
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmploying unnatural promiscuity of sortase to construct peptide macrocycle libraries for ligand discovery†
Yan-Ni
Zhang‡
a,
Xiao-Cui
Wan‡
a,
Yang
Tang‡
b,
Ying
Chen
a,
Feng-Hao
Zheng
a,
Zhi-Hui
Cui
a,
Hua
Zhang
a,
Zhaocai
Zhou
c and
Ge-Min
Fang
 *a
*a
aSchool of Life Sciences, Institutes of Physical Science and Information Technology, Anhui University, Hefei 230601, P. R. China. E-mail: fanggm@ahu.edu.cn
bDepartment of Medical Ultrasound, Department of Stomatology, Shanghai Tenth People's Hospital, Tongji University Cancer Center, Tongji University School of Medicine, Shanghai 200072, P. R. China
cState Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Fudan University, Shanghai 200438, P. R. China
First published on 22nd May 2024
Abstract
With the increasing attention paid to macrocyclic scaffolds in peptide drug development, genetically encoded peptide macrocycle libraries have become invaluable sources for the discovery of high-affinity peptide ligands targeting disease-associated proteins. The traditional phage display technique of constructing disulfide-tethered macrocycles by cysteine oxidation has the inherent drawback of reduction instability of the disulfide bond. Chemical macrocyclization solves the problem of disulfide bond instability, but the involved highly electrophilic reagents are usually toxic to phages and may bring undesirable side reactions. Here, we report a unique Sortase-mediated Peptide Ligation and One-pot Cyclization strategy (SPLOC) to generate peptide macrocycle libraries, avoiding the undesired reactions of electrophiles with phages. The key to this platform is to mine the unnatural promiscuity of sortase on the X residue of the pentapeptide recognition sequence (LPXTG). Low reactive electrophiles are incorporated into the X-residue side chain, enabling intramolecular cyclization with the cysteine residue of the phage-displayed peptide library. Utilizing the genetically encoded peptide macrocycle library constructed by the SPLOC platform, we found a high-affinity bicyclic peptide binding TEAD4 with a nanomolar KD value (63.9 nM). Importantly, the binding affinity of the bicyclic peptide ligand is 102-fold lower than that of the acyclic analogue. To our knowledge, this is the first time to mine the unnatural promiscuity of ligases to generate peptide macrocycles, providing a new avenue for the construction of genetically encoded cyclic peptide libraries.
Introduction
Peptide macrocycles are attractive scaffolds for peptide drug development.1,2 Many peptide drugs entering clinical trials are peptide macrocycles.3 In comparison with linear peptides, peptide macrocycles exhibit many favorable pharmacochemical properties, such as increased affinity for targets, enhanced cell membrane permeability and increased resistance to proteolysis. Additionally, peptide macrocycles have a larger surface than small molecules and are perceived to be more suitable for targeting protein–protein interactions.4–6 Currently, techniques for generating large-scale peptide macrocycle libraries include creating synthetic combinatorial chemical libraries and genetically encoded peptide libraries.7,8 Genetically encoded peptide libraries, such as phage display and mRNA display, can easily provide a diversity of 109–1012 and they are an invaluable avenue for discovering high-affinity peptide ligands.9 New macrocyclization strategies on phage-displayed peptide libraries will expand the structural diversity of genetically encoded libraries and facilitate the discovery of macrocyclic peptide drugs.Traditional phage-displayed peptide macrocycle libraries are disulfide-tethered cyclic peptides formed by cysteine oxidation at several fixed positions.10–16 However, the disulfide bonds are easily exchanged with free sulfhydryl groups and are unstable in reducing environments.17–19 In order to solve this problem, many chemical macrocyclization methods for phage libraries have been developed.20–22 Among them, one of the most widely used approaches is the use of electrophilic reagents to crosslink two or more Cys residues to construct thioether-tethered peptide monocycles or peptide bicycles.23–36 To ensure the efficiency of the macrocyclization reactions, phage libraries are often treated with highly reactive electrophilic reagents such as benzyl bromide derivatives, which sometimes cause undesired side reactions and phage toxicity.23,28,29 The embedding of orthogonal reactive non-canonical amino acids (UAAs) is an alternative approach,37–42 but the incorporation procedure of UAAs is complex. In addition, the yield of phages containing unnatural amino acids is often lower than that containing canonical amino acids.
Enzymatic peptide cyclization is another attractive option for constructing cyclic peptide libraries.43,44 In contrast to chemical macrocyclization, enzymatic strategies can avoid the use of highly reactive electrophiles that are toxic to phages and can achieve efficient peptide cyclization under mild conditions. Strikingly, Urban et al. constructed a lanthiopeptide-displaying phage library by introducing the lanthiopeptide synthetase ProcM into E. coli and discovered a lanthiopeptide analogue targeting urokinase plasminogen activator (uPA).45,46 Recently, Bowers et al. utilized tyrosinase to oxidize tyrosine phenol to an electrophilic o-quinone, which subsequently reacts with cysteine to form side-chain crosslinked peptides for mRNA display.47 Suga et al. introduced the PTM enzymes from laz BGC into flexible in vitro translation (FIT) and established a unique FIT-laz platform for displaying a lactazole-like thiopeptide library.48 However, due to the complexity of the operation, these enzymatic methods have not been widely used. In addition to the above-mentioned peptide-modifying enzymes, there are peptide ligases in nature that can connect two peptides in a sequence-specific manner.49 Sortase (SrtA) is a widely used ligase that can mediate ligation of an N-terminal oligoglycine peptide with another peptide containing C-terminal LPXTG (X can be any natural L-amino acid) and has been used in live-cell protein modification and protein semi-synthesis.50–54 To date, few studies have reported the tolerance of SrtA to unnatural amino acids at the X site of the LPXTG motif, let alone its use in the genetically encoded macrocyclic peptide library.55
Here, we report a sortase-mediated peptide ligation and one-pot cyclization (SPLOC) strategy and its application to genetically encoded phage-displayed cyclic peptide libraries (Fig. 1). We explore the tolerance of sortase to unnatural amino acids at the X site of the LPXTG motif and demonstrate its compatibility with electrophilic unnatural amino acids. The incorporated electrophilic group allows intramolecular cyclization to generate peptide macrocycles. We further employ the SPLOC strategy to construct phage-displayed peptide macrocycle libraries. Using the peptide macrocycle platform, we identify a bicyclic peptide that binds to TEAD4 with a KD value of 63.9 nM and efficiently blocks TEAD4·YAP interaction.56 This represents the first time to mine the unnatural promiscuity of peptide ligase to construct the genetically encoded peptide macrocycle library, and is expected to provide a powerful platform for the discovery of peptide macrocycle ligands.
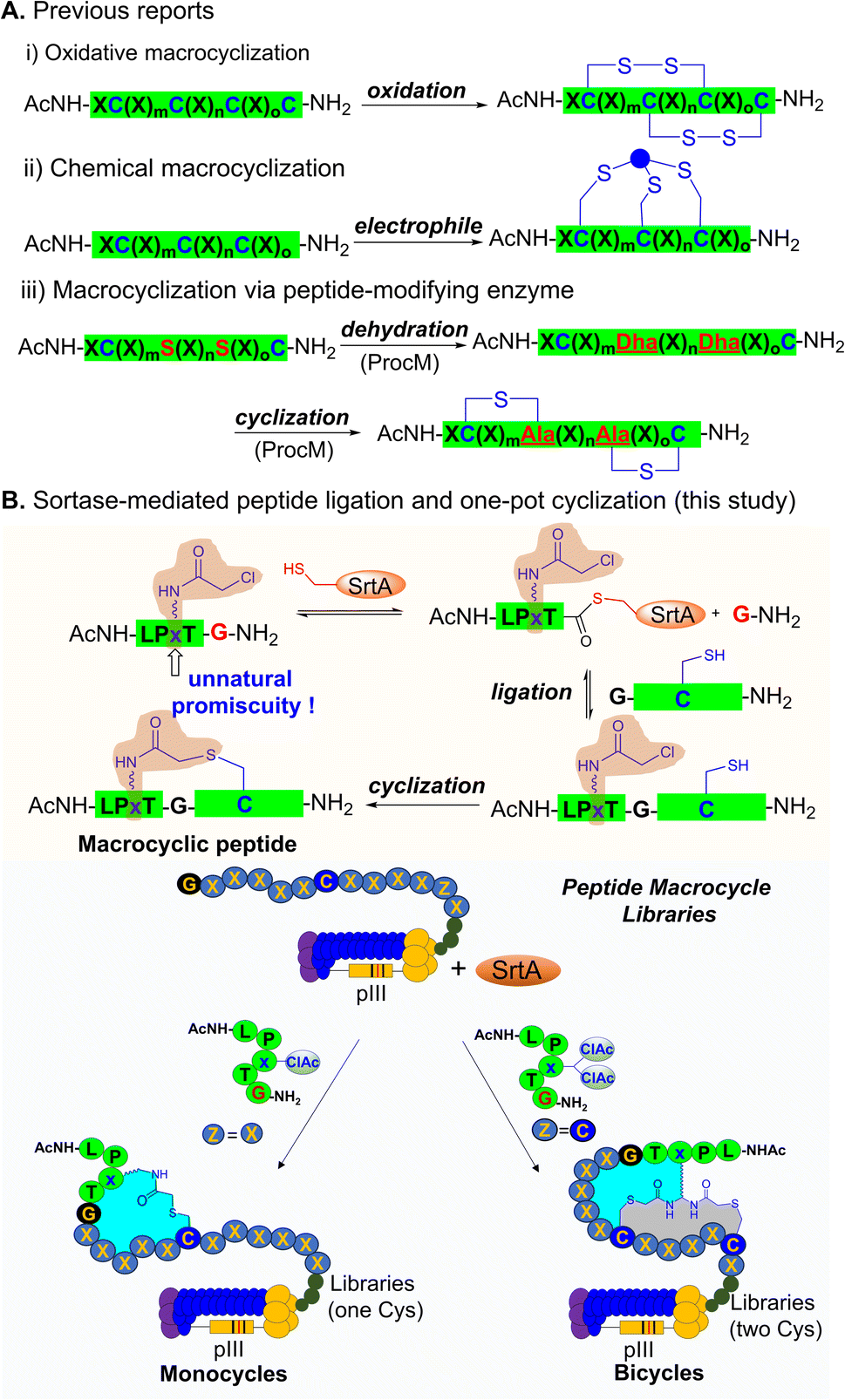 | ||
| Fig. 1 Peptide cyclization for the construction of phage-displayed peptide macrocycle libraries. (A) The previously reported peptide cyclization for phage-displayed libraries. (B) SrtA-mediated peptide ligation and one-pot cyclization (SPLOC) strategy and its application in constructing phage-displayed peptide macrocycle libraries (ClAc: chloroacetyl group). | ||
Results and discussion
Exploring the unnatural promiscuity of sortase for peptide ligation and cyclization
Our study began by investigating the tolerance of sortase to unnatural amino acids at the X site in the pentapeptidic LPXTG sequence and its utility in peptide macrocyclization. In conventional sortase-catalyzed peptide ligation, the X site of the LPXTG motif can be any natural L-amino acid. This natural promiscuity of sortase at the X site inspired us to explore its tolerance to unnatural amino acids. We wondered whether it would be compatible with unnatural amino acids that contain a low-reactive electrophilic group, such as side-chain chloroacetyl-modified lysine (ClAcLys). The chloroacetyl group can undergo efficient intramolecular cyclization with cysteine residues. Accordingly, we synthesized the pentapeptide 1 bearing ClAcLys and a C-terminal glycinamide (Fig. 2A). 1 (50 μM) and N-terminal glycine peptide 2 (50 μM) were dissolved in HEPES (50 mM, pH 8.0) buffer containing 2 mM CaCl2 and 1 mM TCEP (Tris(2-carboxyethyl)phosphine). After adding 5 μM of sortase A (SrtA), we were pleased to observe that 1 and 2 were smoothly converted to the desired cyclic peptide 3 within one hour (Fig. 2D). Note that we observed a small amount of 1 being hydrolyzed by SrtA and the peak of the hydrolysis product of 1 almost coincided with the peak of 3. Luckily, we did not observe enzymatic hydrolysis of the product 3, suggesting that 3 may be not recognized by SrtA. The resistance of 3 to SrtA-mediated hydrolysis would be beneficial to the utility of the SPLOC strategy in the construction of peptide macrocycles.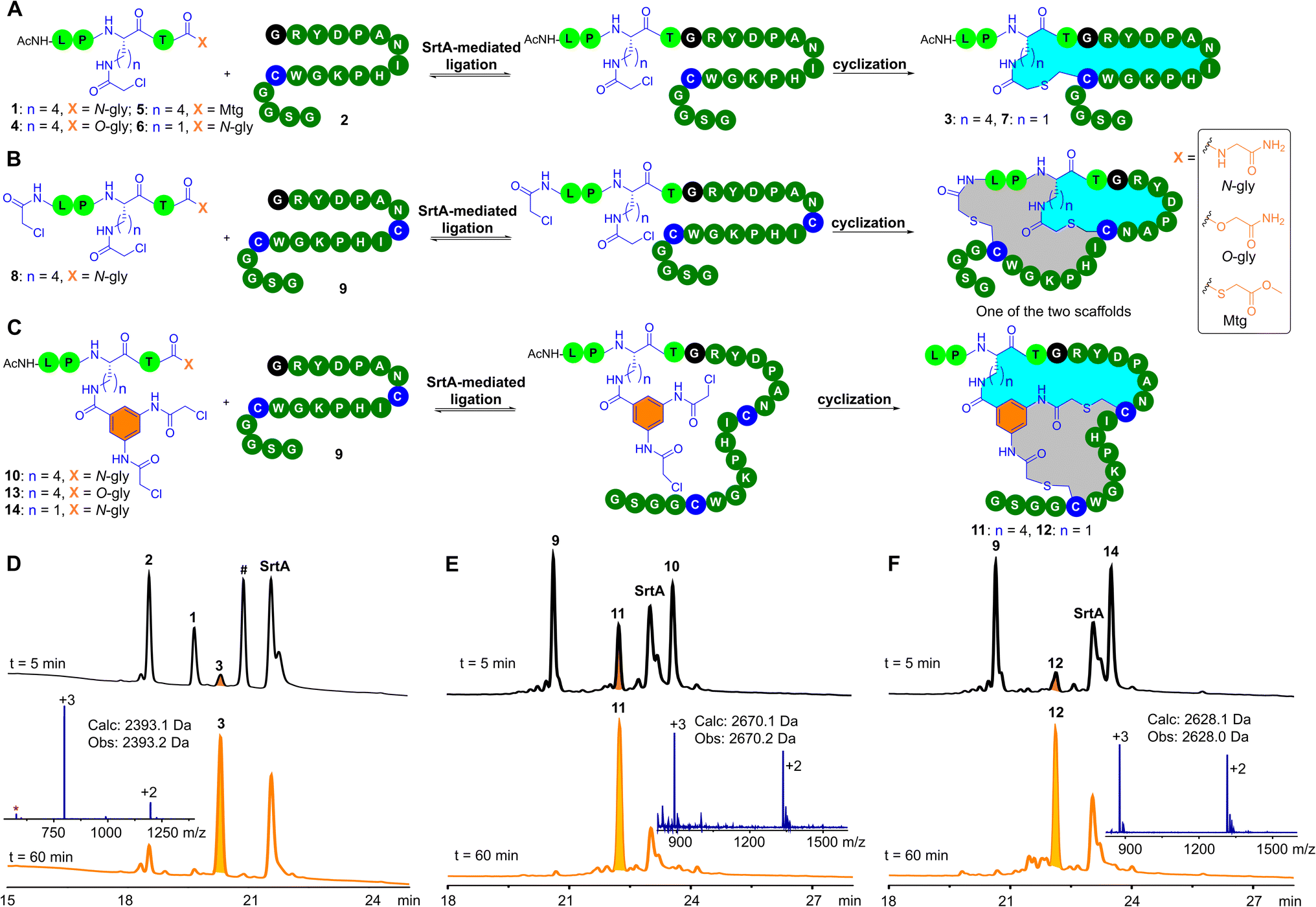 | ||
| Fig. 2 SrtA-mediated ligation and one-pot cyclization (SPLOC). (A) SrtA-mediated ligation and cyclization of pentapeptides with N-terminal glycine peptide 2 to generate the desired monocyclic products. (B) SrtA-mediated ligation and cyclization of pentapeptide 8 with N-terminal glycine peptide 9 to generate bicyclic products of two different scaffolds. (C) SrtA-mediated ligation and cyclization of pentapeptides with N-terminal glycine peptide 9 to generate the desired bicyclic products. (D) HPLC chromatograms (210 nm) of SrtA-mediated ligation and cyclization of 1 with an equivalent amount of 2 (# denotes an uncyclized ligation intermediate; a trace amount of SrtA-mediated hydrolysis of 1 could be observed in the mass spectrum of 3 as indicated by the asterisk). (E) HPLC chromatograms (210 nm) of SrtA-mediated ligation and cyclization of 9 with an equivalent amount of 10. (F) HPLC chromatograms (210 nm) of SrtA-mediated ligation and cyclization of 9 with an equivalent amount of 14. Reaction conditions: 50 μM peptides, 5 μM SrtA, 50 mM HEPES, 100 mM NaCl, 2 mM CaCl2, and 1 mM TCEP, pH 7 or pH 8, 37 °C. Note that the desired macrocyclic peptide product was formed upon addition of SrtA. | ||
The conventional sortase-catalyzed peptide ligation is reversible, and the peptide substrate cannot be completely converted into the ligation product (Fig. S32†).57–59 The product of the conventional sortase-catalyzed ligation can also be hydrolyzed by SrtA.60 We speculated that SrtA does not recognize the cyclic peptide product, and therefore 3 formed by the SPLOC strategy could not be hydrolyzed by SrtA, resulting in irreversible enzymatic ligation of 1 and 2. The ligation of 1 and 2 was completed in 10 minutes, followed by the cyclization between the chloroacetyl group and the cysteine residue. Note that, in the absence of SrtA, the reaction between 1 and 2 did not proceed (ESI, Fig. S32†), demonstrating that peptide ligation and macrocyclization occurred under the catalysis of SrtA.
Given the previously reported SrtA-mediated irreversible ligation of depsipeptides or thioester peptides with N-terminal glycine peptides,57,59 we investigated the reaction of the depsipeptide 4 or the thioester peptide 5 with 2. As expected, upon addition of SrtA, both 4 and 5 react with 2 to generate the desired 3 (ESI, Fig. S33–S34†). In the absence of SrtA, 5 and 2 undergo thioester–thiol exchange to form a thioester-linked byproduct (ESI, Fig. S34†).61 Because of the presence of many cysteine residues in pIII protein, 5 may not be ideal for phage modification. In contrast, 1 and 4 are inert toward thiols in neutral aqueous buffers and therefore are ideal for phage modification.
We further investigated the substrate scope of the SrtA-mediated peptide cyclization for the N-terminal glycine peptide. The arginine residue following the N-terminal glycine residue of 2 was replaced by other natural L-amino acids (except cysteine), resulting in 18 N- terminal glycine peptides. Among these amino acid mutants, 17 mutant peptides (except proline) underwent SrtA-mediated ligation and one-pot cyclization (ESI, Fig. S37–S54†) to generate the desired monocyclic products. Of note, we observed the hydrolysis of 1 catalyzed by sortase A and the appearance of some small peaks after SrtA-mediated ligation and one-pot cyclization. These small peaks are mainly the hydrolysis by-product of 1 and the remaining peptide substrates. SrtA-mediated hydrolysis of 1 may have little effect on the modification of the phage-displayed libraries because the amount of 1 is in large excess relative to the phage particles. Additionally, we replaced ClAcLys of 1 with ClAcDap to obtain 6 (Dap: diaminopropionic acid). The relatively short side chain of Dap may increase the structural rigidity of the formed peptide macrocycles. To our delight, the enzymatic ligation and peptide cyclization of 2 and 6 was completed in one hour, yielding the desired cyclic peptide 7 (ESI, Fig. S35†). Collectively, these results demonstrate that the X site of the LPXTG motif can be unnatural amino acids that contain a electrophilic group, and the electrophilic group can be employed for peptide monocyclization.
Sortase-mediated peptide ligation and bicyclization
We next explored SrtA-mediated peptide bicyclization. The N-terminal amino group of 1 was modified with a chloroacetyl group to afford 8 (Fig. 2B). As expected, upon addition of SrtA, 8 reacted with 9 containing two cysteine residues to form bicyclic peptides. Due to the asymmetry of the chloroacetyl groups in 8, we observed the formation of two bicyclic peptides with the same molecular weight (Fig. 2B, ESI, Fig. S55†). The resultant two bicyclic scaffolds can expand the structural diversity of phage libraries,26 but would increase the workload of identifying bicyclic peptide ligands. To solve this problem, 3,5-bis(2-chloroacetamido)benzoic acid (Cab) was anchored onto the lysine residue of 1 to generate 10 (Fig. 2C). The twofold symmetry of the Cab group ensures the formation of one product. Indeed, 10 reacts with 9 to yield the desired bicyclic peptide 11 (Fig. 2E).Meanwhile, we studied the enzymatic ligation between depsipeptide 13 and 9. Expectedly, 13 reacts with 9 to give the desired 11 (ESI, Fig. S57†). To fine-tune the topological structure of the bicyclic scaffold, we replaced Lys in 10 with Dap to generate 14. Gratifyingly, ligation and one-pot cyclization between 14 and 9 proceeded smoothly to generate the desired 12 (Fig. 2F). In addition, we replaced Cab in 13 with the 1,3-dibromo-methyl benzyl group (Bmb). Although the Bmb analogue of 14 reacts with 9 smoothly to provide the desired bicyclic peptide (ESI, Fig. S59–S60†), TCEP can react with the bromomethylbenzyl moiety.62 During the modification of phage-displayed libraries, this side reaction can be avoided. Specifically, the phage-displayed libraries are first treated with TCEP so that cysteine residues on the phage surface do not form disulfide bonds, and then the excess TCEP is removed by ultrafiltration or PEG precipitation. With the TCEP-treated phage libraries, we could perform sortase-mediated peptide ligation and bicyclization by using the Bmb analogue of 14. It should be noted that, compared to 14, the operation process for the Bmb analogue of 14 is relatively time-consuming.
Sortase-mediated modification of phage display libraries
Encouraged by the success of SrtA-mediated peptide ligation and one-pot cyclization (SPLOC), we decided to combine the SPLOC-based strategy with phage display (Fig. 3A and B). First, we investigated the influence of SrtA-based reaction conditions on phage infectivity. Starting from the M13KE phage vector, a peptide library of GX12C (X is any L-amino acid) was fused to the N-terminus of pIII protein, and a flexible linker (GGSGGSGG) was inserted between pIII protein and the peptide library. After electroporation of the engineered phage vector to cells, a phage library with a diversity of 106 was obtained. To our delight, SrtA and 1 had little effect on the infectivity of the M13KE phage (Fig. 3C). Then, a biotin–avidin pull-down assay was performed to evaluate the efficacy of SrtA-mediated peptide ligation on the phage surface. Biotin-1 was synthesized by coupling biotin to the N-terminal amino group of 1. The phages were dissolved in 50 mM HEPES buffer (100 mM NaCl, 2 mM CaCl2, 1 mM TCEP, pH8) and treated with Biotin-1 (50 μM) for one hour at 25 °C. After pulldown with avidin-coated beads, about 73% of phages were captured (ESI, Fig. S85†), indicating the yield of SrtA-mediated peptide ligation on the phage surface.63 It should be noted that sortase-mediated phage surface modification has been thoroughly demonstrated in previous studies.64–68 As a control, the amount of phage particles without Biotin-1 treatment showed little change before and after biotin capture. Although the biotin capture assay cannot assess the following peptide cyclization, the intramolecular reaction between the chloroacetyl group and cysteine residue has proven to be rapid and efficient in many previous studies.23–36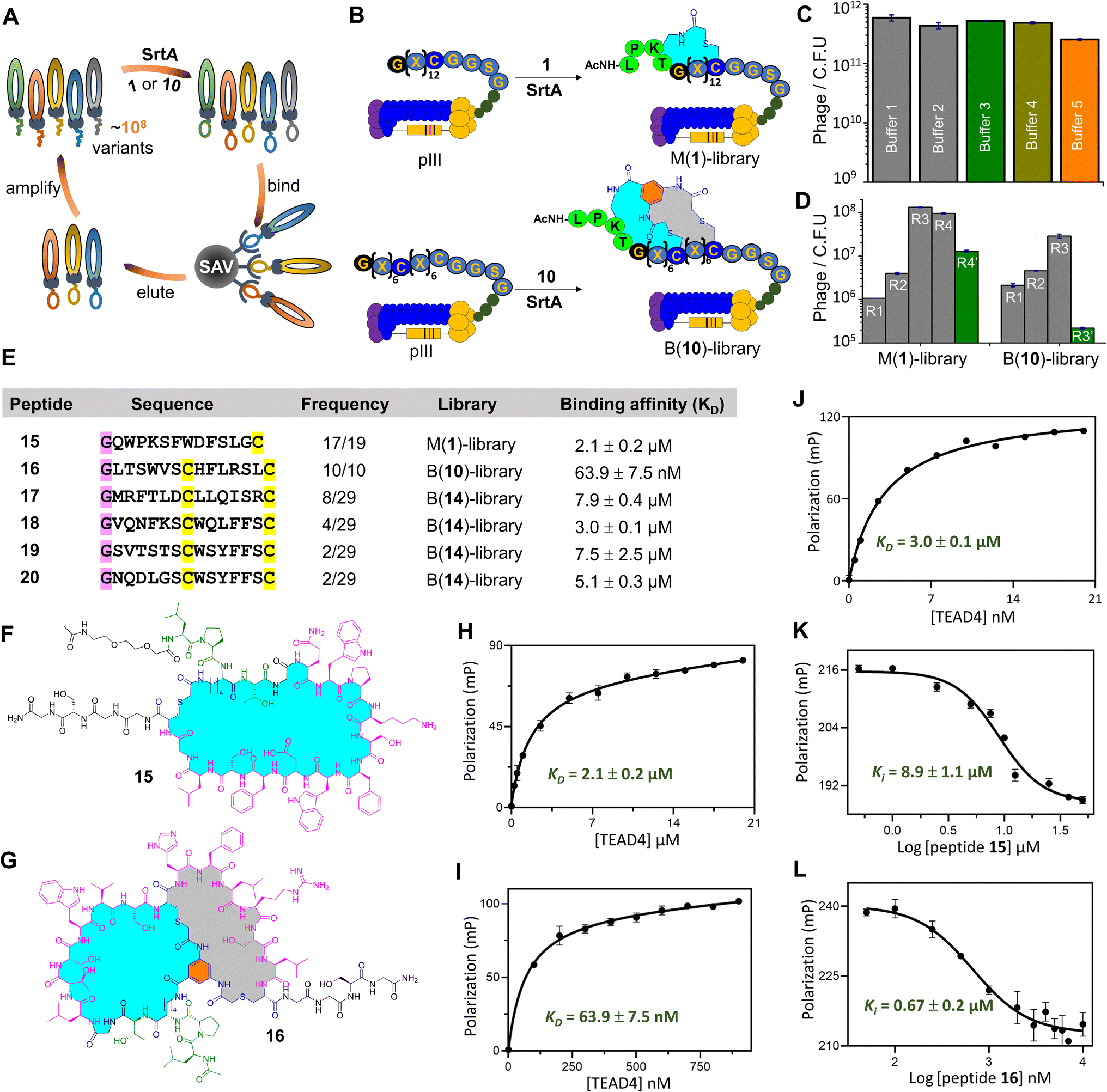 | ||
| Fig. 3 SrtA-mediated phage display of macrocyclic peptides against TEAD4. (A) Scheme for the SrtA-based phage display (SAV: streptavidin-coated beads). (B) Construction of a monocyclic peptide library (M-library) and bicyclic peptide library (B-library) by SrtA-mediated peptide cyclization on the phage surface; M(1)-library is a monocyclic peptide library constructed by macrocyclization of the phage displayed GX12C library with 1; B(10)-library is a bicyclic peptide library constructed by the macrocyclization of the phage-displayed GX6CX6C library with 10. (C) Influence of different buffers on the infectivity of M13KE phages. Buffer 1: PBS (pH 7.4); buffer 2: 50 mM HEPES, 100 mM NaCl, 2 mM CaCl2, 1 mM TCEP, pH 8; buffer 3: 50 mM HEPES, 100 mM NaCl, 2 mM CaCl2, 1 mM TCEP, 5 μM SrtA, pH 8; buffer 4: 50 mM HEPES, 100 mM NaCl, 2 mM CaCl2, 1 mM TCEP, 50 μM peptide 1, pH 8; buffer 5: 50 mM HEPES, 100 mM NaCl, 2 mM CaCl2, 1 mM TCEP, 5 μM SrtA, 50 μM peptide 1, pH 8. (D) Phage tilters after 3–4 rounds of selection against immobilized TEAD4 (R3′ and R4′ are negative selections with solely avidin-coated beads). (E) Enriched peptide sequences after phage display screening targeting TEAD4; B(14)-library is a bicyclic peptide library constructed by macrocyclization of the phage-displayed GX6CX6C library with 14. (F) and (G) Chemical structure of 15 and 16 from the phage biopanning of the M-library and B-library. (H), (I) and (J) Binding affinity of 15, 16 and 18 to TEAD4 measured by the fluorescence polarization method. (K) and (L) Inhibition of TEAD4·YAP by 15 or 16 assessed by the fluorescence polarization assay. | ||
We chose to synthesize the enriched peptide 15 (Fig. 3E and F), and measure its affinity to TEAD4. The dissociation constant (KD) of 15 to TEAD4 was measured to be 2.1 μM in the fluorescence polarization assay (Fig. 3H). In a competition experiment with YAP, 15 inhibited 50% of TEAD4·YAP interaction with an apparent EC50 of 8.9 μM (Fig. 3K). For comparison, the linear version of 15 shows a KD value of 6.3 μM for TEAD4 (ESI, Fig. S79†). To study the influence of the cyclic peptide scaffold on phage biopanning, we used 6 containing ClAcDap (Fig. 2A) to treat the phage library to generate structurally more rigid cyclopeptides. After four rounds of biopanning, phage recovery was only 4-fold higher than in the first round, much lower than that of the phage treated with 1. The observed differences in phage enrichment suggest that the chemical bridge may profoundly affect the result of biopanning.27 Note that, in our platform, different chemical bridges can be easily introduced at the X site of the LPXTG motif to increase the likelihood of finding the ideal peptide ligands.
We synthesized five bicyclic peptides for KD value measurement. Among them, 16 (Fig. 3G) was identified as a good ligand for TEAD4 with a KD value of 63.9 nM (Fig. 3I), superior to the other four bicyclic peptides (Fig. 3E and J). As expected, 16 exhibited concentration-dependent inhibition of TEAD4·YAP interaction with an EC50 of 0.67 μM (Fig. 3L). Moreover, we cut short 16 by truncation of the N-terminal dipeptidic LP sequence and the C-terminal GGSG sequence (ESI, Fig. S68†). Gratifyingly, the KD value of the truncated peptide to TEAD4 is 70.1 nM, comparable to 16 (Supplementary Fig. S79†). Note that the linear version of 16 provides a KD value of 6.5 μM, 102 times higher than that of 16, highlighting the contribution of the bicyclic scaffold to the peptide binding affinity.
Finally, we evaluated the bioactivity of 16 at the cellular level. To confirm the ability of 16 to capture endogenous TEAD4, a biotin was conjugated to the N-terminus of 16 for the streptavidin pull-down assay. After co-incubation with AGS cell lysates, the sample treated with 16-Biotin was captured by streptavidin-coated beads and showed a distinct TEAD4 band on SDS-PAGE (Fig. 4B). In contrast, the biotin-treated sample does not show a TEAD4 band. We then measured the inhibitory activity of 16 on AGS cells. Despite the presence of PDPA (a well-known nanocarrier for intracellular peptide delivery),5416 showed little inhibitory inhibition on AGS cells at 10 μM (ESI, Fig. S92†). Given that TEAD4 is located in the nucleus, a positively charged TAT or NLS (Nuclear Localization Sequence) tag was conjugated to the N-terminus of 16 (Fig. 4A). Gratifyingly, 16-TAT and 16-NLS exhibited cellular inhibitory activity with EC50 values of 13.8 μM and 8.3 μM (Fig. 4C). As a control, TAT and NLS that were not conjugated with 16 exhibit little cytotoxicity to AGS cells, indicating the contribution of 16 to the observed cellular inhibitory activity of 16-TAT and 16-NLS.
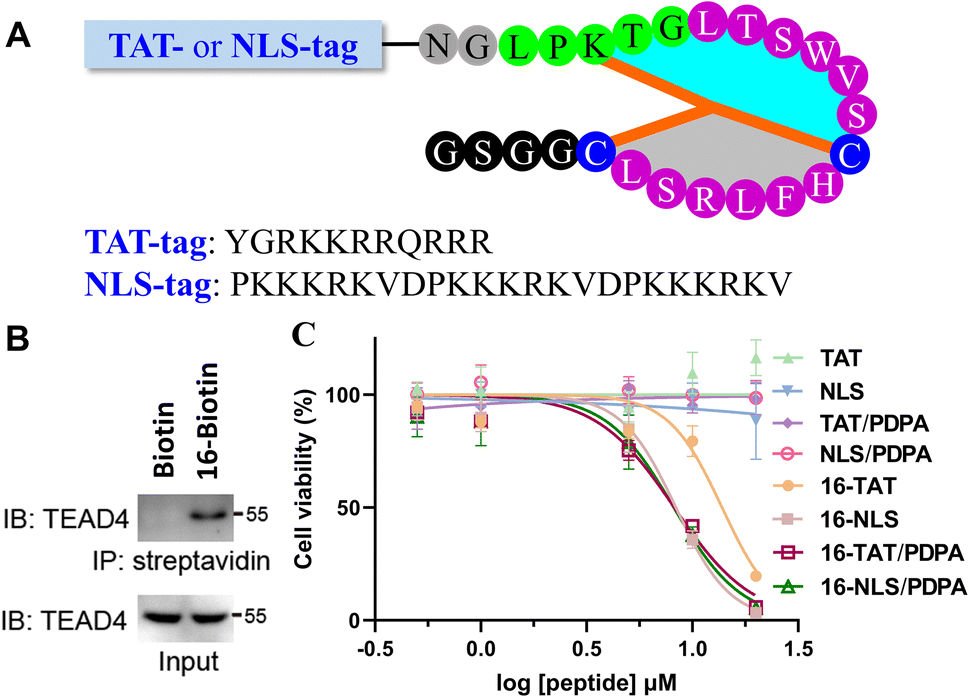 | ||
| Fig. 4 TAT or NLS-modification of the bicyclic peptide 16. (A) Structure of the peptides that contain the N-terminal TAT or NLS tag. (B) Streptavidin–biotin pulldown of endogenous TEAD4 by 16-Biotin (IB: immunoblotting, IP: immunoprecipitation). (C) Cellular inhibitory activity of the peptides to AGS cells. | ||
Conclusions
To summarize, we describe a ligase-based macrocyclization strategy for the construction of phage-displayed peptide macrocycle libraries, named SrtA-mediated peptide ligation and one-pot cyclization (SPLOC). The key to this strategy is to mine the tolerance of sortase to unnatural amino acids at the X site of the LPXTG motif. The electrophilic chloroacetyl group on the side chain of the X residue allows intramolecular cyclization with the cysteine residue to form peptide macrocycles. The SPLOC strategy is combined with phage display to generate a new type of genetically encoded macrocyclic peptide library for peptide ligand discovery. Using TEAD4 as a model target, we identified a potent bicyclic peptide with a KD value of 63.9 nM to TEAD4. This bicyclic peptide can effectively block the TEAD4·YAP interaction and exhibit cellular inhibitory activity. Employing the unnatural promiscuity of peptide ligases to generate peptide macrocycles represents a new direction for the construction of genetically encoded peptide libraries.Data availability
All experimental data that support the results of this study can be found in this article or in the ESI.†Author contributions
The idea of the study was designed by G.-M. Fang. The system of sortase-based phage display was established by Y.-N. Zhang. The sortase-mediated peptide ligation and cyclization was performed by Y.-N. Zhang. The procedure for phage display was established by Y.-N. Zhang and X.-C. Wan. TEAD4-targeting cyclic peptides were synthesized and analysed by Y.-N. Zhang, X.-C. Wan, Y. Chen, Z.-H. Cui, F.-H. Zheng, H. Zhang. The idea of the cellular activity was designed by Z. Zhou. The activity of the peptides was measured by Y. Tang. The manuscript was written and checked by G.-M. Fang and Y.-N. Zhang.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22077002 and 21807001). For the construction of the M13 phage library, ER 2738 cells and the M13KE phage vector were obtained from NEB Ltd. For the construction of the phagemid system, the M13KO7 helper phage, TG1 cells and pCantab 5E vector were obtained from NB Biolab (Chengdu, China). The TEAD4 coding sequence was inserted into the pET-28a vector by the laboratory of Zhaocai Zhou, Fudan University, Shanghai, China. For cellular activity, AGS cells were obtained from the cell library of Chinese Academy of Sciences (Shanghai, China). We thank the staff for providing technical support with using the facility of Institute of Health Sciences & Technology, Anhui University.References
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- D. S. Nielsen, N. E. Shepherd, W. Xu, A. J. Lucke, M. J. Stoermer and D. P. Fairlie, Chem. Rev., 2017, 117, 8094–8128 CrossRef CAS PubMed.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco Jr, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS PubMed.
- E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56, 10294–10323 CrossRef CAS PubMed.
- S. Dong, J.-S. Zheng, Y. Li, H. Wang, G. Chen, Y. Chen, G. Fang, J. Guo, C. He, H. Hu, X. Li, Y. Li, Z. Li, M. Pan, S. Tang, C. Tian, P. Wang, B. Wu, C. Wu, J. Zhao and L. Liu, Sci. China: Chem., 2024, 67, 1060–1096 CrossRef CAS.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288–10317 CrossRef CAS PubMed.
- C. Sohrabi, A. Foster and A. Tavassoli, Nat. Rev. Chem, 2020, 4, 90–101 CrossRef CAS PubMed.
- R. C. Ladner, Trends Biotechnol., 1995, 13, 426–430 CrossRef CAS PubMed.
- R. C. Ladner, A. K. Sato, J. Gorzelany and M. de Souza, Drug Discovery Today, 2004, 9, 525–529 CrossRef CAS PubMed.
- C. Chen, I. R. Rebollo, S. A. Buth, J. Morales-Sanfrutos, J. Touati, P. G. Leiman and C. Heinis, J. Am. Chem. Soc., 2013, 135, 6562–6569 CrossRef PubMed.
- S. Lu, Y. Wu, J. Li, X. Meng, C. Hu, Y. Zhao and C. Wu, J. Am. Chem. Soc., 2020, 142, 16285–16291 CrossRef CAS PubMed.
- H. Dong, J. Li, H. Liu, S. Lu, J. Wu, Y. Zhang, Y. Yin, Y. Zhao and C. Wu, J. Am. Chem. Soc., 2022, 144, 5116–5125 CrossRef CAS PubMed.
- S. Lu, S. Fan, S. Xiao, J. Li, S. Zhang, Y. Wu, C. Kong, J. Zhuang, H. Liu, Y. Zhao and C. Wu, J. Am. Chem. Soc., 2023, 145, 1964–1972 CrossRef CAS PubMed.
- J. Li, H. Liu, S. Xiao, S. Fan, X. Cheng and C. Wu, J. Am. Chem. Soc., 2023, 145, 28264–28275 CrossRef CAS PubMed.
- H.-K. Cui, Y. Guo, Y. He, F.-L. Wang, H.-N. Chang, Y.-J. Wang, F.-M. Wu, C.-L. Tian and L. Liu, Angew. Chem., Int. Ed., 2013, 52, 9558–9562 CrossRef CAS PubMed.
- Y. Guo, D.-M. Sun, F.-L. Wang, Y. He, L. Liu and C.-L. Tian, Angew. Chem., Int. Ed., 2015, 54, 14276–14281 CrossRef CAS PubMed.
- C. Zuo, W.-W. Shi, X.-X. Chen, M. Glatz, B. Riedl, I. Flamme, E. Pook, J. Wang, G.-M. Fang, D. Bierer and L. Liu, Sci. China: Chem., 2019, 62, 1371–1378 CrossRef CAS.
- A. Angelini and C. Heinis, Curr. Opin. Chem. Biol., 2011, 15, 355–361 CrossRef CAS PubMed.
- R. Derda and S. Ng, Curr. Opin. Chem. Biol., 2019, 50, 128–137 CrossRef CAS PubMed.
- S. E. Iskandar, V. A. Haberman and A. A. Bowers, ACS Comb. Sci., 2020, 22, 712–733 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. J. Nuss and R. Derda, Chem. Sci., 2016, 7, 3785–3790 RSC.
- K. F. Tjhung, P. I. Kitov, S. Ng, E. N. Kitova, L. Deng, J. S. Klassen and R. Derda, J. Am. Chem. Soc., 2016, 138, 32–35 CrossRef CAS PubMed.
- S. S. Kale, C. Villequey, X.-D. Kong, A. Zori, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- Y. Yin, Q. Fei, W. Liu, Z. Li, H. Suga and C. Wu, Angew. Chem., Int. Ed., 2019, 58, 4880–4885 CrossRef CAS PubMed.
- X.-D. Kong, J. Moriya, V. Carle, F. Pojer, L. A. Abriata, K. Deyle and C. Heinis, Nat. Biomed. Eng., 2020, 4, 560–571 CrossRef CAS PubMed.
- X. Zheng, Z. Li, W. Gao, X. Meng, X. Li, L. Y. P. Luk, Y. Zhao, Y.-H. Tsai and C. Wu, J. Am. Chem. Soc., 2020, 142, 5097–5103 CrossRef CAS PubMed.
- A. I. Ekanayake, L. Sobze, P. Kelich, J. Youk, N. J. Bennett, R. Mukherjee, A. Bhardwaj, F. Wuest, L. Vukovic and R. Derda, J. Am. Chem. Soc., 2021, 143, 5497–5507 CrossRef CAS PubMed.
- J. Wong, R. Mukherjee, O. Bilyk, J. Miao, V. T. Guzman, M. Miskoizie, Y.-S. Lin, A. Henninot, J. J. Dwyer, S. Kharchenko, A. Iampolska, D. Volochnyuk, L. M. Postovit and R. Derda, Chem. Sci., 2021, 12, 9694–9703 RSC.
- M. Zheng, F. Haeffner and J. Gao, Chem. Sci., 2022, 13, 8349–8354 RSC.
- M. Zheng, F.-J. Chen, K. Li, R. M. Reja, F. Haeffner and J. Gao, J. Am. Chem. Soc., 2022, 144, 15885–15893 CrossRef CAS PubMed.
- T. R. Oppewal, I. D. Jansen, J. Hekelaar and C. Mayer, J. Am. Chem. Soc., 2022, 144, 3644–3652 CrossRef CAS PubMed.
- F.-J. Chen, N. Pinnette, F. Yang and J. Gao, Angew. Chem., Int. Ed., 2023, 62, e202306813 CrossRef PubMed.
- J. He, P. Ghosh and C. Nitsche, Chem. Sci., 2024, 15, 2300–2322 RSC.
- X. S. Wang, P.-H. C. Chen, J. T. Hampton, J. M. Tharp, C. A. Reed, S. K. Das, D.-S. Wang, H. S. Hayatshahi, Y. Shen, J. Liu and W. Liu, Angew. Chem., Int. Ed., 2019, 58, 15904–15909 CrossRef CAS PubMed.
- B. Oller-Salvia and J. W. Chin, Angew. Chem., Int. Ed., 2019, 58, 10844–10848 CrossRef CAS PubMed.
- J. M. Tharp, J. T. Hampton, C. A. Reed, A. Ehnbom, P. C. Chen, J. S. Morse, Y. Kurra, L. M. Pe-rez, S. Xu and W. R. Liu, Nat. Commun., 2020, 11, 1392 CrossRef CAS PubMed.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS PubMed.
- T. Navaratna, L. Atangcho, M. Mahajan, V. Subramanian, M. Case, A. Min, D. Tresnak and G. M. Thurber, J. Am. Chem. Soc., 2020, 142, 1882–1894 CrossRef CAS PubMed.
- J. T. Hampton, T. J. Lalonde, J. M. Tharp, Y. Kurra, Y. R. Alugubelli, C. M. Roundy, G. L. Hamer, S. Xu and W. R. Liu, ACS Chem. Biol., 2022, 17, 2911–2922 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457–5520 CrossRef CAS PubMed.
- J. H. Urban, M. A. Moosmeier, T. Aumüller, M. Thein, T. Bosma, R. Rink, K. Groth, M. Zulley, K. Siegers, K. Tissot, G. N. Moll and J. Prassler, Nat. Commun., 2017, 8, 1500 CrossRef PubMed.
- X. Yang, K. R. Lennard, C. He, M. C. Walker, A. T. Ball, C. Doigneaux, A. Tavassoli and W. A. van der Donk, Nat. Chem. Biol., 2018, 14, 375–380 CrossRef CAS PubMed.
- M. C. Fleming, M. M. Bowler, R. Park, K. I. Popov and A. A. Bowers, J. Am. Chem. Soc., 2023, 145, 10445–10450 CrossRef CAS PubMed.
- A. A. Vinogradov, M. Shimomura, Y. Goto, T. Ozaki, S. Asamizu, Y. Sugai, H. Suga and H. Onaka, Nat. Commun., 2020, 11, 2272 CrossRef CAS PubMed.
- M. Schmidt, A. Toplak, P. JIM Quaedflieg and T. Nuijens, Curr. Opin. Chem. Biol., 2017, 38, 1–7 CrossRef CAS PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
- M. W.-L. Popp and H. L. Ploegh, Angew. Chem., Int. Ed., 2011, 50, 5024–5232 CrossRef CAS PubMed.
- H. Ai, G.-C. Chu, Q. Gong, Z.-B. Tong, Z. Deng, X. Liu, F. Yang, Z. Xu, J.-B. Li, C. Tian and L. Liu, J. Am. Chem. Soc., 2022, 144, 18329–18337 CrossRef CAS PubMed.
- Y. Goto and H. Suga, Acc. Chem. Res., 2021, 54, 3604–3617 CrossRef CAS PubMed.
- S. Jiao, H. Wang, Z. Shi, A. Dong, W. Zhang, X. Song, F. He, Y. Wang, Z. Zhang, W. Wang, X. Wang, T. Guo, P. Li, Y. Zhao, H. Ji, L. Zhang and Z. Zhou, Cancer Cell, 2014, 25, 166–180 CrossRef CAS PubMed.
- D. J. Williamson, M. A. Fascione, M. E. Webb and W. B. Turnbull, Angew. Chem., Int. Ed., 2012, 51, 9377–9380 CrossRef CAS PubMed.
- Y.-M. Li, Y.-T. Li, M. Pan, X.-Q. Kong, Y.-C. Huang, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 2198–2202 CrossRef CAS PubMed.
- C. Zuo, R. Ding, X. Wu, Y. Wang, G.-C. Chu, L.-J. Liang, H. Ai, Z.-B. Tong, J. Mao, Q. Zheng, T. Wang, Z. Li, L. Liu and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202201887 CrossRef CAS PubMed.
- H. E. Morgan, W. B. Turnbull and M. E. Webb, Chem. Soc. Rev., 2022, 51, 4121–4145 RSC.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- Y. Wu, A. Zori, J. Williams and C. Heinis, Chem. Commun., 2020, 56, 2917–2920 RSC.
- S. Ng, M. R. Jafari, W. L. Matochko and R. Derda, ACS Chem. Biol., 2012, 7, 1482–1487 CrossRef CAS PubMed.
- G. T. Hess, J. J. Cragnolini, M. W. Popp, M. K. Allen, S. K. Dougan, E. Spooner, H. L. Ploegh, A. M. Belcher and C. P. Guimaraes, Bioconjugate Chem., 2012, 23, 1478–1487 CrossRef CAS PubMed.
- Y. Ding, H. Chen, J. Li, L. Huang, G. Song, Z. Xi, X. Hua, G. Gonzalez-Sapienza, B. D. Hammock and M. Wang, Anal. Chem., 2021, 93, 11800–11808 CrossRef CAS PubMed.
- G. T. Hess, C. P. Guimaraes, E. Spooner, H. L. Ploegh and A. M. Belcher, ACS Synth. Biol., 2013, 2, 490–496 CrossRef CAS PubMed.
- H. D. Wilson, X. Li, H. Peng and C. Rader, J. Mol. Biol., 2018, 430, 4387–4400 CrossRef CAS PubMed.
- T. Teschke, B. Geltinger, A. Dose, C. Freund and D. Schwarzer, ACS Chem. Biol., 2013, 8, 1692–1697 CrossRef CAS PubMed.
- H. Adihou, R. Gopalakrishnan, T. Förster, S. M. Guéret, R. Gasper, S. Geschwindner, C. C. Garcia, H. Karatas, A. V. Pobbati, M. Vazquez-Chantada, P. Davey, C. M. Wassvik, J. K. S. Pang, B. S. Soh, W. Hong, E. Chiarparin, D. Schade, A. T. Plowright, E. Valeur, M. Lemurell, T. N. Grossmann and H. Waldmann, Nat. Commun., 2020, 11, 5425 CrossRef PubMed.
- O. S. Tokareva, K. Li, T. L. Travaline, T. M. Thomson, J.-M. Swiecicki, M. Moussa, J. D. Ramirez, S. Litchman, G. L. Verdine and J. H. McGee, Nat. Commun., 2023, 14, 6992 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01992j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |