
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosensitizing metal covalent organic framework with fast charge transfer dynamics for efficient CO2 photoreduction†
Wang-Kang
Han‡
a,
Jiayu
Li‡
b,
Ruo-Meng
Zhu
a,
Min
Wei
b,
Shu-Kun
Xia
a,
Jia-Xing
Fu
a,
Jinfang
Zhang
 a,
Huan
Pang
c,
Ming-De
Li
*b and
Zhi-Guo
Gu
*a
a,
Huan
Pang
c,
Ming-De
Li
*b and
Zhi-Guo
Gu
*a
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: zhiguogu@jiangnan.edu.cn
bCollege of Chemistry and Chemical Engineering, Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou 515063, China. E-mail: mdli@stu.edu.cn
cSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
First published on 1st May 2024
Abstract
Designing artificial photocatalysts for CO2 reduction is challenging, mainly due to the intrinsic difficulty of making multiple functional units cooperate efficiently. Herein, three-dimensional metal covalent organic frameworks (3D MCOFs) were employed as an innovative platform to integrate a strong Ru(II) light-harvesting unit, an active Re(I) catalytic center, and an efficient charge separation configuration for photocatalysis. The photosensitive moiety was precisely stabilized into the covalent skeleton by using a rational-designed Ru(II) complex as one of the building units, while the Re(I) center was linked via a shared bridging ligand with an Ru(II) center, opening an effective pathway for their electronic interaction. Remarkably, the as-synthesized MCOF exhibited impressive CO2 photoreduction activity with a CO generation rate as high as 1840 μmol g−1 h−1 and 97.7% selectivity. The femtosecond transient absorption spectroscopy combined with theoretical calculations uncovered the fast charge-transfer dynamics occurring between the photoactive and catalytic centers, providing a comprehensive understanding of the photocatalytic mechanism. This work offers in-depth insight into the design of MCOF-based photocatalysts for solar energy utilization.
Introduction
Solar-driven CO2 conversion into valuable chemical fuels represents an eco-friendly and promising approach toward the carbon-neutral goal.1–4 Particularly, the development of artificial photocatalysts for the CO2 reduction reaction (CO2RR) with both high activity and selectivity is desirable for practical application.5–8 To date, despite the exploration of various photocatalytic materials for the CO2RR, the catalytic performances are severely restricted by their poor visible-light absorption, low charge separation efficiency, and less efficacious catalytic sites.9–11 Thus, there is a strong need to develop better photocatalysts for the CO2RR.Inspired by natural photosynthesis, two crucial functional moieties should be considered for constructing an efficient photocatalytic CO2RR system: photosensitizers (PSs), which are key components responsible for light-harvesting and electron excitation, and catalytic centers (CAT), which accept electrons from PSs for the CO2RR.12–14 Importantly, it is essential to establish an effective channel for electronic communication between PSs and CAT. Nevertheless, conventional photocatalytic systems have often involved the discrete components of PSs and CAT, resulting in a significantly weak interaction between them.15–18 This, in turn, hinders the efficiency of charge transfer, thereby impacting catalytic activity. Additionally, the substantial use of extra PSs in a catalytic system also gives rise to new difficulties for recycling and leads to waste. In light of this, exploring integrated systems that can couple light-harvesting units and CO2 reduction catalytic sites is highly desired but still challenging.
Metal covalent organic frameworks (MCOFs) featuring robust covalent networks and functional metal ions have recently shown great potential in artificial photosynthesis.19–21 The diverse structural design through the assembly of predesigned building units enables the combination of multiple functional moieties within MCOFs, including PSs and CAT.22–24 Besides, the crystalline robust framework can accommodate multiple functional units with precise arrangement, as well as prevent disadvantageous aggregation of active sites. Among various functional moieties, the Ru(II)–polypyridine family displayed excellent light-harvesting capabilities and long-lived excited states for electron transfer,25–27 while the Re(I)-complexes are excellent catalysts for selective CO2 photoreduction.28,29 Moreover, previous studies have demonstrated an improvement of photocatalytic CO2RR efficiency by linking Ru(II)–Re(I) moieties in supramolecular systems.30,31 However, the homogeneous Ru(II)–Re(I) supramolecular catalysts often suffer from instability and difficulty in recycling. With this in mind, we envision that the functionalization of MCOFs with both Ru(II)- and Re(I)-complexes would be very advantageous for the construction of efficient CO2RR heterogeneous photocatalysts, although this concept has not been realized thus far.
In this work, we present a newly designed MCOF as a functional platform to integrate the crucial components for the CO2RR (Scheme 1): (i) the photoactive Ru(II) fragment that functions as a light absorber; (ii) the coordinated Re(CO)3Cl unit serves as a catalytic center to activate CO2; (iii) the bridging unit, 2,2′-bipyrimidine, links the photosensitive and catalytic centers to readily facilitate charge transfer; (iv) the overall covalent framework provides a stable environment for final catalysis. Owing to the synergistic effect of these multiple functional units, the resultant Ru(II)/Re(I) MCOF demonstrated remarkable CO2 photoreduction ability. Detailed insights into the charge-transfer dynamics and photocatalytic reaction pathways were provided through femtosecond transient absorption spectroscopy, DFT calculations, and experimental results.
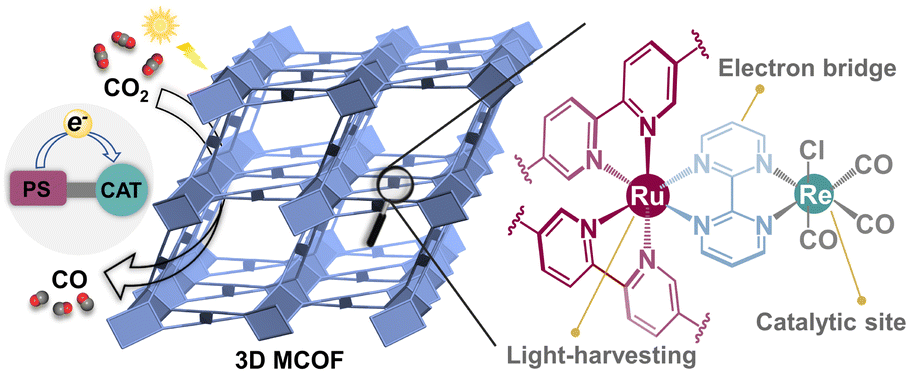 | ||
| Scheme 1 Illustration of a design strategy to synthesize a MCOF-based photocatalyst for the CO2RR. | ||
Results and discussion
To implement our design, an acetal functionalized metal complex building unit [Ru(5,5′-di(1,3-dioxolan-2-yl)-2,2′-bipyridine)2(2,2′-bipyrimidine)](PF6)2, Ru-bpm (Fig. 1a), was first synthesized and characterized by single crystal X-ray diffraction analysis (Fig. S1–S6, ESI†). The acetal groups were introduced to ensure the solubility of Ru-bpm and slow down the polymerization rate during the MCOF crystallization. The model reaction confirmed that Ru-bpm was stable under solvothermal conditions, and the acetal functional group could react with amine to form imine bonds (Fig. S7 and S8, ESI†). When Ru-bpm reacted with 4′,4′′′,4′′′′′,4′′′′′′′-(ethene-1,1,2,2-tetrayl)tetrakis([1,1′-biphenyl]-4-amine) (ETTBA) in a mixture solvent of o-dichlorobenzene (o-DCB), n-butanol (n-BuOH) and 6 M acetic acid (AcOH) at 120 °C for 4 days, crystalline product MCOF-Ru was obtained (Fig. S9, ESI†). The Re(I) moiety was incorporated into MCOF-Ru via a reaction between the reserved coordination sites on 2,2′-bipyrimidine ligands and Re(CO)5Cl to afford MCOF-Ru/Re (Fig. 1a and S10, ESI†).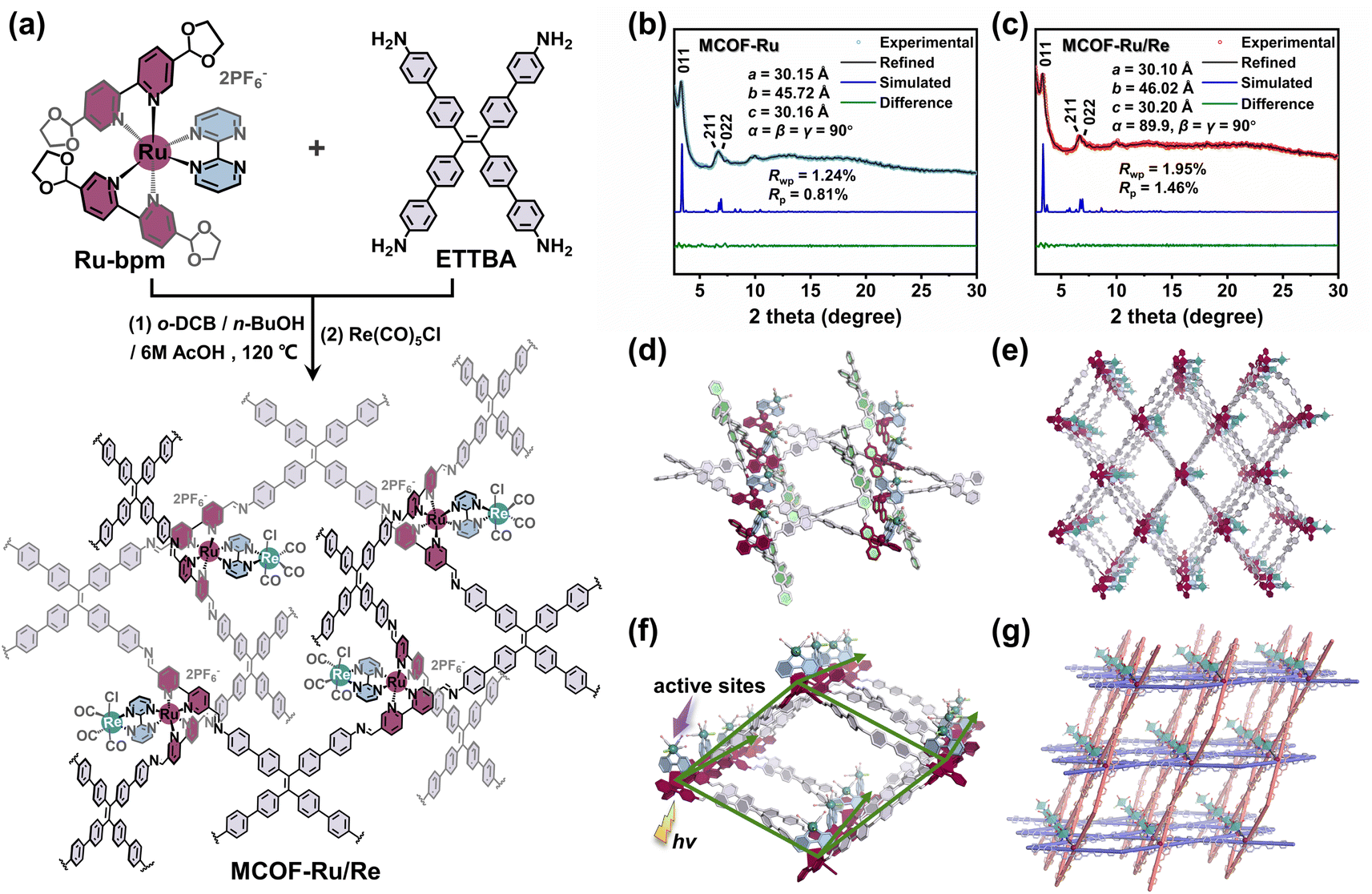 | ||
| Fig. 1 (a) Illustration of the synthetic procedures for MCOFs. PXRD patterns of (b) MCOF-Ru and (c) MCOF-Ru/Re (insets: the refined unit cell parameters). (d) The ETTBA and Ru-based building units are 4,4-connected, (e) affording a 3D framework of MCOF-Ru/Re with lvt topology. (f) 1D rhombic channel shows the Ru photoactive centers and Re catalytic sites. (g) The inclined interpenetration of covalent 2D nets (represented in purple and orange, respectively) to form a 3D framework with Ru(II) ions as templates. | ||
Compared with the Fourier transform infrared (FI-IR) spectra of MCOFs and building units, the signals corresponding to –CH2– (2890–2960 cm−1) and –C–O–C– (1086 cm−1) of Ru-bpm and N–H stretching vibration (3250–3450 cm−1) of ETTBA amine disappeared. Meanwhile, a new peak corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration appeared at around 1643 cm−1 (Fig. S11, ESI†). The FT-TR results indicated that the in situ deprotection and polymerization result in the formation of imine linkages. The FI-IR spectrum of MCOF-Ru/Re exhibited two additional peaks at 2019 and 1886 cm−1, attributed to the C
N stretching vibration appeared at around 1643 cm−1 (Fig. S11, ESI†). The FT-TR results indicated that the in situ deprotection and polymerization result in the formation of imine linkages. The FI-IR spectrum of MCOF-Ru/Re exhibited two additional peaks at 2019 and 1886 cm−1, attributed to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching vibration of the Re(I) moieties (Fig. S12, ESI†).32 Besides, it was further verified by solid-state 13C cross-polarization magic-angle-spinning NMR spectroscopy, which showed a characteristic resonance peak of imine carbon (161.8 ppm for MCOF-Ru, 162.7 ppm for MCOF-Ru/Re) (Fig. S13 and S14, ESI†). Thermogravimetric analysis showed that the resultant MCOFs were thermally stable up to about 320 °C under a N2 atmosphere (Fig. S15, ESI†). The modest adsorption of N2 with a Brunauer–Emmett–Teller (BET) surface area of 328 m2 g−1 for MCOF-Ru and 175 m2 g−1 for MCOF-Ru/Re was mainly attributed to the occupation of space by the bulky PF6− counterions (Fig. S16, ESI†).33 The NLDFT pore size distribution analysis showed the pore size of about 1.82 and 1.46 nm for MCOF-Ru and MCOF-Ru/Re, respectively (Fig. S16, ESI†). Scanning and high-resolution transmission electron microscopies displayed uniform crystalline nanoparticles with clear lattice fringes of MCOFs (Fig. S17–S20, ESI†). The elemental compositions of C, N, Ru, F, P and Re in MCOFs were obtained by X-ray photoelectron spectroscopy (Fig. S21–S25, ESI†). Additionally, elemental mapping images demonstrated the uniform distribution of Ru and Re in MCOF-Ru/Re, providing further evidence for the successful incorporation of the Re moiety (Fig. S26 and S27, ESI†). The Ru and Re contents of MCOF-Ru/Re were quantified as 6.24 and 6.52 wt% by ICP tests, respectively, suggesting that about 56% of the reserved coordination sites on 2,2′-bipyrimidine ligands were converted to Re(I) sites.
O stretching vibration of the Re(I) moieties (Fig. S12, ESI†).32 Besides, it was further verified by solid-state 13C cross-polarization magic-angle-spinning NMR spectroscopy, which showed a characteristic resonance peak of imine carbon (161.8 ppm for MCOF-Ru, 162.7 ppm for MCOF-Ru/Re) (Fig. S13 and S14, ESI†). Thermogravimetric analysis showed that the resultant MCOFs were thermally stable up to about 320 °C under a N2 atmosphere (Fig. S15, ESI†). The modest adsorption of N2 with a Brunauer–Emmett–Teller (BET) surface area of 328 m2 g−1 for MCOF-Ru and 175 m2 g−1 for MCOF-Ru/Re was mainly attributed to the occupation of space by the bulky PF6− counterions (Fig. S16, ESI†).33 The NLDFT pore size distribution analysis showed the pore size of about 1.82 and 1.46 nm for MCOF-Ru and MCOF-Ru/Re, respectively (Fig. S16, ESI†). Scanning and high-resolution transmission electron microscopies displayed uniform crystalline nanoparticles with clear lattice fringes of MCOFs (Fig. S17–S20, ESI†). The elemental compositions of C, N, Ru, F, P and Re in MCOFs were obtained by X-ray photoelectron spectroscopy (Fig. S21–S25, ESI†). Additionally, elemental mapping images demonstrated the uniform distribution of Ru and Re in MCOF-Ru/Re, providing further evidence for the successful incorporation of the Re moiety (Fig. S26 and S27, ESI†). The Ru and Re contents of MCOF-Ru/Re were quantified as 6.24 and 6.52 wt% by ICP tests, respectively, suggesting that about 56% of the reserved coordination sites on 2,2′-bipyrimidine ligands were converted to Re(I) sites.
The crystalline structures of MCOFs were investigated by powder X-ray diffraction (PXRD) analyses together with theoretical simulation. The predominant (011) Bragg diffraction peak was detected at 2θ = 3.37° for MCOF-Ru, which differed greatly from those of its starting monomers, indicating the crystalline nature (Fig. 2b and S28, ESI†). For MCOF-Ru/Re, the same Bragg diffraction peak was also observed (Fig. 2c), confirming the retention of the crystalline structure after the incorporation of the Re(I) moiety. Based on the geometry and connectivity of the building units, several possible structural models were built. The calculated PXRD pattern of the geometrically optimized structure adopting lvt topology with the Pnc2 space group matched well with the experimental one (Fig. 2b and c). The Pawley refinements reproduced PXRD patterns and obtained the unit cell parameters with good agreement factors (Rp = 0.81% and Rwp = 1.24% for MCOF-Ru, and Rp = 1.46% and Rwp = 1.95% for MCOF-Ru/Re). In contrast, the structural models based on the possible two-dimensional (2D) structures with sql topology, 3D ssb topology, or two-fold interpenetrated lvt topology were not in agreement with the experimental PXRD data (Fig. S29–S31†). Thus, all the above results suggested that a non-interpenetrated structure with an lvt net is reasonable.
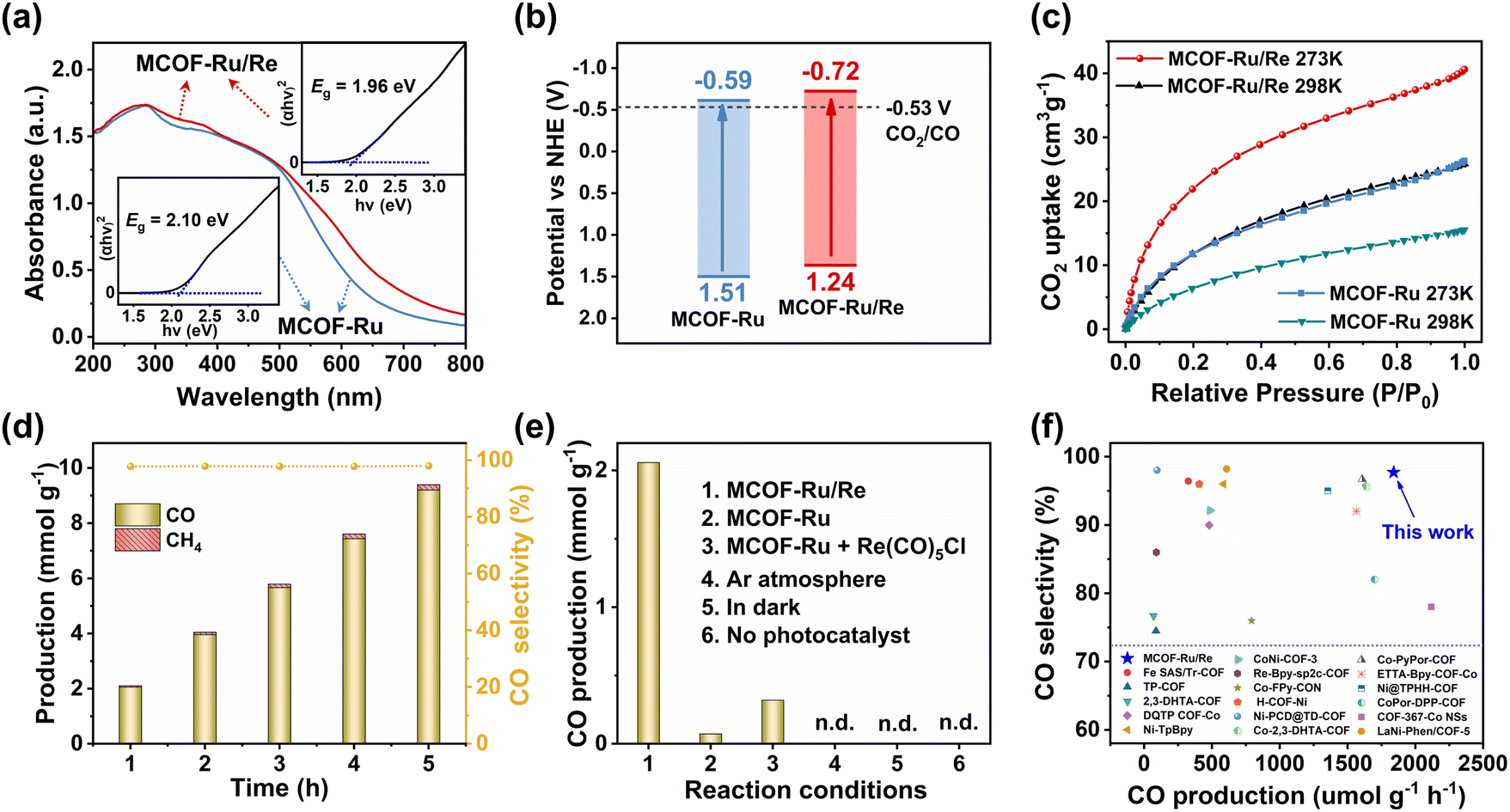 | ||
| Fig. 2 (a) Solid state UV/vis diffuse reflectance spectra. Insets: Tauc plots. (b) Energy level diagram. (c) CO2 sorption isotherms of MCOFs. (d) Time-dependent photocatalytic CO and CH4 production. (e) Control experiments under different catalytic conditions within 1 h. (f) Comparison of photocatalytic activity of MCOF-Ru/Re with reported representative catalysts. | ||
In the framework of MCOF-Ru/Re, the 4-connected Ru building units are covalently linked to quadrilateral ETTBA linkers via imine bonds. Interestingly, one of the linear bipyridine ligands in the Ru building unit is (2,4)-connected with ETTBA linkers, forming a planer rhombic network. Meanwhile, the other bipyridine ligand has spatial inclination relative to this planer network due to the Ru(II) octahedral coordination, connecting to another set of ETTBA linkers (Fig. 1d). This process generates a 3D metalated framework with overall lvt topology, showing one-dimensional (1D) channels (Fig. 1e). Notably, within each rhombic 1D channel, the light-harvesting Ru(II) entities adorn the edges of the rhombic prism, while the Re(I) sites are closely decorated on the side of the Ru(II) center through a 2,2′-bipyrimidine bridging ligand (Fig. 1f). The organized arrangement of the Ru(II) photoactive unit and Re(I) site in the crystalline framework provides a favourable environment for light-harvesting and electron transfer. Besides, the Re(I) site is geometrically oriented toward the inside of 1D channels, making it easily accessible for substrate binding (Fig. 1e and f). Interestingly, the 3D structure of MCOF-Ru/Re can be discerned from an interlocking perspective with the inclined interpenetration of 2D networks (Fig. 1g). By tracing the covalently linked skeleton that is not interrupted by metal ions, we can find two sets of chemically identical 2D COFs with sql topology that are mechanically interdigitated at each crossing point, with the Ru(II) centers serving as points of registry.
The solid-state UV-vis diffuse reflectance spectroscopy showed broad absorption for both MCOFs in the visible-light region (Fig. 2a). And the slight red-shift in absorption on MCOF-Ru/Re compared to that of MCOF-Ru was attributed to the increased electron delocalization after Re(I) chelation into the bridging ligands.32 The bandgap (Eg) of MCOF-Ru/Re was estimated to be 1.96 eV according to the Tauc plot, which was narrower than that of MCOF-Ru (2.10 eV). Thus, the incorporation of Re(I) centers can not only function as potential catalytic sites but also optimize the energy band structure. The energy level was obtained by Mott–Schottky measurements coupled with bandgap calculations, confirming the thermodynamic suitability of the resultant MCOFs for CO2 reduction (Fig. 2b and S32, S33, ESI†).
The CO2 sorption properties of the MCOFs were then studied. As shown in Fig. 2c, the CO2 uptake capacity of MCOF-Ru/Re was 40.6 cm3 g−1 at 273 K and 25.8 cm3 g−1 at 298 K, which were higher than those of MCOF-Ru (26.3 cm3 g−1 at 273 K and 15.4 cm3 g−1 at 298 K). Correspondingly, the initial isosteric heat of adsorption (Qst) was calculated to be 37.5 and 31.3 kJ mol−1 for MCOF-Ru/Re and MCOF-Ru, respectively (Fig. S34, ESI†). The higher Qst value of MCOF-Ru/Re implies its stronger interaction with CO2 molecules. And the introduction of Re(I) sites could improve the affinity for CO2 molecules, favourably promoting the fixation and activation of CO2.34
Based on the above findings, our designed MCOFs integrate the advantages of a robust covalent skeleton, wide visible-light harvesting, abundant catalytic sites, suitable energy levels and strong CO2 adsorption, which motivated us to further explore their potential in the photocatalytic CO2RR. Under the optimized conditions (Fig. S35 and S36, ESI†), MCOF-Ru/Re exhibited an impressive CO2 photoreduction performance with a CO generation rate of up to 1840 μmol g—1 h—1 and 97.7% CO selectivity for a 5 hour reaction period (Fig. 2d). No liquid product was detected in the 1H NMR spectrum after photocatalytic experiments (Fig. S37, ESI†). After separating the above MCOF photocatalyst from the reaction mixture, irradiation of the MCOF free media revealed no further CO production, demonstrating the heterogeneous nature of the reaction. Besides, the ICP-OES test also revealed no detectable leaching of Ru or Re contents in the solution. By contrast, the catalytic activity of MCOF-Ru was negligible (Fig. 2e). The simple physical mixture of MCOF-Ru and Re(CO)5Cl exhibited significantly lower activity with a CO production rate of only 320 μmol g−1 h−1. These findings unambiguously suggested the crucial role of the overall framework with the integration of light-harvesting site and catalytic center for efficient charge transfer. Besides, control experiments showed no release of CO in the absence of either photocatalyst, CO2, or light irradiation (Fig. 2e). The 13CO2 isotope experiment clearly verified that the generated 13CO originated from the 13CO2 source (Fig. S38, ESI†). Moreover, the catalytic activity only showed a slight change after 5 runs (Fig. S39, ESI†). The FT-IR, PXRD and SEM of the recovered catalyst confirmed its stability during the photocatalytic CO2RR (Fig. S40, ESI†).
It should be noted that in most of the traditional photocatalytic systems additional PSs, such as [Ru(bpy)3]Cl2, were substantially used. However, their catalytic efficiencies were assessed solely based on the amounts of catalysts. If the amounts of PSs are considered for calculating catalytic performance (i.e., gtotal = gcatalyst + gphotosensitizer), the activity of MCOF-Ru/Re is superior to that of similar photocatalytic systems (Fig. 2f), such as Fe SAS/Tr-COF17 (327 μmol gtotal−1 h−1), Re-Bpy-sp2c-COF35 (90 μmol gtotal−1 h−1) and TP-COF36 (87 μmol gtotal−1 h−1). Although the CO generation rates of some samples, such as CoPor-DPP-COF37 (1700 μmol gtotal−1 h−1, 82% selectivity), ETTA-Bpy-COF-Co38 (1566 μmol gtotal−1 h−1, 92.7 selectivity) and COF-367-Co NSs16 (2117 μmol gtotal−1 h−1, 78% selectivity) are comparable to that of Ru/Re-MCOF, their CO selectivity remains inferior to that of Ru/Re-MCOF. More detailed comparisons are listed in Table S2.† This suggests that engineering the molecular photosensitizer into the MCOF is an effective strategy for significantly enhancing the utilization of the photosensitizer. In addition, the apparent quantum efficiency of MCOF-Ru/Re at 420 nm was 1.16%, outperforming most of the reported COF-based photocatalysts. The turnover frequency (TOF) of MCOF-Ru/Re was further estimated to be 5.81 h−1, comparable to that of some molecular Ru/Re photocatalysts (Table S3, ESI†). Furthermore, under pure water conditions and without the addition of an extra photosensitizer or sacrificial reagent, MCOF-Ru/Re still achieved CO2 photoreduction with a CO generation rate of 81.7 μmol g−1 h−1 and a selectivity of 90.56% (Fig. S41 and Table S4, ESI†).
To reveal the critical role of the chemical structure in MCOFs for charge separation, photoluminescence (PL) spectroscopy was performed (Fig. 3a). MCOF-Ru showed an emission peak at 570 nm, while the PL intensity was almost completely quenched in MCOF-Ru/Re, indicating that the presence of the Re moiety can promote charge separation and open a new pathway to the non-emissive state. Additionally, the photocurrent response and electrochemical impedance spectroscopic studies also confirmed a notably enhanced efficiency for charge separation in MCOF-Ru/Re compared with MCOF-Ru (Fig. S42 and S43, ESI†).
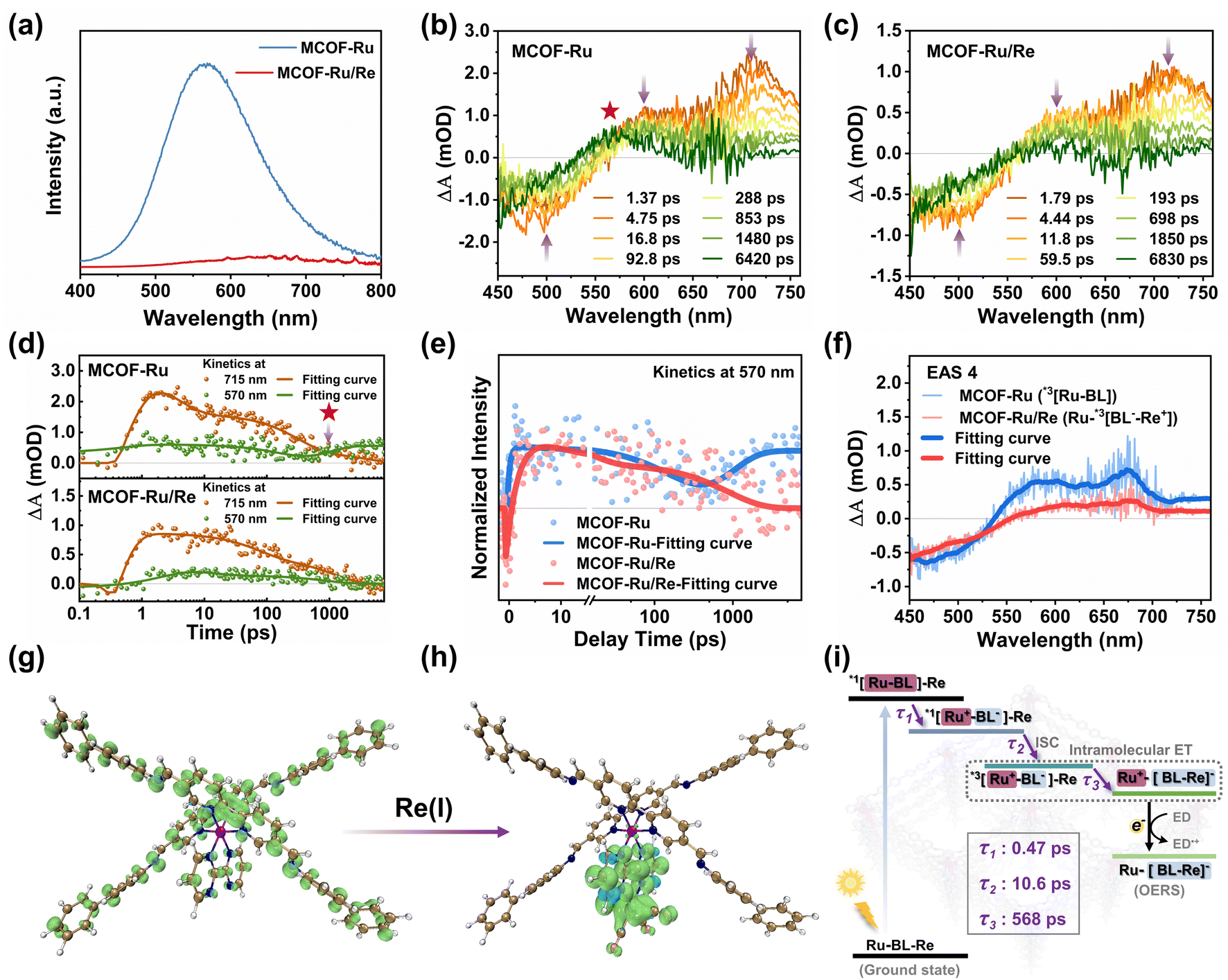 | ||
| Fig. 3 (a) PL emission spectra of MCOF-Ru and MCOF-Ru/Re. fs-TA spectra of (b) MCOF-Ru and (c) MCOF-Ru/Re in glycol, λex = 350 nm. (d) Kinetics analysis of MCOF-Ru and MCOF-Ru/Re at 570 nm and 715 nm. Comparison of (e) kinetics at 570 nm and (f) evolution-associated species (EAS) for MCOF-Ru and MCOF-Ru/Re. Spin density surfaces of the structural fragment for (g) MCOF-Ru and (h) MCOF-Ru/Re in the triplet state (color codes for atoms: C, brown; N, deep blue; Ru, purple; H, light gray). (i) Schematic of photoinduced charge transfer dynamics for MCOF-Ru/Re (Ru-BL-Re represents MCOF-Ru/Re, and BL represents the bridging 2,2′-bipyrimidine ligand). | ||
In the next step, femtosecond transient absorption (fs-TA) spectroscopy was further carried out to uncover the excited state evolution dynamics of MCOF-Ru and MCOF-Ru/Re. For MCOF-Ru, two excited state absorption (ESA) peaks located at 600 and 715 nm appear within 1.37 ps after excitation, which can be assigned to the singlet charge-separated state *1[Ru+–BL−] (Fig. S44, ESI†).39 The negative peak located at 500 nm was attributed to the ground state bleach, according to the steady state UV-vis absorption of MCOF-Ru (Fig. S45, ESI†). Then, the fast drop of the ESA peak at 715 nm generated a broad spectrum from 570–750 nm (Fig. 3b). This process was related to the fast and efficient intersystem crossing (ISC) of Ru moieties within the MCOF and produced a triplet charge-separated state, *3[Ru+–BL−].40,41 Finally, the drop of the triplet charge-separated state was accompanied by the increase of the ESA peak at 570 nm (Fig. 3b). This process can be shown clearer by comparing the kinetics at 570 nm and 715 nm as shown in Fig. 3d. The transient absorption species located at 570 nm could be assigned to the local-excited triple state *3[Ru–BL]. Compared to MCOF-Ru, the fs-TA spectra of MCOF-Ru/Re showed two similar ESA peaks within 1.79 ps (Fig. S46, ESI†). Then, the evolution process to form a broad spectrum was also similar to that of MCOF-Ru (Fig. 3c). Hence, these two ESA peaks and the broad spectrum can be assigned to *1[Ru+–BL−]–Re and *3[Ru+–BL−]–Re, respectively. However, *3[Ru+–BL−]–Re slowly decays to a broad spectrum instead of evolving to a local-excited triple state (Fig. 3c and d). The comparison of kinetics at 570 nm further demonstrates this clear difference in excited-state evolution processes between MCOF-Ru and MCOF-Ru/Re (Fig. 3e). This suggests that the Re center can quench *3[Ru+–BL−]–Re.
To further reveal the transient absorption species in fs-TA spectra, global fitting was used to extract evolution-associated species (EAS) and its dynamics (Fig. S47, ESI†). Four EAS can be obtained from the analysis results of both MCOF-Ru and MCOF-Ru/Re. Both EAS 1 to 3 for MCOF-Ru and MCOF-Ru/Re showed similar spectra, which further confirms the assignment of ESA peaks above. Therefore, EAS 1 to 4 of MCOF-Ru were related to *1[Ru–BL], *1[Ru+–BL−], *3[Ru+–BL−], and *3[Ru–BL], respectively, while EAS 1 to 3 of MCOF-Ru/Re were attributed to *1[Ru–BL]–Re, *1[Ru+–BL−]–Re, and *3[Ru+–BL−]–Re, respectively. However, the long lifetime EAS 4 of MCOF-Ru/Re is kind of different from that of MCOF-Ru (Fig. 3f). According to the quenching process of PL spectra in Fig. 3a, the intramolecular electron transfer (IET) process can be used to describe the evolution process from EAS 3 to EAS 4 for MCOF-Ru/Re. That is the charge located at BL lost connection with the Ru center and is more engaged to the Re center. Besides, the spin-density distributions for the structural fragment of MCOFs were calculated. After introducing the Re(I) center, it is clearly shown that the T1 state was transferred to the Re center and the 2,2′-bipyrimidine ligands within MCOF-Ru/Re, which provides further evidence to support the IET process (Fig. 3g and h). So the produced EAS 4 can be attributed to Ru+–[BL–Re]− with a long lifetime, which is beneficial to the reaction followed.42,43 Overall, Fig. 3i shows the photoinduced charge transfer dynamics of MCOF-Ru/Re. Following rapid charge transfer (τ1 = 0.47 ps) and ISC (τ2 = 10.6 ps) upon photoexcitation, the excited state *3[Ru+–BL−]–Re undergoes an IET process (τ3 = 568 ps) and forms the Ru+–[BL–Re]−. Then, the one-electron reduced species (OERS)44 is generated for the subsequent reaction of CO2 reduction.
To dynamically monitor intermediate species during the CO2RR, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was carried out (Fig. 4a). The observation of CO2 asymmetric stretching around 2337 cm−1 suggested that the initial step of the CO2RR involves CO2 binding to the catalyst.36 The peak at 1270 cm−1 was identified as active *CO2 species. Over time, the noticeable enhancement of the peaks at 1624, 1431, and 1355 cm−1 was assigned to the *COOH intermediate in CO2 photoreduction.17 The peak at 2038 cm−1 was characteristic of the *CO intermediate, contributing to the formation of the final CO product.11 The DFT calculation was further performed to study the free energy changes to gain insight into the CO2 reduction process (Fig. 4b). Initially, the CO2 molecule was activated by the Re site forming the intermediate *CO2 with a ΔG value of 0.72 eV. This step was identified as the potential-determining step for the CO2RR. Then, one of the O sites of *CO2 was protonated to yield *COOH. The formation of *CO follows as *COOH gains a proton and an electron, accompanied by the dissociation of one H2O molecule. Finally, the *CO intermediate preferentially underwent dissociation via pathway I to release CO, rather than further protonating CO* to form CHO* via pathway II. This preference resulted in a high CO selectivity.
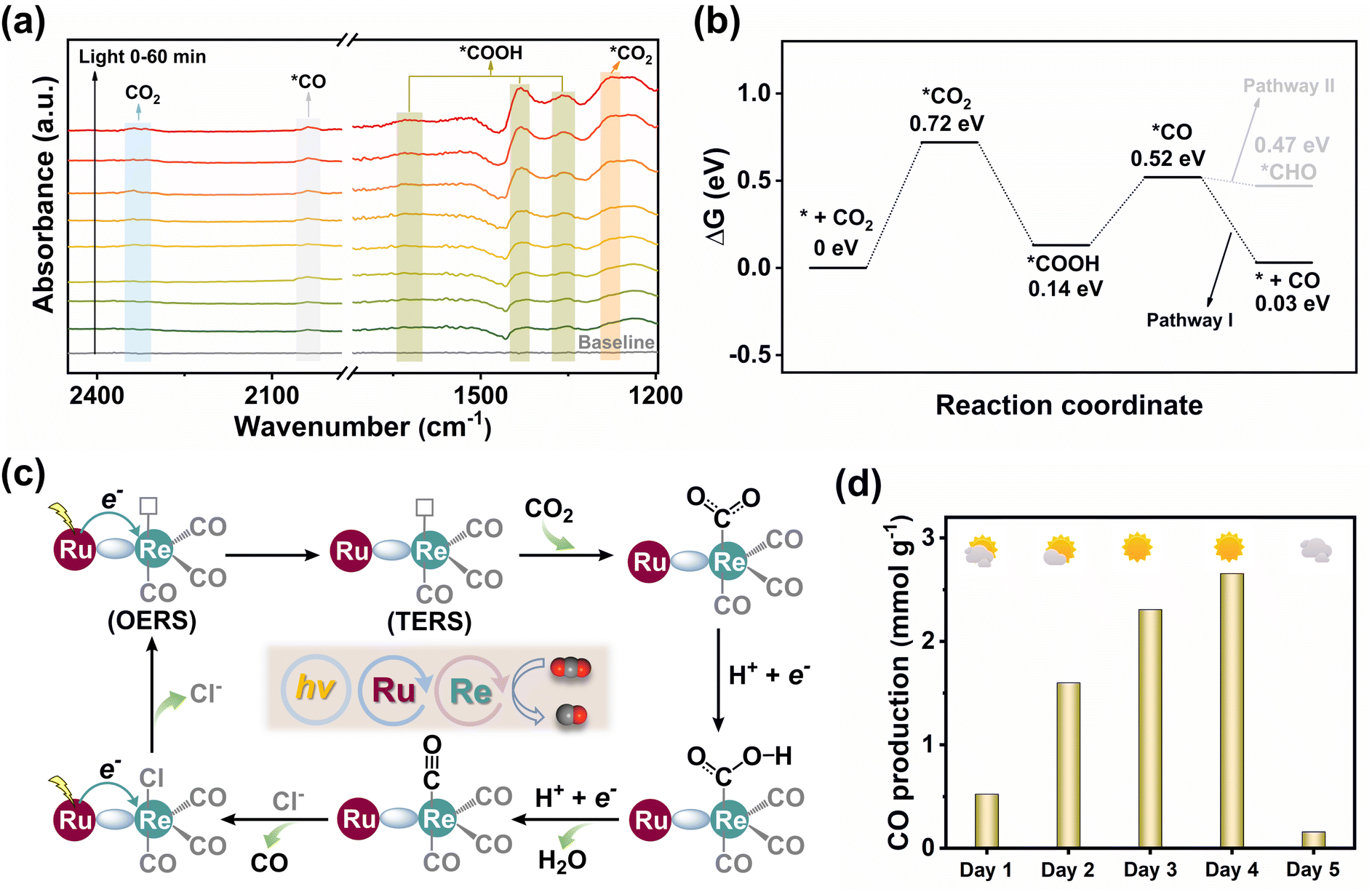 | ||
| Fig. 4 (a) In situ DRIFTS of MCOF-Ru/Re during CO2 photoreduction. (b) Free energy diagram of MCOF-Ru/Re for the CO2RR. (c) Proposed photocatalytic mechanism. (d) Natural sunlight-driven CO2 reduction of MCOF-Ru/Re in 6 hours. | ||
Based on the above analysis, we propose a plausible photocatalytic mechanism (Fig. 4c). Upon light irradiation, the Ru cores within the MCOF generated excited photogenerated electrons, which were migrated to the Re sites, thus forming the one-electron reduced species (OERS). Notably, it is widely acknowledged that following one-electron reduction, the axial Cl− ligand undergoes detachment from the OERS. This intermediate can receive a second electron, forming a two-electron reduced species (TERS).44 Subsequently, the CO2 molecule binds to the coordinatively unsaturated TERS and undergoes protonation, yielding a CO intermediate that can ultimately be released from the Re(I) site. The above mechanism is also consistent with the most reported findings.44–46 In addition to the Ru(II) photoactive units and Re(I) catalytic sites, our MCOF also features two sets of interdigitated covalent organic networks (Fig. 1g). Upon light irradiation, both the Ru(II) center and the covalent skeleton can serve photo-electron generators for CO2 reduction. This hypothesis can be supported by the experimental result, wherein small amounts of CO were still detected when MCOF-Ru was utilized as the photocatalyst (Fig. 2e). On the other hand, the introduction of Re(I) sites in MCOF-Ru/Re can significantly improve the photocatalytic performance, suggesting that the Re(I) sites are the primary catalytic centers for CO2 reduction. However, the intrinsic activity of the MCOF skeleton itself still cannot be completely excluded.
As a more competitive method for practical photocatalysis, natural sunlight was further used to examine the MCOF-Ru/Re catalyzed CO2 reduction. The photocatalytic reaction per day (from 9:00 am to 15:00 pm) was conducted for 5 consecutive days in the outdoor environment of the campus at Jiangnan University (Fig. 4d and S49, ESI†). Under natural sunlight conditions, MCOF-Ru/Re was still effective for CO2 photoreduction, and it was weather dependent. The highest CO production of 2309 μmol g−1 (98% CO selectivity) was achieved under sunny weather on Dec. 7th, 2023. These results suggest the great potential of our strategy in design MCOF artificial photocatalysts for practical applications.
Conclusions
In conclusion, this work presents the pioneering utilization of a MCOF-based photocatalyst incorporating a Ru(II) PS and a Re(I) catalyst for sunlight-driven CO2 reduction. The synergistic collaboration of multiple functional components, including photosensitive and catalytic units, stable covalent framework, and fast charge-transfer dynamics, boosts the overall performance in the photocatalytic CO2RR, even under natural sunlight conditions. The replaceability of both molecular photosensitizers and catalysts in the MCOFs appears to be very promising in the design and development of various artificial photocatalysts.Data availability
Essential data are provided in the main text and the ESI.† Data can be available from the corresponding author upon reasonable request.Author contributions
Z.-G. G., M.-D. L., J. Z. and H. P. supervised the project. W.-K. H., J. L., R.-M. Z., M. W., S.-K. X., and J.-X. F. conducted the experiments. W.-K. H. and J. L. analyzed the data and wrote the paper. All the authors discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075108, 21905116, and 22273057), the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2022B01), the Universities Joint Laboratory of Guangdong, Hong Kong and Macao (2021LSYS009), and the Natural Science Foundation of Guangdong Province (2022A1515011661).Notes and references
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- T. Kong, Y. Jiang and Y. Xiong, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC.
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549–554 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- Y. Yamazaki, M. Miyaji and O. Ishitani, J. Am. Chem. Soc., 2022, 144, 6640–6660 CrossRef CAS PubMed.
- S. Gao, Q. Zhang, X. Su, X. Wu, X. G. Zhang, Y. Guo, Z. Li, J. Wei, H. Wang, S. Zhang and J. Wang, J. Am. Chem. Soc., 2023, 145, 9520–9529 CrossRef CAS PubMed.
- Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS PubMed.
- H. L. Zheng, J. Q. Zhao, Y. Y. Sun, A. A. Zhang, Y. J. Cheng, L. He, X. Bu, J. Zhang and Q. Lin, J. Am. Chem. Soc., 2023, 145, 27728–27739 CrossRef CAS PubMed.
- M. Zhou, Z. Wang, A. Mei, Z. Yang, W. Chen, S. Ou, S. Wang, K. Chen, P. Reiss, K. Qi, J. Ma and Y. Liu, Nat. Commun., 2023, 14, 2473 CrossRef CAS PubMed.
- Z. Wang, Y. Hu, S. Zhang and Y. Sun, Chem. Soc. Rev., 2022, 51, 6704–6737 RSC.
- K. Kosugi, C. Akatsuka, H. Iwami, M. Kondo and S. Masaoka, J. Am. Chem. Soc., 2023, 145, 10451–10457 CrossRef CAS PubMed.
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70 RSC.
- N. Y. Huang, H. He, S. Liu, H. L. Zhu, Y. J. Li, J. Xu, J. R. Huang, X. Wang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 17424–17430 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- Q. Guan, L. L. Zhou and Y. B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- W.-K. Han, Y. Liu, X. Yan and Z.-G. Gu, Mater. Chem. Front., 2023, 7, 2995–3010 RSC.
- H. S. Lu, W. K. Han, X. Yan, C. J. Chen, T. Niu and Z. G. Gu, Angew. Chem., Int. Ed., 2021, 60, 17881–17886 CrossRef CAS PubMed.
- W. K. Han, Y. Liu, X. Yan, Y. Jiang, J. Zhang and Z. G. Gu, Angew. Chem., Int. Ed., 2022, 61, e202208791 CrossRef CAS PubMed.
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y. Q. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204326 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- Y. L. Li, A. J. Li, S. L. Huang, J. J. Vittal and G. Y. Yang, Chem. Soc. Rev., 2023, 52, 4725–4754 RSC.
- N. Y. Huang, J. Q. Shen, X. W. Zhang, P. Q. Liao, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2022, 144, 8676–8682 CrossRef CAS PubMed.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- D. Saito, Y. Yamazaki, Y. Tamaki and O. Ishitani, J. Am. Chem. Soc., 2020, 142, 19249–19258 CrossRef CAS PubMed.
- P. L. Cheung, S. C. Kapper, T. Zeng, M. E. Thompson and C. P. Kubiak, J. Am. Chem. Soc., 2019, 141, 14961–14965 CrossRef CAS PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- Y. Liu, Y. Ma, J. Yang, C. S. Diercks, N. Tamura, F. Jin and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 16015–16019 CrossRef CAS PubMed.
- Y. Z. Cheng, W. Ji, P. Y. Hao, X. H. Qi, X. Wu, X. M. Dou, X. Y. Bian, D. Jiang, F. T. Li, X. F. Liu, D. H. Yang, X. Ding and B. H. Han, Angew. Chem., Int. Ed., 2023, 62, e202308523 CrossRef CAS PubMed.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- X. Wang, X. Ding, T. Wang, K. Wang, Y. Jin, Y. Han, P. Zhang, N. Li, H. Wang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 41122–41130 CrossRef CAS PubMed.
- Y. Wang, T. Sun, T. Zheng, X. Ding, P. Zhang, Q. Xu, T. Li, S. Zhang, K. Wang, L. Xu and J. Jiang, ACS Mater. Lett., 2024, 6, 140–152 CrossRef CAS.
- G. K. Kosgei, M. Y. Livshits, T. R. Canterbury, J. J. Rack and K. J. Brewer, Inorg. Chim. Acta, 2017, 454, 67–70 CrossRef CAS.
- M. Chergui, Acc. Chem. Res., 2015, 48, 801–808 CrossRef CAS PubMed.
- L. S. Forster, Coord. Chem. Rev., 2006, 250, 2023–2033 CrossRef CAS.
- S. Kim, D. Lee, T. Kim, C. H. Kim, H. J. Son, S. O. Kang and J. Y. Shin, J. Phys. Chem. Lett., 2023, 14, 1535–1541 CrossRef CAS PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- A. V. Müller, L. A. Faustino, K. T. de Oliveira, A. O. T. Patrocinio and A. S. Polo, ACS Catal., 2022, 13, 633–646 CrossRef.
- K. Kamogawa, A. Santoro, A. M. Cancelliere, Y. Shimoda, K. Miyata, K. Onda, F. Puntoriero, S. Campagna, Y. Tamaki and O. Ishitani, ACS Catal., 2023, 13, 9025–9032 CrossRef CAS.
- Y. Yamazaki, K. Ohkubo, D. Saito, T. Yatsu, Y. Tamaki, S. Tanaka, K. Koike, K. Onda and O. Ishitani, Inorg. Chem., 2019, 58, 11480–11492 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, characterization and catalytic experiments. CCDC 2323722. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01896f |
| ‡ W. H. and J. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |