
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent progress in the regulation of endogenous molecules for enhanced chemodynamic therapy
Jun
Wang
a,
Yina
Liu
a,
Tingting
Cui
*abc,
Huanghao
Yang
 *a and
Lisen
Lin
*a
*a and
Lisen
Lin
*a
aNew Cornerstone Science Laboratory, MOE Key Laboratory for Analytical Science of Food Safety and Biology, Engineering Technology Research Center on Reagent and Instrument for Rapid Detection of Product Quality and Food Safety in Fujian Province, College of Chemistry, Fuzhou University, Fuzhou, 350108, China. E-mail: ttcui@ciac.ac.cn; hhyang@fzu.edu.cn; lisen.lin@fzu.edu.cn
bDepartments of Diagnostic Radiology, Surgery, Chemical and Biomolecular Engineering, and Biomedical Engineering, Yong Loo Lin School of Medicine and College of Design and Engineering, National University of Singapore, Singapore, 119074, Singapore
cNanomedicine Translational Research Program, NUS Center for Nanomedicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, 117597, Singapore
First published on 7th June 2024
Abstract
Chemodynamic therapy (CDT) is a potential cancer treatment strategy, which relies on Fenton chemistry to transform hydrogen peroxide (H2O2) into highly cytotoxic reactive oxygen species (ROS) for tumor growth suppression. Although overproduced H2O2 in cancerous tissues makes CDT a feasible and specific tumor therapeutic modality, the treatment outcomes of traditional chemodynamic agents still fall short of expectations. Reprogramming cellular metabolism is one of the hallmarks of tumors, which not only supports unrestricted proliferative demands in cancer cells, but also mediates the resistance of tumor cells against many antitumor modalities. Recent discoveries have revealed that various cellular metabolites including H2O2, iron, lactate, glutathione, and lipids have distinct effects on CDT efficiency. In this perspective, we intend to provide a comprehensive summary of how different endogenous molecules impact Fenton chemistry for a deep understanding of mechanisms underlying endogenous regulation-enhanced CDT. Moreover, we point out the current challenges and offer our outlook on the future research directions in this field. We anticipate that exploring CDT through manipulating metabolism will yield significant advancements in tumor treatment.
 Tingting Cui | Tingting Cui received her PhD (2022) from the College of Applied Chemistry and Engineering (Changchun Institute of Applied Chemistry, Chinese Academy of Sciences) at the University of Science and Technology of China. She is now working with Prof Xiaoyuan (Shawn) Chen on Translational Nanomedicine and Therapeutics at the National University of Singapore. Her research interest focuses on designing and constructing multifunctional nanomaterials for stem cell regulation and cancer immunotherapy. |
 Huanghao Yang | HuanghaoYang is a fellow of the Royal Society of Chemistry, who received his PhD from Xiamen University in 2002 and engaged in postdoctoral research at the Hong Kong University of Science and Technology (2002–2004). He joined Fuzhou University in 2008. He has been supported by the National Science Foundation for Distinguished Young Scholars of China in 2011, and the National Award for the Chang Jiang Scholar Program in 2012. His research interests mostly focus on bioanalysis, chemical biology, and nanomedicine. |
 Lisen Lin | Lisen Lin received his PhD from Fuzhou University in 2016 and carried out postdoctoral research at the National Institutes of Health (NIH) from 2016 to 2019. He then joined Fuzhou University in 2019 as a Min Jiang Scholar Professor. His research focuses on the design and synthesis of novel nanomaterials for cancer imaging and therapeutic applications. |
1. Introduction
Cancer has been a severe threat to human life.1 Currently, the main cancer treatments typically involve surgery, radiotherapy, and chemotherapy, yet they offer limited clinical benefits and are usually associated with serious side effects.2 Therefore, there is an urgent need to explore effective and tumor-specific treatment modalities aiming to completely eradicate tumors, prevent tumor recurrence and metastasis, and improve prognosis.3,4 Importantly, cancer cells show elevated reactive oxygen species (ROS) generation compared to non-cancer cells, owing to alterations in genetic and energy metabolism patterns.5 In contrast, excessive ROS can cause irreversible oxidative cell damage, ultimately resulting in cell death.6 Considering the vulnerability of cancer cells to oxidative stress, several ROS-based therapies have emerged,7 including photodynamic therapy (PDT),8 radiotherapy (RT),9 sonodynamic therapy (SDT),10 chemodynamic therapy (CDT),11,12 immunotherapy, etc.13CDT, an emerging efficient ROS-mediated therapeutic modality, was first given by Shi and coworkers in 2016.14 It catalyzes the conversion of intracellular hydrogen peroxide (H2O2) through intratumoral Fenton or Fenton-like reactions, generating a cytotoxic hydroxyl radical (˙OH), which is among the most harmful ROS.15–17 This ROS generation is directly activated by endogenous chemical energy conversion without the need for exogenous stimulations (e.g., laser, ultrasound, or X-rays), thus adequately overcoming the significant limitation of PDT, RT, and SDT, namely low tissue penetration depth.18–20 Moreover, Fenton chemistry triggers the generation of intracellular cytotoxic radicals that are only specifically activated at tumor sites, enabling improved therapeutic effects and fewer side effects of CDT.21–23 Consequently, substantial efforts have been made to engineer various Fenton reaction catalysts with amplified Fenton reaction performance for effectively treating tumors.24–27
Despite extensive attempts, the therapeutic outcome of CDT relying solely on Fenton reaction catalysts remains unsatisfactory.28 The main reason is that tumor cells can construct a robust defense system that allows them to escape from ROS-induced damage by metabolic adaption, inevitably diminishing the antitumor efficacy of CDT.29–31 Mounting evidence indicated that in specific biochemical contexts, aberrant metabolism of tumor cells can cause differential susceptibility of cells to CDT.32,33 A detailed understanding of how endogenous metabolic pathways in tumor cells influence CDT processes is of great therapeutic interest. Herein, we first summarize the mechanisms by which H2O2, iron, lactate, glutathione (GSH), and lipid metabolism impact Fenton chemistry-mediated CDT. Then, the current state-of-the-art designs of CDT nanomedicines integrating metabolic regulation abilities for enhanced tumor therapeutic efficiency will be critically discussed. Last but not least, we discuss the representative challenges and future directions for developing CDT agents. We expect the emergent metabolic reprogramming strategy to push forward the great process of research in the CDT field, which will benefit the clinical treatment of cancers.
2. Pathways controlling CDT
Considering that various endogenous molecules' metabolic processes interfere with the efficiency of Fenton-type reactions that are involved in CDT, researchers are beginning to focus on identifying the metabolic pathways that can impact therapeutic efficacies of CDT. The classifications of currently identified metabolic pathways in tumors, including H2O2, iron, lactate, GSH, and lipid metabolism, are summarized in Fig. 1.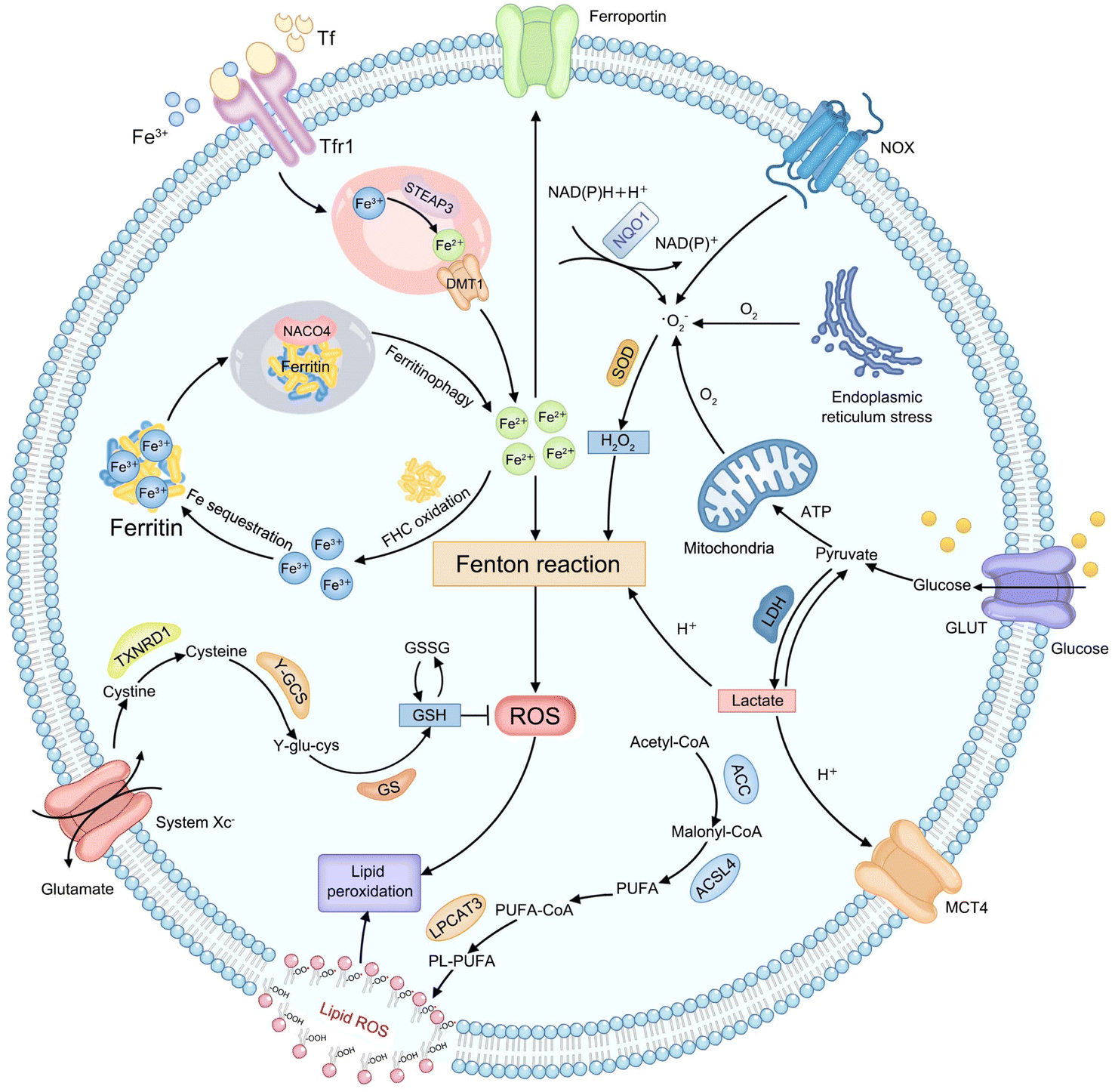 | ||
| Fig. 1 Schematic illustration of endogenous metabolic pathways regulating Fenton chemistry-mediated CDT in tumor cells. | ||
Endogenous H2O2, as an important substrate of the Fenton reaction, can be produced from various cellular metabolic processes in cancer cells. For instance, electrons leaked from the endoplasmic reticulum stress or mitochondrial respiratory chain can be captured by molecular oxygen (O2) to form a superoxide (˙O2−), which is further disproportionated into H2O2 by superoxide dismutase (SOD).34 In addition, the production of H2O2 can also be initiated by activated nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) or nicotinamide adenine dinucleotide (phosphate): quinone oxidoreductase1 (NQO1).35 And then, the ferrous ion (Fe2+) possesses more powerful ˙OH-producing ability compared to other Fenton-reactive metal ions, enabling the strategy of regulating iron metabolism attractive for achieving efficient CDT.36,37 First, the extracellular transferrin (Tf)-ferric ion (Fe3+) (Tf-Fe3+) complex is carried into cells through cell surface Tf receptor protein 1 (Tfr1).38 In the endosome, ferrireductase-active prostate 3 (STEAP3) mediates the transformation of Fe3+ into Fe2+ and divalent metal transporter 1 (DMT1) transports Fe2+ to a cytosolic labile iron pool (LIP).39 Excess Fe2+ is excreted out of cells via ferroportin or is oxidized to Fe3+ by the ferritin heavy chain (FHC) with ferroxidase activity to initiate subsequent Fe sequestration in ferritin, which may be limiting the Fe-driven Fenton reaction,40 while iron deficiency can trigger nuclear receptor coactivator 4 (NCOA4)-dependent autophagic ferritin degradation, namely ferritinophagy, to release free iron on demand.41 Lactate metabolism regulation can interfere with the construction of the acidic tumor microenvironment (TME), which provides a feasible strategy to meet the strong acidity requirements of an effective Fenton reaction.42,43 Glucose is imported into cells by the glucose transporter (GLUT) on the cell membrane and subsequently transformed into pyruvate in the cytoplasm. Then lactate dehydrogenase (LDH) mediates the conversion between pyruvate and lactate, and the monocarboxylic acid transporter (MCT) mediates lactic acid efflux, inducing tumor acidification.44 The cellular antioxidative system plays an indispensable role in maintaining the equilibrium of a reductive environment against excessive ROS.45 Such a redox homeostasis can alleviate oxidative stress which can attack key biomacromolecules and cause irreversible cellular damage.46 As the most abundant low-molecular-weight thiol-containing tripeptide synthesized in the cell cytosol, GSH has long been considered the representative reducing agent for constantly neutralizing cellular oxidative damage and detoxifying lipid ROS.47 The high GSH content in tumor cells can weaken the effect of CDT by directly scavenging ROS.48 For GSH synthesis, extracellular cystine is internalized by cells via the cystine/glutamate antiporter (system Xc−) and reduced into cysteine by thioredoxin reductase 1 (TXNRD1).49 Then cysteine is conjugated with glutamate in the presence of γ-glutamylcysteine synthetase (γ-GCS) to form γ-glutamylcysteine (γ-glu-cys). Finally, GSH synthetase (GS) catalyzes γ-glutamylcysteine and glycine to produce GSH. Lipid metabolism is also closely related to the effect of CDT.50 Typically, acetyl-CoA is first carboxylated to form malonyl-CoA by acetyl-CoA carboxylase (ACC). Free polyunsaturated fatty acid (PUFA) generated from malonyl-CoA can be esterified by activated acyl-CoA synthetase long-chain family member 4 (ACSL4) and is inserted into the cell membrane with the assistance of lysophosphatidylcholine acyltransferase 3 (LPCAT3), eventually improving sensitivity of cells to CDT.51 The above metabolic processes of endogenous molecules provide numerous practical targets for CDT treatment. Next, the mechanism of CDT regulation by each metabolic pathway will be elucidated and discussed in detail, using representative examples in the corresponding section, with the aim of providing references for designing more efficient chemodynamic agents.
2.1 Elevation of H2O2 contents
The H2O2 produced by cell metabolism can be converted into lethal ˙OH by Fenton/Fenton-like reactions.52 Despite higher levels of H2O2 in cancer cells versus non-cancer cells, the endogenously produced H2O2 is still insufficient to assure a satisfactory tumor-killing effect.53 Developing pertinent strategies aimed at promoting the accumulation of intracellular H2O2 is still the primary focus of research on CDT. For example, Yang et al. constructed H2O2-responsive supramolecular polymers (PCSNs) by self-assembly of β-cyclodextrin-ferrocene (CD-Fc) conjugates with platinum(IV) complex modification to overcome this shortcoming (Fig. 2A).54 The hydrophobic Fc in PCSNs was oxidized into water-soluble ferrocenium (Fc+) upon exposure to elevated H2O2 in the tumor. The disruption of host–guest interaction in the nanostructure resulted in dissociation behavior. Meanwhile, the released platinum(IV) prodrug could be reduced into cisplatin(II) to activate NOX-mediated H2O2 replenishment, which could further promote the Fc-initiated Fenton reaction. In vivo experiments confirmed that such a positive feedback loop enabling self-boosting ROS generation dramatically inhibited tumor growth. Wang and co-workers developed amphiphilic polypeptide self-assembling nanomedicine (PtkDOX-NM) for the encapsulation of Fe3+ and β-lapachone (Lap), an o-naphthoquinone that can generate H2O2via futile redox cycles under the catalysis of overexpressed NQO1 (Fig. 2B).55 PtkDOX-NM was formed by co-assembly of two amphiphilic polymers: poly(ethylene glycol) (PEG)-block-poly diisopropylaminoethyl methacrylate block-poly dopamine (PEG-PDPA-PDA) with pH-responsiveness and PEG-block-poly cinnamaldehyde (CA)-Fc polyprodrug (PEG-PCASFc) with ROS-responsiveness, thus achieving dual responses to pH and ROS in the TME. Next, the hydrophobic-to-hydrophilic transition of PDPA responding to the acidity of endo/lysosomes in tumor cells resulted in nanostructure disassembly and Lap release. Lap-induced H2O2 generation could enhance ˙OH production through the Fe3+-catalyzed Fenton reaction, causing the rapid release of the DOX prodrug and the further collapse of nanostructure cleavage of the ROS-responsive thioacetal linker. Their study demonstrated that CDT combined with H2O2 replenishment realizes efficient tumor inhibition with improved survival in mice.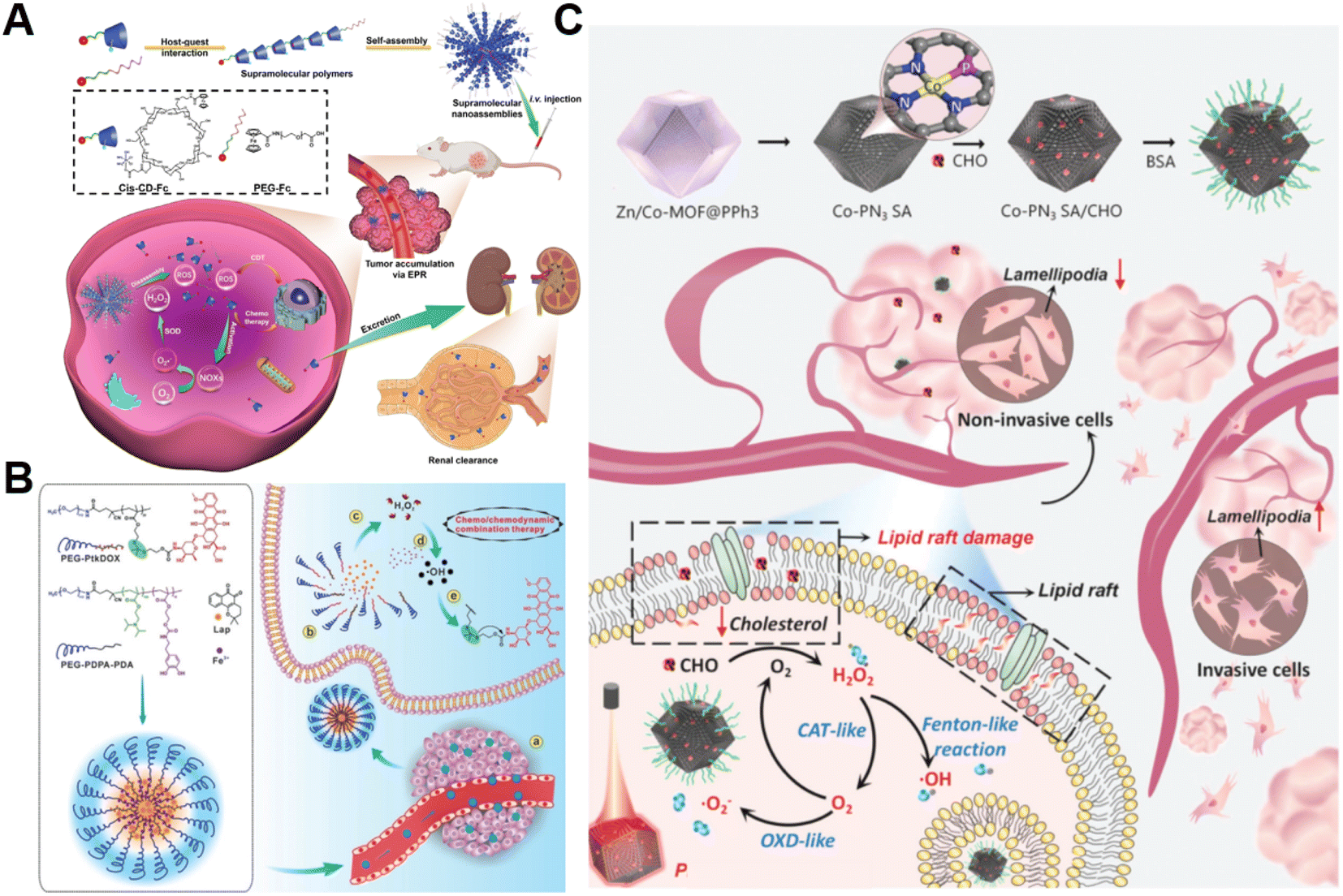 | ||
| Fig. 2 (A) The main synthesis and therapeutic process of PCSNs. Reproduced with permission from ref. 54. Copyright 2021, Wiley-VCH GmbH. (B) Schemes displaying the construction process of PtkDOX-NM and its therapeutic mechanisms. Reproduced with permission from ref. 55. Copyright 2019, Wiley-VCH GmbH. (C) Illustrative diagram of the antitumor mechanism of Co-PN3 SA/CHO. Reproduced with permission from ref. 59. Copyright 2023, Wiley-VCH GmbH. | ||
Additionally, natural enzymes or artificial nanoenzymes have been widely applied to catalyze the transformation of intracellular substances such as glucose,56,57 lactate,58 and cholesterol into H2O2, which can provide an adequate substrate to accelerate the Fenton reaction. In a recent study by Lin's group, multienzyme-like Co-PN3 SA/CHO nanoagents were designed and constructed by utilizing a well-dispersed phosphorus (P)-doped cobalt single atom nanozyme loaded with cholesterol oxidase (CHO) to achieve efficient tumor catalytic therapy for tumor metastasis inhibition (Fig. 2C).59 The as-prepared Co-PN3 SA/CHO could concurrently mimic catalase (CAT), oxidase (OXD), and peroxidase (POD) to produce ˙O2− and highly cytotoxic ˙OH for cancer cell killing. Moreover, CHO effectively catalyzed the oxidization of intratumoral cholesterol to generate H2O2 and cholestenone, promoting the Co-PN3 SA-mediated Fenton reaction. Simultaneously, the CHO-catalytic depletion of cholesterol resulted in the disruption of lipid raft integrity and the inhibition of invasive lamellipodium formation, suppressing tumor progression and metastasis. This work provides a promising strategy to improve therapeutic efficiency through the combination of CHO-mediated cholesterol consumption and Co-PN3 SA-induced ˙OH generation. Notably, bacteria engineered with overexpression of respiratory chain enzyme II (NDH-2 enzyme) have also been utilized to promote intracellular H2O2 content elevation. For example, Fan et al. designed an integrative bioreactor by assembling magnetic Fe3O4 nanoparticles on the surface of NDH-2 enzyme-functionalized nonpathogenic bacterium Escherichia coli MG1655.60 During Escherichia coli MG1655 respiration, NDH-2 enzyme accepted electrons from NADH and subsequently transferred electrons to O2 to generate H2O2. The Fe3O4 nanoparticles converted the sustainably synthesized H2O2 to produce ˙OH through the Fenton-like reaction, achieving H2O2 self-supplying CDT.
2.2 Disruption of iron homeostasis
The conversion of intracellular H2O2 into ˙OH via Fenton chemistry is dependent on Fenton catalysts.61 Fe2+ and its derivatives could serve as excellent Fenton catalysts due to their high catalytic activity in ˙OH generation.62 Generally, tumor cells require more iron ingestion compared to non-tumor cells to maintain rapid proliferation, while coordinating the regulation of iron metabolism to avoid excess iron-induced toxicity.63 The regulation of intracellular iron homeostasis encompassing iron storage/release and import/export has been regarded as a promising approach for reinforcing CDT. For instance, Bu's group prepared a mesoporous silicon nanocarrier loaded with amorphous elemental Fe0 and 2-amino-1,2,4-triazole (AT, a catalase inhibitor) (DMON@Fe0/AT) to integrate redox regulation and iron metabolism disruption for enduring CDT (Fig. 3A).64 Once DMON@Fe0/AT was endocytosed by tumor cells, acidic pH-triggered Fe2+ and AT release from this nanocarrier occurred. The released AT suppressed the catalase activity to promote H2O2 accumulation and the exposed DMON-mediated GSH depletion, thus enhancing the Fe2+-driven Fenton reaction. The elevated intracellular ˙OH induced mitochondrial dysfunction, leading to the downregulation of cell membrane protein ferroportin 1 expression and the further disruption of intracellular iron homeostasis. In short, the as-prepared intelligent biodegradable DMON@Fe0/AT demonstrates a new method for effective CDT treatment by blocking the iron exporter and regulating redox homeostasis.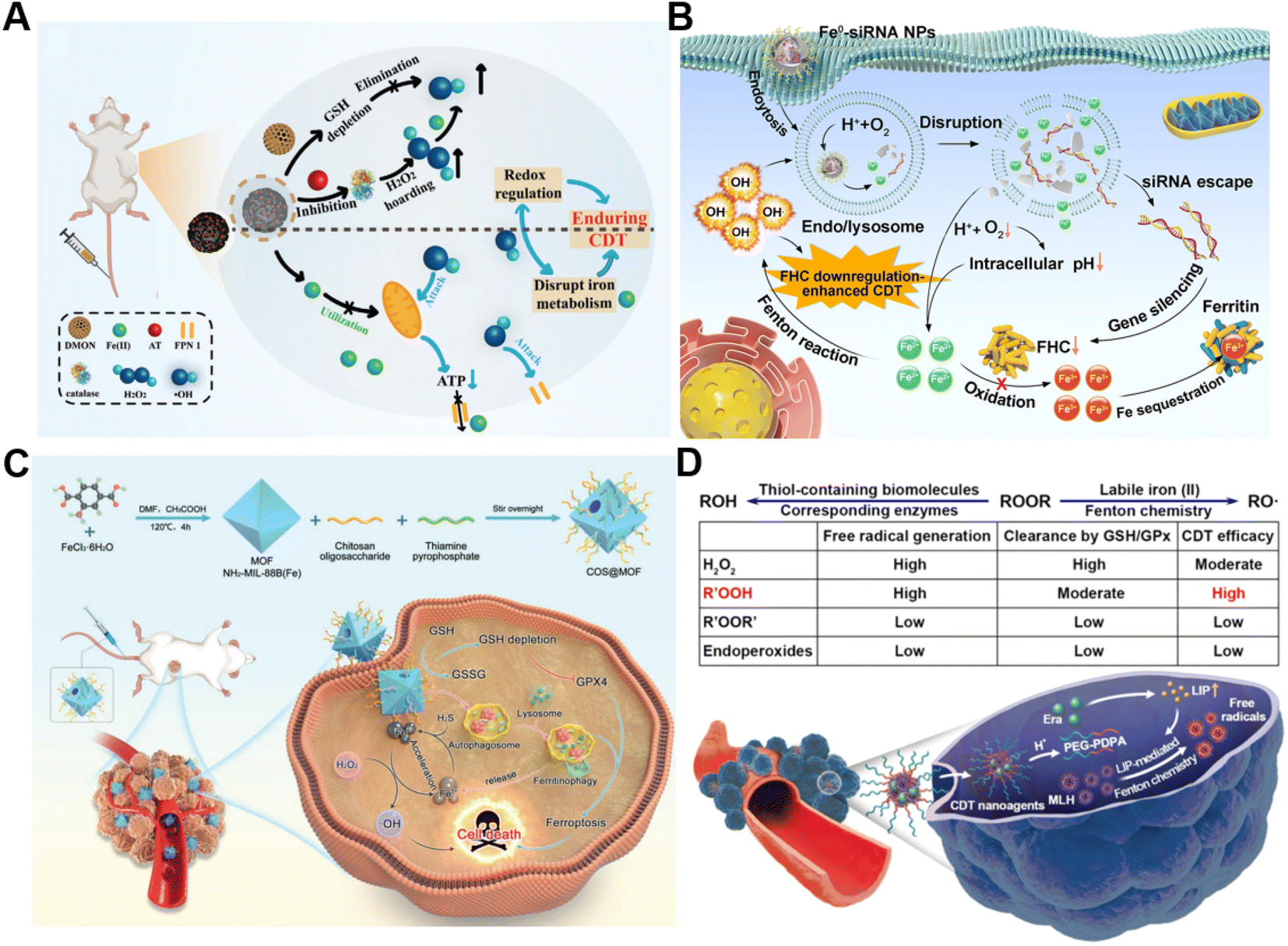 | ||
| Fig. 3 (A) Schematic presentation of synthetic procedure of DMON@Fe0/AT and the corresponding anticancer process. Reproduced with permission from ref. 64. Copyright 2021, Wiley-VCH GmbH. (B) Schematic description of the sequential release of Fe2+ and siRNA from Fe0-siRNA NPs responding to tumor acidity for enhancement of CDT. Reproduced with permission from ref. 65. Copyright 2023, Wiley-VCH GmbH. (C) Illustration of an engineered MOF with amplifying ferritinophagy to cause iron ion overload for improved antitumor therapy. Reproduced with permission from ref. 66. Copyright 2023, Wiley-VCH GmbH. (D) The scheme of factors affecting endogenous labile iron-based CDT outcome of ROOR and the utilization of MLH-loading nanomedicines for CDT. Reproduced with permission from ref. 68. Copyright 2020, American Chemical Society. | ||
Our group recently proposed a Fe0-small interfering RNA (siRNA) composite system (Fe0-siRNA) to realize FHC downregulation-potentiated CDT (Fig. 3B).65 When the Fe0-siRNA nanoparticles (Fe0-siRNA NPs) were internalized into tumor cells and located in mildly acidic lysosomes, they were disassembled in the presence of O2 and released Fe2+ and siRNA. The acidity amplification resulting from O2 depletion could promote further degradation of Fe0-siRNA NPs. The Fe2+ from Fe0-siRNA NPs not only activated the conversion of intracellular H2O2 into highly toxic ˙OH for CDT, but also facilitated endosome escape of FHC siRNA through ˙OH-triggered lipid peroxide (LPO) of endo/lysosomal membranes. More importantly, FHC silencing with siRNA suppressed the transformation of Fe2+ into Fe3+ with less Fenton acitivity and the sequestration of Fe in ferritin, allowing the accumulation of higher reactive Fe2+, as well as increasing the CDT efficiency. In another study, Du et al. decorated a thiamine pyrophosphate (TPP)-etched open cavity metal–organic framework (MOF) with chitosan oligosaccharide (COS) through electrostatic interaction to develop a nanoplatform (termed COS@MOF) enabling hydrogen sulfide (H2S)-activated CDT and ferroptosis for colon cancer treatment (Fig. 3C).66 On the one hand, the upregulated H2S could boost the transformation of Fe3+ into Fe2+ to improve Fenton reaction efficiency. On the other hand, utilization of autophagy inducer COS enabled the degradation of ferritin to promote the release of iron from ferritin and Fe2+ replenishment, further enhancing CDT and ROS-mediated ferroptosis. In another example, Li et al. developed a nanodrug (T10@cLAV) by the integration of transferrin-homing peptide T10 and cross-linked lipoic acid vesicles for Fe2+ self-supplying tumor therapy.67 The T10@cLAV bound to the overexpressed Tf receptor on cancer cells (Tf@T10@cLAV) and was subsequently imported into endosomes/lysosomes. The acidic environment of endosomes/lysosomes could trigger the degradation of Tf, thus inducing the sustained release of iron ions and activating the Fe-driven Fenton reaction. More importantly, cLAV was disrupted with the assistance of GSH and TXNRD1 to release dihydrolipoic acid (DHLA) for DHLA-mediated reduction of Fe3+ to Fe2+, resulting in an increase in Fe2+ levels to further enhance nanocatalytic therapy. Completely unlike traditional treatment strategies involving the introduction of exogenous Fenton-type metal ions, our group first utilized the intracellular labile LIP to provide the continuous catalyst for catalytic generation of toxic free radicals through Fenton-type chemistry (Fig. 3D).68 We developed a pH-sensitive polymer for the co-delivery of the model ROOH molecule and LIP-increasing agent, which can sequentially respond to pH and labile iron in the TME to realize enhanced CDT.
2.3 Interference of lactate metabolism
In addition to the Fenton catalyst activity, the reaction conditions, particularly acidity, also influence the reaction kinetics of the Fenton reaction.69 It has been confirmed that acidic pH favors the Fenton reaction.70 Lactate is considered a metabolic byproduct of glycolysis in tumor cells, leading to elevated tumor acidity.71 Consequently, lactate metabolism regulation is an alternative strategy for enhancing CDT.72 Shi's group reported a pH-responsive and self-augmented nanoplatform (defined as FePt@FeOx@TAM-PEG) fabricated by incorporating a core–shell FePt@FeOx nanocatalyst and pH-responsive tamoxifen (TAM) drug in a poly(styrene-co-maleic anhydride) (PSMA) polymer polymeric matrix and further modifying the surface with PEG (Fig. 4A).73 Once internalized by cancer cells, the nanoplatform disintegrated due to acidic pH-triggered hydrophobic–hydrophilic transitions of TAM and released the FePt@FeOx nanocatalyst. The FePt@FeOx nanocatalyst catalyzed the inherent H2O2 decomposition to produce lethal ˙OH to damage cancer cells. Meanwhile, the liberated TAM could suppress the mitochondrial complex I, thus increasing the intracellular lactate level as well as elevating environmental acidity. This simultaneously amplified ˙OH production and accelerated disassembly of the FePt@FeOx@TAM-PEG nanostructure, inducing more therapeutic cargo release. As a result, this intelligent nanoplatform overcomes the limitation of weak acidic conditions in tumors by introducing metabolism regulators to upregulate lactate content, thereby achieving the enhanced antitumor effect of CDT. Another related study involving the transport modulation of lactate to remodel the reaction environment is worth noting as well. Wang et al. prepared a calcium phosphate-based biomineralized multifunctional nanosystem with the co-delivery of a DOX-Fe2+ complex and MCT4-inhibiting siRNA (siMCT4) (CaP-DOX@Fe2+-siMCT4-PEG-HA) for interfering lactate efflux and enhancing anti-tumor CDT (Fig. 4B).74 The nanosystem was specifically taken up by tumor cells with the aid of hyaluronic acid (HA)-mediated targeting and degraded in the acidic environment of lysosomes due to the pH-triggered hydrolysis of CaP, resulting in the dissociation of the DOX-Fe2+ complex and the release of Fe2+, DOX, and siMCT4. MCT4 silencing by siMCT4 could exacerbate intracellular acidification by blocking lactate efflux, which not only enhanced the Fe2+-driven Fenton reaction, but also decreased adenosine triphosphate (ATP) production for inhibition of DOX efflux, thus significantly improving the treatment efficiency. Typically, Wang's group developed an intelligent bioreactor (Sa@FeS) in which ferrous sulfide (FeS) nanoparticles were anchored on the surface of Salmonella typhimurium strain (Sa) via biomineralization.75 The effective tumor penetration of Sa enabled FeS nanoparticles to mediate photothermally enhanced Fenton catalytic reactions in deep tumor tissues upon 1064 nm laser irradiation. At the same time, the H2S produced by Sa metabolism can facilitate glucose uptake by tumor cells, resulting in the elevation of intracellular lactic acid levels and ultimately boosting FeS-mediated CDT enhancement. Given the limited therapeutic effect of single lactate consumption, simultaneous regulation of multiple metabolic pathways may be a promising way to enhance CDT. Wu et al. proposed a strategy with a dual effect on lactate metabolism based on lactate oxidase (LOX) and syrosingopine (Syr) co-loaded hollow Fe3O4 nanoparticles (denoted as Syr/LOD@HFN), which could combine LOX-mediated lactate consumption and lactate transport chain blockade (Fig. 4C).76 In the tumor TME, LOX could catalyze lactate to generate H2O2in situ while Syr-induced MCT4 downregulation could achieve lactate metabolism blockade, leading to continuous H2O2 production and pH reduction for boosting the Fe3O4-catalyzed Fenton reaction. Owing to the augmented ROS production and the reversion of the lactate-related immunosuppressive TME, this nanoplatform caused severe immunogenic cell death in tumor cells and promoted antitumor immunity. This work reveals that manipulation of multiple lactate metabolic pathways enables a better therapeutic effect than a single regulation strategy.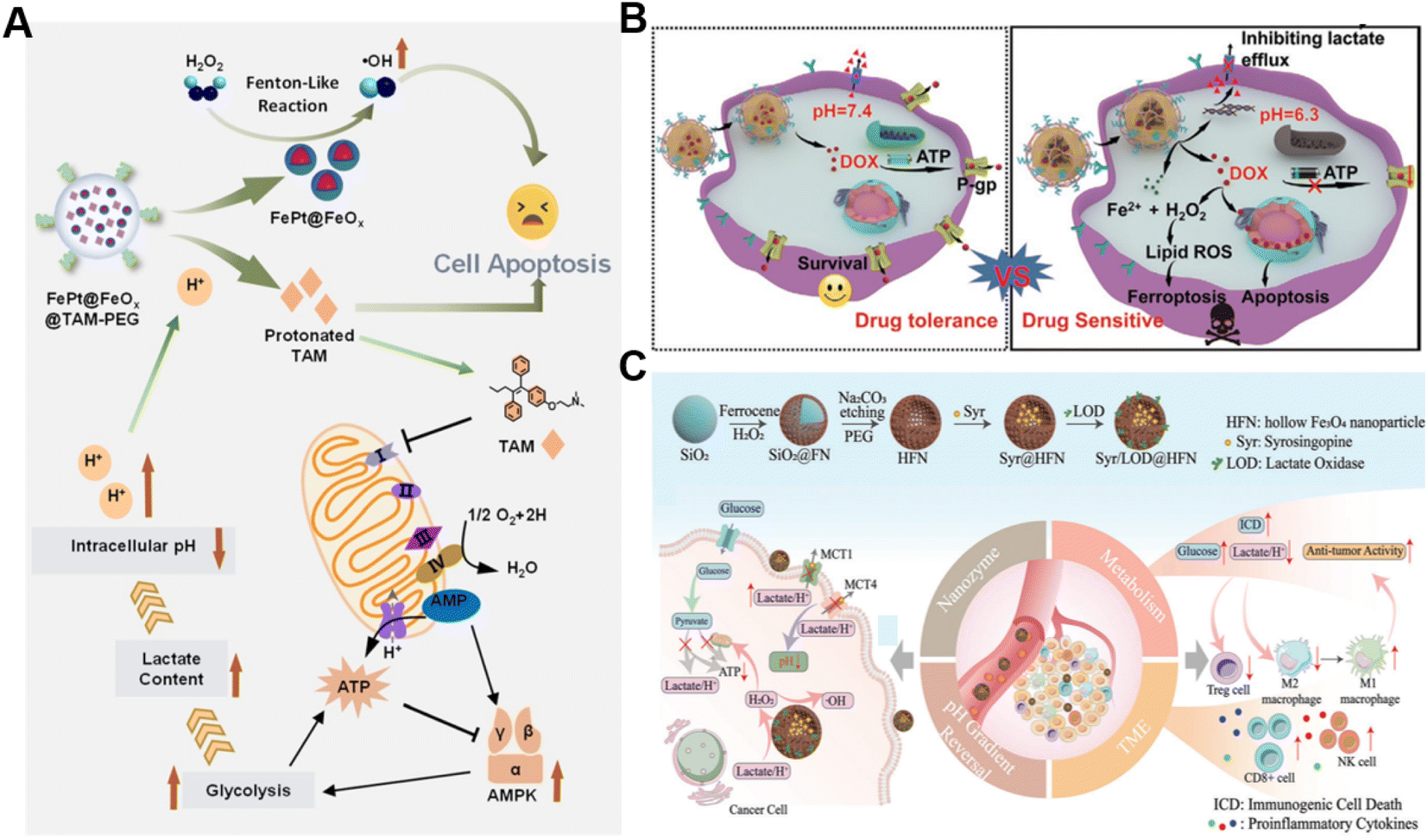 | ||
| Fig. 4 (A) The underlying mechanism of enhanced CDT induced by FePt@FeOx@TAM-PEG. Reproduced with permission from ref. 73. Copyright 2021, Wiley-VCH GmbH. (B) Schematic diagram of CaP-DOX@Fe2+-siMCT4-PEG-HA enabling TME remodeling for improved CDT. Reproduced with permission from ref. 74. Copyright 2021, Wiley-VCH GmbH. (C) Illustration of the construction and application of the Syr/LOD@HFN nanoplatform. Reproduced with permission from ref. 76. Copyright 2023, Wiley-VCH GmbH. | ||
2.4 Depletion of GSH
It is well established that GSH is capable of potent ROS scavenging, thus endowing cancer cells with the ability to resist oxidative stress and weaken the effect of CDT. Generally, metals with oxidation states could consume GSH via undergoing redox reactions with GSH.77 Our group first reported the synthesis of manganese dioxide (MnO2)-coated mesoporous silica nanoparticles for precise magnetic resonance imaging (MRI)-guided self-reinforcing CDT. Upon encountering the high level of GSH, MnO2 NPs were degraded to release Mn2+, which not only initiated CDT by Mn2+-mediated Fenton-like chemistry but also acted as MRI contrast agents to monitor the CDT process.78 Recently, redox-responsive nanosized frameworks coordinated with Cu2+ and 2,3,6,7,10,11-hexahydroxytriphenylene (HPT, a catechol ligand), namely CuHPT, were designed to eliminate drug-resistant cancers by disrupting cellular redox homeostasis (Fig. 5A).79 Experimental results indicated that upon being activated by GSH, CuHPT nanoparticles displayed enhanced CDT with remarkable tumor growth inhibition and negligible side effects via a combination of auto-oxidation and Cu+-catalyzed reactions. In addition to introducing oxidizing agents to consume GSH, numerous electrophilic reagents that can directly react with the thiol group in GSH for reduction of GSH content were utilized.80,81 Guo et al. fabricated a hybrid mesoporous silica/organosilicate nanocomposite (MSN@MON) followed by coating the surface with disulfide bond-containing link poly(acrylic acid) and loading with N,N,N′,N′-tetrakis(2-pyridinyl methyl)-1,2-ethanediamine (TPEN, a metal ion chelator) to obtain MSN@MON-TPEN@PAASH (Fig. 5B).82 Because of GSH-induced disulfide bond cleavage in MSN@MON-TPEN@PAASH, intracellular GSH was partially consumed and the NPs collapsed to release TPEN. The liberated TPEN could chelate with Cu2+ in the Cu–Zn superoxide dismutase, enabling its deactivation and the formation of a TPEN-Cu(II) complex. Then, the TPEN-Cu(II) complex was reduced into TPEN-Cu(I) by remaining GSH, inducing the further decline of antioxidant defense resulting from GSH and ultimately promoting TPEN-Cu(I)-driven Fenton-like reactions for self-reinforced CDT.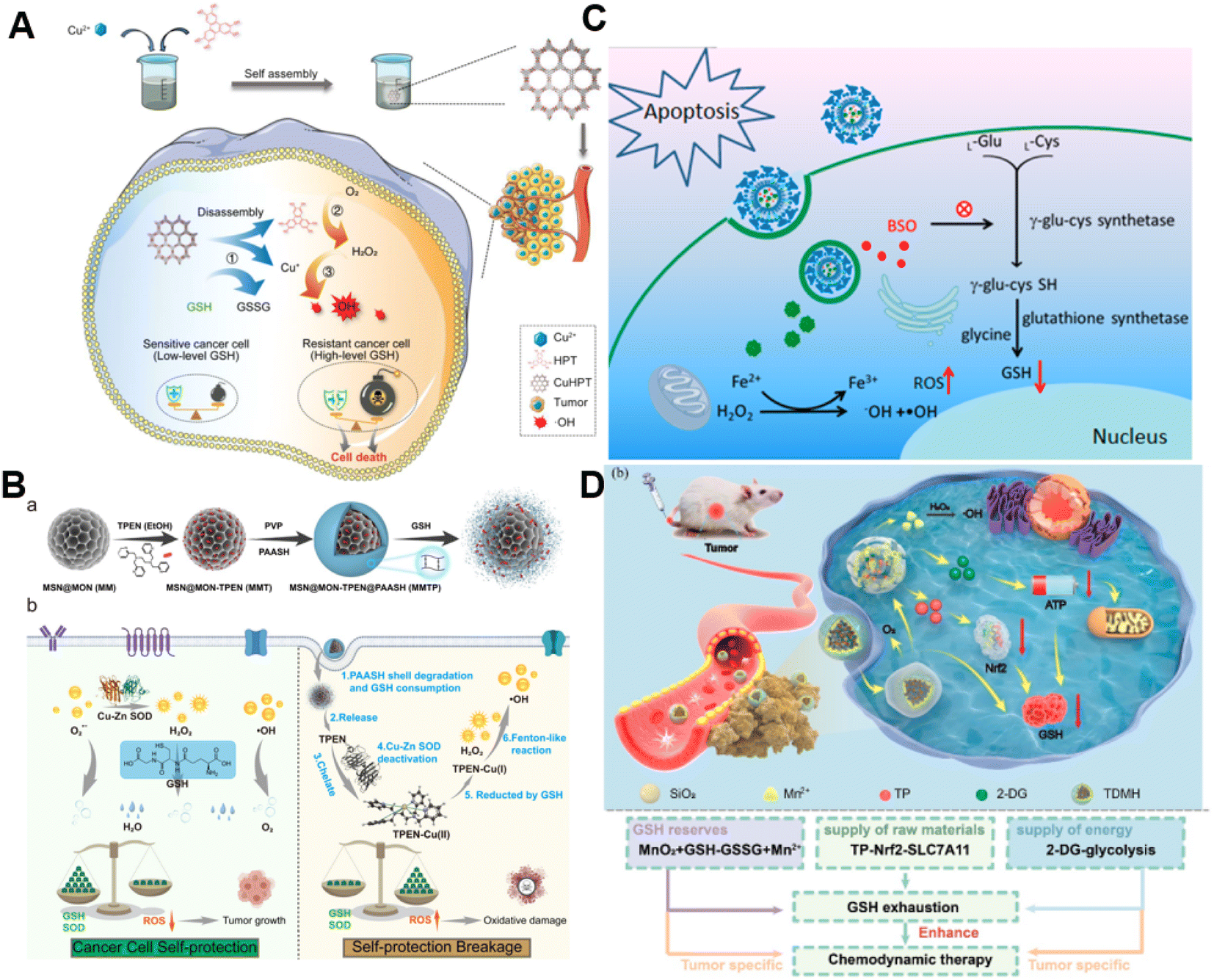 | ||
| Fig. 5 (A) The synthesis of CuHPT and scheme of its antitumor mechanisms against cancer cells. Reproduced with permission from ref. 79. Copyright 2022, American Chemical Society. (B) Illustration for the fabrication procedure and multifunctional application in vivo of MSN@MON-TPEN@PAASH nanoparticles. Reproduced with permission from ref. 82. Copyright 2019, American Chemical Society. (C) A diagram of BSO/GA–Fe(II)@liposome for enhanced CDT via reducing GSH biosynthesis. Reproduced with permission from ref. 84. Copyright 2021, American Chemical Society. (D) Schematic illustration of the use of the TDMH nanomedicine for multistage GSH depletion-augmented CDT. Reproduced with permission from ref. 85. Copyright 2022, American Chemical Society. | ||
Normally, GSH biosynthesis occurs in the cytosol of almost all cells.83 Blocking GSH synthesis using agents has been a good way to decrease GSH levels in cancer cells. Dong and co-workers reported a biocompatible liposomal nanoformulation containing L-buthionine sulfoximine (BSO) and gallic acid-ferrous ion (GA-Fe(II)) complexes to inhibit the synthesis of GSH for combined CDT-RT (Fig. 5C).84 Interestingly, GA boosted the transformation of Fe3+ into Fe2+, promoting the Fenton reaction to generate highly cytotoxic ˙OH. Besides, BSO was capable of limiting the GSH synthesis rate via inhibiting the activity of glutamate-cysteine ligase to reduce GSH levels, thus intensifying the Fenton reaction-enabled cancer cell damage and CDT. Moreover, Huang and co-workers developed a multi-faceted GSH depletion-potentiated CDT system (TP/2-DG@HMnO2@HA, TDMH), in which 2-deoxy-D-glucose (2-DG) and triptolide (TP) were co-encapsulated in hollow mesoporous MnO2 (HMnO2) followed by modifying HA on the nanoparticles for tumor targeting (Fig. 5D).85 When the nanomedicine reached the tumor area, MnO2 participated in a redox reaction with GSH, yielding glutathione disulfide (GSSG) and Mn2+ that could drive the Fenton-like reaction with the assistance of bicarbonate to lead to intracellular GSH consumption as well as ˙OH production. Meanwhile, the disintegration of the HMnO2 structure occurred, triggering the release of TP and 2-DG. TP downregulated solute carrier family 7 member 11 (SLC7A11) expression and decreased cysteine uptake, and its combination with the glycolysis inhibitor 2-DG could significantly reduce the intracellular ATP level and block the regeneration of GSH synthesis. Owing to the synergistic effect of GSH depletion and GSH synthesis inhibition, this nanoplatform efficiently strengthened CDT against tumors.
2.5 Regulation of lipid metabolism
The ROS accumulation induced by Fenton-type reactions can generally cause LPO. PUFAs, a family of membrane lipids possessing two or more carbon–carbon double bonds, are extremely susceptible to ROS-initiated LPO.86,87 Therefore, the CDT susceptibility of cancer cells can be raised by increasing PUFA content in the lipid chain.88 Taking advantage of this strategy, our group constructed a chemodynamic nanoagent (OA@Fe-SAC@EM) capable of modulating PUFA metabolism by loading oleanolic acid (OA) onto a single-atom Fe-anchored hollow carbon nanosphere (Fe-SAC) and further coating with an erythrocyte membrane (EM) for enhanced CDT (Fig. 6A).89 After internalization by tumor cells, Fe-SAC could catalyze intracellular H2O2 to generate ˙OH, which not only mediated CDT but also induced the disruption of the surface EM for the controlled release of OA. Notably, OA caused a dramatically elevated expression of ACSL4, thus increasing membrane unsaturation via enriched PUFAs. As a result, this nanoagent exerted enhanced chemodynamic efficiency by LPO amplification. Another relevant study was developed by Liu and co-workers. They successfully prepared the FeCo/Fe–Co dual-atom nanozyme (FeCo/Fe–Co DAzyme) possessing four-enzyme-like activities and subsequently loaded lipoxygenase (LOX) and phospholipase A2 (PLA2) into these NPs for the fabrication of a six-enzyme co-expressed nanoplatform (FeCo/Fe–Co DAzyme/PL) to enhance catalytic therapy and immunotherapy (Fig. 6B).90 FeCo/Fe–Co DAzyme from this nanomedicine exhibited POD-like and OXD-like activities to catalyze both O2 and H2O2 to generate ˙O2− and ˙OH, as well as exerted glutathione oxidase (GSHOx)-like activity for GSH elimination to reduce ˙OH consumption and enhanced ROS-triggered LPO. Meanwhile, FeCo/Fe–Co DAzyme also in situ converts H2O2 into O2 that could combine with free arachidonic acid (AA) liberated from PLA2-catalyzed phospholipids, producing AA-OOH through the catalysis of LOX. These two processes synergistically induced an LPO self-amplification effect for effectively initiating immunogenic ferroptosis. The resulting systemic immune activation mediated the reprogramming of ACSL4-associated phospholipids and further strengthened the cascade of immunogenic tumor ferroptosis when combined with AA. Therefore, this strategy targeting lipid metabolism provides new insight into the design of ROS-activated immunotherapy. Additionally, Yang and co-workers co-loaded Fenton catalysts hemin and lipoxidase into a pH-responsive self-assembled CaCO3-encapsulated poly(lactic-co-glycolic acid) (PLGA) nanoreactor for cascade catalytic amplification of tumor oxidative stresses.91 Upon entering tumor cells, CaCO3 could be degraded in the acidic microenvironment to release hemin and lipoxidase. Hemin catalyzed the Fenton reaction to convert H2O2 into toxic ˙OH for ROS-initiated LPO. Moreover, lipoxidase further oxidized PUFA to generate lipid hydroperoxides and led to massive lipid hydroperoxide accumulation as well as aggravated membrane LPO, eventually causing tumor cell death.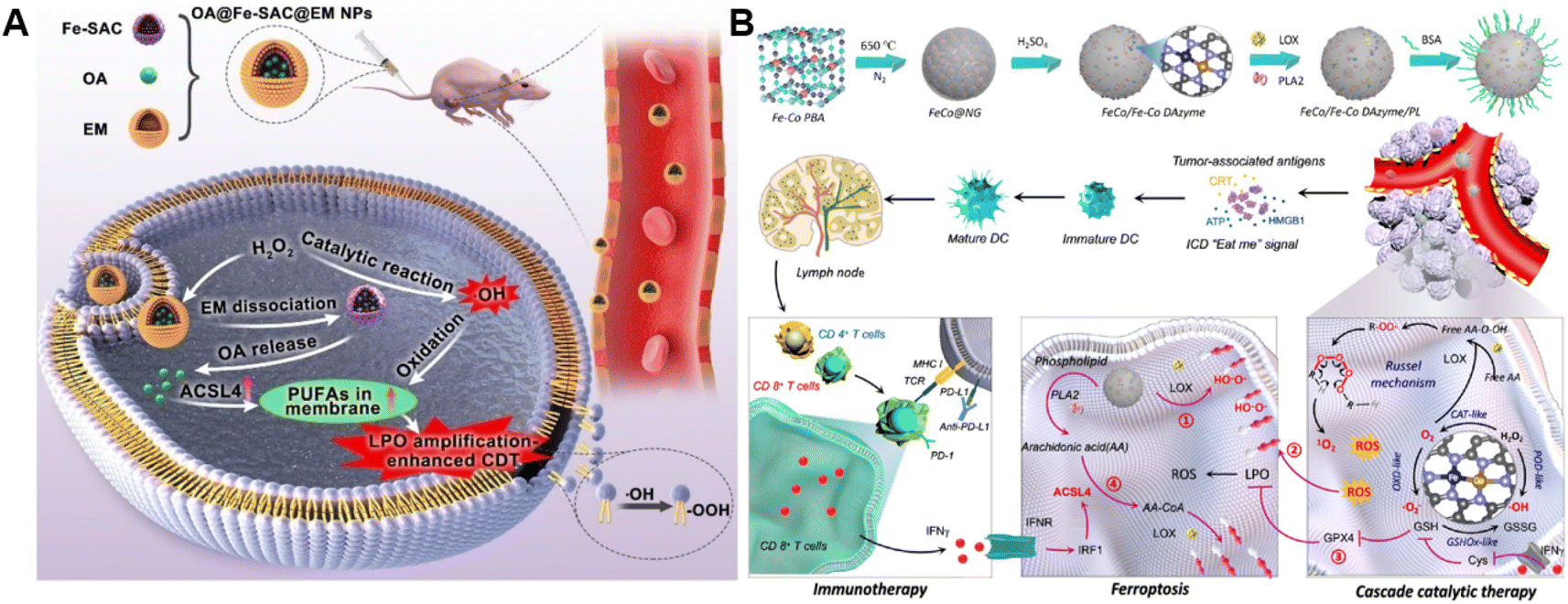 | ||
| Fig. 6 (A) Schematic diagram of the preparation process of OA@Fe-SAC@EM as a self-reinforcing CDT agent via the manipulation of cell membrane unsaturation. Reproduced with permission from ref. 89. Copyright 2023, Wiley-VCH GmbH. (B) Schematic illustration of the design and proposed therapeutic mechanism of FeCo/Fe–Co DAzyme. Reproduced with permission from ref. 90. Copyright 2023, American Chemical Society. | ||
3. Summary and outlook
In this perspective, we focused on the metabolic pathways of endogenous molecules within the TME and discussed the availability of these pathways in modulating Fenton chemistry for antitumor CDT. We briefly outlined the fundamental principles of the regulatory network involving endogenous molecules. We then thoroughly discussed the relevance of Fenton chemistry to H2O2, iron, lactate, GSH, and lipid metabolism. In these sections, we described recent progress in developing chemodynamic agents that modulate endogenous metabolites to strengthen tumor CDT and tried to attract broad interest of researchers from various fields. While interfering with multiple endogenous metabolic regulations has shown promise in boosting the effectiveness of traditional CDT therapies, addressing the following issues is crucial to advancing the development of CDT.Firstly, the endogenous metabolic pathways of cancer cells varied greatly due to the significant difference in tumor types, sizes, locations, and stages. Therefore, selecting appropriate experiment models and developing antitumor strategies with enhanced targeting ability are essential to achieve precise therapy. Secondly, the metabolic network of cancer cells is complex, and there can be crosstalk among different metabolic pathways involving various molecules. Inhibiting metabolic pathways of a single endogenous molecule may lead to the development of compensatory metabolic pathways in tumor cells, conferring resistance to the single intervention and compromising therapeutic efficacy. Undoubtedly, gaining a comprehensive understanding of the interdependencies among different metabolism systems would provide us with new ideas for the design of endogenous regulation-based CDT. Additionally, we have only explored several endogenous metabolic pathways while the abundance of other cellular metabolites has not been exploited for their roles in influencing CDT, where research in this field is much needed. Meanwhile, it is important to consider that most of the metabolic pathways associated with alternative molecules are pervasively present in both normal and cancer cells. Thus, plenty of efforts should be devoted to enhancing the tumor-specificity of these chemodynamic nanomedicines that regulate endogenous metabolites, aiming to mitigate their adverse effects on normal tissues and organs. Despite the existence of certain challenges, growing knowledge about Fenton chemistry and its related endogenous regulators foresees the emergence of novel avenues and significant potential for endogenous regulation-based strategies to enhance CDT in the field of anti-tumor therapy.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this perspective.Author contributions
All authors participated in this review work. J. Wang, T. T. Cui, and L. S. Lin conceived the structure of this perspective. J. Wang, Y. N. Liu, and T. T. Cui prepared the draft. H. H. Yang and L. S. Lin reviewed and refined the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2020YFA0210800), the National Natural Science Foundation of China (22374026 and 22027805), and the Major Project of Science and Technology of Fujian Province (2020HZ06006).Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- P. Gotwals, S. Cameron, D. Cipolletta, V. Cremasco, A. Crystal, B. Hewes, B. Mueller, S. Quaratino, C. Sabatos-Peyton, L. Petruzzelli, J. A. Engelman and G. Dranoff, Nat. Rev. Cancer, 2017, 17, 286–301 CrossRef CAS PubMed.
- J. Mateo, L. Steuten, P. Aftimos, F. André, M. Davies, E. Garralda, J. Geissler, D. Husereau, I. Martinez-Lopez, N. Normanno, J. S. Reis-Filho, S. Stefani, D. M. Thomas, C. B. Westphalen and E. Voest, Nat. Med., 2022, 28, 658–665 CrossRef CAS PubMed.
- S. H. Shin, A. M. Bode and Z. Dong, npj Precis. Oncol., 2017, 1, 12 CrossRef PubMed.
- M. P. Murphy, H. Bayir, V. Belousov, C. J. Chang, K. J. A. Davies, M. J. Davies, T. P. Dick, T. Finkel, H. J. Forman, Y. Janssen-Heininger, D. Gems, V. E. Kagan, B. Kalyanaraman, N.-G. Larsson, G. L. Milne, T. Nyström, H. E. Poulsen, R. Radi, H. Van Remmen, P. T. Schumacker, P. J. Thornalley, S. Toyokuni, C. C. Winterbourn, H. Yin and B. Halliwell, Nat. Metab., 2022, 4, 651–662 CrossRef PubMed.
- X. Jing, F. Yang, C. Shao, K. Wei, M. Xie, H. Shen and Y. Shu, Mol. Cancer, 2019, 18, 157 CrossRef PubMed.
- H. Pelicano, D. Carney and P. Huang, Drug Resistance Updates, 2004, 7, 97–110 CrossRef CAS PubMed.
- E. Ju, K. Dong, Z. Chen, Z. Liu, C. Liu, Y. Huang, Z. Wang, F. Pu, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2016, 55, 11467–11471 CrossRef CAS.
- Q. Dai, L. Wang, E. Ren, H. Chen, X. Gao, H. Cheng, Y. An, C. Chu and G. Liu, Angew. Chem., Int. Ed., 2022, 61, e202211674 CrossRef CAS PubMed.
- J. Fu, T. Li, Y. Zhu and Y. Hao, Adv. Funct. Mater., 2019, 29, 1906195 CrossRef CAS.
- X. Niu, Y. Zhu, C. Ding, J. Ma, P. Wei, Y. Lin, W. Fang, Q. He, C. Li, J. Cheng, J. Zou, L. Lin, X. Chen and D. Kang, Adv. Funct. Mater., 2023, 33, 2306778 CrossRef CAS.
- L.-S. Lin, T. Huang, J. Song, X.-Y. Ou, Z. Wang, H. Deng, R. Tian, Y. Liu, J.-F. Wang, Y. Liu, G. Yu, Z. Zhou, S. Wang, G. Niu, H.-H. Yang and X. Chen, J. Am. Chem. Soc., 2019, 141, 9937–9945 CrossRef CAS PubMed.
- B. Guo, F. Yang, L. Zhang, Q. Zhao, W. Wang, L. Yin, D. Chen, M. Wang, S. Han, H. Xiao and N. Xing, Adv. Mater., 2023, 35, e2212267 CrossRef PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- R. Zhao, Y. Zhu, L. Feng, B. Liu, Y. Hu, H. Zhu, Z. Zhao, H. Ding, S. Gai and P. Yang, Adv. Mater., 2024, 36, e2307115 CrossRef PubMed.
- M. Yu, Z. Ye, S. Liu, Y. Zhu, X. Niu, J. Wang, R. Ao, H. Huang, H. Cai, Y. Liu, X. Chen and L. Lin, Angew. Chem., Int. Ed., 2024, 63, e202318155 CrossRef CAS.
- T. Chen, P. Hou, Y. Zhang, R. Ao, L. Su, Y. Jiang, Y. Zhang, H. Cai, J. Wang, Q. Chen, J. Song, L. Lin, H. Yang and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 15006–15012 CrossRef CAS PubMed.
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS.
- Y. Zhou, S. Fan, L. Feng, X. Huang and X. Chen, Adv. Mater., 2021, 33, e2104223 CrossRef PubMed.
- X. Wang, X. Zhong, Z. Liu and L. Cheng, Nano Today, 2020, 35, 100946 CrossRef CAS.
- S. L. Li, P. Jiang, F. L. Jiang and Y. Liu, Adv. Funct. Mater., 2021, 31, 2100243 CrossRef CAS.
- C. Cao, X. Wang, N. Yang, X. Song and X. Dong, Chem. Sci., 2022, 13, 863–889 RSC.
- Y. Wu, Y. Li, G. Lv and W. Bu, Chem. Sci., 2022, 13, 2202–2217 RSC.
- T. Chen, Z. Chen, Q. Zhou, H. Ding, P. Gong, J. Wang, H. Cai, R. Ao, M. Yu, J. Song, L. Lin and H. Yang, Adv. Funct. Mater., 2022, 32, 2208720 CrossRef CAS.
- Q. Tian, F. Xue, Y. Wang, Y. Cheng, L. An, S. Yang, X. Chen and G. Huang, Nano Today, 2021, 39, 101162 CrossRef CAS.
- D. Jana and Y. Zhao, Explore, 2022, 2, 20210238 CAS.
- Y. Zhu, W. Wang, P. Gong, Y. Zhao, Y. Pan, J. Zou, R. Ao, J. Wang, H. Cai, H. Huang, M. Yu, H. Wang, L. Lin, X. Chen and Y. Wu, ACS Nano, 2023, 17, 3064–3076 CrossRef CAS PubMed.
- S. Zhang, L. Jin, J. Liu, Y. Wang, T. Zhang, Y. Liu, Y. Zhao, N. Yin, R. Niu, D. Xue, Y. Yu and Y. Yang, Adv. Funct. Mater., 2022, 32, 2113397 CrossRef CAS.
- P. Icard and H. Lincet, Biochim. Biophys. Acta, 2012, 1826, 423–433 CAS.
- L. K. Boroughs and R. J. DeBerardinis, Nat. Cell Biol., 2015, 17, 351–359 CrossRef CAS PubMed.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed.
- J. Yang, Y. Zhao, Y. Zhou, X. Wei, H. Wang, N. Si, J. Yang, Q. Zhao, B. Bian and H. Zhao, Biomaterials, 2022, 286, 121565 CrossRef CAS PubMed.
- F. Gao, J.-H. Dong, C. Xue, X.-X. Lu, Y. Cai, Z.-Y. Tang and C.-J. Ou, Small, 2024, 2310248 CrossRef.
- H. Wu, Y. Wang, M. Ying, C. Jin, J. Li and X. Hu, Signal Transduct. Target Ther., 2021, 6, 242 CrossRef CAS PubMed.
- P. A. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- X. R. Xu, H. B. Li, W. H. Wang and J. D. Gu, Chemosphere, 2004, 57, 595–600 CrossRef CAS PubMed.
- J. Wang and K. Pantopoulos, Biochem. J., 2011, 434, 365–381 CrossRef CAS PubMed.
- J. Zheng and M. Conrad, Cell Metab., 2020, 32, 920–937 CrossRef CAS.
- Y. Xie, W. Hou, X. Song, Y. Yu, J. Huang, X. Sun, R. Kang and D. Tang, Cell Death Differ., 2016, 23, 369–379 CrossRef CAS PubMed.
- C. Liang, X. Zhang, M. Yang and X. Dong, Adv. Mater., 2019, 31, e1904197 CrossRef PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascon, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS PubMed.
- X. Chen, H. Zhang, M. Zhang, P. Zhao, R. Song, T. Gong, Y. Liu, X. He, K. Zhao and W. Bu, Adv. Funct. Mater., 2020, 30, 1908365 CrossRef CAS.
- C. S. Hong, N. A. Graham, W. Gu, C. Espindola Camacho, V. Mah, E. L. Maresh, M. Alavi, L. Bagryanova, P. A. L. Krotee, B. K. Gardner, I. S. Behbahan, S. Horvath, D. Chia, I. K. Mellinghoff, S. A. Hurvitz, S. M. Dubinett, S. E. Critchlow, S. K. Kurdistani, L. Goodglick, D. Braas, T. G. Graeber and H. R. Christofk, Cell Rep., 2016, 14, 1590–1601 CrossRef CAS PubMed.
- J. Gu, J. Sun, Y. Liu, G. Chong, Y. Li and H. Dong, Nano Res., 2022, 16, 654–671 CrossRef.
- T. Liu, L. Sun, Y. Zhang, Y. Wang and J. Zheng, J. Biochem. Mol. Toxicol., 2022, 36, e22942 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- G. Wu, J. R. Lupton, N. D. Turner, Y.-Z. Fang and S. Yang, J. Nutr., 2004, 134, 489–492 CrossRef CAS PubMed.
- A. Bansal and M. C. Simon, J. Cell Biol., 2018, 217, 2291–2298 CrossRef CAS PubMed.
- B. Niu, K. Liao, Y. Zhou, T. Wen, G. Quan, X. Pan and C. Wu, Biomaterials, 2021, 277, 121110 CrossRef CAS PubMed.
- L. E. Pope and S. J. Dixon, Trends Cell Biol., 2023, 33, 1077–1087 CrossRef CAS.
- Y. Zou, W. S. Henry, E. L. Ricq, E. T. Graham, V. V. Phadnis, P. Maretich, S. Paradkar, N. Boehnke, A. A. Deik, F. Reinhardt, J. K. Eaton, B. Ferguson, W. Wang, J. Fairman, H. R. Keys, V. Dančík, C. B. Clish, P. A. Clemons, P. T. Hammond, L. A. Boyer, R. A. Weinberg and S. L. Schreiber, Nature, 2020, 585, 603–608 CrossRef CAS PubMed.
- W. Xuan, Y. Xia, T. Li, L. Wang, Y. Liu and W. Tan, J. Am. Chem. Soc., 2020, 142, 937–944 CrossRef CAS PubMed.
- P. Yu, X. Li, G. Cheng, X. Zhang, D. Wu, J. Chang and S. Wang, Chin. Chem. Lett., 2021, 32, 2127–2138 CrossRef CAS.
- K. Yang, G. Yu, Z. Yang, L. Yue, X. Zhang, C. Sun, J. Wei, L. Rao, X. Chen and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 17570–17578 CrossRef CAS PubMed.
- S. Wang, G. Yu, Z. Wang, O. Jacobson, L. S. Lin, W. Yang, H. Deng, Z. He, Y. Liu, Z. Y. Chen and X. Chen, Angew. Chem., Int. Ed., 2019, 58, 14758–14763 CrossRef CAS PubMed.
- C. Fang, Z. Deng, G. Cao, Q. Chu, Y. Wu, X. Li, X. Peng and G. Han, Adv. Funct. Mater., 2020, 30, 1910085 CrossRef CAS.
- Y. Ding, H. Xu, C. Xu, Z. Tong, S. Zhang, Y. Bai, Y. Chen, Q. Xu, L. Zhou, H. Ding, Z. Sun, S. Yan, Z. Mao and W. Wang, Adv. Sci., 2020, 7, 2001060 CrossRef CAS PubMed.
- T. J. Zhou, X. Wan, M. M. Zhang, D. M. Liu, L. L. Huang, L. Xing, Y. Wang and H. L. Jiang, Biomaterials, 2023, 300, 122205 CrossRef CAS.
- Y. Liu, R. Niu, R. Deng, Y. Wang, S. Song and H. Zhang, Adv. Mater., 2024, 36, e2307752 CrossRef PubMed.
- J.-X. Fan, M.-Y. Peng, H. Wang, H.-R. Zheng, Z.-L. Liu, C.-X. Li, X.-N. Wang, X.-H. Liu, S.-X. Cheng and X.-Z. Zhang, Adv. Mater., 2019, 31, 1808278 CrossRef.
- W. Li, Y. Wang and A. Irini, Chem. Eng. J., 2014, 244, 1–8 CrossRef CAS.
- R. Yang, Z. Ouyang, H. Guo, J. Qu, J. Xia, M. Shen and X. Shi, Nano Today, 2022, 46, 101615 CrossRef CAS.
- L. M. Bystrom, M. L. Guzman and S. Rivella, Antioxid. Redox Signal., 2012, 20, 1917–1924 CrossRef PubMed.
- Y. Liu, S. Zhai, X. Jiang, Y. Liu, K. Wang, C. Wang, M. Zhang, X. Liu and W. Bu, Adv. Funct. Mater., 2021, 31, 2010390 CrossRef CAS.
- J. Wang, H. Ding, Y. Zhu, Y. Liu, M. Yu, H. Cai, R. Ao, H. Huang, P. Gong, Y. Liao, Z. Chen, L. Lin, X. Chen and H. Yang, Angew. Chem., Int. Ed., 2023, 62, e202302255 CrossRef CAS PubMed.
- J. Du, M. Zhou, Q. Chen, Y. Tao, J. Ren, Y. Zhang and H. Qin, Adv. Funct. Mater., 2023, 33, 2215244 CrossRef CAS.
- S. Zhang, X. Wu, X. Liao and S. Zhang, J. Am. Chem. Soc., 2024, 146, 8567–8575 CrossRef CAS PubMed.
- L. Lin, S. Wang, H. Deng, W. Yang, L. Rao, R. Tian, Y. Liu, G. Yu, Z. Zhou, J. Song, H.-H. Yang, Z.-Y. Chen and X. Chen, J. Am. Chem. Soc., 2020, 142, 15320–15330 CrossRef CAS PubMed.
- S. Fu, R. Yang, L. Zhang, W. Liu, G. Du, Y. Cao, Z. Xu, H. Cui, Y. Kang and P. Xue, Biomaterials, 2020, 257, 120279 CrossRef CAS PubMed.
- Y. Liu, X. Ji, W. W. L. Tong, D. Askhatova, T. Yang, H. Cheng, Y. Wang and J. Shi, Angew. Chem., Int. Ed., 2018, 57, 1510–1513 CrossRef CAS.
- Q. Cheng, X.-L. Shi, Q.-L. Li, L. Wang and Z. Wang, Adv. Sci., 2024, 11, 2305662 CrossRef CAS PubMed.
- F. Tian, S. Wang, K. Shi, X. Zhong, Y. Gu, Y. Fan, Y. Zhang and M. Yang, Adv. Sci., 2021, 8, e2102595 CrossRef PubMed.
- L. Shi, Y. Wang, C. Zhang, Y. Zhao, C. Lu, B. Yin, Y. Yang, X. Gong, L. Teng, Y. Liu, X. Zhang and G. Song, Angew. Chem., Int. Ed., 2021, 60, 9562–9572 CrossRef CAS PubMed.
- X. Wang, Y. Zhao, Y. Hu, Y. Fei, Y. Zhao, C. Xue, K. Cai, M. Li and Z. Luo, Small, 2021, 17, e2102269 CrossRef PubMed.
- W. Wang, J. Song, W. Yu, M. Chen, G. Li, J. Chen, L. Chen, L. Yu and Y. Chen, Adv. Funct. Mater., 2024, 2400929 CrossRef.
- S. Wu, L. Xu, C. He, P. Wang, J. Qin, F. Guo and Y. Wang, Adv. Sci., 2023, 10, e2300686 CrossRef PubMed.
- J. Lu, Z. Jiang, J. Ren, W. Zhang, P. Li, Z. Chen, W. Zhang, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2022, 61, e202114373 CrossRef CAS PubMed.
- L. S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H. H. Yang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- J. Liu, Y. Yuan, Y. Cheng, D. Fu, Z. Chen, Y. Wang, L. Zhang, C. Yao, L. Shi, M. Li, C. Zhou, M. Zou, G. Wang, L. Wang and Z. Wang, J. Am. Chem. Soc., 2022, 144, 4799–4809 CrossRef CAS PubMed.
- J. R. Winther and C. Thorpe, Biochim. Biophys. Acta, 2014, 1840, 838–846 CrossRef CAS PubMed.
- Y. Li, P. Zhao, T. Gong, H. Wang, X. Jiang, H. Cheng, Y. Liu, Y. Wu and W. Bu, Angew. Chem., Int. Ed., 2020, 59, 22537–22543 CrossRef CAS PubMed.
- Y. Guo, Y. Xu, Q. Bao, C. Shen, D. Ni, P. Hu and J. Shi, ACS Nano, 2021, 15, 16286–16297 CrossRef CAS PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS PubMed.
- Z. Dong, L. Feng, Y. Chao, Y. Hao, M. Chen, F. Gong, X. Han, R. Zhang, L. Cheng and Z. Liu, Nano Lett., 2019, 19, 805–815 CrossRef CAS.
- Y. Huang, S. Wu, L. Zhang, Q. Deng, J. Ren and X. Qu, ACS Nano, 2022, 16, 4228–4238 CrossRef CAS PubMed.
- L. Magtanong, P. J. Ko and S. J. Dixon, Cell Death Differ., 2016, 23, 1099–1109 CrossRef CAS PubMed.
- J. Luo, Y. Li, Y. Li, X. Chen, P. Du, Z. Wang, A. Tian and Y. Zhao, ACS Nano, 2023, 17, 25257–25268 CrossRef CAS PubMed.
- K. D'Herde and D. V. Krysko, Nat. Chem. Biol., 2017, 13, 4–5 CrossRef PubMed.
- Y. Zhu, P. Gong, J. Wang, J. Cheng, W. Wang, H. Cai, R. Ao, H. Huang, M. Yu, L. Lin and X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202218407 CrossRef CAS PubMed.
- Y. Liu, R. Niu, R. Deng, S. Song, Y. Wang and H. Zhang, J. Am. Chem. Soc., 2023, 145, 8965–8978 CrossRef CAS PubMed.
- Z. Yang, Y. Zhu, Z. Dong, W. Li, N. Yang, X. Wang, L. Feng and Z. Liu, Nat. Commun., 2021, 12, 4299 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |