
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermolecular sulfur atom transfer cascade enabled late-stage introduction of sulfilimines into peptides†
Zeyuan
He
a,
Yuyang
Liu
a,
Guangjun
Bao
a,
Yiping
Li
 a,
Xiufang
Zhao
a,
Quan
Zuo
b,
Kai
Li
a,
Wangsheng
Sun
*a and
Rui
Wang
ab
a,
Xiufang
Zhao
a,
Quan
Zuo
b,
Kai
Li
a,
Wangsheng
Sun
*a and
Rui
Wang
ab
aKey Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences & Research Unit of Peptide Science, Chinese Academy of Medical Sciences, 2019RU066, Lanzhou University, Lanzhou 730000, China. E-mail: sunws@lzu.edu.cn
bState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
First published on 19th September 2024
Abstract
Sulfilimines, a privileged class of –S(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– functional groups found in nature, have been exploited as valuable building blocks in organic synthesis and as pharmacophores in drug discovery, and have aroused significant interest in the chemical community. Nevertheless, strategies for late-stage introduction of sulfilimines into peptides and proteins have still met with limited success. Herein, we have developed a method of introducing biological sulfilimine fragments into peptides by an intermolecular sulfur atom transfer cascade reaction, utilizing hydroxylamine condensed with the acid moieties of peptides and varied diaryl disulfides. It provides a convenient, efficient, metal-free and widely applicable method for late-stage modification and functionalization of peptides at their acid sites both in the homogeneous phase and on-resins in SPPS. Moreover, the modified peptides with sulfilimines have been demonstrated as cleavable linkers for peptide conjugates under reducible conditions, providing unique opportunities in peptide therapeutics development and drug discovery.
N– functional groups found in nature, have been exploited as valuable building blocks in organic synthesis and as pharmacophores in drug discovery, and have aroused significant interest in the chemical community. Nevertheless, strategies for late-stage introduction of sulfilimines into peptides and proteins have still met with limited success. Herein, we have developed a method of introducing biological sulfilimine fragments into peptides by an intermolecular sulfur atom transfer cascade reaction, utilizing hydroxylamine condensed with the acid moieties of peptides and varied diaryl disulfides. It provides a convenient, efficient, metal-free and widely applicable method for late-stage modification and functionalization of peptides at their acid sites both in the homogeneous phase and on-resins in SPPS. Moreover, the modified peptides with sulfilimines have been demonstrated as cleavable linkers for peptide conjugates under reducible conditions, providing unique opportunities in peptide therapeutics development and drug discovery.
Introduction
Thanks to the significance of peptides as therapeutics,1 a platform for targeted therapeutics,2 and chemical biology probes,3 conjugations of peptides with functional molecules and conventional drugs have drawn tremendous interest during the past two decades, leading to an arsenal of peptide bioconjugations and modifications.4 Despite the fruitful advances, the development of novel peptide bioconjugation and modification is still urgently needed. Sulfilimines, a privileged class of –S(IV)N– functional groups, that were initially discovered approximately a century ago, have been exploited as valuable building blocks in organic synthesis,5 and as pharmacophores in drug discovery.6 More interestingly and significantly, they were identified by Hudson et al. in 2009 as unique crosslinks that covalently bind hydroxylysine-211 and methionine-93 in the collagen IV network, which is a highly conserved major component of basement membranes and associated with the occurrence of several biofunctions and diseases.7 Later, the same group disclosed the Br-dependent peroxidase catalyzed biochemical formation of sulfilimines between methionine-93 and hydroxylysine-211 within collagen IV, an event critical for basement membrane assembly and tissue development in animals (Fig. 1a).8 Inspired by this discovery, Tang et al. developed sulfilimine-based molecular probes for imaging native HOBr in live cells and Zebrafish.9
 | ||
| Fig. 1 Strategies for the introduction of sulfilimines into peptides and proteins. | ||
These elegant seminal studies have aroused scientists to artificially build sulfilimines between biomolecules, particularly peptides and proteins, and small functional molecules. Chang, Toste, and co-workers developed a methionine selective bio-conjugation by oxidizing methionines to corresponding sulfilimines with oxaziridines,10 which was demonstrated successful in cyclization of peptides, protein functionalization, antibody–drug conjugates (ADCs), and chemoproteomic identification of functional methionines in cells (Fig. 1b).10,11 These studies indicate that NS bonds have significant potential in drug conjugates. More recently, Kozlowski, Jia, and co-workers have developed a biocompatible Chan–Lam coupling reaction, which can introduce N–Ar diaryl sulfilimines into peptides and proteins at the site of an exogenous inserted arylboronic acid (Fig. 1c).12 Nevertheless, a novel and mild alternative that is beyond the above methods to construct sulfilimines between peptides and functional small molecules, which could be of great potential for peptide–drug conjugates (PDCs) construction, is still appealing to the chemical community.
Very recently, we disclosed the late-stage introduction of oxime ethers into peptides using N-alkoxylpeptidylamides that were prepared in situ from amino acids13 and C–S bond formation between tryptophan and thiophenol enabled site-selective functionalization of peptides.14 As a continuation of our ongoing interests in peptide bioconjugation and medicinal chemistry,13–15 and inspired by the intermolecular sulfur atom transfer cascade reaction between aryloxyamides and thiols that was reported by Xiong et al.,16 we sought to explore the reaction between thiols and N-phenoxylpeptidylamides that could be prepared in situ from the acid moiety of peptides to furnish late-stage incorporation of sulfilimines with diverse functional groups into peptides at the position of interest via an intermolecular sulfur atom transfer cascade reaction, and thereby provide a novel alternative for peptide modification and PDC construction (Fig. 1d).
Results and discussion
Our investigation was initiated by using N-phenoxylglycinamide 2a that was prepared by condensation of N-Boc-glycine 1a and O-phenylhydroxylamine, and 4,4′-dichlorodiphenyl disulfide 3a as a model substrate in the presence of CsOAc in DMSO at room temperature for 10 h. To our delight, glycyl sulfilimine 4a was obtained smoothly, albeit with moderate yield (Table 1, entry 1). Encouraged by this result, a series of reaction conditions including reaction media, base, temperature, and other parameters (for a complete list of the conditions screened, see the section ‘Optimization studies’ in the ESI†) were screened to verify optimal reaction conditions. It revealed that base was crucial for the reaction (Table 1, entries 1–4 and Tables S1–S3†) and 0.5 equiv. Cs2CO3 gave the optimal results (Table 1, entry 3). The reaction could be promoted at 37 °C with a significantly shortened reaction time in high isolated yield (Table 1, entry 5, defined as method A). Interestingly, when simple 4-chlorothiophenol monomer 3a′ was used instead of 4,4′-dichlorodiphenyl disulfide 3a, the reaction could proceed smoothly as well, providing glycyl sulfilimine 4a in equivalent yield (Table 1, entry 5 vs. 6), probably because simple thiophenols were easily oxidized to disulfides under air.16b Meanwhile, DMF, the most commonly used solvent in solid-phase peptide synthesis (SPPS), was also well compatible with this reaction (Table 1, entry 5 vs. 7). To further simplify the reaction protocol, a two-step-one-pot procedure was investigated. The reaction proceeded smoothly when 1a, O-phenylhydroxylamine, EDCI and HOBt were stirred for 6 hours in DMF before addition of 3a and Cs2CO3 (Table 1, entry 8, two-step-one-pot procedure defined as method B). The condensation reaction between the carboxyl group and O-phenylhydroxylamine could generate some by-products, which may explain why the yields of the two-step-one-pot procedure are lower than those of the one-step reaction. Unluckily, it failed when 1a, 3a, O-phenylhydroxylamine, EDCI, HOBt and Cs2CO3 were all added together in solution (Table 1, entry 9, one-pot procedure).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Time (h) | Temp. (°C) | Yielde (%) |
| a 2a (0.05 mmol), 3a (0.05 mmol) and base in 1 mL solvent. b 1a (0.1 mmol), NH2OPh (0.1 mmol), EDCI (0.12 mmol) and HOBt (0.12 mmol) in 2 mL DMF at 37 °C for 6 h, then 3a (0.1 mmol) and Cs2CO3 (0.17 mmol) were added and stirred for an additional 6 h. c 1a (0.1 mmol), NH2OPh (0.1 mmol), EDCI (0.12 mmol), HOBt (0.12 mmol), 3a (0.1 mmol) and Cs2CO3 (0.17 mmol) in 2 mL DMF at 37 °C for 6 h. d 0.1 mmol 4-chlorothiophenol 3a′ was used instead of 3a. e Yield was determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. Isolated yield after column chromatography was given in parentheses. | |||||
| 1a | DMSO | CsOAc (1 eq.) | 10 | rt | 61 |
| 2a | DMSO | Cs2CO3 (1 eq.) | 10 | rt | 83 |
| 3a | DMSO | Cs2CO3 (0.5 eq.) | 10 | rt | 90 |
| 4a | DMSO | — | 10 | rt | 0 |
| 5a | DMSO | Cs2CO3 (0.5 eq.) | 6 | 37 | 90 (85) |
| 6a,d | DMSO | Cs2CO3 (0.5 eq.) | 6 | 37 | 90 |
| 7a | DMF | Cs2CO3 (0.5 eq.) | 6 | 37 | 89 |
| 8b | DMF | Cs2CO3 (1.7 eq.) | 6 | 37 | 53 |
| 9c | DMF | Cs2CO3 (1.7 eq.) | 6 | 37 | 0 |

With the optimum conditions in hand, we explored the scope of the reaction (Fig. 2). We first test the universality of the reaction by changing the N-phenoxypeptidylamide substrate scope. To our delight, all the substrates that started from different N-protected amino acids could be involved in the reaction through method A or method B. A wide variety of functional residues, including aromatic amino acid residues, aliphatic amino acid residues, and polar amino acid residues (4a–4h), were tolerated, albeit the presence of reactive side chains like hydroxyl and indole results in lower yield. Large scale (10 mmol) reactions can be carried out using method A or method B to obtain corresponding high and moderate yields (4a). In addition, the reaction can implement late-stage modification of oligopeptides, incorporating special sulfilimines into oligopeptides at their C-terminal with good yields (4i, 4j). Meanwhile, the late-stage introduction of sulfilimines into the side chain of amino acid or oligopeptide was demonstrated by a one-pot reaction using Glu-containing oligopeptides (4k–4n). Significantly, fully deprotected peptides worked well in reaction solution (4n), indicating that O-aryl hydroxamic acid derivatives have the potential to be biorthogonal precursors for incorporation of a disulfide reagent.
 | ||
| Fig. 2 Substrate scope. Method A: 2 (0.05 mmol), 3 (0.05 mmol), and Cs2CO3 (0.025 mmol) in DMSO (1.0 mL) at 37 °C for 6 h. Method B: corresponding amino acids or peptides 1 (0.1 mmol), NH2OPh (0.1 mmol), EDCI (0.12 mmol) and HOBt (0.12 mmol) in 2 mL DMF at 37 °C for 6 h, then 3a (0.1 mmol) and Cs2CO3 (0.17 mmol) were added and stirred for an additional 6 h. Yields of isolated products are given. a 10 mmol scale. b 0.025 mmol scale. c 12 h. d 24 h. | ||
Unfortunately, however, unprotected cysteine is incompatible with this reaction, which may be due to the high reactivity of thiol groups. Subsequently, we explored the substrate scope of diaryl disulfides. Various alkyl-, amino-, Cl-, Br- and CF3-substituted phenyl or dinaphthyl disulfides could participate in the reaction smoothly to give the desired products in moderate to good yields (4o–4v). Compared with electron withdrawing group substitutions such as 4-Cl, 4-Br and 4-CF3 (71–89%), diaryl disulfides possessing electron donating substitutions such as 4-OMe, 4-Me, 4-tBu or 2-naphthyl provided lower yield (51–65%) and needed a longer reaction time. It is particularly noticeable that drugs such as chlorambucil and indomethacin could be introduced as payloads into amino acids (4w, 4x) and oligopeptides (4y) with good yields.
We then turned our attention to examine the practicality of the reaction on-resin (Fig. 3). Allyl protected Glu(OAllyl)–OH or Asp(OAllyl)–OH was inserted at the position of interest during the routine Fmoc SPPS on-resin and thereby site-specific modification could be achieved with orthogonal protection of tert butyl ester and allyl ester. After the routine Fmoc-SPPS on-resin, the N-phenoxypeptidylamide substrate was obtained by coupling with O-phenylhydroxylamine after deallylation of Allyl. The substrate on-resin was reacted with diaryl disulfide substrate 3 in DMF or DMSO for 24–48 hours, followed by cleavage from the resin and purification by semi-preparative HPLC.
 | ||
| Fig. 3 Late-stage sulfilimination of glutamate/aspartate-containing peptides on resin. Pyr, pyroglutamic acid; e, D-Glu. Isolated yield after semi preparative HPLC was given. a 24 h in step (3). b 36 h in step (3). c 48 h in step (3). d 5 equiv. Cs2CO3 was added. e DMSO instead of DMF in step (3). | ||
To our delight, this protocol worked well with some bioactive molecule analogues and bioactive peptide analogues. 5a was obtained in 37% isolated yield after 14 steps from resin loading compared to the 46% isolated yield of the one-step reaction in solution (4n in Fig. 2). Peptide–peptide conjugates (5b), peptide–indomethacin conjugates (5c), peptide–D-biotin conjugates (5d), peptide–flurbiprofen conjugates (5e) and peptide–niflumic acid conjugates (5f) were successfully obtained through this strategy after 14 steps. More importantly, product 5g containing cysteine can be synthesized through this solid-phase synthesis using a Trt-protected cysteine. Conjugate 5g was slowly degraded in solution due to the unprotected thiol group, which is consistent with the subsequent experimental results. Bioactive peptides such as thymopentin (5h), splenopentin (5i), angiotensin II (5j), melittin fragment (5k), argireline (5l) and TAT cell penetrating peptides (5m, 5n) were all applicable to this protocol. Conjugate 5o only achieved 3% yield, possibly due to the poor solubility of substrate coumarin (3o). Furthermore, the success of gonadotropin-releasing hormone (GnRH) derivatives and substance P conjugated with chlorambucil (5p, 5q) further demonstrated the potential application of this reaction to construct PDCs for cancer therapy.
As the connecting bridge between drugs and peptides in PDCs, linkers determine the circulation time and stability of PDCs in vivo.17 To explore the possibility of these sulfilimines as releasable PDC linkers under reducible conditions,5b,10a,10b a series of experiments were then carried out. It was found that sulfilimines could be decomposed into corresponding peptidyl amides and diaryl thioethers under reducible conditions (Fig. 4). Particularly interesting, for example, is that the product peptide conjugate 5i could be deconjugated in a GSH concentration-dependent manner. Specifically, 5i was decomposed slowly (t1/2 ∼ 12 h) in 1 mM GSH of PBS buffer (pH 7.4) solution and decomposed rapidly (t1/2 < 1 h) in 10 mM GSH of PBS buffer (pH 7.4) solution, with complete decomposition within 6 hours (Fig. 4b). In addition, 5i was stable in PBS solution and relatively stable in FBS (Fetal Bovine Serum) solution, with more than 79% existing in its original form after 24 hours (Fig. 4c and S19–S22†). These features provide us with an opportunity to design GSH-sensitive PDCs and prodrugs for therapy.18
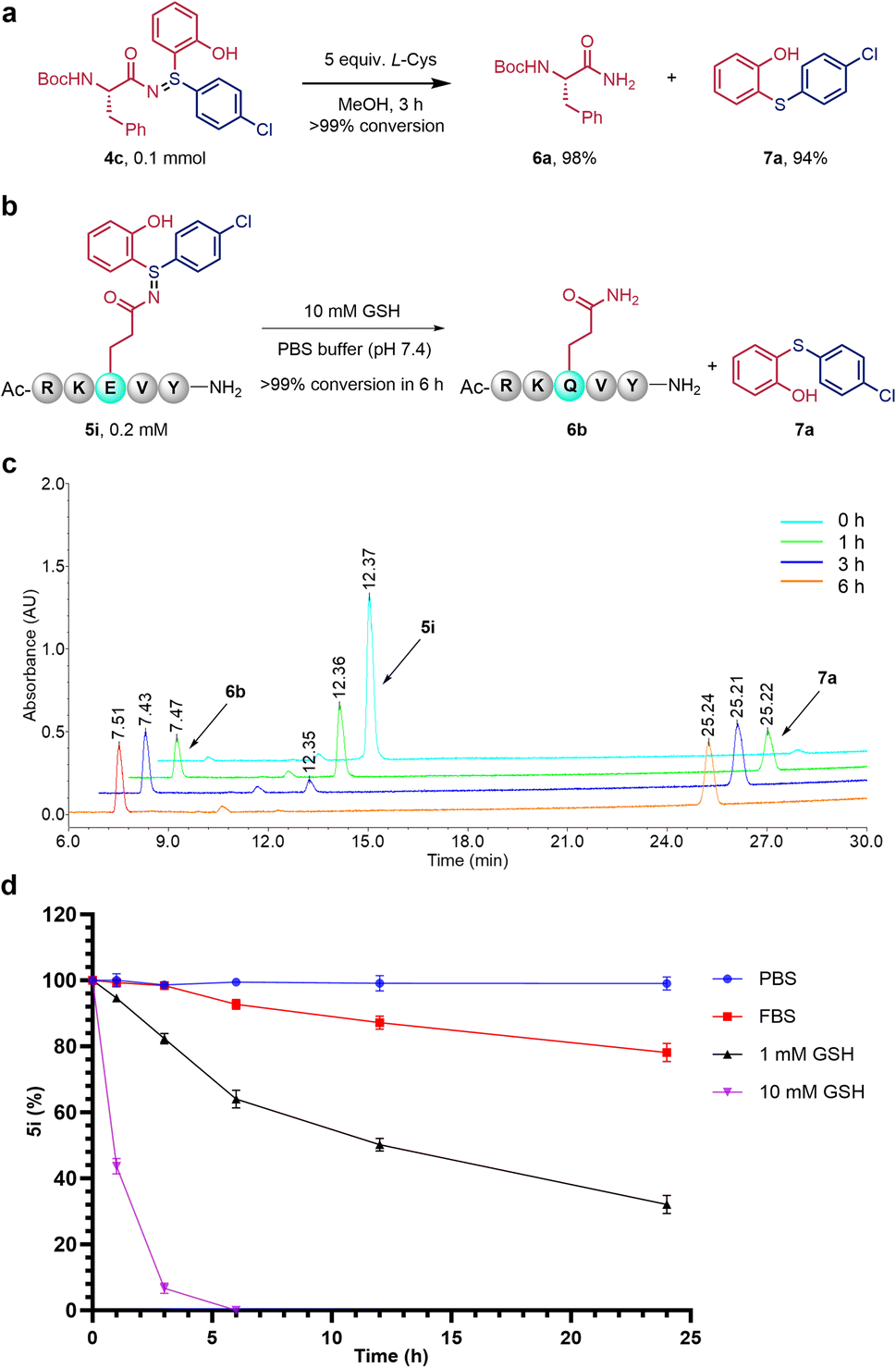 | ||
| Fig. 4 Deconjugation and stability of modified products. (a) Deconjugation of products 4c. The reaction was carried out with 0.1 mmol 4c, 0.5 mmol L-Cys in 1 mL MeOH at room temperature for 3 h, isolated yield of 6a (98%) and 7a (94%) were given. (b) Deconjugation of product 5i. 5i (0.2 mM) could be decomposed into corresponding peptidyl amides (6c) and diaryl thioether (7a) by 10 mM GSH in PBS buffer (pH 7.4) solution. (c) Schematic diagram of the HPLC chromatogram indicates the conversion of peptide 5i in 10 mM GSH solution at different times. (d) Stability of 5i. 5i (0.2 mM) was incubated at 37 °C with PBS buffer (pH 7.4) solution, 95% FBS in PBS buffer solution (pH 7.4), 1 mM GSH in PBS buffer (pH 7.4) solution and 10 mM GSH in PBS buffer (pH 7.4) solution respectively. | ||
Conclusions
In summary, a late-stage introduction of sulfilimines into peptides at a carboxylic acid site has been achieved. This method enabled the introduction of sulfilimines with diverse functional groups, such as drugs, natural products, bioactive handles, and fluorescent tags into peptides both in the homogeneous phase and on-resins in SPPS under mild conditions. Moreover, sulfilimines were demonstrated as cleavable linkers for peptide conjugates under reducible conditions, enabling on-demand control of peptide functions. It might provide a novel tool kit for peptide chemical biology and prodrug discovery. Further applications of this strategy in these areas are currently under investigation in our laboratory.Data availability
All data associated with this study are available in the article and ESI.†Author contributions
W. S. and R. W. conceptualized the project, supervised the work, designed the experiments, and assisted in data analysis. Z. H., Y. L., G. B., Y. L., X. Z., Q. Z. and K. L. performed the experiments and analyzed the data. All authors discussed the results and commented on the manuscript. W. S. and Z. H. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the CAMS Innovation Fund for Medical Sciences (CIFMS) (2019-I2M-5-074, 2021-I2M-3-001, 2021-I2M-1-026), and the Fundamental Research Funds for the Central Universities (lzujbky-2024-ey10).References
-
(a) I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed
; (b) M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed
-
(a) B. M. Cooper, J. Iegre, D. H. O' Donovan, M. Ö. Halvarsson and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1480–1494 RSC
-
(a) C. L. Charron, J. L. Hickey, T. K. Nsiama, D. R. Cruickshank, W. L. Turnbull and L. G. Luyt, Nat. Prod. Rep., 2016, 33, 761–800 RSC
-
(a) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed
-
(a) T. L. Gilchrist and C. J. Moody, Chem. Rev., 1977, 77, 409–435 CrossRef CAS
-
(a) S. Zhou, T. Yan, Y. Li, Z. Jia, B. Wang, Y. Zhao, Y. Qiao, L. Xiong, Y. Li and Z. Li, Org. Biomol. Chem., 2014, 12, 6643–6652 RSC
-
(a) R. Vanacore, A. J. L. Ham, M. Voehler, C. R. Sanders, T. P. Conrads, T. D. Veenstra, K. B. Sharpless, P. E. Dawson and B. G. Hudson, Science, 2009, 325, 1230–1234 CrossRef CAS PubMed
- A. S. McCall, C. F. Cummings, G. Bhave, R. Vanacore, A. Page-McCaw and B. G. Hudson, Cell, 2014, 157, 1380–1392 CrossRef CAS PubMed
- K. Xu, D. Luan, X. Wang, B. Hu, X. Liu, F. Kong and B. Tang, Angew. Chem., Int. Ed., 2016, 55, 12751–12754 CrossRef CAS PubMed
-
(a) S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS PubMed
-
(a) D. Lin, M. Wallace, A. J. Allentoff, D. J. Donnelly, E. Gomes, K. Voronin, S. Gong, R. Y. C. Huang, H. Kim, J. Caceres-Cortes and S. Bonacorsi Jr, Bioconjugate Chem., 2020, 31, 1908–1916 CrossRef CAS PubMed
- T. Meng, L. A. Wells, T. Wang, J. Wang, S. Zhang, J. Wang, M. C. Kozlowski and T. Jia, J. Am. Chem. Soc., 2022, 144, 12476–12487 CrossRef CAS PubMed
- Y. Liu, Z. He, W. Ma, G. Bao, Y. Li, C. Yu, J. Li, R. E, Z. Xu, R. Wang and W. Sun, Org. Lett., 2022, 24, 9248–9253 CrossRef CAS PubMed
- G. Bao, P. Wang, J. Li, X. Song, T. Yu, J. Zhang, Y. Li, Z. He, R. E, X. Miao, J. Xie, J. Ni, R. Wang and W. Sun, CCS Chem., 2024, 6, 1547–1556 CrossRef CAS
-
(a) G. Bao, X. Song, Y. Li, Z. He, Q. Zuo, R. E, T. Yu, K. Li, J. Xie, W. Sun and R. Wang, Nat. Commun., 2024, 15, 6909 CrossRef PubMed
-
(a) F. Xiong, L. Lu, T.-Y. Sun, Q. Wu, D. Yan, Y. Chen, X. Zhang, W. Wei, Y. Lu, W. Sun, J. J. Li and J. Zhao, Nat. Commun., 2017, 8, 15912 CrossRef PubMed
-
(a) M. Alas, A. Saghaeidehkordi and K. Kaur, J. Med. Chem., 2020, 64, 216–232 CrossRef PubMed
-
(a) Y. Xiong, C. Xiao, Z. Li and X. Yang, Chem. Soc. Rev., 2021, 50, 6013–6041 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02166e |
| This journal is © The Royal Society of Chemistry 2024 |