
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermolecular C–C/C–N σ-bond metathesis enabled by visible light†
Rujuan
Li‡
a,
Renqin
Zhan‡
a,
Yatao
Lang
a,
Chao-Jun
Li
 *b and
Huiying
Zeng
*a
*b and
Huiying
Zeng
*a
aThe State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 Tianshui Road, Lanzhou, 730000, P. R. China. E-mail: zenghy@lzu.edu.cn
bDepartment of Chemistry, and FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. West, Montreal, QC H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 12th July 2024
Abstract
Transition-metal-catalyzed double/triple bond metathesis reactions have been well-established due to the ability of transition-metal catalysts to readily interact with π bonds, facilitating the progression of the entire reaction. However, activating σ-bonds to induce σ-bond metathesis is more challenging due to the absence of π bonds and the high bond energy of σ bonds. In this study, we present a novel photo-induced approach that does not rely on transition metals or photosensitizers to drive C–C and C–N σ-bond metathesis reactions. This method enables the cross-coupling of tertiary amines with α-diketones via C–C and C–N single bonds cleavage and recombination. Notably, our protocol exhibits good compatibility with various functional groups in the absence of transition metals and external photosensitizers, resulting in the formation of aryl alkyl ketones and aromatic amides in good to high yields. To gain insights into the mechanism of this pathway, we conducted controlled experiments, intermediate trapping experiments, and DFT (Density Functional Theory) calculations. This comprehensive approach allowed us to elucidate the detailed mechanism underlying this transformative reaction.
Introduction
The recombination of chemical bonds is the most basic research content in the field of organic synthesis. The precise manipulation of chemical bond recombination processes under mild conditions remains a pivotal scientific challenge within this field. It has been consistently posed as one of the most formidable and intriguing subjects for organic synthesis researchers. Metathesis reactions, due to their exceptional atom economy, have garnered substantial attention from the organic chemistry community.1Alkene and alkyne metathesis reactions are among the most common forms of metathesis reactions.2 Transition-metal-catalyzed olefin metathesis and carbonyl–olefin metathesis reactions have been extensively developed to produce the corresponding metathesis products, enabling the reliable, precise, and efficient synthesis of various important chemical compounds (Scheme 1a).3 Similarly, transition-metal-catalyzed alkyne metathesis reactions have also been well-established to yield their respective metathesis products (Scheme 1b).4 Since both double and triple bonds contain π bonds, and transition metal catalysts readily interact with π bonds within these bond types, they facilitate the overall reaction process. Conversely, due to the high bond energy associated with σ bonds and the absence of π bonds, it is more challenging for transition metals to insert into unactivated σ bonds. Therefore, there are enormous challenges of transititon-metal catalyzed σ-bond metathesis. In 1990, Vollhardt group reported a nickel-catalyzed ring-opening dimerization of biphenylenes to generate tetrabenzocyclooctatetraene driven by the strain release of four-membered-rings.5 Up to the present, only a few examples of intramolecular ring-closing metathesis of C–P/C–P,6 C–C/Si–Si,7 C–O/C–O8 form five-membered-ring products which are thermodynamically favorable via organo-transition-metal intermediates. More recently, intermolecular metathesis of C–C/C–I,9 C–C/C–S,10 or C–C/C–C11 catalyzed by transition metals via organometallic intermediates has also been developed (Scheme 1c). To the best of our knowledge, C–C/C–N σ-bond metathesis reaction, especially photo-induced one, has not been reported. Towards our recent endeavor to develop photo-induced catalyst-free reactions,12 herein, we report a visible-light-induced transition-metal- and external-photosensitizer-free C–C/C–N σ-bond metathesis reaction between amines and α-diketones to generate amides and ketones under mild conditions (Scheme 1d).
 | ||
| Scheme 1 Transition-metal catalyzed metathesis via organometallic complexes vs. photo-induced transition-metal-free metathesis via radical intermediates. (a) Transition-metal catalyzed alkene metathesis via organometallic. (b) Transition-metal catalyzed alkyne metathesis via organometallic. (c) Transition-metal catalyzed σ-bond metathesis via organometallic. (d) Visible-light induced catalyst-free σ-bond metathesis via radical intermediates. | ||
Results and discussion
Our investigation commenced with benzil (1a) and triethylamine (2a) as standard substrates, mixed in acetonitrile (1.0 mL) under an argon atmosphere, and irradiated with 405 nm visible light, resulting in the amide product 3aa in 55% yield and ketone 4aa in 7% yield (Table 1, entry 1). Encouraged by this outcome, we explored different solvents (entries 2–7). We found that in polar solvents DMSO or DMF, trace desired product 4aa was observed (entries 2–3). The highest yield of the ketone product was achieved in toluene solvent, along with a moderately generated yield of the amide (entry 6). Subsequently, adjusting the ratio of 1a and 2a to 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 resulted in a higher yield of amides (55%) compared to other ratios (entries 8–10). Increasing the volume of toluene to 1.5 mL improved the yield of 4aa to 51% (entry 11). Lowering the temperature to 15 °C also enhanced the yield (entry 12). Further lowering the temperature did not increase the yield (please see ESI,† part II). Importantly, addition of 75 μL H2O to the reaction system gave the highest yields for both 3aa and 4aa (entry 13). A control experiment conducted in the absence of light showed no product detection (entry 14). The detailed optimization of temperature, the volume of water and the wavelength of light were shown in ESI.†
:1 resulted in a higher yield of amides (55%) compared to other ratios (entries 8–10). Increasing the volume of toluene to 1.5 mL improved the yield of 4aa to 51% (entry 11). Lowering the temperature to 15 °C also enhanced the yield (entry 12). Further lowering the temperature did not increase the yield (please see ESI,† part II). Importantly, addition of 75 μL H2O to the reaction system gave the highest yields for both 3aa and 4aa (entry 13). A control experiment conducted in the absence of light showed no product detection (entry 14). The detailed optimization of temperature, the volume of water and the wavelength of light were shown in ESI.†
|
|
|||
|---|---|---|---|
