Li2Sx, 4 ≤ x ≤ 8; peak I) and their subsequent reduction to short-chain sulfides (peak II). The anodic peaks (peak III and peak IV) originate from the oxidation of short-chain sulfides eventually to S8.54 As shown in Fig. 5(a), the reduction peak of S@GeS2-MoS2/rGO at about 2.0 V shifts significantly to a higher potential compared with that of S@GeS2/rGO and S@MoS2/rGO, demonstrating a promoted conversion from LiPSs to Li2S. And the S@GeS2-MoS2/rGO cathode also shows the lowest oxidation potential in the oxidation process, indicating an enhanced Li2S oxidation reaction. These results demonstrate that the GeS2-MoS2/rGO heterostructure shows fast redox kinetics and high reversibility with the help of rich heterointerfaces and the synergistic effect.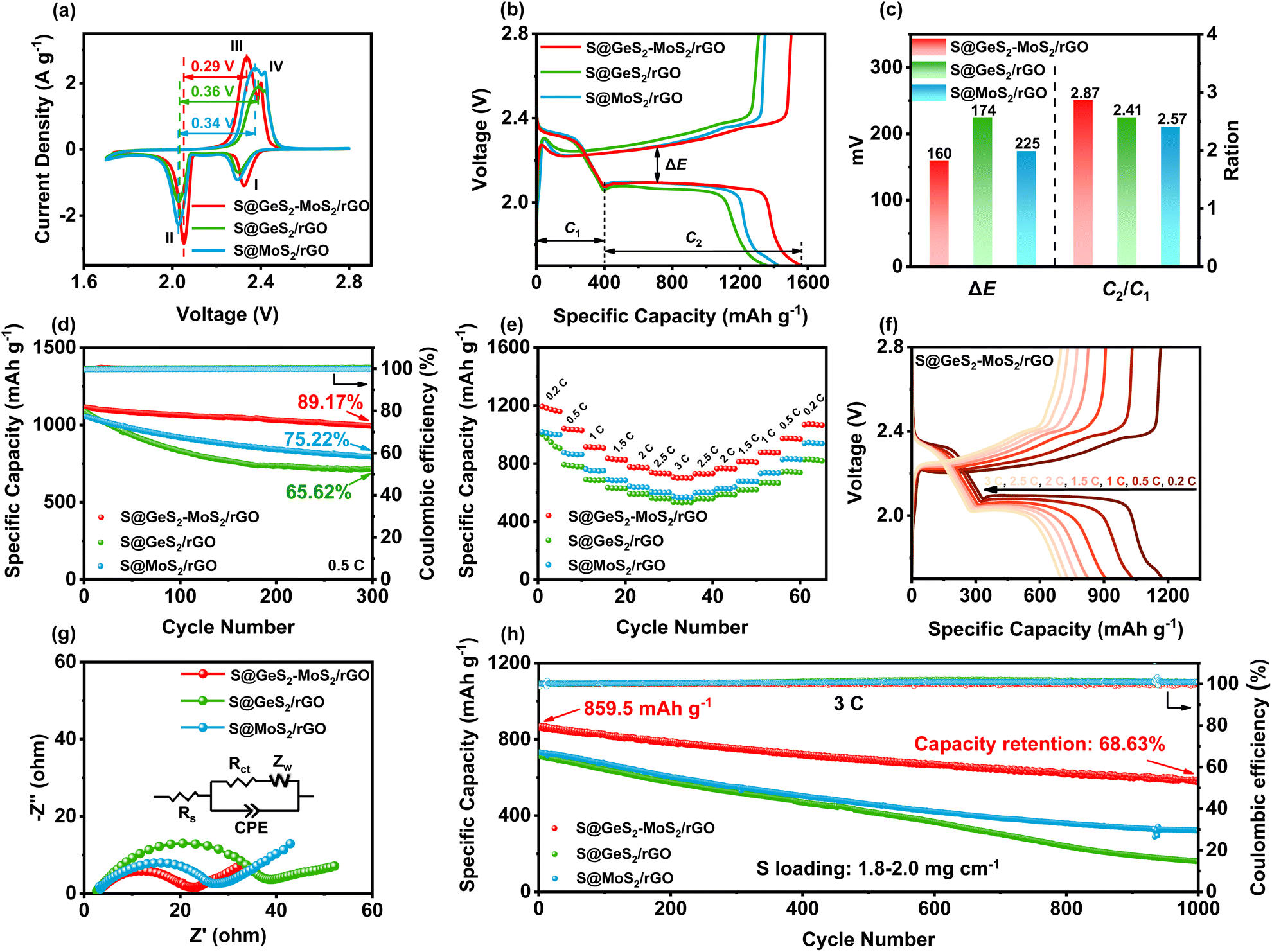 | ||
| Fig. 5 (a) CV curves of different electrodes at a scan rate of 0.1 mV s−1 within 1.7–2.8 V. The S@GeS2-MoS2/rGO cathode shows the lowest potential, indicating enhanced redox kinetics. (b) Galvanostatic discharge/charge profiles of different electrodes at a 0.1C current rate. (c) ΔE and C2/C1 values of galvanostatic discharge/charge profiles. The S@GeS2-MoS2/rGO cathode exhibits the lowest polarization potential and highest C2/C1 value, demonstrating its excellent electrocatalytic activity. (d) Cycling life of different electrodes at 0.5C over 300 cycles. (e) Rate performance of different electrodes with various current densities. The S@GeS2-MoS2/rGO electrode displays the highest capacity retention and rate capacities. (f) Galvanostatic discharge/charge profiles of S@GeS2-MoS2/rGO at various current densities. (g) EIS spectra of different cathodes after 100 cycles. GeS2-MoS2/rGO exhibits a minimal Rct value, which is attributed to the excellent electrical conductivity of the heterostructure. (h) Cycling stability of different electrodes at 3C over 1000 cycles. The S@GeS2-MoS2/rGO electrode shows a high discharge capacity and stable cycling performance, indicating that LiPS shuttling during cycling is effectively suppressed. | ||
The hierarchical nanosheets and strong catalytic heterointerfaces endow the S@GeS2-MoS2/rGO cathode with reduced polarization and stable cycling performance. In Fig. 5(b), the discharge/charge curves of different cathodes at 0.1C (1C = 1675 mA g−1) are compared, and the discharge and charge plateaus are consistent with the CV analysis. The voltage gap between the second discharge and the charge plateaus is the polarization potential (ΔE), denoting a hysteresis in the redox reaction, and the value of ΔE is taken at 50% of the discharge capacity. The S@GeS2-MoS2/rGO cathode exhibits a lower polarization potential (ΔE = 160 mV) than S@GeS2/rGO (ΔE = 225 mV) and S@MoS2/rGO cathodes (ΔE = 174 mV), due to the excellent electrocatalytic activity of GeS2-MoS2/rGO for LiPS conversion.
C 1 and C2 are defined as the capacities of the two discharge plateaus, respectively (Fig. 5(c)). And the ratio C2/C1 can be explained by the catalytic activity of the LiPS conversion reaction. C1 represents the amount of liquid LiPSs produced (S8 → S62− → S42−), and C2 represents the efficiency of reducing LiPSs to Li2S (S42− → Li2S2 → Li2S).55 Therefore, the higher the C2/C1, the better the catalytic ability. The slow kinetics and the shuttle effect caused by the diffusion of liquid LiPSs in the discharge process lead to a decrease in the capacity between the C1 and C2 stages.56 As shown in Fig. 5(c), the C2/C1 of S@GeS2-MoS2/rGO is 2.87, much higher than that of S@GeS2/rGO (2.41) and S@MoS2/rGO (2.57), which further confirmed the superior catalytic activity of the GeS2-MoS2/rGO heterostructure toward the LiPS redox reaction.
The cycling performance of different electrodes is tested at a current of 0.2C (Fig. S15†). Among the three electrodes, the S@GeS2-MoS2/rGO cathode shows the highest capacity and the best cycling stability, with a high capacity retention of 90.10% after 300 cycles. In contrast, the S@GeS2/rGO and S@MoS2/rGO electrodes deliver a lower capacity retention of 79.16% and 84.85%, respectively. The excellent electrochemical performance of the battery with the S@GeS2-MoS2/rGO electrode is mainly attributed to the improved electronic conductivity and rich catalytic heterointerfaces of the GeS2-MoS2/rGO heterostructure.
The cycling performance of various cathodes at 0.5C is displayed in Fig. 5(d). Among the three cathodes, the S@GeS2-MoS2/rGO electrode delivers the highest initial capacity of 1114.5 mA h g−1 at 0.5C and stabilizes at 993.8 mA h g−1 over 300 cycles. The S@GeS2-MoS2/rGO electrode also maintains the highest capacity retention at 89.17%, indicating excellent reaction kinetics and cycling stability. On the other hand, the S@GeS2/rGO and S@MoS2/rGO electrodes show discharge capacities of 714.0 and 794.8 mA h g−1 after 500 cycles with a capacity retention of 65.62% and 75.22%, respectively. The lower capacity retention of these two cathodes is mainly related to the rapid dissolution of LiPSs into the electrolyte. These results demonstrate that the S@GeS2-MoS2/rGO electrode achieves limited LiPS shuttling as well as fast sulfur reaction kinetics because of the rich catalytic heterointerfaces and advanced deposition of Li2S in the GeS2-GeS2-MoS2/rGO heterostructure.
The rate performance of the three cathodes at various current densities in the range of 0.2 to 3C is presented in Fig. 5(e). Clearly, LSBs with the GeS2-MoS2/rGO catalyst deliver the highest rate performance in different cathodes. The discharge capacities of S@GeS2-MoS2/rGO are 1173.3, 1034.6, 909.5, 827.8, 776.7, 732.2 and 700.2 mA h g−1 at 0.2, 0.5, 1, 1.5, 2, 2.5 and 3C, respectively, while the batteries using S@GeS2/rGO and S@MoS2/rGO cathodes show lower capacities. At a current density of 3C, the capacity retention of the S@GeS2-MoS2/rGO electrode is 59.7%, much higher than that of S@GeS2/rGO (56.2%) and S@MoS2/rGO (56.5%) electrodes, indicating a significantly higher sulfur utilization and improved LiPS conversion of the GeS2-MoS2/rGO heterostructure. Fig. 5(f) displays the discharge/charge profiles of the S@GeS2-MoS2/rGO electrode. The potential gap between the discharge and charge plateaus gradually increases with increasing current density. However, even at high current densities of 3C, two distinct discharge plateaus can still be obtained, which indicates the fast reaction kinetics of LiPSs in the GeS2-MoS2/rGO catalyst. Meanwhile, as shown in Fig. S16,† the corresponding discharge/charge voltage profiles of S@GeS2/rGO and S@MoS2/rGO cathodes show a larger polarization compared to S@GeS2-MoS2/rGO.
Electrochemical impedance spectroscopy (EIS) after 100 cycles further demonstrated the improved redox reactions of the GeS2-GeS2-MoS2/rGO heterostructure (Fig. 5(g)). In the equivalent circuit, the spot intersecting the horizontal axis is the interphase-contact resistance (Rs) between the electrolyte and the battery. And the semicircle diameter at low frequencies indicates the charge-transfer resistance (Rct), which is related to the charge transfer between the electrode and the electrolyte on the electrode surface.57 According to the fitting results (Table S5†), the S@GeS2-MoS2/rGO electrode (16.52 Ω) has a smaller Rct compared to the S@GeS2/rGO (33.64 Ω) and S@MoS2/rGO (24.37 Ω) electrodes. The battery using GeS2-GeS2-MoS2/rGO exhibits minimal Rct. This is attributed to the excellent electrical conductivity of the heterostructure, and the uniform precipitation and effective dissolution of Li2S, which is highly exposed to the catalytic surface after cycling.
Ultra-long cycling capabilities were tested at a high current density of 3C to explore the cycling stability of different catalysts (Fig. 5(h)). After 1000 cycles, the discharge capacity of the S@GeS2-MoS2/rGO electrode can be maintained as high as 589.9 mA h g−1, while those with S@GeS2/rGO and S@MoS2/rGO suffered a rapid capacity decay with retained capacities of 158.9 and 321.5 mA h g−1, respectively. The cycling capacity of different heterostructures at different current rates is compared in Table S6.† Most of the cathodes have excellent capacity retention at low current rates. Sulfur can be more easily embedded in the electrode material and form more stable chemical bonds at low current rates, resulting in better capacity retention. The capacity of LSBs decays more significantly at high rates. The dissolution and precipitation rate of electrode materials increase during cycling at high rates, and the migration rate of lithium ions in the electrolyte accelerates, which leads to a lower battery capacity retention. In this work, the S@GeS2-MoS2/rGO electrode delivers a high specific capacity and stable cycling performance, with a capacity retention of 68.63% and coulombic efficiency over 99.6% after 1000 cycles. The high cycling stability of the GeS2-MoS2/rGO battery suggests that the LiPS shuttling is effectively inhibited during electrochemical processes, which is attributed to the rich catalytic heterointerfaces and advanced deposition of Li2S in the heterostructure.
2.5 Low E/S ratio and LSB pouch battery performances
To evaluate the practical applications for commercial LSBs, the electrochemical performances of the S@GeS2-MoS2/rGO electrode under high sulfur loading and in lean electrolyte were explored (Fig. 6(a and b)). With a sulfur loading of 6.5 mg cm−2 and E/S = 10 μL mg−1, the S@GeS2-MoS2/rGO cathode shows an initial discharge capacity of 837.9 mA h g−1 at 0.2C and a high capacity retention of 82.98% after 100 cycles. The cycling result of S@GeS2-MoS2/rGO displays a lower specific capacity when the E/S ratio decreases to 8 μL mg−1, but it still maintains a stable cycling performance. However, when the E/S ratio decreases to 6 μL mg−1, the S@GeS2-MoS2/rGO cathode exhibits an increasing trend in the first few cycles due to insufficient wetting of the electrode surface. This is attributed to the gradual infiltration and activation of the low amount of electrolyte in the highly loaded sulfur-active material. Even so, the S@GeS2-MoS2/rGO cathode still maintains a high stable cycling performance after 100 cycles, with a capacity retention rate of 79.57%. These results suggest that the GeS2-MoS2/rGO catalyst shows superiority in achieving good sulfur electrochemistry with low electrolyte usage.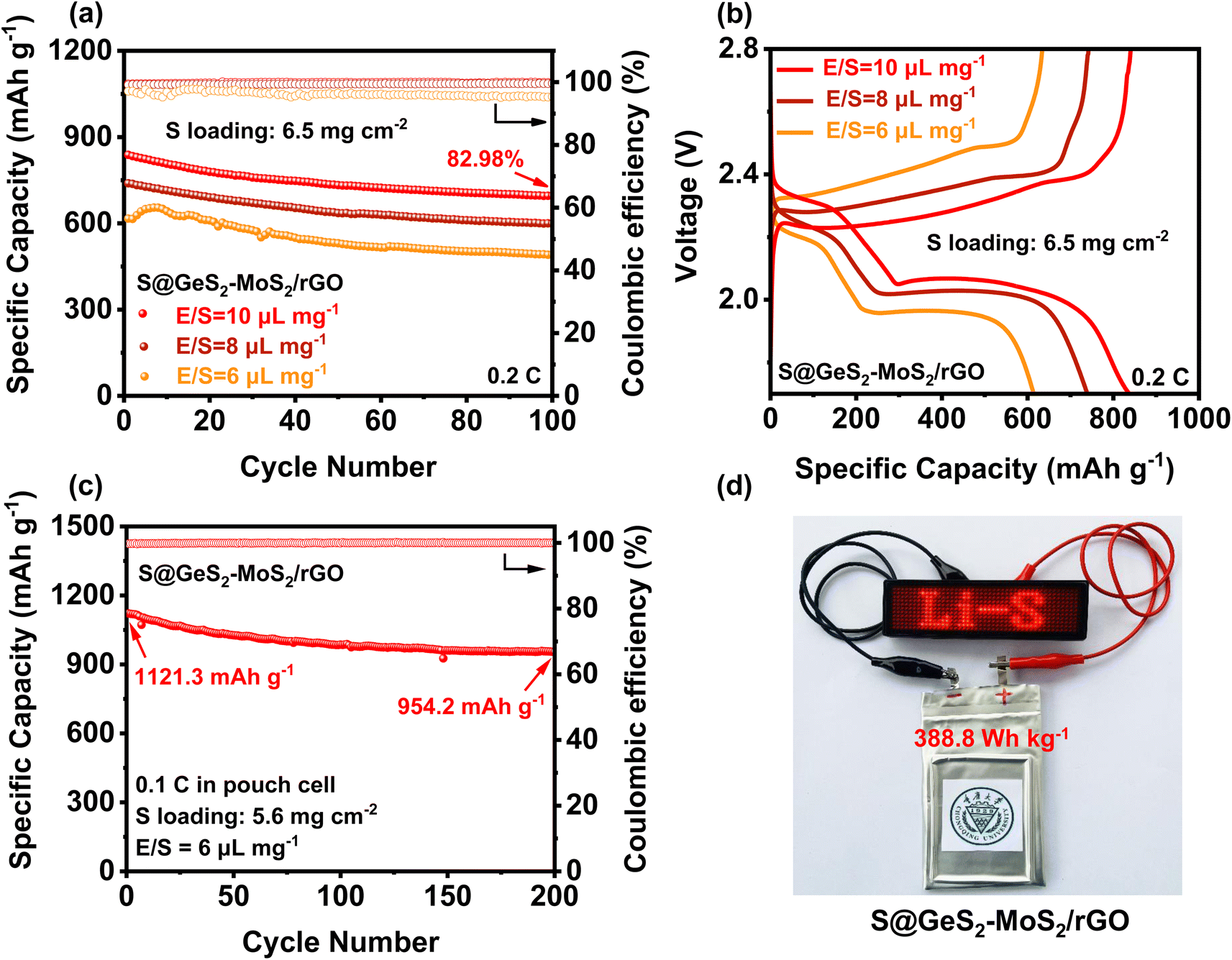 | ||
| Fig. 6 (a) Cycling performances of the S@GeS2-MoS2/rGO cathode with different E/S ratios at 0.2C. The S@GeS2-MoS2/rGO cathode obtains stable cycling and high capacity retention after 100 cycles, showing excellent sulfur utilization at low electrolyte usage. (b) Galvanostatic discharge/charge profiles of the S@GeS2-MoS2/rGO electrode with different E/S ratios at 0.2C. (c) Cycling stability of a pouch cell with the S@GeS2-MoS2/rGO electrodes at 0.1C. The S@GeS2-MoS2/rGO cathode maintains a high and stable capacity, suggesting potential electrochemical performance in practical applications. (d) Optical photograph of a pouch cell based on S@GeS2-MoS2/rGO electrodes charging a “Li–S” shaped LED. | ||
To further approach the practical applicability of LSBs, pouch LSB cells with the S@GeS2-MoS2/rGO cathode were also fabricated and investigated (Fig. 6(c, d) and S17†). The result in Fig. 6(c) reveals a stable cycling stability at 0.1C, achieving a high initial capacity of 1121.3 mA h g−1 and an excellent discharge capacity of 954.2 mA h g−1 after 200 cycles. More importantly, the pouch cell can attain a practical specific energy of 388.8 W h kg−1. Even after 200 cycles, the energy density is still over 330 Wh kg−1. Besides, the charged light-emitting diode (LED) can be easily lit (Fig. 6(d)). The excellent electrochemical performance of the pouch battery can be attributed to the outstanding synergetic effect and rich heterointerfaces of the GeS2-MoS2/rGO heterostructure. All these results suggest that the GeS2-MoS2/rGO heterostructure has the potential to help LSBs reach practical applications.
3 Conclusion
The transformation of S8 into Li2S is found to be cross-executed rather than stepwise under practical working conditions for LSBs. Advancing the Li2S deposition can reduce the accumulation of liquid polysulfides and therefore increase the stability of the LSBs. Therefore, we designed a hierarchical petal-spherical GeS2-MoS2 “butterfly” material to accelerate the conversion of LiPSs and the deposition of Li2S simultaneously. The rich catalytic heterointerfaces and the lattice-matching nature between Fdd2 GeS2 and Fmm Li2S enhance the adsorption of LiPSs and guide the Li2S growth in a 3D model, thus always providing transport channels for electrons and ions and improving the redox reaction kinetics. The above advantages realized the advanced deposition of Li2S from 20% to about 80% SOC in the discharge process, thereby achieving robust LSBs. The designed cathodes show excellent long-term cycling performance with a capacity retention of 68.63% at 3C over 1000 cycles. A high initial capacity of 837.9 mA h g−1 is achieved at a high sulfur loading of 6.5 mg cm−2 and a low E/S ratio of 10 μL mg−1. Moreover, a pouch LSB battery using S@GeS2-MoS2/rGO electrodes can attain a practical specific energy of 388.8 W h kg−1. Based on these fascinating advantages, this work provides a useful avenue for designing heterostructural catalysts for batteries and other advanced energy storage.
4 Methods
4.1 Materials
First, 2.0 g of GeO2 powder was heated in a mixed atmosphere of H2 and N2 at a volume ratio of 1:8 for 4 h at 700 °C to produce the precursor Ge powder. And graphene oxide (GO) was synthesized from natural graphite by the modified Hummers' method.58 Second, 0.1 g GO and 1.0 g Ge powder were evenly ground, dispersed in 20.0 mL of deionized water (DI), and stirred in a water bath at 50 °C to form a homogeneous solution. The obtained solution was freeze-dried for 24 h in a vacuum at −50 °C to obtain the dried Ge/GO composite. Finally, GeS2 mixed with reduced graphite oxide (rGO) samples (GeS2/rGO) was obtained by adding sulfur powder to Ge/rGO composites (molar ratio of 2:1) and calcining at 500 °C for 4 h under N2 conditions. Typically, 1.5 g (NH4)6Mo7O24·4H2O and 3.0 g CH3CSNH2 were dissolved in 100 mL DI and then 0.6 g polyvinyl pyrrolidone (PVP) was added. After the solution was mixed evenly, 0.05 g GO was added and stirred in a water bath at 50 °C for 4 h. After that, the solution was transferred to an oven and heated at 180 °C for 24 h. The black precipitate was collected by washing with DI and freeze-drying for 24 h. At last, MoS2/rGO composites were obtained by annealing in a N2 atmosphere at 500 °C for 4 h.
The synthesis of GeS2-MoS2/rGO is similar to that of MoS2/rGO. First, 1.5 g (NH4)6Mo7O24·4H2O, 3.0 g CH3CSNH2 and 0.6 g PVP were added to 100 mL of DI. Then 0.05 g GO and 0.5 g Ge were added to the mixed solution and stirred in a water bath at 50 °C for 4 h. Next, the mixture was heated in an oven at 180 °C for 24 h and then freeze-dried to obtain a dry black powder. Finally, an appropriate amount of sulfur powder was added to the black powder and calcined at 500 °C for 4 h to obtain GeS2-MoS2/rGO samples.
4.2 Electrode preparation
Three different cathodes were synthesized by mixing sulfur powder and the prepared samples (GeS2/rGO, MoS2/rGO and GeS2-MoS2/rGO) in a mass ratio of 7:3. The mixture was heated to 155 °C under a flowing N2 atmosphere for 12 h. After cooling, the powder obtained was ball-milled uniformly. The active materials (S@GeS2/rGO, S@MoS2/rGO and S@GeS2-MoS2/rGO) were mixed with conductive carbon black (Super P) and polyvinylidene fluoride (PVDF) (8:1:1 by mass) in N-methyl-2-pyrrolidone (NMP, 99.5%) solution to prepare the working electrodes. The prepared homogeneous slurry was coated on a piece of aluminum foil and vacuum dried at 60 °C overnight. The diameter of each composite cathode was 12 mm and the average surface loading was 2.0 mg cm−2. And pieces of lithium foil were used as the anodes and Celgard 2400 films were used as separators to assemble coin-type (LIR2032) batteries. The electrolyte was 1.0 M lithium bis(trifluoromethanesulfonyl) imide (LiTFSI, 99%) in a solvent mixture of 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) (1:1 by volume) with 2 wt% LiNO3. The coin batteries were assembled in an Ar-filled glove box (H2O and O2 < 1.0 ppm) and 40 μL mg−1 of electrolyte was used for each battery (the electrolyte-to-sulfur ratio was 11.8 μL mg−1).
4.3 Li2S6 adsorption test
The adsorption experiment was carried out in a glove box fitted with an Ar atmosphere. Li2S6 solution was prepared by mixing sulfur powder with Li2S (99.9%, Alfa Aesar) in a 5:1 molar ratio, which was dissolved in DOL and DME (1:1 volume ratio) solution and stirred for 12 h. After that, 20 mg of samples (GeS2/rGO, MoS2/rGO and GeS2-GeS2-MoS2/rGO) were added to the Li2S6 solution (2 mM, 4 mL) and left for several hours. After 12 h, the liquid supernatant from the bottle was extracted as a sample for ultraviolet-visible (UV-vis) testing.
4.4 Li2S nucleation test
Li2S8 catholyte was prepared by mixing sulfur powder and Li2S (7:1 molar ratio) and dissolved in tetraglyme solution and stirred overnight. Different samples (GeS2/rGO, MoS2/rGO and GeS2-MoS2/rGO) were prepared as working electrodes, and pieces of Li foil as counter electrodes. And the coin-type batteries were assembled with a Celgard 2400 membrane as the separator. 20 μL of Li2S8 catholyte was added into the cathode side, and 20 μL of the above electrolyte without Li2S8 was used as the anolyte drop to the anode side. The batteries were first galvanostatically discharged to 2.06 V at 0.012 mA, then discharged potentiostatically at 2.05 V until the current was below 0.01 mA. The nucleation rate and the specific capacity of Li2S deposition were evaluated by Faraday's law.59 After the nucleation test, the cathodes were disassembled and washed in a tetraglyme solution in an Ar-filled glove box to observe the morphology of Li2S. Theoretical equations of the current–time transients of four classic electrochemical deposition models (2D instantaneous nucleation (eqn (4)) and 2D progressive nucleation (eqn (5)) are based on Bewick, Fleischman, and Thirsk models; 3D instantaneous nucleation (eqn (6)) and 3D progressive nucleation (eqn (6)) are based on Scharifker–Hills models):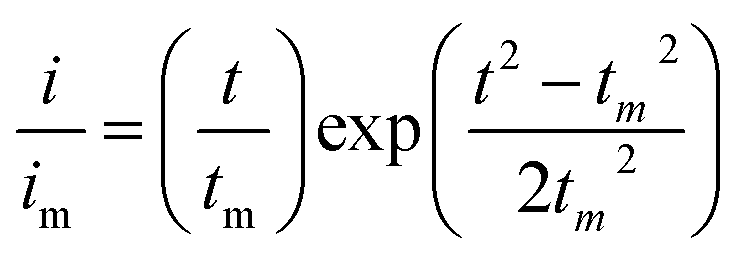 | (4) |
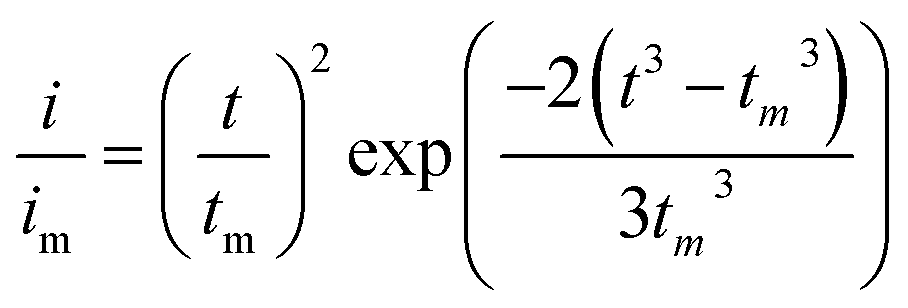 | (5) |
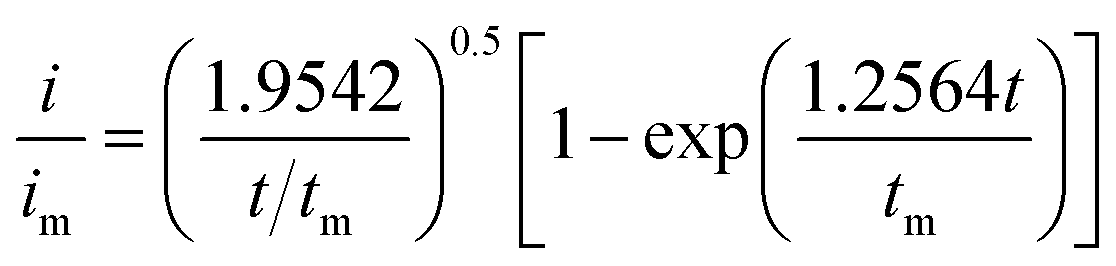 | (6) |
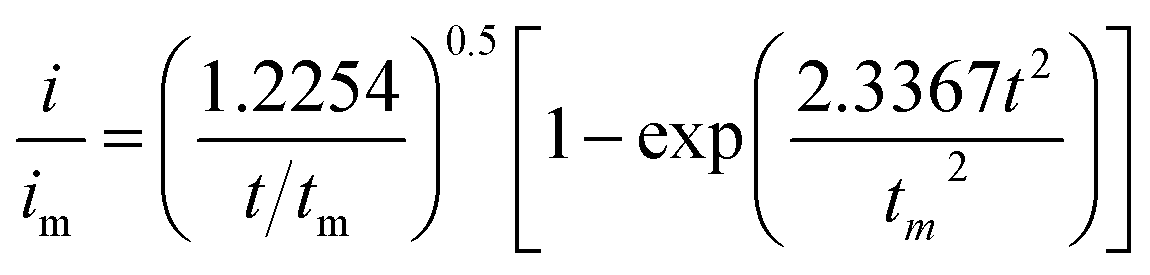 | (7) |
4.5 Li2S dissolution test
The dissolution of Li2S on the three electrodes was tested in an assembled CR2032 coin cell, where the different electrodes were used as working electrodes and lithium foil was used as the counter electrode. A 20 μL solution of 2.0 mol L−1 Li2S8 and 1.0 mol L−1 LiTFSI in tetraglyme was applied as catholyte, and 20 μL of control electrolyte without Li2S8 was used as anolyte. The above assembled batteries were first galvanostatically discharged to 1.80 V at 0.1 mA and then followed by 1.80 V at 0.01 mA, so that LiPSs were completely converted to solid Li2S. Then, the batteries were charged potentiostatically at 2.40 V until the current was below 0.01 mA for the oxidization process from solid Li2S to liquid LiPSs.4.6 Symmetric battery test
Symmetric batteries were assembled in the same way as LSBs. Two electrodes with the same active materials (GeS2/rGO, MoS2/rGO, or GeS2-GeS2-MoS2/rGO) served as the working and counter electrodes. 40 μL of 0.5 M Li2S6 solution prepared by the adsorption test was used as the electrolyte. They were assembled into a typical CR2032 coin cell with a polypropylene (PP) membrane as the separator. For comparison, symmetric batteries with GeS2-MoS2/rGO electrodes and the above solution without Li2S6 were also assembled and tested. CV curves were obtained by using an electrochemical workstation in a voltage window of −1.0 to 1.0 V at a scan rate of 20 mV s−1.4.7 Theoretical calculations
Density functional theory (DFT) calculations were performed using the projector-augmented wave pseudopotentials (PAW) in the Vienna ab initio simulation package (VASP) software.60,61 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) function was employed to handle the exchange-correlation energy. The weak intermolecular interactions between atoms are finely described by the DFT-D3 correction method in Grimme's scheme.62 The plane-wave basis set with a cutoff energy of 500 eV was set. Heterointerfaces were built at a relatively low crystal parameter mismatch (less than 5.0%).63 In the vertical direction, a 20 Å vacuum layer was established for all surfaces regardless of the periodic layer effect. A k-point grid of 1 × 1 × 1 determined by the Gamma-centered Monkhorst–Pack method in the Brillouin zone was used for GeS2 (311), MoS2 (002), and the mixed heterostructure, respectively. The geometry optimization was considered convergent when the force change was below 0.02 eV Å−1. The electron energy was considered self-consistent when the energy change was less than 10−5 eV. The U correction was adopted for the Mo atom in this system. The adsorption energies (Ead) of LiPSs were calculated by using the following equation:64,65| Ead = Etotal − ELi2Sx − Esub | (8) |
4.8 Pouch battery assembly and measurements
Both the S@GeS2-MoS2/rGO cathode and lithium anode were cut into pieces (6 × 4 cm). The sulfur loading of the cathode in the pouch cell was 5.6 mg cm−2. The thickness of the lithium belt anode was 0.5 mm. And the E/S ratio was 6 μL mg−1. The separator (Celgard 2400) was sandwiched between the tailored S@GeS2-MoS2/rGO cathode and lithium anode. The electrochemical performances of pouch batteries are tested under the same conditions as those of coin cells. The energy density of the pouch battery is calculated using eqn (9):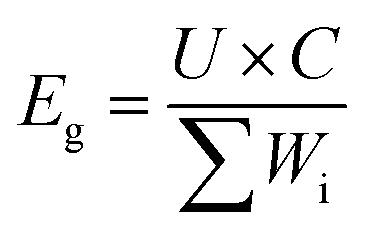 | (9) |
4.9 Material characterization
X-ray diffraction (XRD) patterns were recorded using an X-ray diffractometer (Rigaku D/max 2200 pc) with Cu (Kα) radiation (λ = 1.54 Å) at 40 kV and 40 mA. For in situ XRD analysis, the in situ XRD cell was cycled at 1.7–2.8 V at 0.02C with a Neware battery test system. Thermogravimetric (TG) analysis was carried out by using a thermogravimetric analyzer (SHIMADZU, DTG-60AH) to obtain S loadings at a heating rate of 10 °C min−1 over a temperature range of 30 to 500 °C. Electronic conductivity of different samples was investigated using a CHI1140C workstation with a constant voltage of 1.0 V. All samples were pressed into compact discs and loaded into the cuvette for constant voltage testing, ultimately obtaining the parameters of the current over a period of time. XANES and EXAFS data reduction and analysis were processed using Athena software. The nitrogen adsorption isotherms were obtained by the Brunauer–Emmett–Teller method (BET, BSD-PM1/2, BSDINSTRU MENT). An ICP-OES (Agilent 5110) tester is used to analyze the Mo and Ge amounts in the GeS2-MoS2/rGO heterostructure. Atomic force microscopy (AFM) (Dimension Icon, Bruker, USA) was used to characterize the particle size and thickness distribution of GeS2-MoS2/rGO. The ultraviolet-visible (UV-vis) absorption spectra were measured in the range of 350–700 nm on a PerkinElmer Lambda 750 spectrophotometer. X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Scientific instrument (ESCALAB 250XI) with Al (Kα) radiation. Scanning electron microscopy (SEM) images were collected with a field emission scanning electron microscope (JEOL JSM-7800F, 5/10 kV). Transmission electron microscopy (TEM), energy dispersive X-ray (EDX) and high resolution transmission electron microscopy (HRTEM) images were observed on a Tecnai G2F20 TWIN and JEM-2100F.4.10 Electrochemical characterization
The galvanostatic discharge/charge was tested on a LAND battery tester (1.7–2.8 V). Cyclic voltammetry (CV) measurements were conducted and electrochemical impedance spectroscopy (EIS) data were obtained using a CHI 660D workstation (scan rate of 0.1 mV s−1) and a Princeton 1260A impedance analyzer (amplitude of 10 mV and frequency range of 10−2 to 105 Hz), respectively. All tests were conducted at room temperature. The capacity was calculated based on the mass of sulfur.Data availability
Data are available from the authors upon reasonable request.Author contributions
Xun Jiao: writing the original draft, methodology, investigation. Xiaoxia Tang: methodology, investigation. Jinrui Li: methodology. Yujiao Xiang: investigation. Cunpu Li: data curation, formal analysis, funding acquisition, project administration, writing–review & editing, conceptualization, supervision. Cheng Tong: data curation, formal analysis, funding acquisition, project administration, writing–review & editing, conceptualization, supervision. Minhua Shao: writing–review & editing. Zidong Wei: data curation, project administration, formal analysis, writing–review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 22075033 and U21A20312), the Fundamental Research Funds for the Central Universities (2023CDJXY-045), and the Postdoctoral Fellowship Program of CPSF (GZB20230909).Notes and references
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Z. Ye, Y. Jiang, L. Li, F. Wu and R. Chen, Adv. Mater., 2022, 34, 2109552 CrossRef CAS PubMed.
- M. Zhao, B.-Q. Li, X.-Q. Zhang, J.-Q. Huang and Q. Zhang, ACS Cent. Sci., 2020, 6, 1095–1104 CrossRef CAS PubMed.
- Y. Liu, M. Zhao, L.-P. Hou, Z. Li, C.-X. Bi, Z.-X. Chen, Q. Cheng, X.-Q. Zhang, B.-Q. Li and S. Kaskel, et al. , Angew. Chem., Int. Ed., 2023, 62, e202303363 CrossRef CAS PubMed.
- K. Wu, H. He, Q. Xue, C. Zhang, X. Qi, A. Cabot and X. Hu, Chem. Eng. J., 2023, 466, 142988 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Y. Song, W. Cai, L. Kong, J. Cai, Q. Zhang and J. Sun, Adv. Energy Mater., 2020, 10, 1901075 CrossRef CAS.
- Y. Zhong, Q. Wang, S.-M. Bak, S. Hwang, Y. Du and H. Wang, J. Am. Chem. Soc., 2023, 145, 7390–7396 CrossRef CAS PubMed.
- X. Zhang, H. Xie, C.-S. Kim, K. Zaghib, A. Mauger and C. Julien, Mater. Sci. Eng., R, 2017, 121, 1–29 CrossRef.
- T. Tang and Y. Hou, Small Methods, 2020, 4, 1900001 CrossRef CAS.
- M. Zhang, W. Chen, L. Xue, Y. Jiao, T. Lei, J. Chu, J. Huang, C. Gong, C. Yan and Y. Yan, et al. , Adv. Energy Mater., 2020, 10, 1903008 CrossRef CAS.
- Z. Zhu, Y. Zeng, Z. Pei, D. Luan, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2023, 62, e202305828 CrossRef PubMed.
- S. Zhou, J. Shi, S. Liu, G. Li, F. Pei, Y. Chen, J. Deng, Q. Zheng, J. Li, C. Zhao, I. Hwang, C.-J. Sun, Y. Liu, Y. Deng, L. Huang, Y. Q. G.-L. Xu, J.-F. Chen, K. Amine, S.-G. Sun and H.-G. Liao, Nature, 2023, 621, 75–81 CrossRef CAS PubMed.
- W. Hua, T. Shang, H. Li, Y. Sun, Y. Guo, J. Xia, C. Geng, Z. Hu, L. Peng, Z. Han, Z. Chen, L. Wei and W. Ying, Nat. Catal., 2023, 6, 174–184 CrossRef CAS.
- R. Wang, J. Qin, F. Pei, Z. Li, P. Xiao, Y. Huang, L. Yuan and D. Wang, Adv. Funct. Mater., 2023, 33, 2305991 CrossRef CAS.
- M. Wang, Z. Bai, T. Yang, C. Nie, X. Xu, Y. Wang, J. Yang, S. Dou and N. Wang, Adv. Energy Mater., 2022, 12, 2201585 CrossRef CAS.
- L. Huang, S. Shen, Y. Zhong, Y. Zhang, L. Zhang, X. Wang, X. Xia, X. Tong, J. Zhou and J. Tu, Adv. Mater., 2022, 34, 2107415 CrossRef CAS PubMed.
- M. Li, J. Lu, J. Shi, S.-B. Son, D. Luo, I. Bloom, Z. Chen and K. Amine, J. Am. Chem. Soc., 2021, 143, 2185–2189 CrossRef CAS PubMed.
- H. Ye, M. Li, T. Liu, Y. Li and J. Lu, ACS Energy Lett., 2020, 5, 2234–2245 CrossRef CAS.
- T. Wu, J. Qi, M. Xu, D. Zhou and Z. Xiao, ACS Nano, 2020, 14, 15011–15022 CrossRef CAS PubMed.
- A. Vizintin, L. Chabanne, E. Tchernychova, I. Arčon, L. Stievano, G. Aquilanti, M. Antonietti, T.-P. Fellinger and R. Dominko, J. Power Sources, 2017, 344, 208–217 CrossRef CAS.
- Z.-X. Chen, Q. Cheng, X.-Y. Li, Z. Li, Y.-W. Song, F. Sun, M. Zhao, X.-Q. Zhang, B.-Q. Li and J.-Q. Huang, J. Am. Chem. Soc., 2023, 145, 16449–16457 CrossRef CAS PubMed.
- L. C. Gerber, P. D. Frischmann, F. Y. Fan, S. E. Doris, X. Qu, A. M. Scheuermann, K. Persson, Y.-M. Chiang and B. A. Helms, Nano Lett., 2016, 16, 549–554 CrossRef CAS.
- Y. Cao, S. Gu, J. Han, Q.-H. Yang and W. Lv, Chem. Rec., 2022, 22, e202200124 CrossRef CAS.
- R. Wang, C. Luo, T. Wang, G. Zhou, Y. Deng, Y. He, Q. Zhang, F. Kang, W. Lv and Q.-H. Yang, Adv. Mater., 2020, 32, 2000315 CrossRef CAS PubMed.
- Y. Yao, H. Wang, H. Yang, S. Zeng, R. Xu, F. Liu, P. Shi, Y. Feng, K. Wang and W. Yang, et al. , Adv. Mater., 2020, 32, 1905658 CrossRef CAS PubMed.
- R. Xu, H. Tang, Y. Zhou, F. Wang, H. Wang, M. Shao, C. Li and Z. Wei, Chem. Sci., 2022, 13, 6224–6232 RSC.
- F. Feng, S. Han, Q. Lu and Q. Yun, Energy Mater. Devices, 2023, 1, 9370008 CrossRef.
- Y. Pan, L. Gong, X. Cheng, Y. Zhou, Y. Fu, J. Feng, H. Ahmed and H. Zhang, ACS Nano, 2020, 14, 5917–5925 CrossRef CAS.
- T. Xiong, X. Yao, D. Adekoya, H. Yang and M.-S. Balogun, J. Mater. Sci. Technol., 2023, 145, 14–24 CrossRef CAS.
- J. Liu and J. Zhang, Chem. Rev., 2020, 120, 2123–2170 CrossRef CAS PubMed.
- Z. Sun, X. Cao, M. Tian, K. Zeng, Y. Jiang, M. H. Rummeli, P. Strasser and R. Yang, Adv. Energy Mater., 2021, 11, 2100110 CrossRef CAS.
- C. Ye, Y. Jiao, H. Jin, A. D. Slattery, K. Davey, H. Wang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 16703–16707 CrossRef CAS PubMed.
- Z. A. Ghazi, X. He, A. M. Khattak, N. A. Khan, B. Liang, A. Iqbal, J. Wang, H. Sin, L. Li and Z. Tang, Adv. Mater., 2017, 29, 1606817 CrossRef.
- H. Chen, C. Keiser, S. Du, H.-J. Gao, P. Sutter and E. Sutter, Phys. Chem. Chem. Phys., 2017, 19, 32473–32480 RSC.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- H. Shan, J. Qin, Y. Ding, H. M. K. Sari, X. Song, W. Liu, Y. Hao, J. Wang, C. Xie, J. Zhang and X. Li, Adv. Mater., 2021, 33, 2102471 CrossRef CAS PubMed.
- Y. Yan, H. Li, C. Cheng, T. Yan, W. Gao, J. Mao, K. Dai and L. Zhang, J. Energy Chem., 2021, 61, 336–346 CrossRef CAS.
- Z. Ye, Y. Jiang, T. Yang, L. Li, F. Wu and R. Chen, Advanced Science, 2022, 9, 2103456 CrossRef CAS PubMed.
- L. Luo, J. Li, H. Yaghoobnejad Asl and A. Manthiram, ACS Energy Lett., 2020, 5, 1177–1185 CrossRef CAS.
- J.-L. Yang, D.-Q. Cai, X.-G. Hao, L. Huang, Q. Lin, X.-T. Zeng, S.-X. Zhao and W. Lv, ACS Nano, 2021, 15, 11491–11500 CrossRef CAS PubMed.
- Y. Ren, A. Bhargav, W. Shin, H. Sul and A. Manthiram, Angew. Chem., Int. Ed., 2022, 61, e202207907 CrossRef CAS.
- B. Yan, X. Li, Z. Bai, X. Song, D. Xiong, M. Zhao, D. Li and S. Lu, J. Power Sources, 2017, 338, 34–48 CrossRef CAS.
- A. Fu, C. Wang, F. Pei, J. Cui, X. Fang and N. Zheng, Small, 2019, 15, 1804786 CrossRef.
- W. Yao, W. Zheng, J. Xu, C. Tian, K. Han, W. Sun and S. Xiao, ACS Nano, 2021, 15, 7114–7130 CrossRef CAS PubMed.
- Z. Li, J. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2015, 54, 12886–12890 CrossRef CAS PubMed.
- H. Yao, G. Zheng, P.-C. Hsu, D. Kong, J. J. Cha, W. Li, Z. W. Seh, M. T. McDowell, K. Yan, Z. Liang, V. K. Narasimhan and Y. Cui, Nat. Commun., 2014, 5, 3943 CrossRef CAS PubMed.
- W.-G. Lim, S. Kim, C. Jo and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 18746–18757 CrossRef CAS PubMed.
- Z. Li, Y. Zhou, Y. Wang and Y.-C. Lu, Adv. Energy Mater., 2019, 9, 1802207 CrossRef.
- F. Y. Fan, W. C. Carter and Y.-M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS PubMed.
- L. Kong, J.-X. Chen, H.-J. Peng, J.-Q. Huang, W. Zhu, Q. Jin, B.-Q. Li, X.-T. Zhang and Q. Zhang, Energy Environ. Sci., 2019, 12, 2976–2982 RSC.
- D.-Q. Cai, J.-L. Yang, T. Liu, S.-X. Zhao and G. Cao, Nano Energy, 2021, 89, 106452 CrossRef CAS.
- M. Wang, Y. Song, Z. Sun, Y. Shao, C. Wei, Z. Xia, Z. Tian, Z. Liu and J. Sun, ACS Nano, 2019, 13, 13235–13243 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- J. Xu, W. Zhang, H. Fan, F. Cheng, D. Su and G. Wang, Nano Energy, 2018, 51, 73–82 CrossRef CAS.
- W. Li, Z. Gong, X. Yan, D. Wang, J. Liu, X. Guo, Z. Zhang and G. Li, J. Mater. Chem. A, 2020, 8, 433–442 RSC.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- B. Yan, Y. Li, L. Gao, H. Tao, L. Zhang, S. Zhong, X. Li and X. Yang, Small, 2022, 18, 2107727 CrossRef CAS PubMed.
- Y. Han, Y. Dai, J. Wang, D. Shu and B. Sun, Appl. Surf. Sci., 2011, 257, 7831–7836 CrossRef CAS.
- Y. Wang, Z.-K. Liu, L.-Q. Chen and C. Wolverton, Acta Mater., 2007, 55, 5934–5947 CrossRef CAS.
- S. Hussain, X. Yang, M. K. Aslam, A. Shaheen, M. S. Javed, N. Aslam, B. Aslam, G. Liu and G. Qiao, Chem. Eng. J., 2020, 391, 123595 CrossRef CAS.
- Y. Wang, R. Chen, X. Luo, Q. Liang, Y. Wang and Q. Xie, ACS Appl. Nano Mater., 2022, 5, 8371–8381 CrossRef CAS.
- Y. Han, Y. Dai, D. Shu, J. Wang and B. Sun, Appl. Phys. Lett., 2006, 89, 144107 CrossRef.
- D. Li, R. Li, D. Zhou, F. Zeng, W. Yan and S. Cai, Appl. Surf. Sci., 2022, 586, 152765 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02420f |
| This journal is © The Royal Society of Chemistry 2024 |