
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin of copper dissolution under electrocatalytic reduction conditions involving amines†
Yani
Guan
 a,
Justus
Kümper
b,
Sonja D.
Mürtz
b,
Simran
Kumari
a,
Peter J. C.
Hausoul
b,
Regina
Palkovits
*bc and
Philippe
Sautet
*ad
a,
Justus
Kümper
b,
Sonja D.
Mürtz
b,
Simran
Kumari
a,
Peter J. C.
Hausoul
b,
Regina
Palkovits
*bc and
Philippe
Sautet
*ad
aDepartment of Chemical and Biomolecular Engineering, University of California Los Angeles, Los Angeles, CA 90095, USA. E-mail: sautet@ucla.edu
bChair of Heterogeneous Catalysis and Technical Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
cInstitute for Sustainable Hydrogen Economy (INW-2), Forschungszentrum Jülich, Am Brainergy Park 4, 52428 Jülich, Germany
dDepartment of Chemistry and Biochemistry, University of California Los Angeles, Los Angeles, CA 90095, USA
First published on 19th August 2024
Abstract
Cu dissolution has been identified as the dominant process that causes cathode degradation and losses even under cathodic conditions involving methylamine. Despite extensive experimental research, our fundamental and theoretical understanding of the atomic-scale mechanism for Cu dissolution under electrochemical conditions, eventually coupled with surface restructuring processes, is limited. Here, driven by the observation that the working Cu electrode is corroded using mixtures of acetone and methylamine even under reductive potential conditions (−0.75 V vs. RHE), we employed Grand Canonical density functional theory to understand this dynamic process under potential from a microscopic perspective. We show that amine ligands in solution directly chemisorb on the electrode, coordinate with the metal center, and drive the rearrangement of the copper surface by extracting Cu as adatoms in low coordination positions, where other amine ligands can coordinate and stabilize a surface copper–ligand complex, finally forming a detached Cu–amine cationic complex in solution, even under negative potential conditions. Calculations predict that dissolution would occur for a potential of −1.1 V vs. RHE or above. Our work provides a fundamental understanding of Cu dissolution facilitated by surface restructuring in amine solutions under electroreduction conditions, which is required for the rational design of durable Cu-based cathodes for electrochemical amination or other amine involving reduction processes.
1. Introduction
The electrochemical reductive amination is a green way for the conversion of biomass,1–3e.g. levulinic acid,4,5 which is a longer-chain ketone. Thus, it is of great importance to rationally design catalysts that are effective and stable under reaction conditions.Cu demonstrates high efficiency in facilitating electrochemical amination2,3,6–11 reactions, where Mürtz et al.3 named copper as a green cathode material for the reductive amination of acetone (the first representative of levulinic acid's homologous series) with methylamine (a simple primary amine) as nitrogen source and obtained N-methylpropan-2-amine in a yield of 65% (±2%). Copper is also used for a large ensemble of electroreduction reactions, such as CO and CO2 electroreduction into C1 and C2+ products,12–14 where amines could be a co-reactant, for example in the case of the electroreduction of amine-captured CO2.15 Numerous studies have shown that under electrochemical reduction conditions, Cu suffers from instability and the arrangement of Cu surface atoms has a profound effect on performance.16–18 Even though several experimental instruments, such as liquid cell transmission electron microscopy (TEM)19–21 and in situ electron microscopy,22 can monitor the morphological evolution of electrocatalysts under reaction conditions,22–25 where the specific morphological features26,27 are responsible for a given activity and selectivity, the transient Cu-cation species in solution are straggling to be fully grasped since experimental conclusions are largely drawn from samples that have been removed from the electrochemical environment and evaluated in the absence of an applied potential. However, these redox processes play a role in Cu-reconstruction, including dissolution and re-deposition.28 During the cell startup, when the working electrode experiences cathodic potentials, a dissolution–redeposition of Cu species in solution occurs and induces changes in the catalyst morphology and total exposed surface.22,29,30 This kind of changes are in need to be explored in order to design durable catalysts.
Here, after a 30 minute electrolysis of an acetone/methylamine mixture, the concentration of Cu in the solution was measured, showing that at −0.75 V vs. RHE, the concentration can still hold a value of around 13 ppm. To probe the underlying mechanism, grand canonical DFT was used to understand the reconstruction and dissolution of copper by complexation with soluble amine ligands. These ligands initially adsorb on the Cu surface in electroreduction conditions and then enable transient restructuring and dissolution of Cu by forming coordination complexes with Cu cations. NH3 is considered as a model amine in our calculations but, in addition, ligand effects are explored by performing calculations for two other amines as ligands. Our study offers essential insights into the dynamic process of the Cu surface during electrochemical reduction, vital for designing durable Cu-based cathodes for a large array of processes.
2. Results and discussion
2.1 Experimental observations
Building upon the research conducted by Mürtz et al.,3 we utilized the reductive amination technique to synthesize N-methylpropan-2-amine from acetone and methylamine. The electrolysis setup involved Cu‖Pb electrodes with a N-324 membrane (Cu 99.98% from Manufacture Aldrich), using 0.5 M KH2PO4 (pH = 8.3) as the electrolyte (the following nomenclature will be used: cathode‖anode, with ‖ being the N-324 membrane). Additionally, a cooling phase lasting 18 minutes was implemented to achieve the desired temperature of 10 °C before starting the electrolysis process. This procedure is crucial for forming the intermediate N-methylpropan-2-imine, as its equilibrium with acetone and methylamine is temperature dependent (Scheme 1). | ||
| Scheme 1 The equilibrium reaction of acetone and methylamine is a condensation and yields N-methylpropan-2-imine. The imine can be reduced and results in N-methylpropan-2-amine. | ||
Subsequently, a current density of −40 mA cm−2 was applied for electrolysis, yielding an amine amount of 3.9% (±0.2%) as measured by quantitative 1H-NMR spectroscopy (depicted as the first set of bars in Fig. 1), representing a 61% decrease compared to a prior study.3 Notably, corrosion was evident on the copper electrode surface (Fig. S1†), and that corrosion was initiated by methylamine in the reaction solution (Fig. S2†). Initially, after an 18 minute cooling period, copper concentration in solution reached approximately 84 ppm measured via ICP-OES. Employing a cathodic potential around −0.16 V vs. RHE stabilized the working electrode during cooling, where at this stabilizing potential no electrochemical hydrogenation occurred as indicated by the absence of faradaic currents in CV measurements (Fig. S3†). Consequently, the copper concentration decreased to 19 ppm in the acetone–methylamine mixture, leading to an amine yield of 30% (±2%) after subsequent electrolysis. This was still 35% lower than that in the prior work3 but 26% higher than that obtained from the initial test (illustrated in the second set of bars in Fig. 1). The reduced copper concentration and increased amine yield underscore the feasibility of stabilizing the cathode in the presence of methylamine. To assess performance without a cooling phase, a third electrolysis experiment was conducted, yielding an amine of 51% (±2%), albeit still lower than the reported value,3 yet exhibiting the highest yield.
 | ||
| Fig. 1 Examining the influence of the cooling interval and its modification on the electrochemical reductive amination of acetone with methylamine. The resulting yields of N-methylpropan-2-imine (YImine), N-methylpropan-2-amine (YAmine) and isopropanol (YAlcohol) are depicted. Additionally, the conversion of acetone (X) is illustrated. Carbon balance (C.B.): 80–81%. Experimental conditions include Cu‖Pb electrode with N-324 membrane (was previously used at least 3 times with Pb as anode); current density (j) of −40 mA cm−2; Faraday equivalents (Feq.) = 1; solvent: 0.5 M KH2PO4 (pH 8.3); substrates concentrations: acetone: 2.4 M, methylamine: 2.9 M; temperature (T) maintained at 10 °C; pH at 10 °C: 12.9; anolyte: 25% H3PO4. | ||
These observations suggest that the cathode surface underwent dissolution during the cooling phase, inhibiting the electrochemical reduction of the imine while promoting water splitting.
As it was observed that a cathodic potential stabilized the copper electrode during the cooling period, the relationship between copper dissolution and applied potential will be investigated next. Four distinct potentials ranging from 0.25 V to −1.25 V vs. RHE were employed for a duration of 30 minutes, followed by measuring the concentration of dissolved copper (depicted in Fig. 2). Generally, enhanced stability of copper is observed at −1.25 V vs. RHE, evidenced by the lower concentration of Cu in the reaction solution post-electrolysis compared to potentials of −0.75 V, −0.25 V, and 0.25 V vs. RHE. This suggests that the stability of copper in the presence of methylamine is contingent upon the applied potential, indicating a critical minimum cathodic potential below which the copper electrode experiences stronger stabilization.
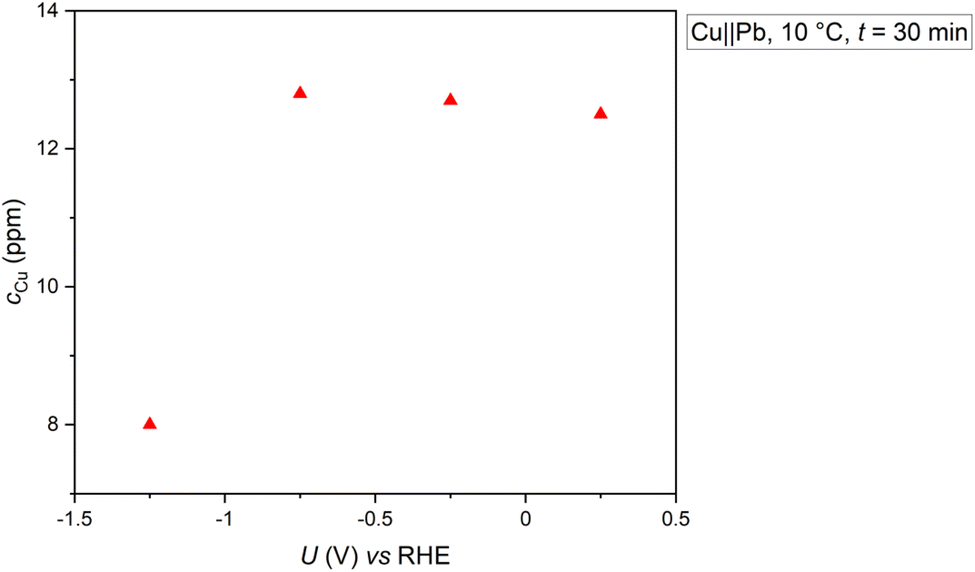 | ||
| Fig. 2 The effect of the applied potential on copper dissolution from the cathode in the presence of an acetone/methylamine mixture after 30 minutes of electrolysis is examined. Experimental conditions include Cu‖Pb electrodes with a usage of a N-324 membrane (previously utilized at least 3 times with Pb as anode); solvent: 0.5 M KH2PO4 (pH 8.3); substrate concentrations: acetone: 2.4 M, methylamine: 2.9 M; temperature (T) maintained at 10 °C; pH at 10 °C: 12.9; anolyte: 25% H3PO4. | ||
2.2 Theoretical investigations
The above experimental measurements trigger the question: why does dissolution happen in electroreduction potential (e.g. at −0.75 V vs. RHE)? Here, we computationally determined the mechanism of evolution of the Cu surface in the presence of amine ligands in water and formation of dissolved Cu complexes at different potential conditions using grand canonical density functional theory31–34 (GC-DFT). Since we consider a negative potential vs. RHE, we modeled a reduced Cu surface, in the absence of O species, as previously experimentally shown in similar reducing conditions.35,36 The study was performed using the PBE37 exchange correlation functional with dDsC van der Waals correction38 and including implicit solvation effects through the VASPsol39 framework, as reported in the Methods section. A Cu(843) surface presenting step and kink sites and a flat Cu(111) surface were considered to assess the role of low coordination site atoms (kink sites) and close-packed high coordination domains, respectively. Methylamine, NH2–CH3, chemisorbs on Cu surfaces by forming strong Cu–N bonds. The –CH3 group in methylamine introduces additional geometric degrees of freedom. For simplicity, our starting model amine is NH3, replacing CH3 with H. In Section 2.2.4, the ligand's effects by itself will be discussed again using other four different amines.In the simulations, the mechanism of Cu dissolution was then determined from the energetics of formation and transformation of metal–amine complexes, on the Cu surface and their detachment in solution, with various amine counts, under different potential conditions. The formation Gibbs free energy of surface amine complexes is calculated with a reference to the Gibbs free energy of bare Cu slab Gslab and of amine ligands in solution Gligand, (formula (1) where Gslab–ligands is the Gibbs free energy of the restructured Cu slab with chemisorbed amine ligands), while the formation of the dissolved complex is expressed with a reference to the chemical potential of Cu μCu and to the Gibbs free energy of ligands Gligand in solution (formula (2)). We give here the example of the formation of a dissolved Cu(NH3)42+ complex, but other cases will be also described later. Detailed mathematical derivation including entropy terms in the Gibbs free energy are provided in ESI Note 2.1.†
| Gformation = Gslab–ligands − Gslab − n × Gligand | (1) |
| Gformation = (GCu(NH3)42+ − 2eU) − μCu − 4 × Gligand | (2) |
Additional method information can be found in Section 4.
 | ||
| Fig. 3 (A) Three different chemisorption sites on Cu(843), including kink site (blue), (100) step site (orange) and (111) terrace site (green). Cu atoms of the upper terrace are in brown for clarity. Free energy of adsorption of methylamine (B) or NH3 (C) on the three different sites of Cu(843) as a function of potential. pH equals 12.9. | ||
The stronger chemisorption of NH3 makes the kink site a good candidate for the dissolution process and mechanism.40,41 We start with one NH3 molecule adsorbed on the kink site (Fig. 4A – structure 1a). We were not able to locate a stable configuration with two NH3 molecules bound to the kink atom. However, upon one NH3 adsorption, the kink Cu atom can be displaced on the terrace as an adatom with low metallic coordination of 3 (Fig. 4A – structure 1b). The energy required to form that NH3 capped Cu adatom is low (+0.1 to +0.3 eV depending on the potential, Fig. 4C), and this process creates a new kink atom, previously a neighbor of the former kink site. In addition, the formed Cu adatom can accept a second NH3, in a stabilizing process if the potential is less negative than −0.7 V vs. RHE (Fig. 4A – structure 2). A third NH3 can bind forming a pseudo-threefold Cu(NH3)3 surface complex (Fig. 4A – structure 3). The binding of the fourth NH3 is slightly more difficult and adopts a “butterfly” C2v geometry (Fig. 4A – structure 4a).
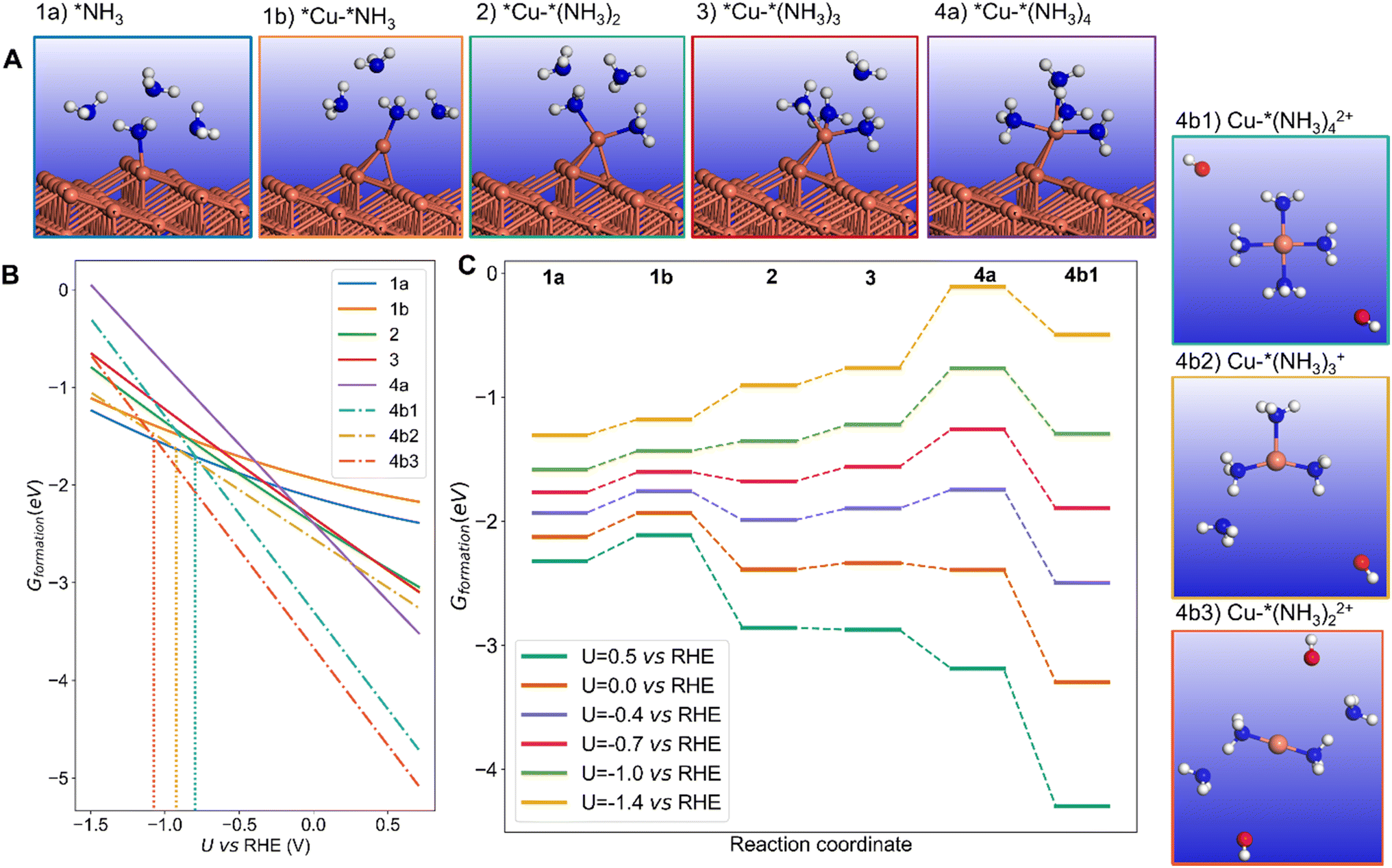 | ||
| Fig. 4 Dissolution of the Cu(843) surface in the presence of NH3 in water as a function of the potential, initiated at the kink site. (A) mechanisms and intermediates during the dissolution; 1a: adsorption of one NH3 on the Cu kink site, 1b: diffusion of the NH3-capped kink site Cu atom forming a Cu adatom on the nearby terrace; adsorption of a second (2), third (3) and fourth (4a) NH3 on the formed Cu adatom; (4b) Cu(NH3)42+, Cu(NH3)3+, and Cu(NH3)22+ complexes formation and detachment. (B) Free energy of the NH3 induced surface or dissolved intermediates described in (A). The vertical blue dashed line describes the threshold potential above which the dissolved complex becomes most stable. (C) Potential dependent reaction pathway for NH3 adsorption, surface complex formation and detachment, where labels refer to (A). pH equals 12.9. | ||
The surface complex can finally be detached to form a Cu–amine complex in solution,42–45 in a stabilizing process (Fig. 4A –structures 4b1, 4b2 and 4b3). Complexes with multiple number of ligands or Cu oxidation states can be formed.43,45–47 It is known that Cu2+ with four NH3 ligands adopts a (quasi)-planar structure.43–45 Besides, Cu2+ can also be complexed by two NH3 ligands in a linear structure. Cu+ should also be considered, where it is reported that this cation forms a tetrahedron configuration with four NH3 or a planar structure with three NH3.42,48 Our detailed explorations of Cu+/Cu2+ with a maximal number of four NH3 ligands is reported in ESI Note 2.4.† Our calculations show that for Cu2+, the square planar complex is more stable than the tetrahedron by 0.15 eV. Besides, the linear two-ligand complex Cu(NH3)22+ is 0.37 eV more stable than the square planar structure. With regard to Cu+, if one NH3 in the tetrahedron complex moves away, it forms a planar structure with three NH3, which is 0.07 eV more stable. Therefore, a 4-NH3 square planar Cu2+, a 3-NH3 planar Cu+, and 2-NH3 linear Cu2+ structures were chosen to represent the final dissolved complexes. From the calculations, Cu(NH3)22+ appears as the most stable dissolution product in our conditions, and dissolution is thermodynamically favored vs. the NH3 covered Cu surface until a potential of −1.1 V vs. RHE. However other complexes could also be formed, Cu(NH3)3+ and Cu(NH3)42+ being more stable than NH3 covered Cu surface until −0.9 V and −0.8 V vs. RHE respectively. These threshold stability potentials are dependent on the pH, where lower pH would shift the values to more negative potential, extending the potential range where dissolution occurs. For example, at pH = 10 (compared to 12.9 on Fig. 4), the threshold potentials would be more negative by about 0.1 V.
We will discuss here the formation of the Cu complex with the maximal number of NH3 ligands (four) as illustrated in Fig. 4C (structure 4b). The overall free energy profile for complexation and dissolution depends on the applied potential. At positive (+0.5 V vs. RHE) or zero potential the process is very easy and thermodynamically limited by the formation of the NH3 capped Cu adatom. At negative potential, the process becomes limited by the formation of the Cu(NH3)4 surface complex. At −0.7 V vs. RHE, the formation of that Cu(NH3)4 surface complex represents a reasonable thermodynamic barrier of 0.51 eV. It can be underlined that forming the four-ligand surface complex Cu(NH3)4 is not mandatory, since starting from the surface Cu(NH3)3 complex, the approach of the fourth NH3 can be concerted with the detachment of the complex from the surface. In that case, the thermodynamic barrier is only 0.21 eV at −0.7 V vs. RHE. For a potential more negative than −0.8 V vs. RHE, the calculations show that the dissolution process forming Cu(NH3)42+ is not thermodynamically favored, and NH3 adsorption on the kink site becomes more favorable than the formation and detachment of the Cu(NH3)42+ complex. This is clearly seen in Fig. 4B, which represents the free energy of the various intermediates as a function of the potential. For more positive potential the dissolved Cu(NH3)42+ complex is more stable than chemisorbed NH3 (structure 1a), while at more negative potential, the chemisorbed NH3 is more stable. Note that other complexes (Cu(NH3)22+, Cu(NH3)3+) can be stable on a slightly more negative potential zone. We assume here that the various processes have low activation energy, which is typically the case for adsorption and metal atom diffusion at metal surfaces. The dissolution of the kink Cu atom generates a new kink atom on the surface, and the process can continue. It should be underlined that in the absence of an NH3 ligand, the displacement of the kink Cu atom on the terrace as an adatom is uphill by ∼1 eV so NH3 is crucial for that restructuring (ESI Note 2.2 and Fig. S5†).
At pH 12, the Pourbaix diagram of Cu in water shows the formation of Cu(OH)2 at a potential more positive than +0.4 vs. RHE, while the Cu surface is stable at more negative potentials.49 Our simulations show the formation of OH adsorbates on the Cu(843) surface at a potential more positive than 0.25 V vs. RHE (ESI Fig. S7 and S8†). The presence of NH3 therefore results in a considerable potential shift for the stability domain of the Cu electrode, favoring dissolution by formation of the Cu(NH3)n complex until a potential of −0.8 V to −1.1 V vs. RHE, depending on the Cu complex formed. The implication is that Cu electrocatalysts cannot be used in a stable manner for electroreduction in the presence of amines.
 | ||
| Fig. 5 Dissolution of the Cu(111) surface in the presence of NH3 in water, featuring the case where the vacancy is compensated by the concomitant migration of a Cu atom from the bulk, as a function of the potential. (A) mechanisms and intermediates during the dissolution; 1a: adsorption of one NH3 on the Cu surface, 1b: diffusion of the NH3-capped Cu atom forming a Cu adatom on the surface, where the accompanying vacancy formation is compensated by the diffusion of a Cu atom from the bulk to the vacancy site; adsorption of a second (2), third (3) and fourth (4a) NH3 on the formed Cu adatom; (4b) Cu(NH3)42+, Cu(NH3)3+, and Cu(NH3)22+ complexes formation and detachment. (B) Free energy of the NH3 induced surface or dissolved intermediates described in (A). The vertical dashed line describes the threshold potential above which the dissolved complex becomes most stable. (C) Potential dependent reaction pathway for NH3 adsorption, surface complex formation, and detachment, where labels refer to (A). pH equals 12.9. | ||
The potential thresholds for the stability of the dissolved Cu(NH3)22+ is −1.1 V, for that of Cu(NH3)3+ is −0.9 V and for that of Cu(NH3)42+ is −0.8 V vs. RHE, which are the same values as that for Cu(843).This is due to the fact that the binding energy of the kink atom on Cu(843) is equal to the binding energy of Cu in the bulk. In the case of the Cu(111) surface, if the vacancy is filled by a bulk atom, then the dissolution process is equivalent to the detachment of a bulk atom.
Therefore, the dissolution of an atom from a (111) terrace of Cu is possible, if we assume that the formed vacancy can be refilled in a concomitant way by a Cu atom from the bulk. It is somewhat less favorable, however, than the dissolution of a kink atom. In contrast, if the vacancy at the (111) surface is maintained, then the formation of the surface complex and its dissolution become highly endergonic.
 | ||
| Fig. 6 Chemical bond strength as quantified by the –ICOHP between extracted Cu in the surface complex and the three bonded surface Cu sites, as a function of the number of NH3 ligands, on Cu(843) (A1) or Cu(111) (B1). Chemical bond strength as quantified by the -ICOHP between extracted Cu in the surface complex and N atoms of the NH3 ligands, as a function of the number of NH3 ligands, on Cu(843) (A2) or Cu(111) (B2). Labels on the x-axis refer to structures in Fig. 4 for Cu(843) and Fig. 5 for Cu(111). | ||
If we consider the kinked Cu(843) surface, the initial adsorption of one NH3 molecule is stabilized if one H is replaced by a methyl or ethyl group (Fig. 7B) since these electron donor substituents increase the reactivity of the amine. If we now look at the energy of the final CuL4 dication (where L is the amine ligand), we see on the right part of Fig. 7B that it is also markedly stabilized by electron donor substituents, as well known. The bonding stability between N and Cu2+ is following the sequence: ethylamine ≈ methylamine > NH3. Ethylamine is very comparable with methylamine, which is because even though ethylamine has a slightly larger alkyl group, providing a greater electron-donating inductive effect compared to methylamine, its longer carbon chain has slightly higher steric hindrance compared to methylamine. On the thermodynamic aspect, dissolution is more favorable for substituted amines and the thermodynamic potential threshold (the crossing points in Fig. 7B) is even more negative than that of NH3, in the order ethylamine ≈ methylamine < NH3.
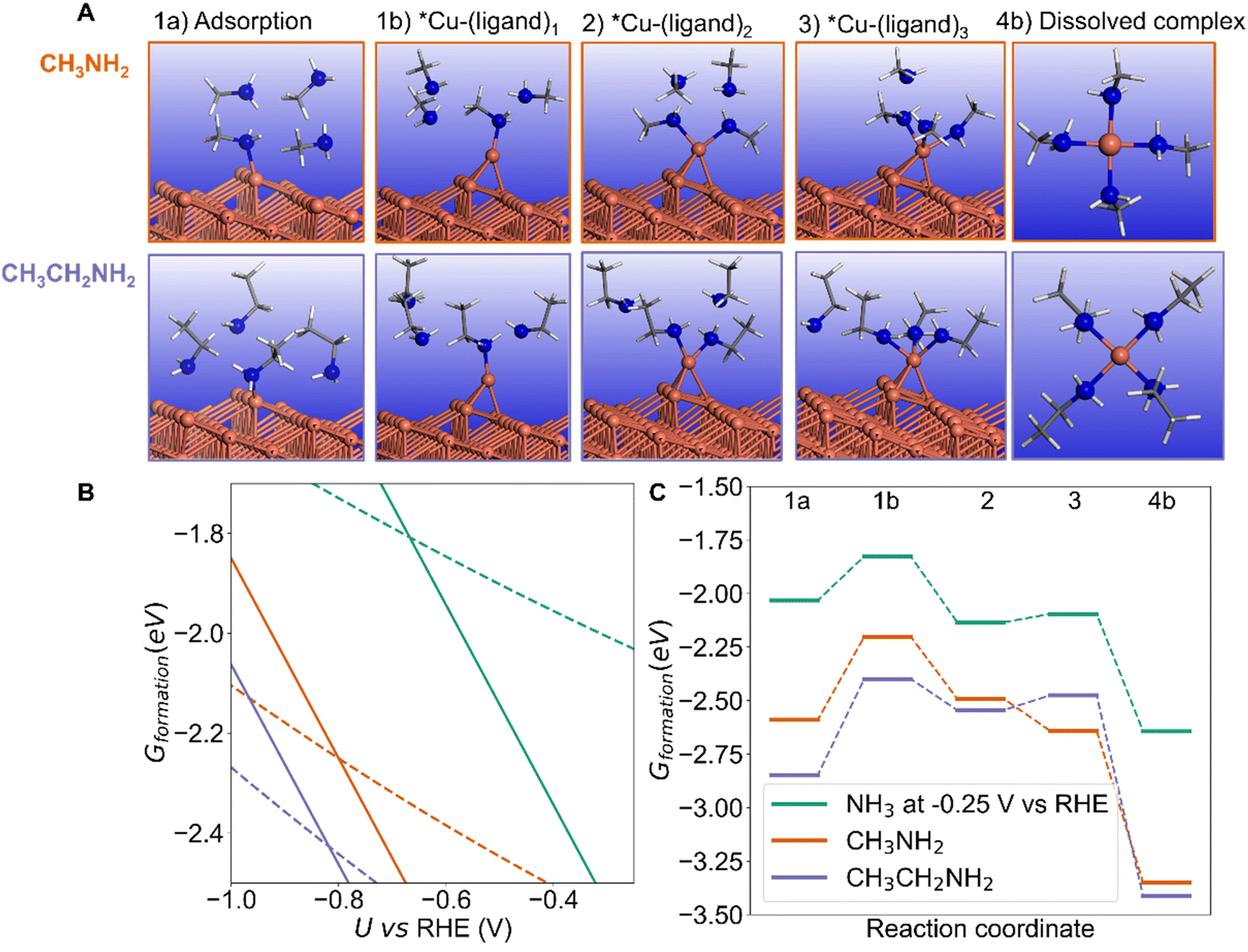 | ||
| Fig. 7 Influence of the type of amines on the structure and energy of the intermediate surface complexes on Cu(843) and of the dissolved dication, using three amines as ligands, including NH3, methylamine, and ethylamine. (A) structure of the adsorbed amine on the kink site (1a), of the displaced adatom with one ligand (1b), with two ligands (2), three ligands (3), and the corresponding detached complex (4b). (B) energy diagram comparing ligand adsorption on the kink atom (structure 1a, dashed lines) and dissolved complex (4b, solid lines) for the three considered ligands: NH3 (green), methylamine (orange) and ethylamine (purple). (C) Free energy profile at 0 V vs. RHE for each ligand type for the adsorption of one ligand on the kink atom of Cu(843) (1a), the formation of the surface complex with one ligand (1b), two ligands (2), three ligands (3), and finally the dissolved dication CuL4 complex (4b). pH equals 12.9. | ||
However, the stability of the intermediate surface Cu complexes does not follow the same trend. The initial extraction of the Cu kink atom with one ligand to form an amine-capped Cu adatom (from structure 1a to 1b) is slightly uphill in energy at −0.25 V vs. RHE for NH3 and methylamine (0.20 eV and 0.39 eV resp.) but more difficult for ethylamine: 0.45 eV (Fig. 7C). From there, the formation of the surface complex with 2 and 3 ligands from 1b is exothermic for NH3, methylamine and ethylamine. Calculations therefore show that the formation of the surface complex is more difficult for the substituted ethylamine even at U = −0.25 V vs. RHE. Changing the potential provides a consistent trend: the dissolution is easier at a less negative potential, and always more difficult at a more negative potential (ESI Fig. S11–S13†). Calculations hence show that the dissolution process can be kinetically controlled by changing the substituent on the amine, with a slower dissolution for substituted amines. Experiments with NH3, methylamine and ethylamine at a potential −0.25 V show that dissolution occurs in each case but that the amount of Cu in solution is lower for the substituted amines (Fig. S14†). Secondary or tertiary amines were not included in our study since they cannot be used for the synthesis of secondary amines from carbonylic compounds in the considered amination reaction under alkaline conditions in water.2,3,10
3. Conclusions
In complement to observations of the dissolution of copper (Cu) and its negative impact on the performance for the reductive amination of acetone with methylamine as nitrogen sources, even under reductive potential conditions (−0.75 V vs. RHE), this study employs Grand Canonical density functional theory to delve into this dynamic phenomenon from a microscopic perspective. The findings illustrate that amine ligands in solution directly chemisorb onto the electrode, coordinating with the metal center and driving surface rearrangement by moving Cu atoms into low coordination adatom positions. Subsequently, other ligands stabilize the formed copper–ligands complex on the Cu surface, eventually leading to the dissolution of a Cu–amine cationic complex, even under negative potential conditions. Calculations indicate that dissolution is predicted to be thermodynamically favored in the case of ammonia ligands at a potential of −1.1 V vs. RHE or higher, with a small dependence of this threshold potential on the nature of the complex formed, Cu(NH3)42+, Cu(NH3)22+ or Cu(NH3)+. Besides, the electron-donating group in the amine ligands will contribute to the thermal stability of the complex, but can also partially hinder the formation of the surface complex as shown for ethylamine. The dissolution of the Cu atom is facilitated by the weakening of the Cu–Cu bond, itself promoted by the multiple coordination of amine ligands on the Cu adatom. This work significantly advances our fundamental understanding of Cu dissolution facilitated by surface restructuring in amine solutions under electroreduction conditions. Such insights are crucial for the rational design of durable Cu-based cathodes for electrochemical amination and other amine-involving reduction processes.4. Methods
4.1 Experimental measurements
The electrolysis experiments were performed in a divided cell, whereby the cathode and anode chamber were separated by a Nafion-324 membrane. The Nafion-324 membrane was used at least three times with lead as anode before these experiments were performed. The half-cells had a cylindrical geometry (r = 1 cm, h = 1 cm). The cathode compartment was cooled by cryostat (Julabo CORIO CD-200F) to 10 °C from the back of the working electrode. A three-electrode setup consisting of the working (cathode), counter (anode), and reference electrode was used. The cathode was copper, while lead was the anode. As reference electrode, a 1 M Hg/HgO electrode was applied. The galvanostatic or potentiostatic electrolysis experiments were performed by using a Metrohm Autolab PGSTAT 302N potentiostat/galvanostat. Before each electrolysis, copper and lead were freshly wet sanded using a 1000 and 2000 grid. | (3) |
Methylamine: 1H-NMR (400 MHz, DMSO-d6) δ2.17 (s, 1H).
Acetone: 1H-NMR (400 MHz, DMSO-d6) δ2.06 (s, 6H).
N-Methylpropan-2-imine: 1H-NMR (400 MHz, DMSO-d6) δ2.87 (s, 3H), 1.81 (d, J = 49.1 Hz, 6H).
N-Methylpropan-2-amine: 1H-NMR (400 MHz, DMSO-d6) δ 2.58–2.45 (m, 1H), 2.16 (s, 3H), 0.92 (d, J = 6.3 Hz, 6H).
Isopropanol: 1H-NMR (400 MHz, DMSO-d6) δ 3.76 (hept, J = 6.1 Hz, 1H), 1.02 (d, J = 6.1 Hz, 6H).
Signals used for the calculation of yield, conversion and C.B. are printed in bold.
| Substance/material | Manufacture | Purity |
|---|---|---|
| Acetone | Chemsolute | 99.50% |
| Ammonium hydroxide | Honeywell Fluka | 29% |
| Copper | Aldrich | 99.98% |
| Dimethylsulfoxide-d6 | Deutero | 99.80% |
| 1,4-Dioxane | Emsure | 99.50% |
| Ethylamine | Aldrich Chemistry | 69% |
| Methylamine | Merck | 40% |
| Monopotassium phosphate | Fluka | 99% |
| Nafion N-324 (0.15 mm, Teflon fabric reinforced) | Thermo scientific | |
| Phosphoric acid | Merck | 85% |
| Potassium hydroxide | Chemsolute | 85% |
| 1,3,5-Trioxane | Aldrich | 99% |
4.2 Theoretical calculations
Vienna ab initio simulation package (VASP) was used to carry out all the periodic DFT calculations.50 The Perdew–Burke–Ernzerhof (PBE) parametrization of the generalized gradient approximation (GGA) of the exchange-correlation functional was employed, along with the dDsC dispersion correction to account for van der Waals interaction.37,38 The cut off energy was 400 eV. The interactions between the atomic cores and electrons were described by the projector augmented wave (PAW) method.51 All structures were optimized under vacuum firstly until the force and energy on each atom was less than 0.01 eV Å−1 and 10−6 eV. Then geometry optimization based on implicit solvation provided by VASPsol39 was used to reach the force and energy on each atom less than 0.02 eV Å−1 and 10−6 eV.The Cu(843) was modeled by an around 3-layer (1 × 1) supercell with a cell dimension of 8.08 Å × 8.44 Å. The bottom layer (0–2 Å region) was fixed as the bulk region. Meanwhile, a 3-layer (3 × 3) supercell of Cu(111) termination was used, where the bottom one layer was fixed. A vacuum slab of 15 Å thickness was added in the Z direction. When it comes to solvation optimization and Surface Charging, all structures are symmetrized and the box thickness in the Z direction is 60 Å for the implicit solvation region. To be specific, five layers were used with adsorbates on both sides. The Brillouin zone was sampled using 5 × 5 × 1 Gamma-centered k-point grids for structure optimization. The COHP analysis was performed using the LOBSTER program with the pbeVaspFit2015 basis set,52 where self-consistent calculation used a denser k-point grid of 8 × 8 × 1. The Gibbs free energies for adsorbates on the surface are obtained from the DFT energies with ZPE and entropy corrections determined from frequency calculations performed for all the considered structures using the Harmonic Oscillator approximation.
| nsurface = Nsurface − Nsurface, neutral |
| Esurface = Esurface,raw + εfermishiftnsurface |
| Ω(U) = Esurface − nsurfaceμelectron |
| μelectron = qUvac = −eUvac |
| −eUvac = εF + εfermishift |
For the metallic systems, the potential-dependent grand canonical energy, Ω(U), exhibits a quadratic behaviour around the potential of zero charge (U0) in the vacuum scale:

| USHE = Uvac − 4.44 |
The linearized Poisson Boltzmann implicit solvation model implemented in VASPsol53 is used to represent the polarizable electrolyte region. The dielectric constant of water, 78.4, and the Debye screening length corresponding to 0.1 M concentration of electrolytes, 9.6 Å, were used. The surface slab is symmetrized along the z-axis to avoid asymmetric potential in the implicit solvation region. Here the implicit solvent thickness is set to 60 Å for the symmetrized slab.
Data availability
Data for this article, including experimental data (yield, conversion, carbon balance and copper concentration) and theoretical data (calculated structures) are available at Zenodo at https://doi.org/10.5281/zenodo.13241433.Author contributions
Yani Guan performed calculations and drafted the manuscript. Justus Kümpe conducted the experimental work and drafted the experimental part of the manuscript. Sonja D. Mürtz and Simran Kumari assisted with data analysis and manuscript writing. Peter J. C. Hausoul aided in data analysis and manuscript revision. Regina Palkovits conceived the experiments, oversaw the work at RWTH Aachen and reviewed the manuscript. Philippe Sautet conceived the calculations, supervised the study, and edited and finalized the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Y. G., S. K. and P. S. acknowledge support from the National Science Foundation award 2140374. Computational resources for this work were provided by the UCLA-shared cluster Hoffman2 and the Expanse cluster through the allocation CHE170060 at the San Diego Supercomputing Center through ACCESS. J. K., S. M., P. H. and R. P. gratefully acknowledge the German Research Foundation for funding this project within PA1689/17-1 as well as in the frame of NFDI4Cat (ID 441926934). Part of this work was supported by the Cluster of Excellence Fuel Science Center (EXC 2186, ID: 390919832) funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities. J. K., S. M., P. H. and R. P. also acknowledge funding by the Federal Ministry of Food and Agriculture granted by the Agency for Renewable Resources (FNR, 2220NR101X). S. M. thanks Cusanuswerk e.V. for funding.References
- C. Fernández-Dacosta, V. Stojcheva and A. Ramirez, J. CO2 Util., 2018, 23, 128–142 CrossRef.
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC.
- S. D. Mürtz, N. Kurig, F. J. Holzhäuser and R. Palkovits, Green Chem., 2021, 23, 8428–8433 RSC.
- S. V. Vassilev, D. Baxter, L. K. Andersen and C. G. Vassileva, Fuel, 2010, 89, 913–933 CrossRef CAS.
- Y. M. Bar-On, R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6506–6511 CrossRef CAS PubMed.
- Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed.
- X. Yang, Q.-L. Yang, X.-Y. Wang, H.-H. Xu, T.-S. Mei, Y. Huang and P. Fang, J. Org. Chem., 2020, 85, 3497–3507 CrossRef CAS PubMed.
- T. Ghosh, H. Kaizawa, S. Funato, M. A. Rahman, N. Sasaki, T. Nokami, M. Abe and T. Nishikata, ChemElectroChem, 2024, 11, e202300636 CrossRef CAS.
- P. J. L. Broersen, T. de Groot, D. F. Bruggeman, E. S. Caarls, J. A. Trindell, D. Anastasiadou, M. C. Figueiredo, G. Rothenberg and A. C. Garcia, ChemCatChem, 2024, 16, e202301370 CrossRef CAS.
- T. Pienemann and H.-J. Schäfer, Synthesis, 1987, 1987, 1005–1007 CrossRef.
- T. Kim, D. I. Park, S. Kim, D. Yadav, S. Hong, S. H. Kim, H. J. Yoon and K. Jin, Chem. Commun., 2023, 59, 4818–4821 RSC.
- R. Chen, X. Zu, J. Zhu, Y. Zhao, Y. Li, Z. Hu, S. Wang, M. Fan, S. Zhu, H. Zhang, B. Ye, Y. Sun and Y. Xie, Adv. Mater., 2024, 36, 2314209 CrossRef CAS PubMed.
- P. Trogadas, L. Xu and M.-O. Coppens, Angew. Chem., 2024, 136, e202314446 CrossRef.
- X. Meng, H. Huang, X. Zhang, L. Hu, H. Tang, M. Han, F. Zheng and H. Wang, Adv. Funct. Mater., 2024, 34, 2312719 CrossRef CAS.
- K. Shen, D. Cheng, E. Reyes-Lopez, J. Jang, P. Sautet and C. G. Morales-Guio, Joule, 2023, 7, 1260–1276 CrossRef CAS.
- D. Cheng, A. N. Alexandrova and P. Sautet, J. Phys. Chem. Lett., 2024, 1056–1061 CrossRef CAS PubMed.
- D. Cheng, Z. Wei, Z. Zhang, P. Broekmann, A. N. Alexandrova and P. Sautet, Angew. Chem., 2023, 135, e202218575 CrossRef.
- Z. Zhang, J. Li and Y.-G. Wang, Acc. Chem. Res., 2024, 57, 198–207 CrossRef CAS PubMed.
- N. De Jonge and F. M. Ross, Nat. Nanotechnol., 2011, 6, 695–704 CrossRef CAS PubMed.
- F. M. Ross, Science, 2015, 350, aaa9886 CrossRef PubMed.
- M. L. Taheri, E. A. Stach, I. Arslan, P. A. Crozier, B. C. Kabius, T. LaGrange, A. M. Minor, S. Takeda, M. Tanase, J. B. Wagner and R. Sharma, Ultramicroscopy, 2016, 170, 86–95 CrossRef CAS PubMed.
- V. Beermann, M. E. Holtz, E. Padgett, J. F. De Araujo, D. A. Muller and P. Strasser, Energy Environ. Sci., 2019, 12, 2476–2485 RSC.
- G.-Z. Zhu, S. Prabhudev, J. Yang, C. M. Gabardo, G. A. Botton and L. Soleymani, J. Phys. Chem. C, 2014, 118, 22111–22119 CrossRef CAS.
- S. F. Tan, S. W. Chee, Z. Baraissov, H. Jin, T. L. Tan and U. Mirsaidov, J. Phys. Chem. Lett., 2019, 10, 6090–6096 CrossRef CAS PubMed.
- A. Impagnatiello, C. F. Cerqueira, P.-E. Coulon, A. Morin, S. Escribano, L. Guetaz, M.-C. Clochard and G. Rizza, ACS Appl. Energy Mater., 2020, 3, 2360–2371 CrossRef CAS.
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- J. Vavra, G. P. L. Ramona, F. Dattila, A. Kormányos, T. Priamushko, P. P. Albertini, A. Loiudice, S. Cherevko, N. Lopéz and R. Buonsanti, Nat. Catal., 2024, 1–9 Search PubMed.
- S. J. Raaijman, N. Arulmozhi and M. T. M. Koper, ACS Appl. Mater. Interfaces, 2021, 13, 48730–48744 CrossRef CAS PubMed.
- S. Popovic, M. Bele and N. Hodnik, ChemElectroChem, 2021, 8, 2634–2639 CrossRef CAS.
- X. Fu, D. Cheng, C. Wan, S. Kumari, H. Zhang, A. Zhang, H. Huyan, J. Zhou, H. Ren, S. Wang, Z. Zhao, X. Zhao, J. Chen, X. Pan, P. Sautet, Y. Huang and X. Duan, Adv. Mater., 2023, 35, 2301533 CrossRef CAS PubMed.
- S. Kumari, T. Masubuchi, H. S. White, A. Alexandrova, S. L. Anderson and P. Sautet, J. Am. Chem. Soc., 2023, 145, 5834–5845 CrossRef CAS PubMed.
- Elucidation of the Active Site for the Oxygen Evolution Reaction on a Single Pt Atom Supported on Indium Tin Oxide, https://pubs.acs.org/doi/epdf/10.1021/acs.jpclett.3c00160, accessed 2 October 2023.
- D. Cheng, Z. Wei, Z. Zhang, P. Broekmann, A. N. Alexandrova and P. Sautet, Angew. Chem., 2023, 135, e202218575 CrossRef.
- A. A. Permyakova, J. Herranz, M. El Kazzi, J. S. Diercks, M. Povia, L. R. Mangani, M. Horisberger, A. Pătru and T. J. Schmidt, ChemPhysChem, 2019, 20, 3120–3127 CrossRef CAS PubMed.
- S. B. Scott, T. V. Hogg, A. T. Landers, T. Maagaard, E. Bertheussen, J. C. Lin, R. C. Davis, J. W. Beeman, D. Higgins, W. S. Drisdell, C. Hahn, A. Mehta, B. Seger, T. F. Jaramillo and I. Chorkendorff, ACS Energy Lett., 2019, 4, 803–804 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Gautier, S. N. Steinmann, C. Michel, P. Fleurat-Lessard and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930 RSC.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CrossRef CAS PubMed.
- B. Eren, R. S. Weatherup, N. Liakakos, G. A. Somorjai and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 8207–8211 CrossRef CAS PubMed.
- M. Pavelka and J. V. Burda, Chem. Phys., 2005, 312, 193–204 CrossRef CAS.
- T. E. Da-yang, J. J. Fifen, A. Malloum, S. Lahmar, M. Nsangou and J. Conradie, New J. Chem., 2020, 44, 3637–3653 RSC.
- J. Hu, Q. Chen, H. Hu, Z. Jiang, D. Wang, S. Wang and Y. Li, J. Phys. Chem. A, 2013, 117, 12280–12287 CrossRef CAS PubMed.
- A. Bérces, T. Nukada, P. Margl and T. Ziegler, J. Phys. Chem. A, 1999, 103, 9693–9701 CrossRef.
- R. J. Deeth and L. J. A. Hearnshaw, Dalton Trans., 2005, 3638 RSC.
- N. R. Texler and B. M. Rode, J. Phys. Chem., 1995, 99, 15714–15717 CrossRef CAS.
- H. D. Pranowo, A. H. Bambang Setiaji and B. M. Rode, J. Phys. Chem. A, 1999, 103, 11115–11120 CrossRef CAS.
- J. Gambino, J. Robbins, T. Rutkowski, C. Johnson, K. DeVries, D. Rath, P. Vereecken, E. Walton, B. Porth, M. Wenner, T. McDevitt, J. Chapple-Sokol and S. Luce, in 2008 15th International Symposium on the Physical and Failure Analysis of Integrated Circuits, 2008, pp. 1–4 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01944j |
| This journal is © The Royal Society of Chemistry 2024 |