DOI:
10.1039/D4SC02116A
(Edge Article)
Chem. Sci., 2024,
15, 10519-10528
Helical polycyclic hydrocarbons with open-shell singlet ground states and ambipolar redox behaviors†
Received
31st March 2024
, Accepted 28th May 2024
First published on 4th June 2024
Abstract
Organic π-conjugated polycyclic hydrocarbons (PHs) with an open-shell diradical character are attracting increasing interest due to their promising applications in organic electronics and spintronics. However, most of the open-shell PHs synthesized thus far are based on planar π-conjugated molecules. Herein, we report the synthesis and characterization of two new quinodimethane-embedded expanded helicenes H1 and H2. The helical structures of both molecules were revealed using X-ray crystallographic analysis. It was elucidated in detailed experimental and theoretical studies that they possess an open-shell singlet biradical structure in the ground state and show a small energy gap and amphoteric redox behavior. Both compounds can also be easily oxidized or reduced into relatively stable charged species. The dianions of H1 and H2 exhibit similar electronic structures to the respective isoelectronic structures of their all-benzenoid helical analogues according to NMR measurements and theoretical calculations. Moreover, the structures of the dication and dianion of H2 were identified by X-ray crystallographic analysis, revealing the effect of electron transfer on their backbones and aromaticity. This study thus opens up new avenues for both helical polycyclic π-systems and diradicaloids.
Introduction
Organic π-conjugated open-shell polycyclic hydrocarbons (PHs) have attracted a great deal of interest because of their unique optical, electronic, and magnetic properties, which make them promising candidates suitable for functional materials in organic electronic and spintronic devices.1 According to broken symmetry density functional theory (DFT) calculations, any molecule with a nonzero diradical character in the ground state can be classified as a diradicaloid (diradical-like molecule). It has been demonstrated that the physical properties of open-shell singlet diradicaloids closely correlate with their diradical character.2 However, PHs with a larger diradical character are expected to show higher reactivity; therefore, instability remains a key obstacle for their practical applications. Over the past few decades, various families of stable diradicaloids based on indenofluorenes,3 zethrenes,4 anthenes,5 bisphenalenyl,6 and extended quinodimethanes (QDMs)7 through thermodynamic and kinetic stabilizing strategies have been synthesized to elucidate the interaction of two unpaired electrons and the properties originating from their unique electronic structures. So far, however, most reported open-shell PHs are derived from planar π-conjugated skeletons, and there are only a few examples based on helical systems. For example, cethrene,8 diindeno-fused bischrysen,9 5,14-diaryldiindeno[2,1-f:1′,2′-j]picene,10para-QDM embedded double [6]helicene,11 and dinor[7]helicene12 were developed by the groups of Juríček, Feng, Wu, Chi, and Stępień, respectively, and these demonstrated an open-shell singlet diradical character and unique electronic properties. Despite these encouraging advances, structural diversity of helical singlet diradicaloids is still limited, which hampers detailed understanding of their electronic structures and properties and the exchange interaction between radicals on the 3D framework. On the other hand, there has been very limited exploration of the effects of electron transfer in these systems, which are of great fundamental importance to understand their electronic and structural responses. In this context, here, we designed and synthesized two novel open-shell helical π-conjugated PHs (H1 and H2) in which one m-QDM or π-extended p-QDM unit is fused by two phenanthrene rings. We also demonstrated that the recovery of Clar's13 aromatic sextet rings (the hexagons shaded in blue color in Fig. 1) in the diradical canonical form can serve as a major driving force to form diradicals. For both molecules, two additional aromatic sextet rings can be gained from their closed-shell forms to their diradical forms, and thus, a moderate diradical character can be expected for both molecules. Therefore, they are expected to show significant magnetic characters. Considering their potential open-shell diradical character, bulky and electron deficient 2,6-dichlorophenyl groups are attached onto two cyclopenta (CP) rings to kinetically block the reactive sites. In addition, two-electron oxidation/reduction of the two diradicals may induce aromaticity switching. In particular, their dianions could be regarded as isoelectronic structures of the corresponding all-benzenoid expanded helicenes because of two CP rings in the dianion being aromatic and equal to the benzene ring. Therefore, their aromaticity in neutral and charged forms was discussed, which is also of great interest to understand their unique electronic structures. These characteristics arising from both their helical non-planar structure and aromaticity tunability also make them appealing for photoswitchable applications.14 In this work, we disclose their synthesis, (anti)aromaticity and magnetic, photophysical and electrochemical properties, together with in-depth elucidation of their redox states.
 |
| | Fig. 1 Representative resonance structures of H1/H2 and their charged species. The structures of all-benzenoid expanded helicenes are also shown. | |
Results and discussion
Synthesis
The synthesis of H1 and H2 was mainly based on a Suzuki coupling and intramolecular Friedel–Crafts alkylation followed by an oxidative dehydrogenation strategy (Scheme 1). First, palladium-catalyzed Suzuki coupling between equimolar amounts of 1-iodo-2-bromobenzene 3 and boronic acid 4 followed by desilylation gave the biphenyl 5 in 86% yield. Subsequent intramolecular cyclization provided the phenanthrene monobromide 6 in 82% yield. Next, palladium-catalyzed Suzuki–Miyaura borylation of 6 gave the key intermediate pinacol borate 7 in 76% yield. Suzuki coupling between 7 and 8 or 11 (ref. 15) produced the dialdehyde intermediates 9 and 12, respectively. Treatment of 9/12 with 2,6-dichlorophenylmagnesium chloride reagent afforded the corresponding diols, which were subjected to a BF3·OEt2-mediated Friedel–Crafts alkylation reaction to generate the dihydro-precursors 10/13. Finally, deprotonation of 10/13 by KOBut (to generate dianions) followed by oxidization by p-chloranil provided the final target compounds H1 and H2 in 87% and 85% yield, respectively. The bulky 2,6-dichlorophenyl groups are essential to kinetically block the reactive sites, and also make the final products soluble. Both H1 and H2 are stable and can be purified by normal silica gel column chromatography.
 |
| | Scheme 1 Synthetic route for H1 and H2: (a) (1) Pd(PPh3)4, K2CO3, THF/H2O; (2) K2CO3, MeOH/DCM; (b) PtCl2, DCE; (c) B(pin)2, Pd(dppf)Cl2, dioxane; (d) Pd(PPh3)4, K2CO3, THF/H2O; (e) (1) 2,6-dichloro-1-bromobenzene, isopropylmagnesium chloride, THF; (2) boron trifluoride etherate, DCM; (f) (1) potassium tert-butoxide, 18-crown-6, THF; (2) p-chloranil. | |
X-ray crystallographic analysis
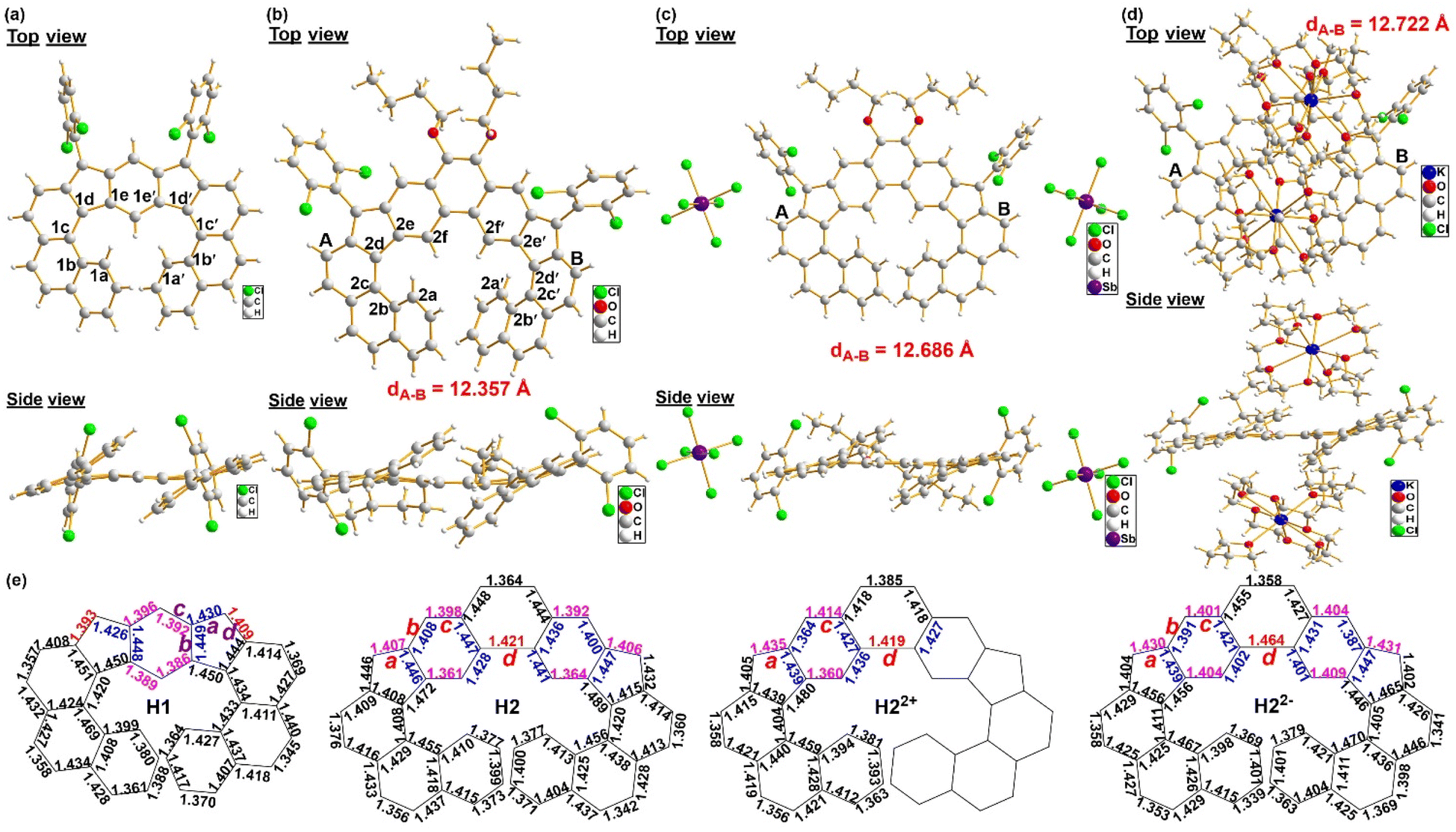
Single crystals suitable for X-ray crystallographic analysis were obtained for H1 and H2 by a slow solvent diffusion method.16H1/H2 show a helical π-conjugated structure (Fig. 2a and b). The extents of twisting in the helical structures of H1/H2 were obtained by analyzing the corresponding torsion angles, and the geometries of H1/H2 deviate from C2 symmetry due to the helical twisting, which introduces chirality and asymmetry in the molecule, breaking the C2 symmetry. As a result, both molecules adopt a C1 symmetry, which is reflected by the unequal torsion angles of the two cyclopenta-fused [5]helicene substructures. As defined by the selected dihedral angles, the angles of torsion are 21.19° for C1b–C1c–C1d–C1e, 24.08° for C1b′–C1c′–C1d′–C1e′ (Fig. 2a), 21.49° for C2b–C2c–C2d–C2e and 22.17° for C2b′–C2c′–C2d′–C2e′ (Fig. 2b). In H1, bonds a (1.430 Å) and d (1.409 Å) are longer than those in typical olefins (1.33–1.34 Å), and there is also large bond length alternation between bonds b (1.449 Å) and c (1.392 Å) in the central benzenoid rings (Fig. 2e), indicating that the molecule still remains a major s-indacene sub-structure after annulation with two respective aromatic phenanthrene rings. In H2, bonds a (1.407 Å), c (1.398 Å) and d (1.421 Å) are significantly longer than those in typical olefins, while bond b (1.408 Å) is much shorter than a typical C(sp2)–C(sp2) bond (∼1.47 Å) (Fig. 2e), indicating that the quinoidal character of the central extended para-quinodimethane unit is diminished by the emergence of the diradical contribution. The above bond length alternation together with the spin populations (Fig. S5†) demonstrates the significant contributions of their diradical resonance structures in the ground state. In the two molecules, the slightly but distinctly different length exo-methylene bond a is in accordance with their different singlet diradical character. Indeed, our spin-unrestricted DFT calculations (LC-UBLYP/6-31G(d,p)) predict a diradical character (y0) of 37.5% and 49.9% for H1 and H2, respectively, in agreement with the bond length analysis.
 |
| | Fig. 2 Top view and side view of the X-ray crystallographic structure of H1 (a), H2 (b), [H22+·2SbCl6−] (c) and [2K+·H22−] (d). (e) Selected bond lengths (in Å) of the backbone of H, H2, [H22+·2SbCl6−] and [2K+·H22−]. | |
Interestingly, both H1 and H2 contain paired enantiomers (P- and M-type) in the crystals (Fig. S19†), but our attempted resolution using various chiral columns was not successful so far, presumably due to the low racemization energy barrier (Fig. S2†). The racemization processes of H1 and H2 were found to proceed through the transition states with face-to-face oriented terminal aromatic rings (Fig. S2†). The transition states adopt a Cs symmetric structure, which is often observed in the transition states of other classical [n]helicenes with n ≥ 5.17 The calculated racemization barriers from C2-symmetric P- to M-forms for H1 and H2 were 6.1 and 7.4 kcal mol−1 at 298 K, respectively, which are much lower than that of classical [n]helicene (n ≥ 5, ΔG‡ ≥ 24.4 kcal mol−1).17 On the other hand, there is no π–π interaction between adjacent molecules in the 3D packing structures of H1/H2, and each molecule is connected to adjacent molecules through intermolecular [C–Cl⋯π] halogen bonds and [C–H⋯π] interactions (Fig. S22†).
Magnetic properties
The diradical nature of H1 and H2 was explored by variable temperature (VT) proton NMR spectroscopy (Fig. 3). Based on their calculated diradical characters and singlet–triplet gaps, one would expect the need for lower temperatures to observe clear NMR signals. As such, some peaks of H1 showed broadened 1H NMR signals in THF-d8 at room temperature, while decreasing the temperature led to sharpened peaks and finally the peaks became clear at 243 K (Fig. 3a). This is strong evidence that the molecule has an open-shell singlet ground state and the NMR broadening is due to the thermal population of triplet species at higher temperatures.18 For H2, its 1H NMR spectra in THF-d8 at room temperature showed a completely flat baseline for the aromatic region, while the aliphatic region displayed well-resolved butoxyl resonances (Fig. S1†). Upon cooling, some peaks did begin to come up while the aromatic signals still do not gain full resolution even after cooling down to 173 K (Fig. 3b). The aliphatic region in H2 become broad as the temperature was lowered (Fig. S1†). All of this indicates that the unresolved spectrum of H2 is a result of two main factors—moderate diradical character and slow configurational flipping at lower temperature (the calculated racemization barrier between the P and M forms of H2 is 7.4 kcal mol−1 at 298 K, Fig. S2†). Similar to the computations, these different results suggest that H1 and H2 have very different singlet–triplet energy gaps.
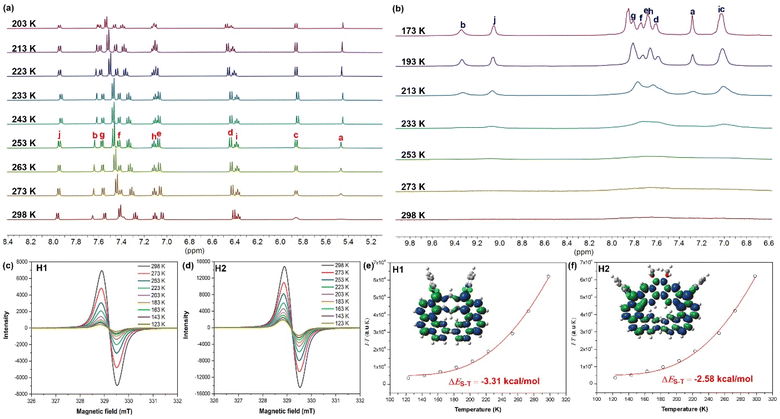 |
| | Fig. 3 VT 1H NMR spectra (aromatic region) of H1 (a) and H2 (b) in THF-d8 (refer to Scheme 1 for labelling). VT ESR spectra and fitted I × T–T curves by using the Bleaney–Bowers equation of compounds H1 (c and e) and H2 (d and f) in crystalline solid state. Inset is the calculated spin density distribution of the singlet diradical form. | |
The magnetic properties of both prepared samples were further investigated by variable temperature electron spin resonance (VT ESR) measurements. Strong ESR signals were observed in the crystalline solid at room temperature, and the ESR intensity of H1 and H2 decreased with decreasing temperature (Fig. 3c and d), in excellent agreement with their VT NMR data. The ESR signal intensity recorded at different temperatures could be fitted by the Bleaney–Bowers equation19 to provide a singlet–triplet gap of ΔES–T = −3.31 and −2.58 kcal mol−1 for H1 and H2, respectively (Fig. 3e and f). Furthermore, broken-symmetry DFT calculations (UCAM-B3LYP/6-31(d,p)) also predict that the energies of the open-shell singlet diradical states in H1/H2 are lower than that of the triplet diradical states, with a singlet–triplet energy gap (ΔES–T) of −1.78 and −1.47 kcal mol−1 for H1 and H2, respectively, indicating that they possess an open-shell singlet ground state, which is consistent with the above experimental results.
Optical and electrochemical properties
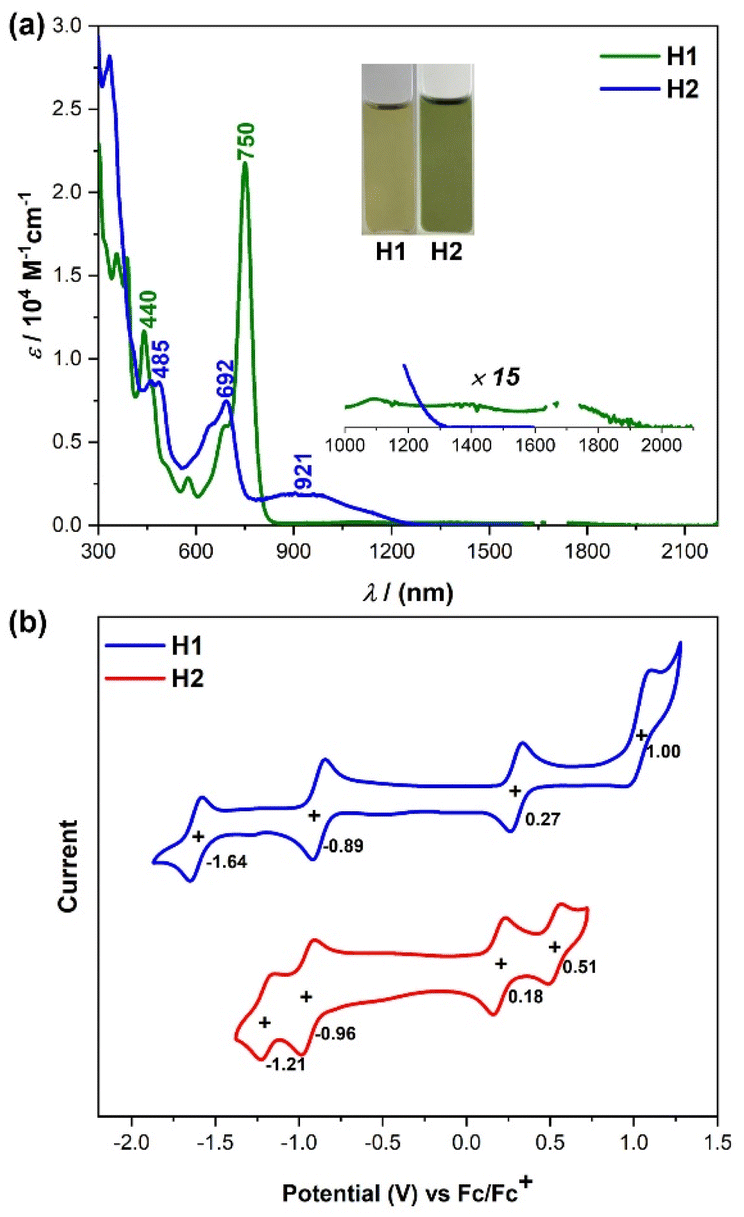
The optoelectronic properties of H1 and H2 were investigated by UV-vis absorption spectroscopy and time-dependent density functional theory (TD DFT) calculations. The new compounds show specific colors in a solution of dichloromethane (see the inset photos in Fig. 4a). These colors originate from intense absorptions in the visible region. The UV-vis absorption spectra of H1 and H2 in dichloromethane at room temperature are depicted in Fig. 4a. H1 shows a well-resolved absorption band with a maximum (λmax) at 750 nm (Fig. 4a), which is significantly redshifted as compared to that of the indeno[2,1-b]fluorene derivative (λmax = 638 nm)3b due to π-extension. TD DFT calculations suggest that this band originates from the HOMO−1 → LUMO electronic transition (f = 0.4254) (Fig. S6 and Table S1†). There is another weak broad absorption in the 900–2000 nm region, which mainly originates from the HOMO → LUMO electronic transition (f = 0.0265) according to TD DFT calculations (Fig. S6 and Table S1†). The electronic absorption spectrum for H2 in dichloromethane displays an intense absorption band with λmax at 692 nm and a weak broad absorption in the region of 750–1200 nm (Fig. 4a). Consistent with experimental results, TD DFT calculations predicted a broad absorption at 1362 nm (HOMO → LUMO transition, f = 0.2269) and an intense band at 768 nm (HOMO−1 → LUMO transition, f = 0.4161) (Fig. S10 and Table S5†). In addition, the optical energy gaps (Eoptg) of H1 and H2 on the basis of the lowest energy absorption onset were roughly estimated to be 0.63 and 0.92 eV, respectively.
 |
| | Fig. 4 (a) UV-vis-NIR spectra of H1 and H2 in DCM solutions. Inset shows photos of the solutions. (b) Cyclic voltammograms of H1 and H2 in DCM solutions. | |
To investigate the electrochemical properties of both radicals, cyclic voltammetry and differential pulse voltammetry measurements were carried out in a typical three-electrode electrochemical cell in a solution of tetrabutylammonium hexafluorophosphate (0.1 M) in dry DCM with a scan rate of 100 mV s−1 (Fig. 4b). H1 displayed one reversible oxidation wave and one irreversible oxidation wave with half-wave potential (Eox1/2) at 0.26 and 1.00 V, and two reversible reduction waves with half-wave potential (Ered1/2) at −0.89 and −1.64 V (vs. Fc/Fc+, Fc = ferrocene, Fig. 4b). H2 exhibited two reversible oxidation waves with Eox1/2 = 0.18 and 0.51 V and two reversible reduction waves with Ered1/2 = −0.96 and −1.21 V (vs. Fc/Fc+, Fig. 4b). By combining these results with the observed absorption edge, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of H1 and H2 were determined to be −5.00 and −3.99 eV for H1, and −4.92 and −3.92 eV for H2. Accordingly, the electrochemical energy gaps (EECg) are estimated to be 1.01 and 1.00 eV for H1 and H2, respectively, which are consistent with the trend observed in their optical energy band gaps.
Chemical oxidation and reduction
The good redox reversibility from the electrochemical data indicates that both compounds can be easily converted into stable charged species, thus offering the opportunity to investigate optical properties and aromaticity. The first one-electron oxidation of H1 and H2 with the oxidant NO·SbF6 gave rise to the corresponding radical cation species. The electronic absorption spectra for H1˙+ and H2˙+ display a weak broad absorption band in the region of 1500–3000 nm and multiple moderate bands in the visible and near-infrared (NIR) region (Fig. 5a and b). Further oxidation of H1 was not observed even when excess oxidant was added. The second oxidation of H2 with the oxidant NO·SbF6 generated a stable dication species, which exhibits strong shoulder peaks with λmax at 775 and 882 nm (Fig. 5b). The one-electron and two-electron reductions of H1 and H2 with freshly prepared sodium anthracenide in dry THF gave their radical anions and dianions, respectively (Fig. 5c and d). The radical anion of H1 shows an intense absorption band in the NIR region (λmax = 892 and 1572 nm, respectively, Fig. 5c), while the radical anion of H2 displays long wavelength absorption with a maximum beyond 2200 nm (Fig. 5d). The dianions of H1 and H2 exhibit an intense absorption band in the short-wavelength range (λmax = 474 nm for H12− and 468 nm for H22−, Fig. 5c and d). The observed spectra are in agreement with the TD DFT calculations (Fig. S7–S9/S11–S14 and Tables S2–S4/S6–S9†).
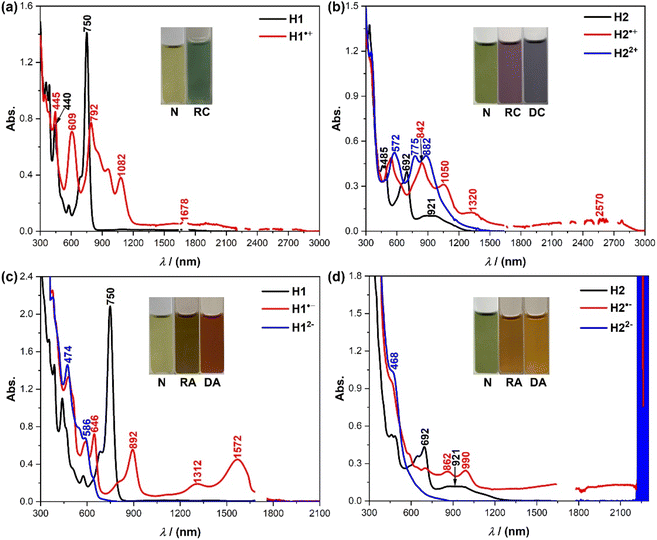 |
| | Fig. 5 (a) UV-vis-NIR absorption spectra of H1 and its radical cation (RC) in DCM. (b) UV-vis-NIR absorption spectra of H2, its radical cation (RC) and dication (DC) in DCM. (c) UV-vis-NIR absorption spectra of H1, its radical anion (RA) and dianion (DA) in THF. (d) UV-vis-NIR absorption spectra of H2, its radical anion (RA) and dianion (DA) in THF; the background absorbance at ca. 2250 nm may arise from the overtone of CH vibration of the solvent. Insets show photos of the solutions. | |
The dication of H2 and the dianions of H1 and H2 were in situ generated, and all showed sharp 1H NMR spectra at room temperature (Fig. 6 and S3†), thus indicating that all of these charged species possess a closed-shell singlet ground state. The 1H NMR spectra of the dication and dianion of H2 clearly show that protons b and j are shielded in the dicationic state, whereas protons a, b and j are highly deshielded in the dianionic state (Fig. 6). Similarly, the 1H NMR spectra of the dianion of H1 clearly show that most of the protons on the backbone appear at low field although the molecule contains two negative charges (Fig. S3†). Single crystals of the dication and dianion of H2 suitable for X-ray crystallographic analysis were successfully obtained.16 The X-ray diffraction characterization confirmed the formation of [H22+·2SbCl6−] (Fig. 2c) and [2K+·H22–] (Fig. 2d), respectively. The dication [H22+·2SbCl6−] has an axisymmetric structure with two counter cations located at the termini of the conjugated backbone, while the dianion [2K+·H22–] adopts a C1 symmetry with two counter cations located on the top and bottom of the H2 molecular plane. In the crystal structure of [2K+·H22–], both K+ ions are solvent-separated from the H22– core, avoiding direct metal ion–π interactions (Fig. 2d). The K+ ion is axially coordinated to an 18-crown-6 ether molecule (K⋯Ocrown, 2.783–2.832 Å) and capped by two THF molecules (K⋯OTHF, 2.670–2.693 Å), with all K⋯O distances being close to those previously reported.20 The lengths of bond a in the dication and dianion of H2 are 1.435 and 1.430 Å (Fig. 2e), respectively, which are much larger than that of a neutral molecule. A comparison of selected angles (Fig. 2) reveals that H2 exhibits structural deformation upon the addition or loss of two electrons. The torsion angle for C2b–C2c–C2d–C2e decreases from 21.49° in H2 to 12.90° in H22+ and 18.01° in H22−, respectively. Thus, the central helicene axis is compressed upon two-electron oxidation or reduction. In order to balance the resulting intramolecular steric hindrance, the helicene core (dA–B, Fig. 2) is lengthened from 12.357 Å in H2 to 12.686 Å in H22+ and 12.722 Å in H22−, respectively.
 |
| | Fig. 6
1H NMR spectra (aromatic region) of H2 (in CD2Cl2), H22+ (in CD2Cl2), and H22− (in [D8]THF) (refer to Scheme 1 for labelling). | |
DFT calculations
To deeply understand the NMR spectra and the difference of the (anti)aromaticity of individual rings at different redox states, nucleus independent chemical shift (NICS)21 and anisotropy of the induced current density (ACID)22 calculations were conducted at the CAM-B3LYP/6-31G(d,p) level of theory based on similar previous computational studies on large polycyclic systems.23 For the neutral compound H1, the three rings in the s-indacene unit have positive NICS(1)zz values (57.81–70.93, i.e., antiaromatic), the neighbouring benzenoid rings have also positive NICS(1)zz values (8.58), while the terminal naphthalene rings have negative NICS(1)zz values (−10.86 to −27.22, i.e., aromatic) (Fig. 7a). The ACID plot shows clockwise diatropic ring current flow along the terminal naphthalene units (aromatic), counter-clockwise paratropic ring current for the three rings in the s-indacene unit (antiaromatic), and there is counter-clockwise paratropic ring current for the benzenoid rings near the s-indacene unit (antiaromatic) (Fig. 7b), in accordance with the NICS(1)zz results. Accordingly, proton a in H1 appears at high field due to the shielding effect from the anti-aromatic s-indacene unit, while proton b in H1 appears at low field due to the deshielding effect from the aromatic naphthalene ring (Fig. 3a). For H2, the terminal phenanthrene rings have negative NICS(1)zz values (−12.75 to −29.14) and the CP rings have positive NICS(1)zz values (21.75), while the central benzenoid ring has a NICS(1)zz value of 0.87 (nearly non-aromatic) (Fig. 7a). The ACID plot displays clockwise diatropic ring current flow along the terminal phenanthrene units and counter-clockwise paratropic ring current at the CP rings, while there is almost no clear ring current for the central benzenoid ring (Fig. 7d). Accordingly, proton a in H2 appears at high field due to the shielding effect from the anti-aromatic CP ring, while proton b in H2 appears at low field due to the deshielding effect from the aromatic phenanthrene ring (Fig. 3b). NICS and ACID calculations of the dication H22+ reveal the aromatic character of the terminal benzene rings, while the nearby benzene rings are nearly non-aromatic (Fig. 7a and e). The two CP rings are anti-aromatic with counter-clockwise paratropic ring current and positive NICS(1)zz values (44.64), while the central benzenoid ring is nearly non-aromatic. Accordingly, proton b in H22+ is shielded by the antiaromatic CP rings (Fig. 6). The ACID plot and NICS values of the dianion H22− (Fig. 7a and f) disclose that all rings are aromatic with negative NICS(1)zz values (−9.83 to −28.72), and π electrons are mainly delocalized along the periphery with a clockwise current flow. Accordingly, protons a, b and j in H22− are deshielded by the whole aromatic backbone (Fig. 6). For H12−, each individual ring has negative NICS(1)zz values (−15.89 to −28.61, Fig. 7a), and a diatropic ring current circuit along the periphery is formed in the calculated ACID plot (Fig. 7c). In addition, NICS and ACID calculations of the corresponding all-benzenoid analogues show that the π-electrons can also form clockwise diatropic ring currents along the periphery of the backbone (Fig. S15†). Thus, the dianions of H1 and H2 can be regarded as the isoelectronic structures of the corresponding all-benzenoid expanded helicenes. All of this suggests that in dianions a global electron delocalization is obtained, leading to global aromaticity in the whole structure, while in the case of the neutral structures and dication the current-density pathways are composed of local aromatic and antiaromatic units. On the other hand, the calculated electrostatic potential maps (Fig. S16 and S17†) suggest that the charges are well delocalized along the entire backbone to minimize Coulomb repulsion. Hirshfeld charge distribution (Fig. S18 and S19†) shows that the head carbons of CP rings in H22+ and H22− have the highest charge densities, while the negative charges of H12− are primarily distributed along the periphery with C18 and C31 atoms having the largest negative charge densities.
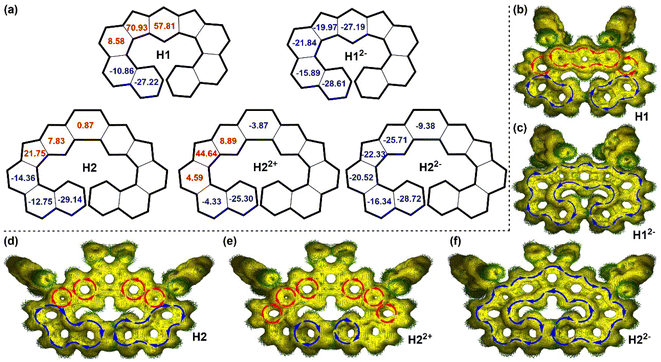 |
| | Fig. 7 (a) NICS(1)zz values (the numbers in the rings) of H1, H12−, H2, H22+ and H22− calculated at the CAM-B3LYP/6-31G(d,p) level. ACID plots of H1 (b), H12− (c), H2 (d), H22+ (e), and H22− (f) calculated at the CAM-B3LYP/6-31G(d,p) level. The red and blue arrows indicate the counterclockwise (paratropic) and clockwise (diatropic) current flow, respectively. | |
Conclusions
In summary, we have synthesized two helical π-conjugated diradicaloids H1 and H2 by fusion of a QDM unit with two phenanthrene rings at the termini, and their electronic structure and geometry in the ground state were investigated by various experiments assisted by DFT calculations. Single crystal X-ray analysis discloses that H1 and H2 have a helical molecular configuration. Due to the anti-aromatic character and quinoidal character of the central units, they exhibit open-shell diradical character and magnetic activity. Moreover, the dication of H2 is experimentally accessible by chemical oxidation with NO·SbF6, thus displaying a closed-shell ground electron state and a local aromatic character. The dianions of H1 and H2 were also obtained, showing similar electronic structures to their respective isoelectronic structures, the all-benzenoid expanded [9]helicene and [11]helicene. The successful X-ray structural characterization of the charged products reaffirms the structural flexibility of H2 driven by electron transfer processes and reveals a notable helical core compression upon two-electron oxidation or reduction. As a result of their intrinsic chirality, electronic and magnetic properties, as well as redox activities, these molecules could hold promise for chirality-induced spin selectivity, field-effect transistors, energy storage, and spintronics. This study not only opens up a new avenue for helical polycyclic π-systems, but also provides important insights into the construction of stable chiral diradicaloids for organic-based magneto-optical devices and magnetic chiral dichroism.
Computational details
All geometries were fully optimized using the B3LYP or CAM-B3LYP functionals, in combination with the 6-31G(d,p) basis set.24,25 Natural orbital occupation number (NOON) calculations were done by a spin unrestricted LC-BLYP/6-31G(d) method and the diradical character (y0) was calculated according to Yamaguchi's scheme: y0 = 1 − (2T/(1 + T2)), and T = (nHONO − nLUNO)/2 (where nHONO is the occupation number of the HONO, and nLUNO is the occupation number of the LUNO).26 Calculations of single point energies, molecular orbitals and time-dependent DFT (TD-DFT) for all DFT-optimized structures were obtained by using the B3LYP/6-31G(d,p). Given that hybrid long-range corrected functionals like CAM-B3LYP are well suited for describing electron delocalization in large polycyclic systems23 and provide magnetic properties consistent with those obtained at the CCSD(T) level,27 we chose the CAM-B3LYP/6-31G(d,p) level of theory for NICS and ACID calculations. NICS values were calculated using the standard GIAO procedure.21,28 An ACID plot was calculated by using the method developed by Herges.22,29 Electrostatic potential maps and Hirshfeld charges were calculated at the B3LYP/6-31G(d,p). In the study of racemization processes, the molecular geometries of all stationary points were optimized at the B3LYP level of DFT with the 6-31G(d) basis set with the IEPCM model for solvation of THF at 298 K. Harmonic vibration frequency calculations at the same level were performed to verify all stationary points as local minima (with no imaginary frequency) and transition states (with one imaginary frequency). IRC calculations30 were also performed to check transition states.
Data availability
The experimental procedures, characterizations, spectral analysis and DFT computational details are available in the ESI.†
Author contributions
Q. Jiang and W. Zeng supervised and designed the project. Q. Jiang, H. Tang, Y. Peng and Z. Hu synthesized and characterized the compounds. All authors discussed the results and contributed to the manuscript writing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Q. Jiang acknowledges the construct program of applied characteristic discipline in Hunan Province and the High-level Talent Program from Hunan University of Science and Engineering for financial support. W. Zeng is thankful for the financial support from the National Natural Science Foundation of China (Grant No. 22175061), Excellent Youth Foundation of Hunan Scientific Committee (Grant No. 2022JJ10025), Key research and development projects of Hunan Province (Grant No. 2022GK2035) and Key research and development projects of Xiangtan City (Grant No. ZX-ZD20221006).
Notes and references
-
(a) X. Hu, W. Wang, D. Wang and Y. Zheng, J. Mater. Chem. C, 2018, 6, 11232 RSC;
(b) Y. Tobe, Top. Curr. Chem., 2018, 376, 1 CrossRef CAS PubMed;
(c) T. Kubo, Chem. Lett., 2015, 44, 111 CrossRef;
(d) Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC;
(e) M. Abe, Diradicals, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed;
(f) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857 RSC;
(g) A. Rajca, Chem. Rev., 1994, 94, 871 CrossRef CAS;
(h) T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186 RSC;
(i) Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582 CrossRef CAS PubMed.
- K. Yamaguchi, Chem. Phys. Lett., 1975, 33, 330 CrossRef CAS.
-
(a) C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977 CrossRef CAS PubMed;
(b) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076 CrossRef CAS PubMed;
(c) J. Dressler, Z. Zhou, J. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. Spisak, M. Nakano, M. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 48, 15363 CrossRef PubMed;
(d) S. Qiu, C. Wang, S. Xie, X. Huang, L. Chen, Y. Zhao and Z. Zeng, Chem. Commun., 2018, 54, 11383 RSC;
(e) D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349 CrossRef CAS PubMed.
-
(a) P. Hu and J. Wu, Can. J. Chem., 2017, 95, 223 CrossRef CAS;
(b) W. Zeng, T. Y. Gopalakrishna, H. Phan, T. Tanaka, T. S. Herng, J. Ding, A. Osuka and J. Wu, J. Am. Chem. Soc., 2018, 140, 14054 CrossRef CAS PubMed;
(c) Z. Sun, S. Lee, K. H. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R.-W. Li, K.-W. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 18229 CrossRef CAS PubMed;
(d) W. Zeng, Z. Sun, T. S. Herng, T. P. Goncalves, T. Y. Gopalakrishna, K.-W. Huang, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 8615 CrossRef CAS PubMed;
(e) Y. Li, W. K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K. W. Huang, R. D. Webster, J. T. L. Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913 CrossRef CAS PubMed;
(f) Z. Sun, W. Fan, Y. Han, W. Yuan, Y. Ni, J. Wang, H. Wei, Y. Zhao, Z. Sun and J. Wu, Chem. Sci., 2023, 14, 7922 RSC;
(g) C. Zong, S. Yang, Y. Sun, L. Zhang, J. Hu, W. Hu, R. Li and Z. Sun, Chem. Sci., 2022, 13, 11442 RSC;
(h) C. Zong, X. Zhu, Z. Xu, L. Zhang, J. Xu, J. Guo, Q. Xiang, Z. Zeng, W. Hu, J. Wu, R. Li and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 16230 CrossRef CAS PubMed.
-
(a) A. Konishi, Y. Hirao, H. Kurata, T. Kubo, M. Nakano and K. Kamada, Pure Appl. Chem., 2014, 86, 497 CAS;
(b) A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430 CrossRef CAS PubMed;
(c) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021 CrossRef CAS PubMed.
-
(a) A. Shimizu, T. Kubo, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Hirao, K. Matsumoto, H. Kurata, Y. Morita and K. Nakasuji, J. Am. Chem. Soc., 2010, 132, 14421 CrossRef CAS PubMed;
(b) T. Kubo, M. Sakamoto, M. Akabane, Y. Fujiwara, K. Yamamoto, M. Akita, K. Inoue, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6474 CrossRef CAS PubMed;
(c) A. Shimizu, M. Uruichi, K. Yakushi, H. Matsuzaki, H. Okamoto, M. Nakano, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, Angew. Chem., Int. Ed., 2009, 48, 5482 CrossRef CAS PubMed;
(d) T. Kubo, A. Shimizu, M. Sakamoto, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 6564 CrossRef CAS PubMed;
(e) A. Shimizu, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, M. Uruichi and K. Yakushi, Chem. Commun., 2012, 48, 5629 RSC.
-
(a) Q. Jiang, T. Tao, H. Phan, Y. Han, T. Y. Gopalakrishna, T. S. Herng, G. Li, L. Yuan, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2018, 57, 16737 CrossRef CAS PubMed;
(b) J. Casado, Top. Curr. Chem., 2017, 375, 73 CrossRef PubMed;
(c) C. KFrederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977 CrossRef PubMed;
(d) X. Shi, P. M. Burrezo, S. Lee, W. Zhang, B. Zheng, G. Dai, J. J. Chang, T. López Navarrete, K.-W. Huang, D. Kim, J. Casado and C. Chi, Chem. Sci., 2014, 5, 4490 RSC;
(e) J. E. Barker, J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, E. T. Strand, L. N. Zakharov, S. N. MacMillan, C. J. Gómez-García, M. Nakano, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2020, 142, 1548 CrossRef CAS PubMed;
(f) S. Dong, T. S. Herng, T. Y. Gopalakrishna, H. Phan, Z. L. Lim, P. Hu, R. D. Webster, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2016, 55, 9316 CrossRef CAS PubMed;
(g) Z. Zeng and J. Wu, Chem. Rec., 2015, 15, 322 CrossRef CAS PubMed;
(h) M. A. Majewski, P. J. Chmielewski, A. Chien, Y. H. Tadeusz Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stepien, Chem. Sci., 2019, 10, 3413 RSC;
(i) H. Hayashi, J. E. Barker, A. C. Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S.-I. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142(48), 20444 CrossRef CAS PubMed;
(j) J. Guo, Z. Li, X. Tian, T. Zhang, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62, e202217470 CrossRef CAS PubMed;
(k) J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143(43), 18272 CrossRef CAS PubMed.
-
(a) P. Ravat, T. Šolomek, D. Haussinger, O. Blacque and M. Juríček, J. Am. Chem. Soc., 2018, 140, 10839 CrossRef CAS PubMed;
(b) T. Šolomek, P. Ravat, Z. Mou, M. Kertesz and M. Juríček, J. Org. Chem., 2018, 83, 4769 CrossRef PubMed;
(c) P. Ravat, T. Šolomek, M. Rickhaus, D. Haussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183 CrossRef CAS PubMed.
-
(a) J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y.-Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Müllen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280 CrossRef CAS PubMed;
(b) J. Ma, K. Zhang, K. S. Schellhammer, Y. Fu, H. Komber, C. Xu, A. A. Popov, F. Hennersdorf, J. J. Weigand, S. Zhou, W. Pisula, F. Ortmann, R. Berger, J. Liu and X. Feng, Chem. Sci., 2019, 10, 4025 RSC.
- Y.-C. Hsieh, C.-F. Wu, Y.-T. Chen, C.-T. Fang, C.-S. Wang, C.-H. Li, L.-Y. Chen, M.-J. Cheng, C.-C. Chueh, P.-T. Chueh and Y.-T. Wu, J. Am. Chem. Soc., 2018, 140, 14357 CrossRef CAS PubMed.
- Q. Jiang, Y. Han, Y. Zou, H. Phan, L. Yuan, T. S. Herng, J. Ding and C. Chi, Chem.–Eur. J., 2020, 26, 15613 CrossRef CAS PubMed.
- A. Borissov, P. J. Chmielewski, C. J. Gómez García, T. Lis and M. Stępień, Angew. Chem., Int. Ed., 2023, 62, e202309238 CrossRef CAS PubMed.
-
E. Clar, The Aromatic Sextet, Wiley, New York, 1972 Search PubMed.
-
(a) M. Mauksch and S. Tsogoeva, Chem.–Eur. J., 2021, 27, 14660 CrossRef CAS PubMed;
(b) M. Orozco-Ic, R. R. Valiev and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 6404 RSC;
(c) M. Orozco-Ic, L. Soriano-Agueda, S. Escayola, D. Sundholm, G. Merino and E. Matito, J. Org. Chem., 2024, 89, 2459 CrossRef CAS PubMed;
(d) H. Kubo, D. Shimizu, T. Hirose and K. Matsuda, Org. Lett., 2020, 22, 9276 CrossRef CAS PubMed;
(e) E. Cherni, B. Champagne, S. Ayadi and V. Liégeois, Phys. Chem. Chem. Phys., 2019, 21, 14678 RSC;
(f) M. Orozco-Ic, J. Barroso, N. D. Charistos, A. Muñoz-Castro and G. Merino, Chem.–Eur. J., 2020, 26, 326 CrossRef CAS PubMed;
(g) C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827 CrossRef CAS PubMed;
(h) G. I. Warren, J. E. Barker, L. N. Zakharov and M. M. Haley, Org. Lett., 2021, 23, 5012 CrossRef CAS PubMed.
- X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052 CrossRef CAS PubMed.
- Deposition numbers 2057406 (H1), 2057410 (H2), 2342019 (H22+) and 2342022 (H22−) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.†.
- J. Barroso, J. L. Cabellos, S. Pan, F. Murillo, X. Zarate, M. A. Fernandez-Herrera and G. Merino, Chem. Commun., 2018, 54, 188 RSC.
-
(a) S. Dong, T. S. Herng, T. Y. Gopalakrishna, H. Phan, Z. L. Lim, P. Hu, R. D. Webster, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2016, 55, 9316 CrossRef CAS PubMed;
(b) S. Dong, T. Y. Gopalakrishna, Y. Han, H. Phan, T. Tao, Y. Ni, G. Liu and C. Chi, J. Am. Chem. Soc., 2019, 141, 62 CrossRef CAS PubMed;
(c) T. Xu, Y. Han, Z. Shen, X. Hou, Q. Jiang, W. Zeng, P. W. Ng and C. Chi, J. Am. Chem. Soc., 2021, 143, 20562 CrossRef CAS PubMed;
(d) A. Ong, T. Tao, Q. Jiang, Y. Han, Y. Ou, K.-W. Huang and C. Chi, Angew. Chem., Int. Ed., 2022, 61, e202209286 CrossRef CAS PubMed;
(e) Z. Li, X. Hou, Y. Han, W. Fan, Y. Ni, Q. Zhou, J. Zhu, S. Wu, K.-W. Huang and J. Wu, Angew. Chem., Int. Ed., 2022, 61, e202210697 CrossRef CAS PubMed;
(f) T. Xu, X. Hou, Y. Han, H. Wei, Z. Li and C. Chi, Angew. Chem., Int. Ed., 2023, 62, e202304937 CrossRef CAS PubMed;
(g) W. Kueh, X. Shi, T. W. Phua, H. Kueh, Y. C. Liau and C. Chi, Org. Lett., 2022, 24, 5935 CrossRef CAS PubMed;
(h) J.-J. Shen, Y. Han, S. Dong, H. Phan, T. S. Herng, T. Xu, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2021, 60, 4464 CrossRef CAS PubMed;
(i) Q. Jiang, H. Wei, X. Hou and C. Chi, Angew. Chem., Int. Ed., 2023, 62, e202306938 CrossRef CAS PubMed;
(j) Q. Jiang, Y. Han, Y. Zou and C. Chi, J. Mater. Chem. C, 2023, 11, 15160 RSC.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CAS.
-
(a) Z. Zhou, X.-Y. Wang, Z. Wei, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 14969 CrossRef CAS PubMed;
(b) Z. Zhou, R. K. Kawade, Z. Wei, F. Kuriakose, O. Ungor, M. Jo, M. Shatruk, R. Gershoni-Poranne, M. A. Petrukhina and I. V. Alabugin, Angew. Chem., Int. Ed., 2020, 59, 1256 CrossRef CAS PubMed;
(c) S. N. Spisak, A. Yu. Rogachev, A. V. Zabula, A. S. Filatov, R. Clérac and M. A. Petrukhina, Chem. Sci., 2017, 8, 3137 RSC;
(d) Z. Zhou, L. Fu, Y. Hu, X.-Y. Wang, Z. Wei, A. Narita, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2020, 59, 15923 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef CAS PubMed.
-
(a) L. J. Karas, S. Jalife, R. V. Viesser, J. V. Soares, M. M. Haley and J. I. Wu, Angew. Chem., Int. Ed., 2023, 62, e202307379 CrossRef CAS PubMed;
(b) I. Casademont-Reig, R. Guerrero-Avilés, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Angew. Chem., Int. Ed., 2021, 60, 24080 CrossRef CAS PubMed;
(c) D. W. Szczepanik, M. Solà, M. Andrzejak, B. Pawełek, K. Dyduch, M. Kukułka, T. M. Krygowski and H. Szatylowicz, J. Comput. Chem., 2017, 38, 1640 CrossRef CAS PubMed;
(d) D. Inostroza, V. García, O. Yañez, J. J. Torres-Vega, A. Vásquez-Espinal, R. Pino-Rios, R. Báez-Grez and W. Tiznado, New J. Chem., 2021, 45, 8345 RSC;
(e) M. Orozco-Ic and G. Merino, Chemistry, 2021, 3, 1381 CrossRef CAS;
(f) M. Orozco-Ic, M. Dimitrova, J. Barroso, D. Sundholm and G. Merino, J. Phys. Chem. A, 2021, 125, 5753 CrossRef CAS PubMed;
(g) I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Molecules, 2020, 25, 711 CrossRef CAS PubMed;
(h) M. Orozco-Ic and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 22487 RSC.
- F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 258 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS PubMed;
(c) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS;
(d) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS;
(e) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, J. Chem. Theory Comput., 2021, 17, 1457 CrossRef CAS PubMed.
-
(a) S. Yamanaka, M. Okumura, M. Nakano and K. Yamaguchi, J. Mol. Struct., 1994, 310, 205 Search PubMed;
(b) K. Kamada, K. Ohta, A. Shimizu, T. Kubo, R. Kishi, H. Takahashi, E. Botek, B. Champagne and M. Nakano, J. Phys. Chem. Lett., 2010, 1, 937 CrossRef CAS.
- H. Fallah-Bagher-Shaidaei, S. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863 CrossRef CAS PubMed.
-
(a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS;
(b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic studies, computational analysis and crystal structures. CCDC 2057406, 2057410, 2342019 and 2342022. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02116a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hui
Tang
*a,
Hui
Tang