| Entry | Solvent | Yieldb (3aa)/% | Yieldb (4aa)/% |
| a General conditions: 1a (0.1 mmol), 2a (0.1 mmol) in solvent (1.0 mL) were irradiated with 405 nm LED (3 W × 2) for 24 h under an argon atmosphere at 25 °C. b Yields were determined by 1H NMR using dibromomethane as the internal standard; isolated yields were shown in brackets. c 1a (0.12 mmol). d 1a (0.15 mmol). e 1a (0.17 mmol). f Toluene (1.5 mL). g 15 °C. h 75 μL H2O was added as the co-solvent. i In the dark. | |||
| 1 | CH3CN | 55 | 7 |
| 2 | DMF | 46 | Trace |
| 3 | DMSO | 22 | Trace |
| 4 | THF | 46 | 20 |
| 5 | CH3OH | 34 | 36 |
| 6 | Toluene | 48 | 40 |
| 7 | DCM | 28 | 11 |
| 8c | Toluene | 45 | 40 |
| 9d | Toluene | 55 | 40 |
| 10e | Toluene | 50 | 32 |
| 11df | Toluene | 55 | 51 |
| 12dfg | Toluene | 58 | 55 |
| 13dfgh | Toluene | 78 (67) | 66 (58) |
| 14dfghi | Toluene | n.p. | n.p. |

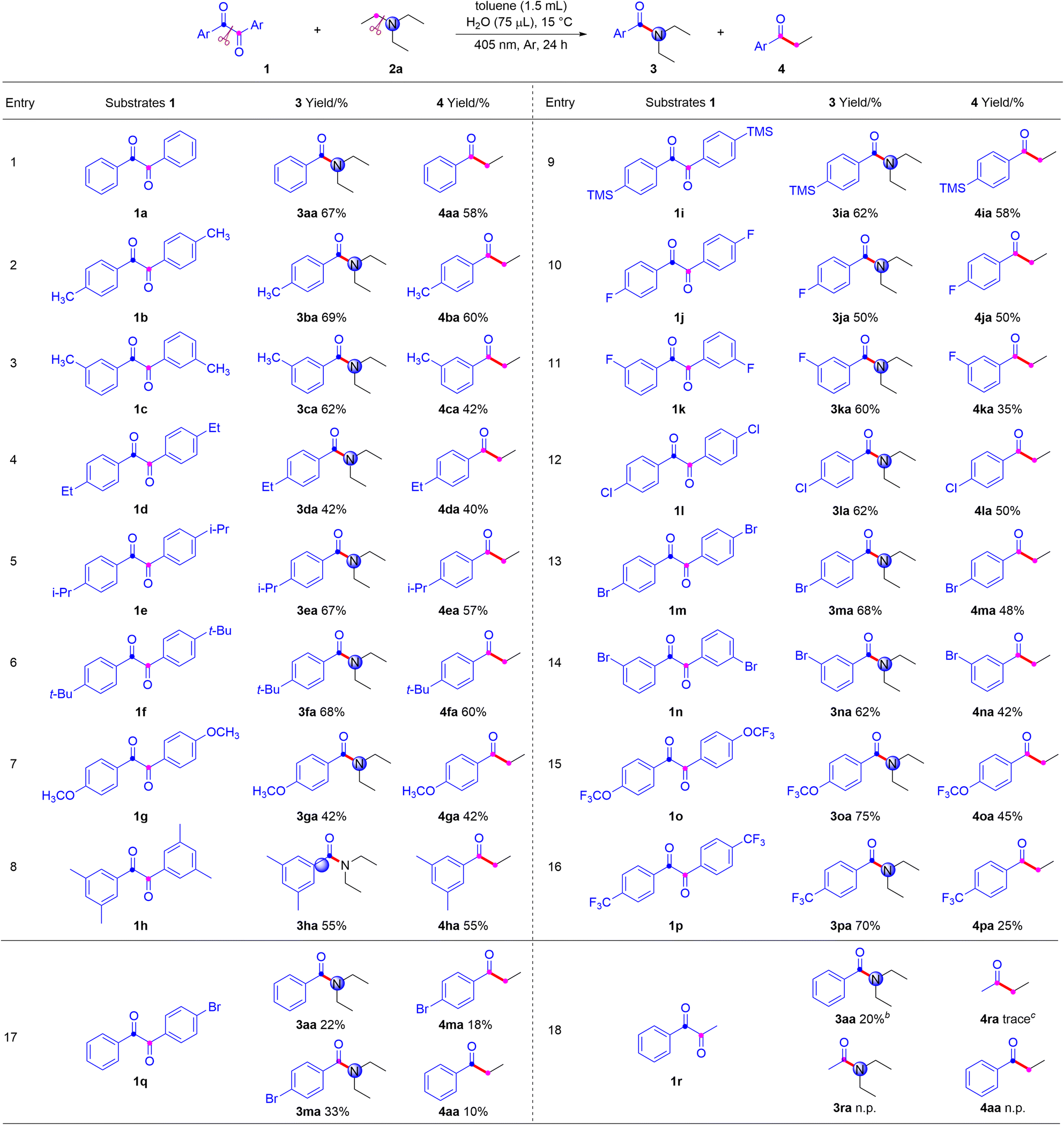
With the optimized reaction conditions in hand, the scope of α-diketones was explored with triethylamine at 15 °C under an argon atmosphere in toluene (1.5 mL) and water (75 μL) under 405 nm light irradiation. As shown in Table 2, when weak electron-donating groups were attached on the aryl ring of α-diketones, both the corresponding amides 3aa–3fa and ketones 4aa–4fa were obtained in good to high yields. Notably, the good yields were achieved regardless of the methyl substituent was located at the meta- or para-position of the aromatic ring. The substrates bearing a strong electron-donating group, such as methoxyl, were smoothly converted to the desired products 3ga and 4ga in moderate yields. Multiple substituted substrate also reacted smoothly, producing the corresponding amide 3ha and ketone 4ha in good yield. It is worthy noting that the reaction demonstrated good tolerance for the trimethylsilyl group, resulting in good yields of corresponding amide 3ia and ketone 4ia. Furthermore, substrates containing photosensitive C–X bonds, including fluoro, chloro, and bromo, remained intact and were successfully converted into amides (3ja–3na) and ketones (4ja–4na) in moderate to good yields under light irradiation. The substrates containing trifluoromethoxy group and strong electron-withdrawing group, such as trifluoromethyl, were efficiently transformed into the desired products 3oa–3pa and 4oa–4pa. In addition, we explored unsymmetric diketones and found that diaryldiketone (1q) could yield two amide products and two ketone products, whereas alkylaryl diketone (1r) only gave one amide product and one ketone product.
| a Reaction conditions: 1 (0.15 mmol), 2a (0.1 mmol), H2O (75 μL), and toluene (1.5 mL) were irradiated by 405 nm LED (3 W × 2) for 24 h under argon atmosphere. Isolated yields were given. b Yields were determined by 1H NMR using dibromomethane as the internal standard. c Yields were determined by GC-MS. |
|---|
|
Additionally, the scope of amines was also investigated under standard conditions. As shown in Table 3, irrespective of the increase in the carbon chain length of tertiary alkyl amines, the corresponding amides were consistently obtained in good to high yields. However, the yields of ketones were lower in comparison. This observation may be attributed to Norrish II-type degradation of ketones under light irradiation, leading to a reduced ketone yield. To test this proposal, the long chain aryl alkyl ketone 4ac was individually investigated under standard reaction conditions for 24 hours, resulting in the detection of a 20% yield of the Norrish II-type degradation product aryl methyl ketone, with 70% of the starting material 4ac remaining (please see ESI, Scheme S2† for details). This result helps to explain the lower yield of ketones obtained. When N-methylpiperidine (2g) was used as a substrate, only amide 3ag and ketone 4ag were obtained, with no detection of piperidine ring-opening products. Moreover, when N-methyl diethylamine (2h) and N-benzyl diethylamine (2i) were employed as starting materials, two types of C–N bonds were cleaved, generating two amides (3ba and 3ah, 3ba and 3ai respectively) and their corresponding ketones in moderate yields.

In order to gain insight into the reaction mechanism, radical trapping experiments were conducted using 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) as radical quencher under standard conditions (Scheme 2a). The yields of metathesis product 3ba and 4ba were significantly decreased upon the addition of TEMPO. HRMS analysis revealed the detection of TEMPO adduct 5 and intermediate 6 (Scheme 2a, please see ESI, Fig. S1† for details), indicating that the benzoin radical and compound 6 might be intermediates in the reaction. To further support this observation, analogues of compound 6, compound 7 was synthesized (please see ESI, Scheme S3† for details) and explored under standard conditions, resulting in the formation of product 3aa in 46% yield and 8 in 57% yield (Scheme 2b). This result further demonstrated that α-hydro-β-diethylamino ketone 7 was the key intermediate of the reaction.
 | ||
| Scheme 2 Mechanistic experiments. (a) Radical capture experiment. (b) Intermediates experiment. | ||
Furthermore, density functional theory (DFT) calculations were conducted to elucidate the metathesis reaction mechanism, as depicted in Fig. 1. Upon absorption of light, benzil (R1) undergoes a transition to the S1 state (R1-S1), subsequently reaching the T1 state (R1-T1) through intersystem crossing. Spin population analysis reveals that the single electron in the triplet state of benzil predominantly resides on the oxygen atom of R1-T1 (Fig. 1b). The triplet state benzil readily abstracts an alpha-hydrogen atom from triethylamine, characterized by a low energy barrier of 0.6 kcal mol−1. The resulting free radical intermediate IM1 returns to the ground state through intersystem crossing and then undergoes free radical coupling to form a C–C bond, yielding intermediate IM2. Furthermore, IM2 can also be photoexcited to reach the S1 state (IM2-S1) (UV-vis absorption for the IM2 analogues, intermediate 7, has significant absorbance persisting above 400 nm at system concentrations, please see the Fig. S2 in ESI†), followed by intersystem crossing to the T1 state (IM2-T1). The triplet can further attack the nitrogen atom viaTS3, leading to the formation of the amide product P1 and the triplet intermediate IM4via C–N bond cleavage. IM4 is then converted into the stable ketone product P2 through intersystem crossing and tautomerization of enol to ketone. Notably, computational results illustrate that water significantly accelerates the transformation of the enol intermediate IM5 into the ketone product P2. With two water molecules present, the reaction barrier is reduced from 56.3 kcal mol−1 to 22.3 kcal mol−1.
 | ||
| Fig. 1 DFT calculation. (a) Potential energy surfaces illustrating metathesis reaction of benzil (R1) and triethylamine (R2), with relative free energies given. Spin multiplicities were marked at the upper left corner of each structure's name. (b) The Mulliken atomic spin densities on the corresponding atoms of R1-T1 were given (isovalue: 0.02). | ||
Based on the aforementioned experimental results and DFT calculations, a plausible mechanism is proposed in Scheme 3. The benzil (1a) is irradiated by light, leading to the formation of the triple excited-state T1 with double radical properties, which abstracts an alpha-hydrogen atom from triethylamine to form radical A and the corresponding carbon radical of amines B. Subsequently, the cross-coupling of B and A facilitates the formation of a new C–C bond, generating intermediate C. Then, intermediate C is once again irradiated by visible-light, causing the cleavage of the C–C and C–N bonds via an ‘olefin-metathesis- like’ four-membered ring transition state D. This process forms new C–C and C–N bonds, ultimately generating amide product 3aa and enol E, which quickly tautomerizes to form ketone product 4aa.
 | ||
| Scheme 3 Possible mechanism. | ||
Conclusions
In conclusion, we have introduced a novel intermolecular C–C and C–N σ-bond metathesis reaction between amine and α-diketones without the need for any transition-metal or external photosensitizer reagents. To the best of our knowledge, in comparison to the limited existing examples of σ-bond metathesis, this work shows the first examples of photo-induced metathesis. The method demonstrated excellent tolerance towards various functional groups, including methoxy, alkyl, various halogens, trimethylsilyl, and trifluoromethyl. This method provides a potentially more environmentally friendly route for synthesizing aryl alkyl ketones and aromatic amides. Furthermore, this strategy offers a novel approach for addressing challenging σ-bond metathesis reactions.Data availability
All experimental procedures, mechanistic investigations, characterisation data, NMR spectra and computational data can be found in the article or in the ESI.†Author contributions
R. Zhan and R. Li conducted the experiments and and analyzed the data. Y. Lang performed DFT calculations. R. Li, R. Zhan and Y. Lang co-write the manuscript. H. Zeng and C.-J. Li conducted general guidance, project directing, and manuscript revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (21971093), the Fundamental Research Funds for the Central Universities (lzujbky-2021-sp53), the International Joint Research Centre for Green Catalysis and Synthesis (2016B01017), The Science and Technology Major Program of Gansu Province of China (22ZD6FA006) and the 111 project for support of our research. We also thank the Canada Research Chair (Tier I) foundation, the E.B. Eddy Endowment Fund, the CFI, NSERC, and FQRNT to C.-J. Li.Notes and references
-
(a) Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef
; (b) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed
-
(a) B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693–699 CrossRef CAS PubMed
-
(a) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC
-
(a) A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794–2819 CrossRef
- H. Schwager, S. Spyroudis and K. P. C. Vollhardt, J. Organomet. Chem., 1990, 382, 191–200 CrossRef CAS
-
(a) Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed
- N. Ishida, W. Ikemoto and M. Murakami, Org. Lett., 2012, 14, 3230–3232 CrossRef CAS PubMed
-
(a) T. Biberger, S. Makai, Z. Lian and B. Morandi, Angew. Chem., Int. Ed., 2018, 57, 6940–6944 CrossRef CAS
-
(a) Y. H. Lee and B. Morandi, Nat. Chem., 2018, 10, 1016–1022 CAS
- T. Delcaillau, P. Boehm and B. Morandi, J. Am. Chem. Soc., 2021, 143, 3723–3728 CrossRef CAS PubMed
- J. Zhu, R. Zhang and G. Dong, Nat. Chem., 2021, 13, 836–842 CrossRef CAS
-
(a) Y. Wang, Y. Lang, C.-J. Li and H. Zeng, Chem. Sci., 2022, 13, 698–703 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02412e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |