
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal control over phosphorescence or thermally activated delayed fluorescence in a metal–organic framework†
Huili
Sun
a,
Qiangsheng
Zhang
b,
Liuli
Meng
a,
Zhonghao
Wang
a,
Yanan
Fan
a,
Marcel
Mayor
 acd,
Mei
Pan
*a and
Cheng-Yong
Su
a
acd,
Mei
Pan
*a and
Cheng-Yong
Su
a
aMOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, IGCME, GBRCE for Functional Molecular Engineering, School of Chemistry, Sun Yat-Sen University, Guangzhou 510006, China. E-mail: panm@mail.sysu.edu.cn
bHainan Provincial Key Laboratory of Fine Chem, School of Chemistry and Chemical Engineering, Hainan University, Haikou, P. R. China
cDepartment of Chemistry, University of Basel, Switzerland
dInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Germany
First published on 1st May 2024
Abstract
By integrating a tailor-made donor–acceptor (D–A) ligand in a metal–organic framework (MOF), a material with unprecedented features emerges. The ligand combines a pair of cyano groups as acceptors with four sulfanylphenyls as donors, which expose each a carboxylic acid as coordination sites. Upon treatment with zinc nitrate in a solvothermal synthesis, the MOF is obtained. The new material combines temperature-assisted reverse intersystem crossing (RISC) and intersystem crossing (ISC). As these two mechanisms are active in different temperature windows, thermal switching between their characteristic emission wavelengths is observed for this material. The two mechanisms can be activated by both, one-photon absorption (OPA) and two-photon absorption (TPA) resulting in a large excitement window ranging from ultraviolet (UV) over visible light (VL) to near infrared (NIR). Furthermore, the emission features of the material are pH sensitive, such that its application potential is demonstrated in a first ammonia sensor.
Introduction
In the past few decades, there has been significant attention focused on thermal activated delayed fluorescence (TADF) and phosphorescence materials due to their ability to produce light from triplet excitons for a variety of applications.1–4 These applications include, among others, luminescent sensors,5,6 biological imaging probes,7,8 information encryption,9,10 and organic light-emitting diodes (OLEDs).11,12 However, TADF fluorophores and phosphorescence based on organic small molecules typically face quenching issues when exposed to air, which is often seen in emitters due to the involvement of T1 states.13 On the other hand, metal–organic frameworks (MOFs) are well-known for their crystal rigidity, structural tunability, and photostability.14–17 Due to their rigid framework, which prevents aggregation of the emitting species and suppresses non-radiative deactivation, MOFs have shown to be a promising platform to address these challenges and can lead to new photoluminescence (PL) properties.Generally, improving the intersystem crossing (ISC) process from the singlet to the triplet state can lead to phosphorescence, while improving the reverse intersystem crossing (RISC) from the triplet to the singlet state can result in TADF.18,19 The spatial separation of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) gives rise to charge transfer (CT) excited states, which serve as the basis for TADF. Molecular design strategies like spatial frontier orbital separation in donor–acceptor (D–A) or D–A–D type molecules are typically applied to achieve this.20–22 TADF materials can also be obtained by reducing the energy gap between singlet and triplet states (ΔEST), introducing heavy atoms to increase spin–orbit coupling, improving structural rigidity, and so on.23–25 Additionally, these strategies can also promote the ISC process from the singlet to the triplet state, leading to triplet state phosphorescence in some materials.26–29 Based on the mechanisms described above, phosphorescence and TADF can potentially be obtained simultaneously in single-component materials.4,19,30,31 However, the controlled switching between ISC-related phosphorescence and RISC-related TADF remains challenging.
Despite advances in TADF and phosphorescence research, current materials mainly trigger TADF and phosphorescence through single photon absorption (OPA) of ultraviolet (UV) excitation. The exploration of TADF and phosphorescence excited by visible light (VL) and near-infrared (NIR) sources remains largely unexplored, although it may possess advantages such as deep penetration and low energy requirement, making it more suitable for potential applications such as biological imaging, in tissue medical applications and many more.32 Two-photon excited fluorescence (TPEF) is a promising field that utilizes low energy NIR excitation through the two-photon absorption (TPA) pathway to generate high-energy emissions, with significant practical applications in optoelectronics and biology.33 Many materials with TPEF potential have been developed. One design concept involves constructing D–A molecules to achieve efficient TPA.34,35
Based on the above considerations, we present here a new D–A type organic ligand consisting of a central benzene core decorated with two nitrile groups in para positions as electron acceptors, and four thiophenyls as electron donating substituents. The intention behind the ligand design is to spatially separate the frontier orbitals by the rigid arrangement of the donor and acceptor substituents at the central benzene core. In addition, the ISC via n–π* transition might be facilitated by the heavy atom character of the sulfur-based substituents. Furthermore, the four thiophenyl substituents are decorated with carboxylic acids in para positions acting as coordination sites.
Coordination with Zn(II) ions leads to the formation of a MOF structure named LIFM-SHL-1, with an intramolecular packing of the ligands promoting the formation of triplet state excitons, resulting in temperature-dependent TADF and phosphorescence. Controlled switching between these relaxation mechanisms becomes possible due to differences between their optimal temperature ranges. Most excitingly, LIFM-SHL-1 displays full-band sensitivity including UV, VL, and NIR light excited TADF and phosphorescence, which was validated by the herein-reported experiments and supported by theoretical calculations.
Results and discussion
The synthesis of the D–A type organic ligand exposing four carboxylic groups DTST (4,4′,4′′,4′′′-((3,6-dicyanobenzene-1,2,4,5-tetrayl)tetrakis(sulfanediyl))tetrabenzoic acid) is described in the ESI (Fig. S1–S3†). Exposing the ligand DTST and Zn(NO3)2·6H2O to solvothermal conditions (100 °C for 24 h) provided the metal–organic framework (MOF) LIFM-SHL-1 ({Zn(DTST)(DMF)(H2O)}n, DMF = N,N-dimethylformamide) by self-assembly as yellow, bar-shaped crystals. The phase purity of LIFM-SHL-1 was confirmed by in situ variable temperature (VT) powder X-ray diffraction (VT-PXRD). Thermogravimetric (TG) curve analysis revealed the thermal stability of LIFM-SHL-1 up to 400 °C, exceeding the one of the parent ligand DTST (∼350 °C) (Fig. S4†). Single crystal structure analysis of LIFM-SHL-1 revealed that the three-dimensional coordination polymer crystallized (Table S1†).Single crystal structural analysis showed that LIFM-SHL-1 exhibits two distinct coordination modes. In the first mode, the Zn2+ ion connects with three carboxyl O atoms derived from three different neighboring DTST ligands, and simultaneously coordinates with one DMF and one H2O molecule. In the second mode, the Zn2+ ion connects in a similar manner with the surrounding DTST ligands and also coordinates with one H2O molecule, resulting in the formation of triangular biconical and tetrahedral geometries, respectively (Fig. 1a and S5†). The coordination polymer is forming a self-assembled supramolecular layer composed of Zn(II) ions and DTST ligands. The adjacent ligands have C–H⋯π and C–H⋯S interactions with the distances of 2.88–2.89 Å and 3.08–3.11 Å (Fig. 1b and c). Hirshfeld surface analysis of LIFM-SHL-1 indicates that C–H⋯π and C–H⋯S contribute 15.5%, and 6.7% of the total weak interactions, respectively (Fig. 1d).
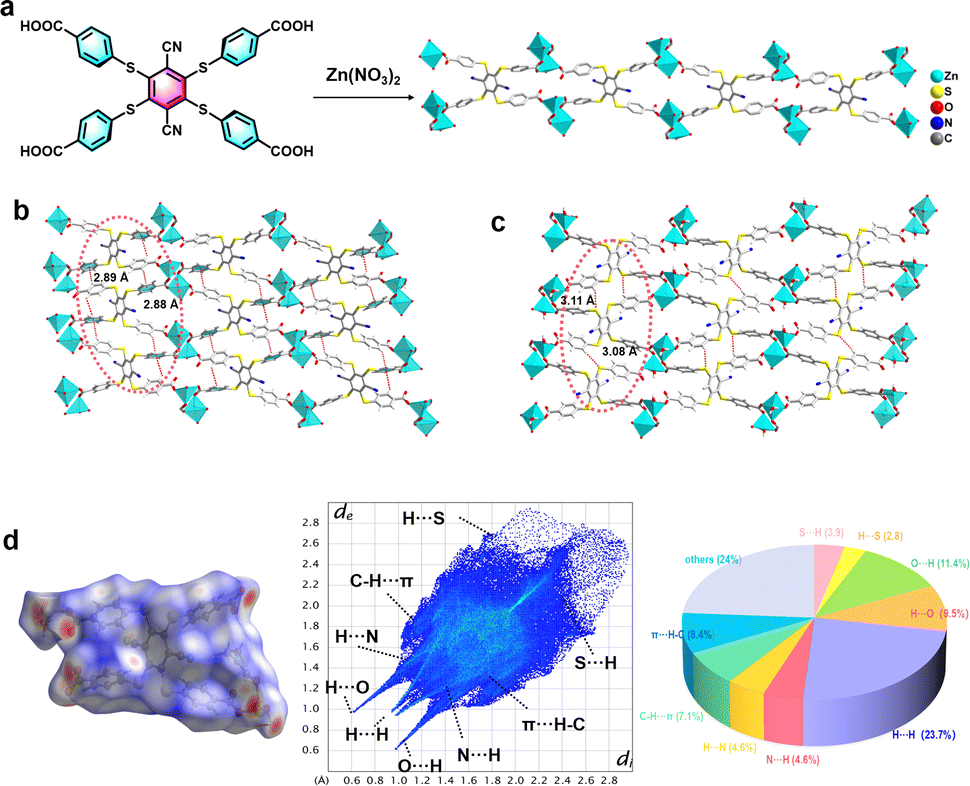 | ||
| Fig. 1 (a) Synthesis and solid-state structure of the Zn2+ ions and DTST ligands (color indication: Zn, nattier blue; O, red; C, gray; H, white; N, blue; and S, yellow) in LIFM-SHL-1: (b) packing among the LIFM-SHL-1 by C–H⋯π interaction and (c) C–H⋯S interaction view along the a-axis; (d) Hirshfeld surface analysis of LIFM-SHL-1 in the crystalline state. | ||
In addition to the C–H⋯π and C–H⋯S interactions mentioned above, the coordination polymer LIFM-SHL-1 also exhibits a robust network of hydrogen bonds with O–H⋯O and C–H⋯N distances of 1.71 and 2.38 Å, respectively. The intermolecular bonding pattern leads to the formation of an extensive three-dimensional network through the packing of hydrogen bond interactions between uncoordinated carboxyl oxygen atoms and coordinated water molecules (Fig. S6†). Furthermore, as depicted in Fig. S7,† we can observe weak interactions between adjacent benzene rings within the same ligand by the interaction region indicator (IRI) analysis. The accumulation of diverse weak interactions of comparable strength is essential to preserve the rigid conformation of LIFM-SHL-1. We hypothesize that the constriction of the subunits' movements in the solid state is restricting non-radiative relaxation pathways from the excited state and thereby might favor optically active ones.
Of particular interest were the photophysical properties of the parent ligand DTST and its coordination polymer LIFM-SHL-1. Both emitters were investigated in the solid-state. DTST and LIFM-SHL-1 exhibit weak, almost structureless and broad absorption bands (Fig. S8†). While the absorption of DTST reaches out to about 530 nm, the one of LIFM-SHL-1 consists of two broad bumps which are bathochromically shifted closing at about 560 nm. It thus seems that the organic ligand is the main photosensitive component in the coordination polymer, and its coordination in the MOF material impacts its excited state energy levels. While the absorption band in the range of 300–400 nm was attributed to a ligand based π–π* electron transition, the one observed in the 400–560 nm range was attributed to a metal to ligand n–π* electron transition. Noteworthy is the extension of the absorption by combining the D–A ligand design with its intramolecular packing in the MOF material, resulting in an absorption range exceeding 550 nm capable of capturing photons across multiple energy levels. To further confirm this point, we calculated the absorption spectrum of the LIFM-SHL-1 fragment (Fig. S8b†). The molecular orbital diagrams representing the π–π* and n–π* transitions at 389 nm and 491 nm for LIFM-SHL-1, respectively, are shown in Fig. S8c.† The absorption band at 389 nm is assigned to the HOMO−1 (π)–LUMO−2 (π*) transition, whereas the absorption band at 491 nm has HOMO (n)–LUMO+1 (π*) character.
To gain a better understanding of the photophysical properties of the emitters, we conducted an analysis of temperature-dependent PL spectra under ambient air conditions. For DTST, a single broad emission peak at 610 nm was observed (PLQY, 7.6%) (Fig. S9a†). As the temperature increases from 77 K to 300 K, both the PL intensity and transient PL decay lifetime at 610 nm decrease (Fig. 2a–c). Transient PL decay characteristics were recorded at 100 K and provided an excited state lifetime of 5.74 μs (Table S2†), pointing at phosphorescence as the underlying mechanism.
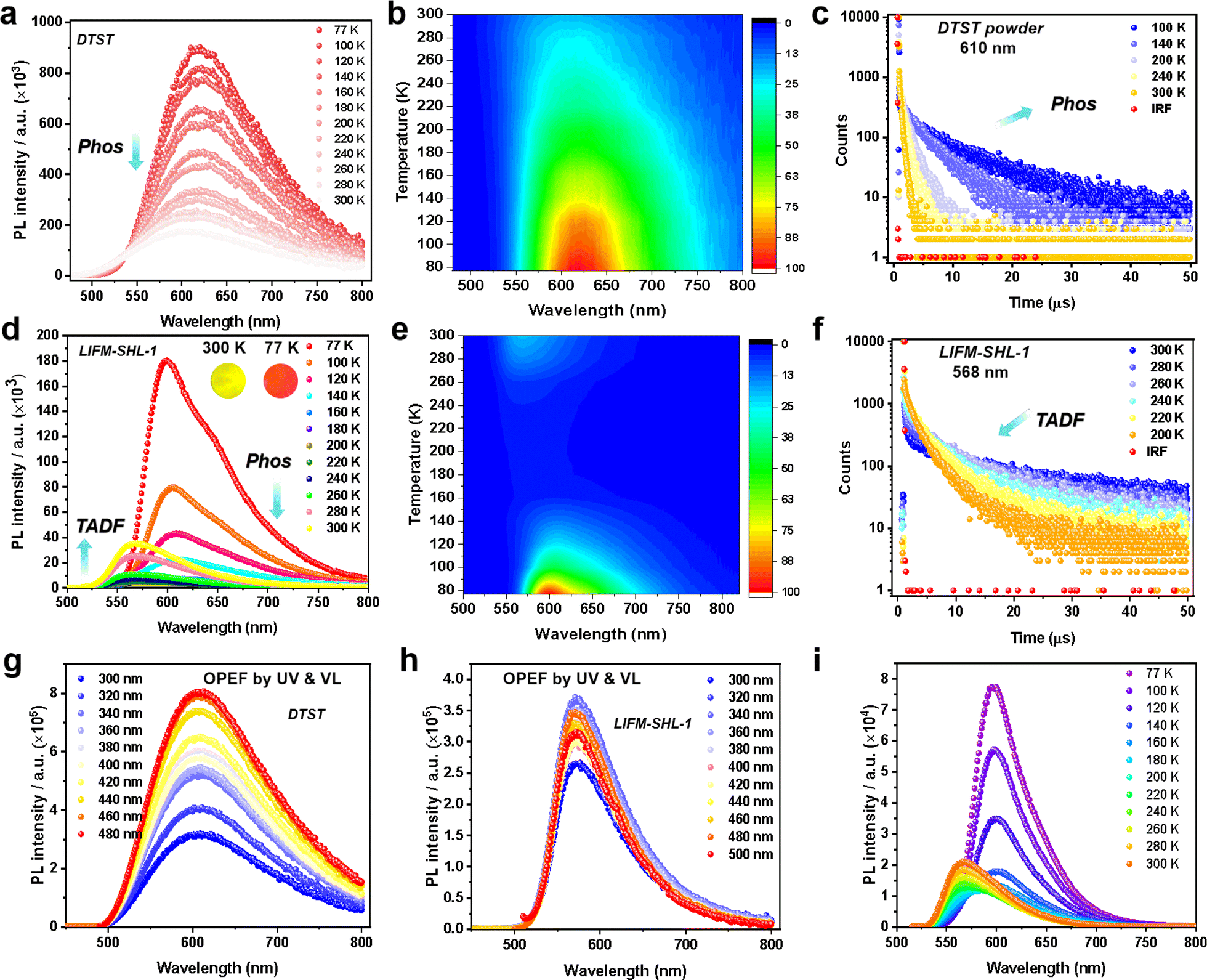 | ||
| Fig. 2 (a) Temperature-dependent steady-state PL spectra, and (b) 2D contours of DTST; (c) decay curves of DTST from 100 K to 300 K at 610 nm; (d) temperature-dependent steady-state PL spectra (the insets show the photoluminescence color under UV light), and (e) 2D contours of LIFM-SHL-1 from 77 K to 300 K; (f) decay curves of LIFM-SHL-1 from 200 K to 300 K at 568 nm; UV to visible light excited OPEF emissions of (g) DTST, and (h) LIFM-SHL-1 in the solid state at room temperature; (i) temperature-dependent steady-state PL spectra of LIFM-SHL-1 at the excitation of 500 nm. | ||
Interestingly, a different temperature dependence of the steady state PL was observed for the ligand integrated in the MOF LIFM-SHL-1 (Fig. 2d–f), displaying a switching between two emission bands. At 100 K, the spectrum is dominated by a broad emission band at 600 nm, resembling the one already observed for the ligand. Its reddish emission with CIE coordinates of (0.60, 0.39) is depicted as the inset of Fig. 2d. The recorded transient PL decay characteristics at 600 nm confirmed an excited state lifetime of 4.37 ms at 100 K (Table S3†). As the temperature increased from 100 K to 200 K, the intensity of the emission at 600 nm gradually diminished (Fig. 2d and e). We thus attribute the emission band at 600 nm to the phosphorescence originating from the DTST ligand. This interpretation is further supported by the transient PL decay curves, illustrating a decrease in lifetime at 600 nm as the temperature increased (Fig. S10 and Table S3†).
Surprisingly, with the phosphorescence diminishing between 200 K and 300 K, another broad emission at 568 nm appears with a yellowish color (CIE coordinates 0.50, 0.49; inset of Fig. 2d) in LIFM-SHL-1. This emission exhibits an opposite temperature-dependence trend, namely an increase of both the PL intensity and decay lifetime with rising temperature, the characteristic features of a TADF mechanism (Fig. 2d–f, S11 and Table S4†). The recorded transient PL decay characteristics at 568 nm confirmed the TADF hypothesis with an excited state lifetime of 7.92 μs and a PLQY of 8.64% at room temperature (Table S4†).
The structural robustness and inertness of LIFM-SHL-1 in the temperature range between 80 K and 300 K was corroborated by VT-PXRD (Fig. S12†). Thus, we hypothesize that the recorded difference in the emission characteristics of LIFM-SHL-1 between low and room temperature points at temperature dependent variations in the effectiveness of non-radiative decay pathways of triplet excited states. At 77 K the access to non-radiative decay pathways is limited as the loss of vibrational energy is minimized, which promotes phosphorescent emission. Conversely, at higher temperatures, alternative relaxation pathways become more significant, leading to TADF emission. The decay lifetime analysis revealed that LIFM-SHL-1 shows a delayed component of 92%, compared to 8% of the prompt component, and exhibits an efficient rate constant of RISC (KRISC, 1.57 × 106 s−1, Table S5†) from the triplet to the singlet energy state.
In general, the photoluminescence mechanism of LIFM-SHL-1 involves a thermally controlled triplet exciton cycle channel, with a threshold temperature of 200 K. Below the threshold temperature, the ISC from singlet to triplet transition and phosphorescence emission dominates. Above 200 K, the RISC channel of the triplet exciton is active and becomes more dominant with increasing temperature.
Furthermore, we investigated the luminescence properties of DTST in the solution state. The ligands in 2-methyltetrahydrofuran solution (2-MTHF, 50.0 μM) showed very weak green fluorescence upon excitation at RT, which is explained by the PL spectra presented in Fig. S13.† The transient PL decay lifetime at 530 nm was measured as 2.80 ns (Fig. S14 and Table S6†). Upon decreasing the temperature to 77 K, DTST exhibited bright green emission, the lifetime was found to be as long as 15.78 ms (Fig. S14 and Table S6†). These observations indicate that under low-temperature solution conditions, due to its ultra-low radiation rate, it can exhibit persistent phosphorescence without TADF properties. This suggests that DTST cannot exhibit TADF properties in both solid and solution states. In contrast to pure ligands, the conformation of ligands within MOF might experience slight alterations, along with variations in intermolecular interactions. These differences could potentially result in a narrower energy gap between singlet and triplet states in LIFM-SHL-1, thereby promoting rapid RISC processes. This makes LIFM-SHL-1 more prone to TADF properties.
Due to the long-wavelength absorption features reaching out into the visible region of LIFM-SHL-1 discussed above, the wavelength-dependent one-photon excitation fluorescence (OPEF) of DTST and LIFM-SHL-1 was of particular interest (Fig. 2g and h). As we can see, the VL excited spectra from 400 to 500 nm are consistent with that of the UV excitation from 300 to 400 nm, for which DTST shows the phosphorescence dominated reddish emission, while LIFM-SHL-1 manifests the TADF dominated yellowish emission. Meanwhile, the temperature-dependent emission spectra of LIFM-SHL-1 at the excitation of 500 nm also exhibit an intensity increasing trend from 200 to 300 K (Fig. 2i), further supporting the TADF character of the mechanism. Obviously, the excited state of the MOF can be populated by both UV and VL radiation.
Furthermore, the photoluminescence of DTST and LIFM-SHL-1 can also be excited by two-photon absorption (TPA) of NIR light. The characteristic emission can be observed at the NIR excitation wavelength from 750 nm to 860 nm. The wide range of two-photon excitation fluorescence (TPEF, Fig. 3a and b) was achieved with a femtosecond laser. In more detail, we investigated the TPEF of DTST and LIFM-SHL-1 (Fig. 3c–i, S15 and S16†) upon irradiation of 800 nm, which exhibits similar characteristics to the OPEF spectra (Fig. 3c), indicating that their mechanisms are the same. Furthermore, a laser power-dependent analysis of the TPEF emission spectra displayed an increase in intensity with the excitation power (Fig. 3d, S15 and S16†). The linear log(PL intensity)–log(excitation power) relationship was established and its linear fit had a slope of approximately 2.0, corroborating the two-photon absorption process for the emissions (Fig. S15 and S16†). The TPEF cross-sections were measured with laser excitation wavelength from 740 to 860 nm (Fig. 3e, see the ESI for details†). When using an 800 nm laser as a two-photon excitation source, and LIFM-SHL-1 have the maximum emission intensity and TPEF cross-sections (6959.16 GM/13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 825.44 GM) (Fig. 3f and Table S7†). LIFM-SHL-1 exhibits multifold increment in both TPEF and TPA cross-sections compared to DTST under excitation at 800 nm/760 nm. This enhanced response was attributed to the limited conformational flexibility of organic molecules fixed in the MOF material, which restricts molecular vibrations and movements and thereby leads to an attenuation of the non-radiative energy loss. Additional metal–ligand charge transfer transitions in MOFs further increases the two-photon absorption capability of the system.
825.44 GM) (Fig. 3f and Table S7†). LIFM-SHL-1 exhibits multifold increment in both TPEF and TPA cross-sections compared to DTST under excitation at 800 nm/760 nm. This enhanced response was attributed to the limited conformational flexibility of organic molecules fixed in the MOF material, which restricts molecular vibrations and movements and thereby leads to an attenuation of the non-radiative energy loss. Additional metal–ligand charge transfer transitions in MOFs further increases the two-photon absorption capability of the system.
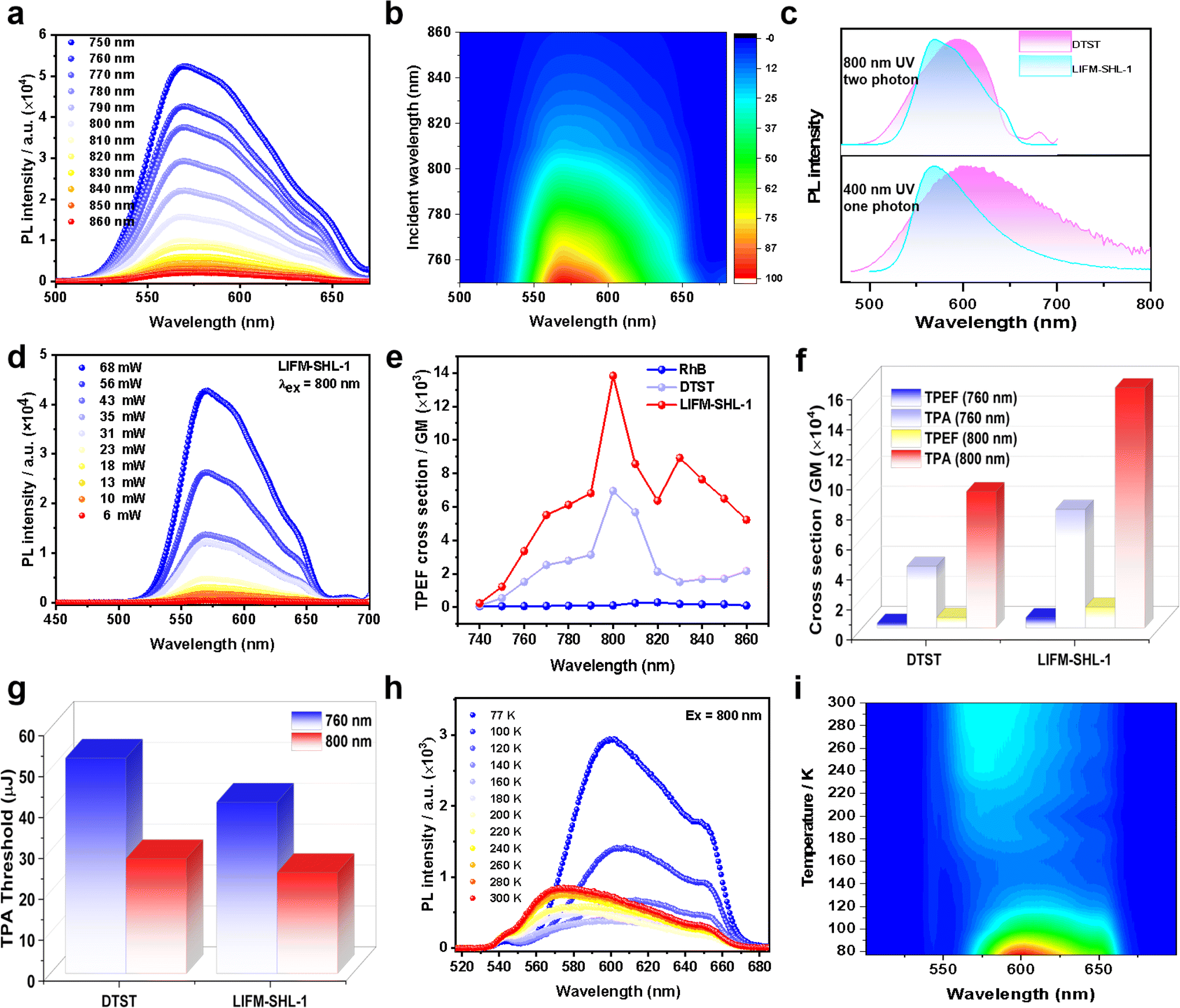 | ||
| Fig. 3 (a) Emission spectra under different two-photon excitation wavelengths, and (b) 2D contours of LIFM-SHL-1; (c) the OPEF and TPEF spectra of ligand DTST and LIFM-SHL-1 excited at 400 and 800 nm; (d) power-dependent emission spectra of LIFM-SHL-1 (λex = 800 nm); (e) TPEF cross-sections of DTST and LIFM-SHL-1 under different excitation wavelengths; (f) TPEF and TPA cross-sections and (g) TPA threshold of DTST and LIFM-SHL-1 under 760 nm and 800 nm; (h) temperature-dependent steady-state photoluminescence spectra at the excitation wavelength of 800 nm (temperature range: 77–300 K) and (i) 2D contours of LIFM-SHL-1. | ||
By plotting the PL intensity vs. the excitation intensity (Fig. S15e, h and S16e, h†), a pronounced change in slope was observed and from the turning point on PL intensity–excitation intensity curves, we can obtain the samples TPEF thresholds. The thereby obtained TPEF thresholds of the samples are summarized in Fig. 3g. Interestingly, the recorded TPEF threshold of LIFM-SHL-1 is lower than the one of DTST for both excitation wavelengths (760 and 800 nm), which is an additional documentation of the improved TPEF performance of the dense framework and close packing arrangement of the emitter in LIFM-SHL-1.
Temperature-dependent TPEF measurements of LIFM-SHL-1 under ambient conditions with an incident laser of 800 nm displayed similar temperature-dependent PL trends as already reported under UV excitation. We are thus confident that the same excited states are populated by both processes, one and two photon absorptions. As both, the TADF and phosphorescence features of LIFM-SHL-1 can be observed in two-photon excitation and favored by the choice of the temperature at which the experiment is performed (Fig. 3h and i), temperature-controlled transition between the ISC and RISC energy cycle mechanism can be performed with both, one and two photon modes in LIFM-SHL-1.
To get an in-depth understanding of the photophysical properties of DTST and LIFM-SHL-1, time-dependent density functional theory (TD-DFT) calculations were performed (Fig. 4, and see the ESI for details†). The excitation energies, transition density plots of holes and particles, and spin–orbital coupling matrix elements (SOCME, ξ) were calculated to further elucidate the underlying transition characters.
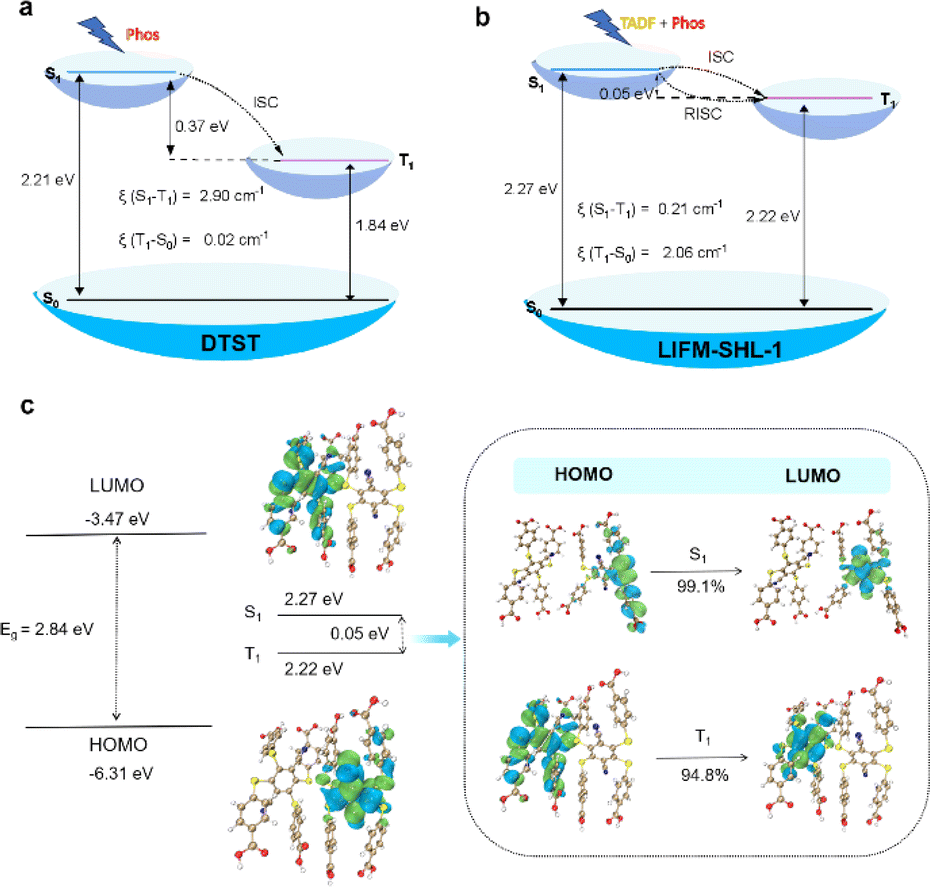 | ||
| Fig. 4 (a and b) Energy level diagram, SOC coefficients (ξ) for (a) DTST and (b) LIFM-SHL-1 fragments; (c) distribution of frontier molecular orbitals (FMOs) and nature transition orbitals (NTOs) of LIFM-SHL-1 fragments. | ||
The TD-DFT calculations for the DTST revealed a vertical excitation energy of 2.60 eV (476 nm) for S0 → S1, which closely approximates the absorption peak value of DTST in its solid state (300–500 nm). The emission energy for T1 → S0 was determined to be 673 nm, thereby confirming that the phosphorescence peak at 610 nm in the DTST can be attributed to T1 → S0. The study also investigated the electronic transition behavior of DTST in Fig. S17 and S18.† Consistent with previous reports, the HOMO and LUMO are spatially well separated, indicating a preference for the charge-transfer state in the excited state. The analysis of the frontier molecular orbitals (FMOs) for DTST shows that the HOMO is mainly localized on the four sulfur atoms of the electron donor, while the LUMO is predominantly located on the dicyanobenzene moiety, which acts as the electron acceptor due to its strong electron-deficient nature. This leads to significant charge transfer from the sulfur-rich electron-donor to the dicyanobenzene-based electron-acceptor during the transition from the ground state to the lowest excited state. Moreover, the FMOs are dispersed over the sulfur atoms with heavy atom effects and n–π* transition characteristics. In the context of electronic transitions, the probability of ISC can be assessed based on the energy gap ΔE(S,T) between singlet and triplet excited states. It can be seen that the energy gap between S1 and T1 is 0.37 eV (Fig. 4a and Table S8†). Specifically, we found that the ξ(S1–T1) value was 2.90 cm−1. Moreover, perturbation theory suggests that small singlet–triplet splitting energy ΔE(Sm–Tn) and high SOC constants (ξ) between Sm and Tn can effectively promote the ISC process.36 The analysis further highlights the ISC process between S1 and T1 in DTST.
Then, LIFM-SHL-1 fragments were selected as the research objects to explore their electronic transition behaviors (Fig. 4b and c). As depicted in Fig. 4c, the HOMO and LUMO of LIFM-SHL-1 fragments were distributed across different ligand molecules. Specifically, the hole density of S1 was localized on the two sulfur atoms and part of the phenyl ring adjacent to the acceptor units, while T1 was positioned on the four sulfur atoms and part of the central phenyl ring. The excited-state energies of S1 and T1 were calculated to be 2.27 and 2.22 eV, respectively (Fig. 4b and Table S9†). The excited-state energy levels of T1 and S1 were almost identical, and the ΔE(S1, T1) is 0.05 eV. The analysis further highlights the rapid RISC process between S1 and T1 in LIFM-SHL-1 fragments. Meanwhile, we found that the ξ(S1–T1) value was 0.21 cm−1. These calculations suggest that the small ΔEST of LIFM-SHL-1 compared to DTST favors the RISC between S1 and T1 states in the MOF. Accordingly, the observed TADF property is absent in DTST. The localization of the HOMO and LUMO, coupled with the well-separated NTOs and identical excited-state- energies of T1 and S1 indicate a strong potential for charge transfer, which is essential for efficient light emission applications.
To characterize the electronic properties of LIFM-SHL-1 in more detail, we performed density functional theory (DFT) calculations. We investigated the band structure, density of states distribution, and transition characteristics of the frontier molecular orbitals of LIFM-SHL-1 separately. LIFM-SHL-1 shows that the HOMO and the LUMO were mainly located on the DTST ligand, as shown in Fig. S19a.† This indicates that light emission, including TADF, and phosphorescence primarily originate from the DTST unit inside the framework. The calculated band structure and density of states (DOS) of LIFM-SHL-1 provided more detailed information about the behavior of excited state electrons, as shown in Fig. S19b and c.† The total/partial electronic density of states (TDOS and PDOS) indicated that LIFM-SHL-1 exhibits a narrow band gap of 1.367 eV. This small band gap promotes the reduction of the singlet state, which is beneficial for singlet-to-triplet ISC.
The photophysical process of LIFM-SHL-1, which exhibits ligand-centered emission, can be explained based on the theoretical calculation results. Combining the results of our experimental data and theoretical calculations, we propose a possible photophysical pathway for LIFM-SHL-1. Upon excitation by higher-energy photons, electrons in the ground state of LIFM-SHL-1 are excited to the S1 energy level. Subsequently, the excitons in S1 rapidly undergo ISC to populate T1. At lower temperatures, vibrational relaxation (VR) of the excited states is suppressed, and ISC efficiency is enhanced, resulting in strong phosphorescent emission dominated by T1 → S0. As the temperature increases, a portion of the population in T1 gains energy to get converted to S1, leading to increased fluorescence intensity and an elongated decay lifetime at 568 nm. However, the rise in temperature also increases the VR process, which contributes to the decrease in phosphorescence intensity and completes the photocycle. Our proposed photophysical pathway provides a comprehensive understanding of the behavior of LIFM-SHL-1 in different temperature regimes, a crucial requirement to further optimize and tune the feature for potential applications in optoelectronic devices and sensors.
In addition, LIFM-SHL-1 displays chemical robustness like, e.g., structural tolerance for acidic to alkaline aqueous solutions. Specifically, the framework retains its high crystallinity after being immersed in a pH range spanning from 1.0 to 12.0 for a period of 12 hours, as demonstrated by PXRD and FT-IR spectra measurement (Fig. S20 and S21†). Upon addition of ammonia to the aqueous solution, there was a gradual increase in both the intensity and lifetimes of the emission spectrum. And with the increase of pH from 6.7 to 11.9, the emission wavelength is continuously red shifted, and the lifetime is also prolonged with the increase of pH (Fig. 5a, S22–S24, and Table S10†). The new emission exhibits a longer lifetime (2.03 μs) than the original one (1.40 μs), and it can be inferred that its property is converted from TADF to phosphorescence (Table S10†). On this basis, hydrochloric acid (HCl) was added to adjust the pH of the solution; as the pH was decreased, the intensity of the emission color returns to its original emission state (Fig. S25†). Excitingly, this phenomenon can be repeated many times further illustrating the structural integrity of the MOF. Building on these findings, HCl was introduced to the solution in order to manipulate its pH. As the current working hypothesis, the change of optical properties may be caused by pH dependent hydrogen bond interactions within the framework of LIFM-SHL-1 and its small guest molecules.37
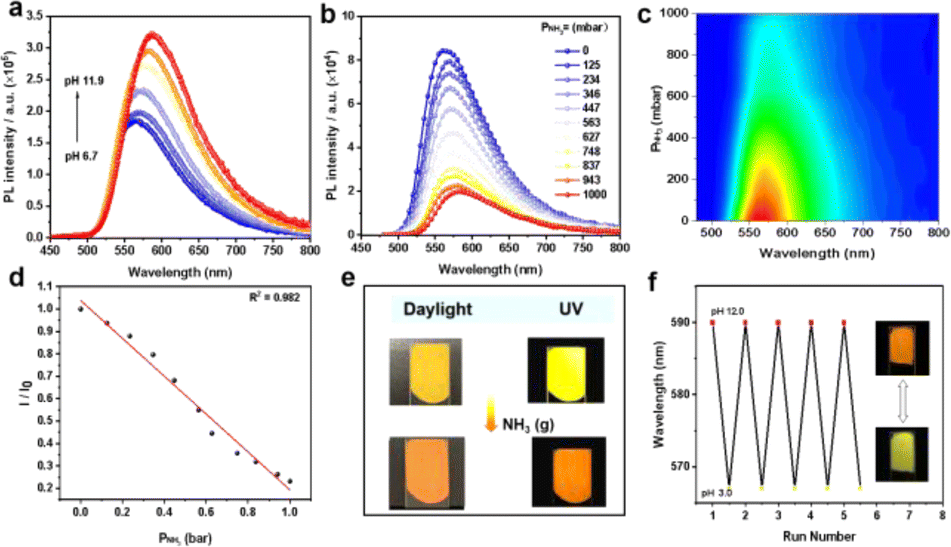 | ||
| Fig. 5 (a) Emission spectra of LIFM-SHL-1 in aqueous dispersions with pH varying from 6.7 to 11.9; (b) photoluminescence spectra of LIFM-SHL-1 exposed to various fractions of ammonia and (c) 2D contours of the relationship between luminescence intensity and pressure of ammonia; (d) variation of the emission intensity with the pressure of ammonia; (e) response of the LIFM-SHL-1@PVDF membrane to NH3 in daylight and photoluminescence color; (f) luminescence switching through alternately immersing LIFM-SHL-1@PVDF in pH 12.0 and pH 3.0 ammonia solutions. | ||
For comparison, the optical response of the free ligand DTST to pH was also studied. As shown in Fig. S26,† as the pH was increased from 6.7 to 10.0, the intensity of the emission spectrum continues to decrease, and the emission color remains unchanged.
The significant changes observed in the emission spectra and lifetimes of LIFM-SHL-1 in the presence and absence of aqueous ammonia prompted us to investigate its potential as an ammonia sensor. To this end, we recorded the NH3 concentration-dependent luminescence spectra of LIFM-SHL-1 under 365 nm UV light excitation. The emission intensity exhibited a clear quenching effect with increasing NH3 concentration (Fig. 5b and c), accompanied by a noticeable red shift in the emission color. The observed response was linear in the range of 0–1000 mbar (Fig. 5d), with a high correlation coefficient (R2 = 0.982). To demonstrate its potential for practical applications, we prepared a LIFM-SHL-1@PVDF composite membrane (PVDF = poly(vinylidene fluoride)) for sensing gaseous NH3. Upon exposure to NH3, the fluorescence of the membrane exhibited a rapid and visible color change from yellow to orange, which was accompanied by a natural color change from yellow to dark yellow (Fig. 5e and S27a†). This indicates that the membrane has potential application value in portable and visually detectable ammonia sensors in real-world applications. The membrane can also be used as a base-triggered fluorescence switch. The fluorescence color changes can be repeated by alternately immersing the membrane in pH 12.0 and pH 3.0 aqueous solutions, the emission colors show good reproducibility at each pH (Fig. 5f). In addition, the LIFM-SHL-1@PVDF film obtained possesses the potential for application in food spoilage detection, as the observable color change in an amine atmosphere is discernible to the naked eye (Fig. S27b†).
Conclusions
A new MOF material LIFM-SHL-1 is presented consisting of an aromatic donor–acceptor structure based on thioether and nitrile substituents, which is fixed in the MOF material and thereby displays promising changes in its optical properties. The fixation in the solid-state structure of the MOF results in two parallel emission mechanisms with different complementing temperature windows, allowing the selection of the preferred emission behavior by temperature. Below the threshold temperature of 200 K, emission occurs via direct triplet radiation (phosphorescence), while TADF sets in above the threshold temperature and becomes even more intensive with increasing temperature. The interpretation of the temperature triggered mechanism change is supported by DFT calculations which identify a fast ISC process from S1 to T1 as the origin of the phosphorescence. A temperature assisted RISC process results in the conversion of the T1 triplet state into the S1, from which the TADF occurs. The broad absorption spectrum of the MOF material allows excitation of the system in a broad window, ranging from UV over VL to NIR, with two photon absorption excitation processes in the last case. The two-photon excitation TADF at room temperature is not only fundamentally interesting, but also expands the application potential of TADF materials. As preliminary proof of concept, LIFM-SHL-1 has been integrated as a gas sensing material, while its application potential in optoelectronic devices, biological imaging, and beyond will be explored in future.Data availability
All experimental and characterisation data, as well as photophysical and NMR spectra are available in the ESI.†Author contributions
H. S. and Q. Z. performed calculations and theoretical analysis. L. M., Z. W., and Y. F. performed spectral data analysis. M. M., M. P. and C. S. supervised the study. H. S. wrote the manuscript with contributions of all authors, and all authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSFC (22171291, 92261114, 21821003), and NKRD Program of China (2021YFA1500401).Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T. Hua, J. Miao, H. Xia, Z. Huang, X. Cao, N. Li and C. Yang, Adv. Funct. Mater., 2022, 32, 2201032 CrossRef CAS.
- G. Xia, C. Qu, Y. Zhu, J. Ye, K. Ye, Z. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 9598–9603 CrossRef CAS PubMed.
- M. Li, W. Xie, X. Cai, X. Peng, K. Liu, Q. Gu, J. Zhou, W. Qiu, Z. Chen, Y. Gan and S. J. Su, Angew. Chem., Int. Ed., 2022, 61, e202209343 CrossRef CAS PubMed.
- T. Wang, J. De, S. Wu, A. K. Gupta and E. Zysman-Colman, Angew. Chem., Int. Ed., 2022, 61, e202206681 CrossRef CAS PubMed.
- F. Li, C. Qian, J. Lu, Y. Ma, K. Y. Zhang, S. Liu and Q. Zhao, Adv. Opt. Mater., 2022, 10, 2101773 CrossRef CAS.
- F. Fang, L. Zhu, M. Li, Y. Song, M. Sun, D. Zhao and J. Zhang, Adv. Sci., 2021, 8, 2102970 CrossRef CAS PubMed.
- T. Li, D. Yang, L. Zhai, S. Wang, B. Zhao, N. Fu, L. Wang, Y. Tao and W. Huang, Adv. Sci., 2017, 4, 1600166 CrossRef PubMed.
- B. Xu, Z. Song, M. Zhang, Q. Zhang, L. Jiang, C. Xu, L. Zhong, C. Su, Q. Ban, C. Liu, F. Sun, Y. Zhang, Z. Chi, Z. Zhao and G. Shi, Chem. Sci., 2021, 12, 15556–15562 RSC.
- Y. Yang, Y. Liang, Y. Zheng, J. A. Li, S. Wu, H. Zhang, T. Huang, S. Luo, C. Liu, G. Shi, F. Sun, Z. Chi and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201820 CrossRef CAS PubMed.
- W. Song and J. Y. Lee, Adv. Opt. Mater., 2017, 5, 1600901 CrossRef.
- K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z. Lu, S. Zhang and C. Yang, J. Am. Chem. Soc., 2018, 140, 8877–8886 CrossRef CAS PubMed.
- B. Bie, L. Guo, M. Zhang, Y. Ma and C. Yang, Adv. Opt. Mater., 2021, 2101992 Search PubMed.
- H. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- Z. Wei, Z. Y. Gu, R. K. Arvapally, Y. P. Chen, R. N. Mcdougald Jr, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H. C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed.
- H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Adv. Opt. Mater., 2016, 4, 1015–1021 CrossRef CAS.
- J. Yu, Y. Cui, H. Xu, Y. Yang, Z. Wang, B. Chen and G. Qian, Nat. Commun., 2013, 4, 2719 CrossRef PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- L. Yu, Z. Wu, C. Zhong, G. Xie, Z. Zhu, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700588 CrossRef.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- P. L. Dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS PubMed.
- R. Pashazadeh, P. Pander, A. Lazauskas, F. B. Dias and J. V. Grazulevicius, J. Phys. Chem. Lett., 2018, 9, 1172–1177 CrossRef CAS PubMed.
- J. Chen, H. Liu, J. Guo, J. Wang, N. Qiu, S. Xiao, J. Chi, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202116810 CrossRef CAS PubMed.
- D. Zhang, C. Jiang, Z. Wen, X. Feng and K. Li, Chem.–Eur. J., 2022, 28, e202202305 CrossRef CAS PubMed.
- H. Mieno, R. Kabe, M. D. Allendorf and C. Adachi, Chem. Commun., 2018, 54, 631–634 RSC.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- J. Yuan, R. Chen, X. Tang, Y. Tao, S. Xu, L. Jin, C. Chen, X. Zhou, C. Zheng and W. Huang, Chem. Sci., 2019, 10, 5031–5038 RSC.
- P. Xue, P. Wang, P. Chen, B. Yao, P. Gong, J. Sun, Z. Zhang and R. Lu, Chem. Sci., 2017, 8, 6060–6065 RSC.
- S. A. Elgadi, D. M. Mayder, R. Hojo and Z. M. Hudson, Adv. Opt. Mater., 2023, 2202754 CrossRef CAS.
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 17651–17655 CrossRef CAS PubMed.
- R. Medishetty, J. K. Zareba, D. Mayer, M. Samoc and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- J. Yu, Y. Cui, C. Wu, Y. Yang, B. Chen and G. Qian, J. Am. Chem. Soc., 2015, 137, 4026–4029 CrossRef CAS PubMed.
- X. Zhang, Y. Xiao, J. Qi, J. Qu, B. Kim, X. Yue and K. D. Belfield, J. Org. Chem., 2013, 78, 9153–9160 CrossRef CAS PubMed.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- K. Xing, R. Fan, F. Wang, H. Nie, X. Du, S. Gai, P. Wang and Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 22746–22756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2251129. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02030h |
| This journal is © The Royal Society of Chemistry 2024 |