
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic meso-β regulation of porphyrins: squeezing the band gap into the near-infrared I/II region†
Chulin
Qu
 a,
Xinxin
Gong
a,
Yufen
Sun
a,
Hu
Gao
a,
Fangjian
Cai
a,
Yue
Zhao
a,
Fan
Wu
*ab and
Zhen
Shen
*a
a,
Xinxin
Gong
a,
Yufen
Sun
a,
Hu
Gao
a,
Fangjian
Cai
a,
Yue
Zhao
a,
Fan
Wu
*ab and
Zhen
Shen
*a
aState Key Laboratory of Coordination Chemistry, Collaborative Innovation Center of Advanced Microstructures, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: wufan@nnu.edu.cn; zshen@nju.edu.cn
bSchool of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China
First published on 3rd June 2024
Abstract
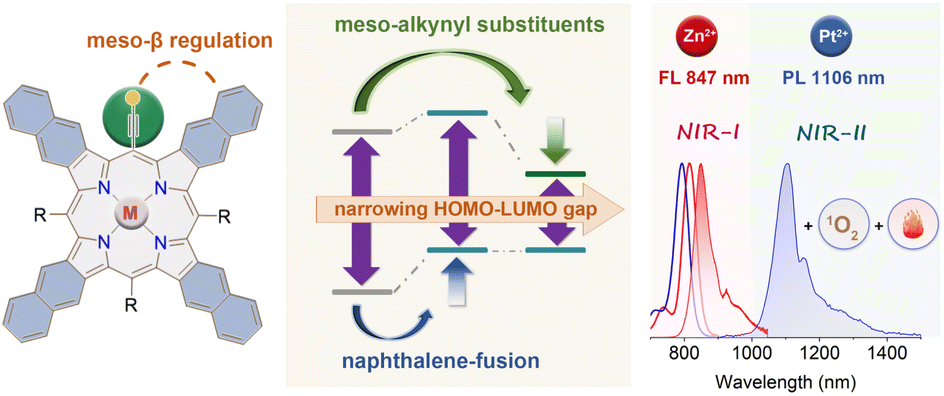
The development of novel near-infrared (NIR) materials with extremely small energy gaps and high stability is highly desirable in bioimaging and phototherapy. Here we report an effective strategy for narrowing the energy gaps of porphyrins by synergistic regulation of meso/β substituents. The novel NIR absorbing/emitting meso-alkynyl naphthoporphyrins (Zn-TNP and Pt-TNP) are synthesized via the retro-Diels–Alder reaction. X-ray crystallography analysis confirms the highly distorted structures of the complexes. Both compounds exhibit intense Q bands around 800 nm, while Zn-TNP shows deep NIR fluorescence at 847 nm. Pt-TNP displays NIR-II room temperature phosphorescence peaking at 1106 nm with an extremely large Stokes shift of 314 nm, which are the longest wavelengths observed among the reported platinum porphyrinoids. Furthermore, Pt-TNP shows remarkable photostability and a notable capacity for synchronous singlet oxygen and heat generation under NIR light irradiation, demonstrating potential in combined photodynamic/photothermal therapy. A theoretical analysis reveals the progressive lifting of the HOMO by the β-fused benzene ring, the decrease of the LUMO upon meso-alkynyl substitution, and energy-releasing pathways varying with metal ions. This dual regulation approach demonstrates great promise in designing innovative multifunctional NIR porphyrin materials.
Introduction
Organic near-infrared (NIR) materials are in high demand for bio-imaging and phototherapy due to their great advantages including low cytotoxicity, deep tissue penetration, and minimal damage to normal tissues.1–5 As important pigments of life, porphyrinoids can be readily functionalized via inner-core coordination and peripheral modifications. The common tetrapyrrolic porphyrins are 18π aromatic conjugated molecules displaying intense Soret bands and weak Q bands in the visible region, but they lack near-infrared (NIR) absorption. Therefore, considerable effort has been devoted to obtaining NIR-absorbed porphyrinoids by modifying the structure, which narrows the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap. Among various designing strategies, such as developing π-extended,6 MO-mixing7,8 and donor–acceptor9 systems, pyrrolic β-aromatic-fusion10 stands out as an exceptional approach for producing rigid porphyrins with intensified Q bands in NIR-absorption and emission. The Q bands of porphyrins can undergo a gradual red-shift by the tetra-fusion of benzene11–13 (600–700 nm), naphthalene14–16 (700–800 nm), and anthracene17–20 (>800 nm), accompanied by a notable intensity increase.21 Despite having the longest NIR absorption and emission bands, the anthracene-fused (anthro)porphyrins were easily photooxidized under ambient light,22 which severely restricted their practical uses.While considerable research has been conducted on expanding the porphyrinoid π-system via pyrrolic β-functionalization, little attention has been paid to decreasing the HOMO–LUMO gap by adjusting meso-substituents. Most porphyrins have meso-aryl substituents, resulting in a minor change in the HOMO–LUMO gap, because both electron-withdrawing and electron-donating groups in phenyl always cause a simultaneous rise or decrease in the HOMO and LUMO. Kobayashi et al. reported the design of stable, NIR-absorbing phthalocyanines by rationally controlling both the central core and the α-substituent, demonstrating the efficiency of multipart regulation in energy-gap tuning.23–25Meso-alkynyl groups have been incorporated into porphyrinoids26 to facilitate intermolecular coupling and create nanoscale tape and rings27–29 with a high degree of conjugation. In this work, combined with π-extension at β-positions, we highlight the additional HOMO–LUMO gap narrowing ability of alkynyl groups at meso positions (Fig. 1). Upon the meso-β double regulation, our newly synthesized meso-alkynyl naphthalene-fused (naphtho)porphyrin complexes Zn-TNP and Pt-TNP demonstrate not only a more pronounced red-shift in both absorption and emission (>800 nm), but also significantly higher photostability than their NIR-absorbing meso-aryl anthroporphyrin counterparts. Notably, the platinum coordinated Pt-TNP is a rare example of a porphyrinoid emitter that displays phosphorescence in the desirable NIR-II (1000–1400 nm) window with a maximum of 1106 nm at room temperature, while presenting an ultra-large Stokes shift exceeding 300 nm. Furthermore, Pt-TNP exhibits significant singlet oxygen generation and photothermal conversion capacity under NIR laser irradiation, making it an ideal agent for multifunctional phototherapy.30
 | ||
| Fig. 1 Molecular design of target NIR-absorbing/emitting porphyrin complexes. | ||
Results and discussion
The synthesis route is presented in Fig. 2a. 4,9-Dihydro-4,9-ethano-2H-benz[f]isoindole 1 bearing a bicyclo[2.2.2]octadiene (BCOD) unit was synthesized via a reported method.31 The condensation of 1 and 3-(triisopropylsilyl)propiolaldehyde 2 (ref. 32) was catalyzed by BF3·OEt2 in ultra-dry CH2Cl2 under an N2 atmosphere, followed by oxidation with p-chloranil (TCQ), and afforded free-base porphyrin 3 in 9% yield.33 Adding a methanol solution of Zn(OAc)2 to the CHCl3 solution of 3 afforded the zinc-coordinated porphyrin Zn-3 in 93% yield. The platinum complex Pt-3 was synthesized in 93% yield via the reaction between 3 and PtCl2 in refluxing acetic acid. The naphthoporphyrins Zn-TNP and Pt-TNP were quantitatively obtained by employing the retro-Diels–Alder method,34 heating precursors Zn-3 and Pt-3 in solid states, respectively, at 300 °C under vacuum.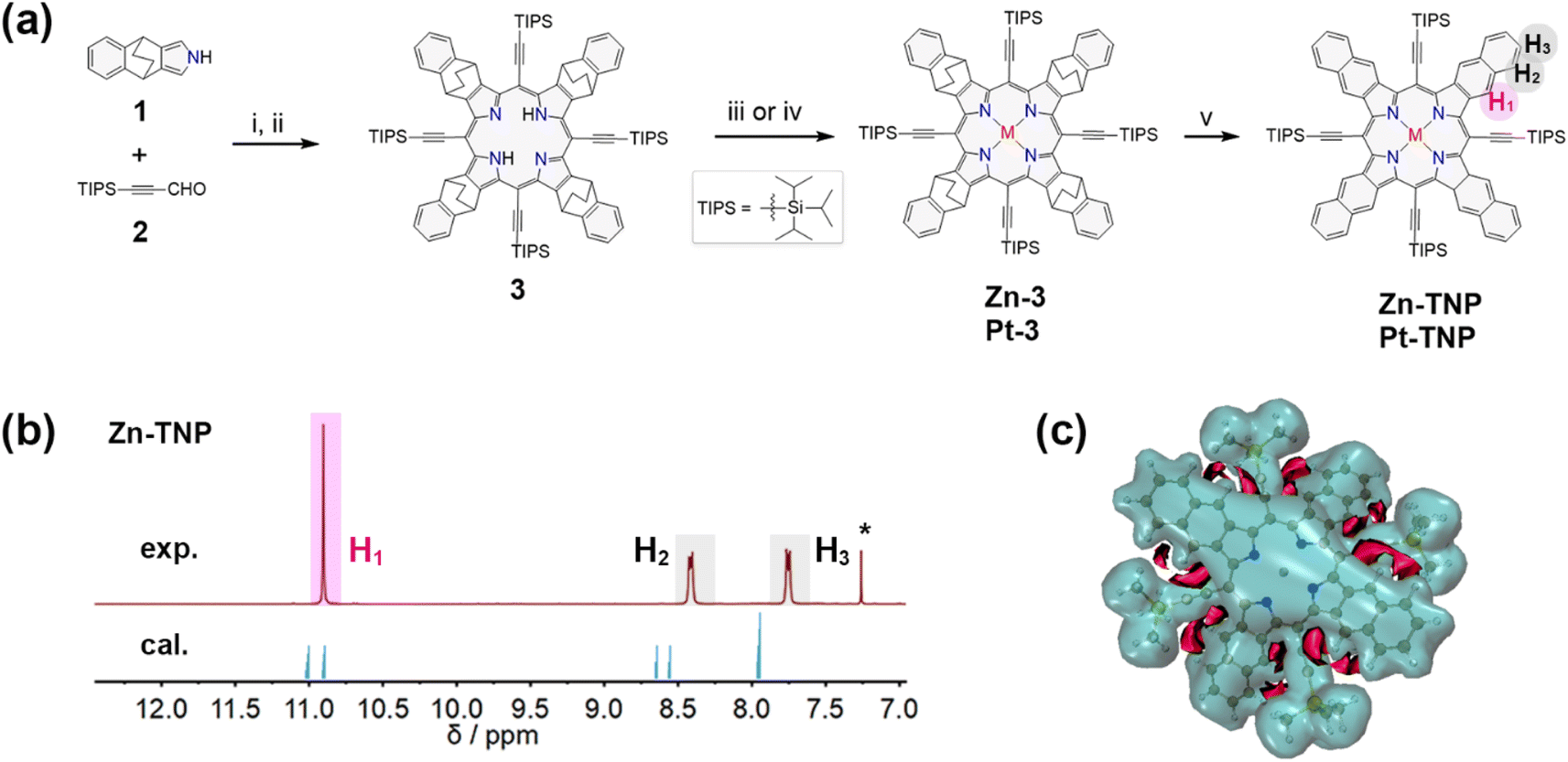 | ||
| Fig. 2 (a) Synthesis of BCOD and π-extended porphyrins: (i) BF3·OEt2, CH2Cl2, −78 °C, 2 h; (ii) TCQ, r.t., 30 min; (iii) Zn(OAc)2·2H2O, CHCl3/MeOH, r.t., 3 h; (iv) PtCl2, NaOAc, AcOH, 118 °C, N2, 24 h; (v) 300 °C, 30 min, in vacuo. (b) 1H NMR spectra of Zn-TNP: experimental data (above) measured in CDCl3 and simulated data (below). (c) 3D ICSS map of Zn-TNP (isovalue = 1.5). The shielding areas are transparent blue and deshielding areas are solid red. | ||
The successful synthesis of the desired fused porphyrins was confirmed by HR-ESI mass spectrometry and MALDI-TOF mass spectrometry (Fig. S1–S10†), and nuclear magnetic resonance (NMR) spectroscopy (Fig. S12–S16†). The resonance signals of protons on the fused naphthalenes of Zn-TNP (Fig. 2b) are divided into three groups: H1 at δ = 10.9 ppm, H2 at δ = 8.41 ppm and H3 at δ = 7.75 ppm, which shift more downfield than those of the meso-tetraphenyl[2,3]tetranaphthoporphyrin zinc complex (Zn-TPTNP) reported by Ito et al.14,35 The NMR computational simulation of Zn-TNP shows consistency with the experimental results, with H1 at δ = 10.9/11.0 ppm, H2 at δ = 8.56/8.66 ppm, and H3 at δ = 7.95/7.97 ppm. To further explain the downfield proton signals on Zn-TNP, particularly the significantly shifted H1, the 3D isochemical shielding surface (ICSS) calculation36,37 was performed (Fig. 2c). The overall aromaticity of Zn-TNP is indicated by the large shielding area (blue) enclosing the entire molecule. Enhanced deshielding (red) is observed in the area between the alkynyl groups and fused naphthalenes of Zn-TNP, which mainly surrounds the H1 protons. Consequently, the presence of alkynyl groups induces a more significant downfield shift in the signal of H1, while the signals of H2 and H3 also undergo a slight downfield shift.
The solid structures of Zn-TNP and Pt-TNP were examined using single-crystal X-ray diffraction. The single crystals of Zn-TNP were obtained by slow diffusion of hexane into its CH2Cl2 solution containing a small amount of Et4NCl. As a consequence of Cl− axial coordination, the Zn–N bond lengths range from 2.072(5) to 2.137(6) Å, which are longer than those of common four-coordinated Zn porphyrins (2.039–2.040 Å).38 The Zn(II) ion is located at 0.384 Å above the 4N plane, and the length of the Zn–Cl bond is 2.335(2) Å. The single crystals of Pt-TNP were obtained by slow evaporation of its toluene solution for a month. The bond length of Pt–N is 2.0073(14) Å, which is identical to those of the reported (5,10,15,20-tetraphenyl-21H,23H-porphinato)platinum(II).39 The bond length alternations in crystal structures are analyzed by the harmonic oscillator model of aromaticity40 (HOMA). The HOMA values of Zn-TNP and Pt-TNP for the 18π-circuit (containing 16 atoms as a macrocyclic internal cross) are 0.88 and 0.90, respectively, which are typical for aromatic porphyrinoids.41Zn-TNP and Pt-TNP have skeleton lengths of 16.957 Å and 17.179 Å, respectively, determined by the distance between the terminal carbon atoms on the fused naphthalenes (Fig. 3a). Both Zn-TNP and Pt-TNP exhibit highly distorted conformations, which are further investigated using the clothes-line diagrams (Fig. 3b) and normal-coordinate structural decomposition (NSD) method42,43 (Tables S3 and S4†). Zn-TNP has a saddled conformation (B2u = 1.89), while Pt-TNP has a distinct hybrid conformation that is primarily saddled (B2u = 2.59) and partly ruffled (B1u = 1.15). The single crystal packing structure of Zn-TNP displays a wave-shaped extension along the a and c directions. The formation of the “wave” is constructed by the face-to-face π-stacking between the fused naphthalenes in Zn-TNP molecules. And the Zn-TNP wave layers are separated by the Et4N+ counterions. The packing structure of Pt-TNP is arranged in an interleaved manner, showing slipped π–π interaction of the fused naphthalenes (Fig. 3c).
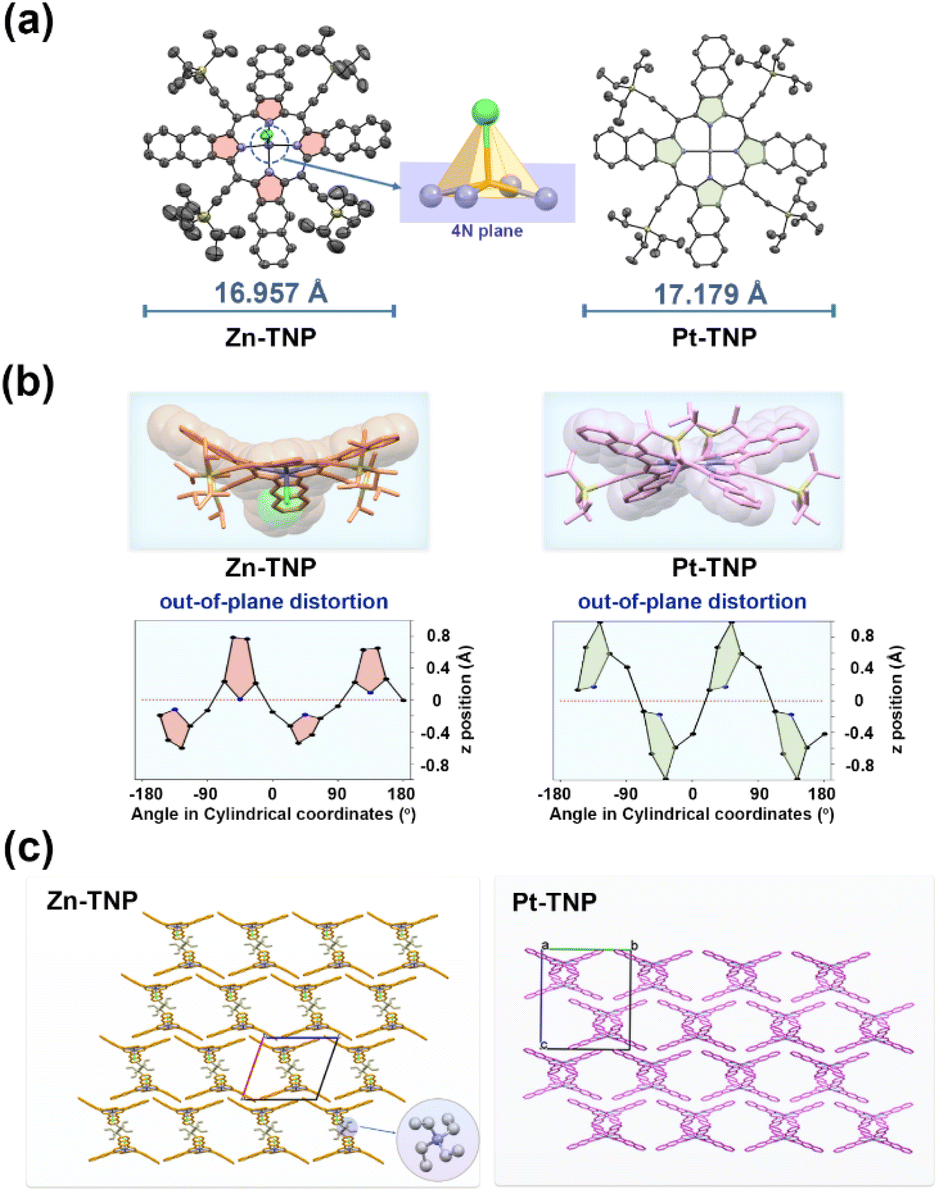 | ||
| Fig. 3 (a) Top views of single crystal structures of Zn-TNP and Pt-TNP using an ORTEP diagram with thermal ellipsoids drawn at the 50% probability. (b) Side views and out-of-plane distortions of Zn-TNP and Pt-TNP. (c) Packing diagrams of Zn-TNP and Pt-TNP. In the packing views, meso-substituents are omitted for clarity. | ||
The absorption, luminescence, and magnetic circular dichroism (MCD) spectra of the new porphyrin compounds were recorded in toluene to investigate the photophysical properties (Fig. 4a and b). The precursors Zn-3 and Pt-3 display typical intense Soret bands at 487 and 466 nm, respectively, which are red-shifted in comparison to the Soret band of meso-tetraphenylporphyrin at ca. 410 nm (Fig. S21 and S22†). The Soret bands of Zn-TNP and Pt-TNP are further red-shifted to 549 nm and 520 nm, respectively. The Q bands of the Zn-3 and Pt-3 are weak and located in the visible region. After naphthalene-fusion, the Q bands of Zn-TNP and Pt-TNP significantly strengthen with more than 3-fold intensity and shift to the NIR region, peaking at 816 and 792 nm, respectively. The MCD spectrum of Zn-TNP shows derivative-shaped Faraday A1 terms with a pair of opposite signs at the absorption of B00 (549 nm) and Q00 (816 nm), respectively, indicating the presence of a highly symmetrical structure and degenerate excited states.44 The Faraday A1 terms of Zn-TNP show a negative-to-positive sign sequence in ascending energy, indicating that the energy difference between HOMO and HOMO−1 (ΔHOMO) is larger than that between LUMO and LUMO+1 (ΔLUMO).45 The MCD pattern of Pt-TNP is identical to that of Zn-TNP. Compounds 3 and Zn-3 show fluorescence bands at 742 nm and 679 nm (Fig. S25 and S26†), respectively, while Zn-TNP shows a red-shifted fluorescence band at 847 nm (Fig. 4b, purple line). The Pt complexes exhibit room temperature phosphorescence (RTP). Pt-3 shows phosphorescence emission at 823 nm with a shoulder at 930 nm (Fig. S27†). For Pt-TNP, the RTP was detected in the range from 1000 to 1400 nm in deaerated toluene, showing the maximum at 1106 nm (Fig. 4b, red line). The phosphorescence band of Pt-TNP is located in the NIR-II region and more red-shifted than that of any other reported Pt-coordinated porphyrins.21,46 Moreover, Pt-TNP has a very large Stokes shift of 314 nm, making it a potential NIR-II phosphorescence probe for biomedical imaging. In air-saturated toluene, the RTP of Pt-TNP could also be observed with reduced intensity, and the singlet oxygen (1O2) generation capacity of Pt-TNP was revealed by the finding of a 1O2 phosphorescence peak at 1270 nm (Fig. 4c, black line). The 1O2 generation abilities of Pt-TNP and Zn-TNP were compared utilizing 1,3-diphenylisobenzofuran (DPBF) as a singlet oxygen scavenger (Fig. 4d, S34 and S35†). Under laser irradiation (808 nm, 10 mW cm−2), the absorbance of DPBF in toluene exhibited no significant change without the addition of porphyrin compounds, but decreased significantly in 21 s with the presence of Pt-TNP. Furthermore, Pt-TNP exhibited excellent photostability, whereas Zn-TNP was unstable under irradiation and underwent rapid photobleaching (Fig. 4d and S36†). Previous research has demonstrated that zinc porphyrins can undergo photo-oxidation upon light exposure.47,48 The easier photodegradation of Zn-TNP may be attributed to its better oxygen affinity than Pt-TNP (Fig. S11, S18 and S19†), while the detailed mechanism still requires further investigation.49,50 The photothermal conversion performance of Pt-TNP and Zn-TNP was investigated in toluene under 808 nm (0.35 W cm−2) laser irradiation. After 6 minutes of irradiation, the Pt-TNP solution had a temperature increase of over 30 °C, whereas the Zn-TNP solution only increased by around 4 °C due to photoinstability (Fig. 4e and S33†). The heating–cooling measurements of Pt-TNP demonstrated excellent repeatability after three cycles (Fig. 4f and S30†). A time constant (τ) of 120.05 was obtained by linearly fitting time (t) as a function of the negative natural logarithm of the driving force temperature (−ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ) at the cooling stage (Fig. S32†). The calculated photothermal conversion efficiency for the Pt-TNP solution was 78%.
θ) at the cooling stage (Fig. S32†). The calculated photothermal conversion efficiency for the Pt-TNP solution was 78%.
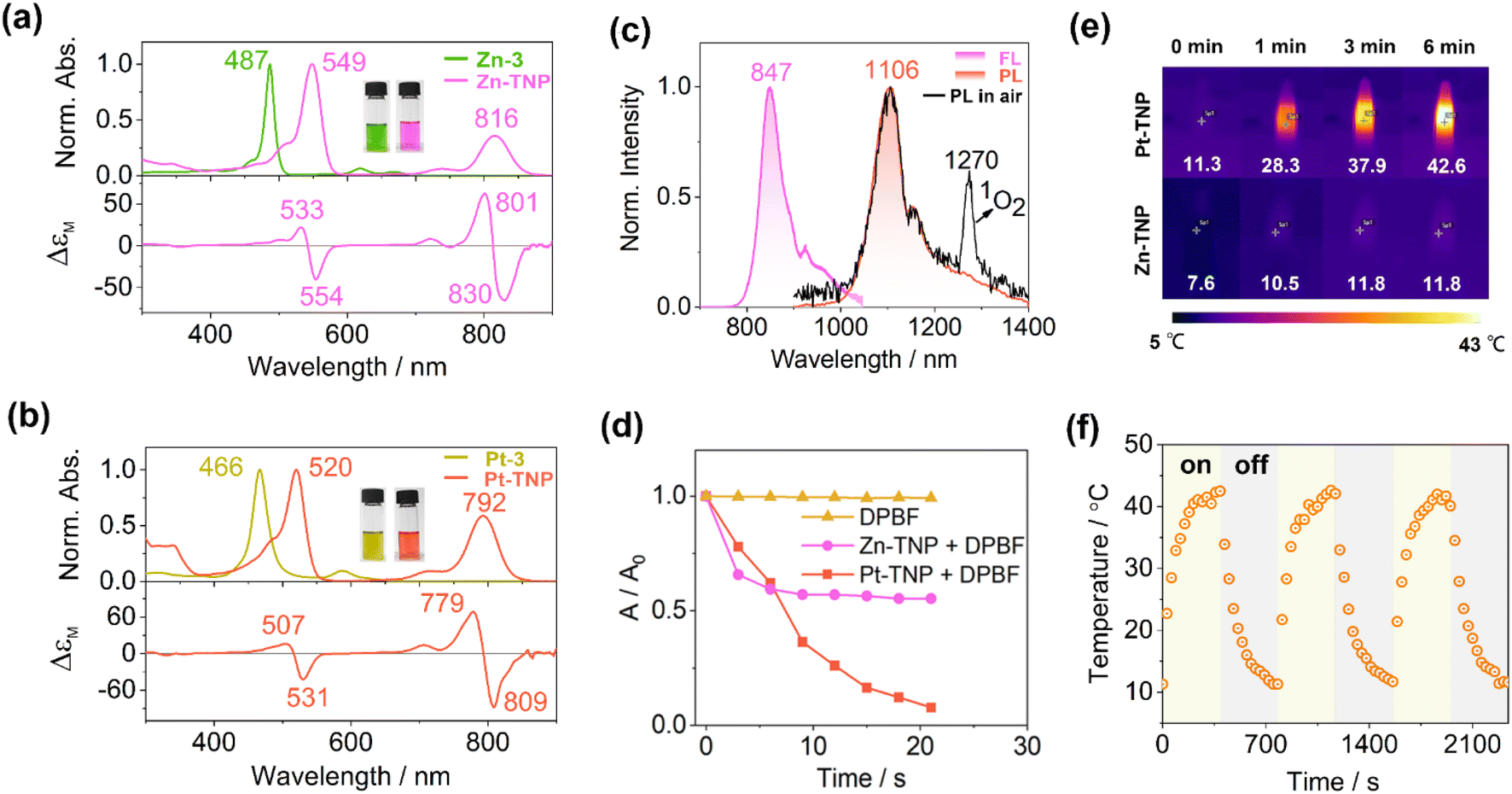 | ||
| Fig. 4 (a and b) Absorption spectra of Zn-3, Zn-TNP, Pt-3 and Pt-TNP in toluene, and the MCD spectra of Zn-TNP and Pt-TNP in toluene. Inset: solution photographs of Zn-3, Zn-TNP, Pt-3, and Pt-TNP. (c) Fluorescence spectrum of Zn-TNP (λex = 549 nm, purple line) in toluene, and phosphorescence spectra of Pt-TNP in deaerated and air-saturated toluene (λex = 520 nm, red line and black line). (d) The absorption changes at 415 nm of DPBF, Zn-TNP + DPBF, and Pt-TNP + DPBF in toluene under 808 nm (10 mW cm−2) laser irradiation within 21 s. (e) Thermal images of Pt-TNP and Zn-TNP (30 μmol) under 808 nm laser irradiation (0.35 W cm−2). (f) Heating–cooling cycles for Pt-TNP (30 μmol) under 808 nm laser irradiation (0.35 W cm−2). | ||
DFT calculations were performed on a series of Pt porphyrins to evaluate the effects of meso- and β-substituents on their energy gaps (Fig. 5a and S39†). The meso-phenyl, β-non-fused (N = 0) platinum porphyrin Pt-TPP has degenerate LUMO/LUMO+1 and near-degenerate HOMO/HOMO−1, showing a large HOMO–LUMO gap of 3.05 eV. The naphthalene-fusion (N = 2) of Pt-TPP considerably lifts its a1u-type HOMO−1 but has little effect on the a2u-type HOMO, resulting in a HOMO/LUMO switch and a huge HOMO/HOMO−1 splitting with a largely reduced HOMO–LUMO gap of 2.24 eV in Pt-TPTNP (Fig. S48†). By replacing the phenyl group with either the electron-withdrawing pentafluorophenyl group or the electron-donating 4-N,N-dimethylphenyl group, the naphthoporphyrin undergoes a simultaneous increase or decrease in both HOMO and LUMO energy levels, resulting in a very small change of the HOMO–LUMO gap. In contrast, the meso-alkynyl Pt-TNP (where triisopropylsilylethynyl is replaced by trimethylsilylethynyl) exhibits not only a significantly elevated HOMO consistent with its meso-phenyl counterpart, but also a substantially reduced LUMO due to the presence of electron-withdrawing alkynyl groups. The meso-β double regulation of Pt-TNP efficiently reduces the HOMO–LUMO gap to 1.92 eV, with the electron density extending to the fused naphthalenes on the HOMO and meso-alkynyl groups on the LUMO. It is noteworthy that the extended electron-density distributions towards alkynyl groups are exclusively observed in the LUMOs, but not in the HOMOs; therefore, conjugative extension could potentially play a substantial role in the large energy decreases of the LUMOs. Additional computational analysis reveals that replacing trimethylsilyl groups with H has a negligible impact on the HOMO/LUMO energy levels. By introducing electron-withdrawing groups like –CF3 and –CN into the alkynyl groups, the HOMO–LUMO gaps are further reduced to 1.80 eV and 1.66 eV, respectively (Fig. S40†). This energy-gap decrease is accompanied by a further extension of the electron density towards the terminal group.
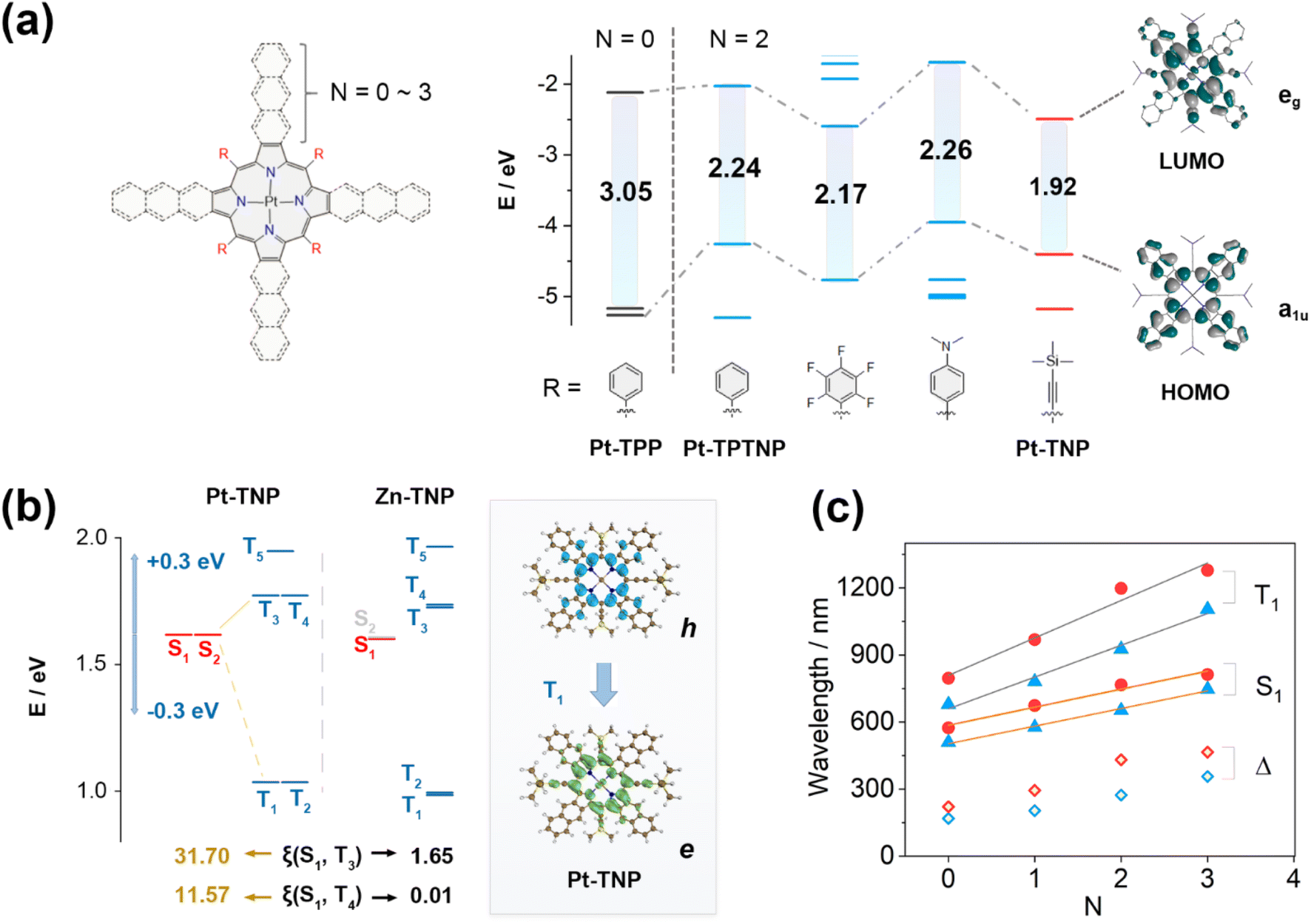 | ||
| Fig. 5 (a) Frontier molecular orbital diagrams of Pt-TNP and other Pt–porphyrins for comparison. (b) Schematic diagram of the calculated energy gaps, spin–orbit coupling constants between S1 and Tn (n = 3–4) of Pt-TNP and Zn-TNP, and the hole and electron distribution in the S0–T1 transition of Pt-TNP. (c) Plots of absorption and phosphorescence peaks of meso-phenyl (blue triangle) and meso-alkynyl (red circle) Pt–porphyrins versus linearly fused benzene numbers (N) in β positions ranging from 0 to 3. Stokes shifts are presented as blue and red rhombuses. | ||
The optimized structures of Pt-TNP and Zn-TNP are distorted, but they exhibit varying degrees and patterns of distortion compared to the single-crystal structures (Fig. S53†). These distortions may be caused by steric effects and further influenced by the crystal-packing forces. We successfully optimized Zn-TNP for both saddled (sad-Zn-TNP) and ruffled (ruf-Zn-TNP) conformations. The total energies, HOMO–LUMO energy levels, and energy gaps of sad-Zn-TNP and ruf-Zn-TNP are nearly identical, suggesting that the distortion patterns have minimal impact on the electronic structures and optical properties. DFT calculations are also conducted on a variety of meso-free/β-fused and β-free/meso-substituted porphyrins, all of which exhibit completely planar conformations due to reduced steric hindrance. The significant HOMO-lifting effect of aromatic fusion and the LUMO-reducing effect of alkynyl substitution, which can be controlled by the degree of π-extension, were further validated on the planar porphyrin molecules (Fig. S38†).
Time-dependent (TD) DFT calculations were performed to further investigate the influence of excited states caused by different substituents and coordinated metal ions (Fig. 5b and c). The energy levels of the S1 and Tn states of Pt-TNP and Zn-TNP are similar, and both compounds exhibit degenerate S1/S2 and T1/T2 states. Zn-TNP and Pt-TNP have large S1–T1 gaps (ΔES1–T1) of 0.62 eV and 0.58 eV, respectively, while their T3 and T4 states have small S1–Tn gaps (ΔES1–Tn < 0.3 eV),51 which promote the intersystem crossing (ISC) activities. The S1–Tn spin–orbit coupling matrix elements (SOCMEs) of Zn-TNP are smaller than 2.4 cm−1. In contrast, the S1–T3 and S1–T4 SOCMEs for Pt-TNP have large values of 31.70 and 11.57 cm−1, respectively. The energy can efficiently pass through the ISC process between S1 and T3 of Pt-TNP due to the narrow energy gap (ΔES1–T3 = 0.15 eV) and large SOCME. Subsequently, the energy undergoes a rapid internal conversion (IC) to T1 and deactivates to the ground state S0 with phosphorescence emission. The T1 state of Pt-TNP can react with oxygen to generate singlet oxygen, and a significant amount of energy can be released as heat through internal conversion. The electron and hole distributions of Pt-TNP indicate that both S0–S1 and S0–T1 are π–π* transitions with negligible involvement of a central metal (Fig. 5b and S75†). The Sr index, which indicates the degree of electron–hole overlap, is 0.86 for S1 and 0.79 for T1, demonstrating that the two states are locally excited (LE).
The wavelength variation trends for the longest absorption (S0–S1) and phosphorescence (S0–T1) peaks of a series of Pt porphyrins are investigated (Fig. 5c). The molecules are divided into two groups: meso-phenyl (blue triangle) and meso-alkynyl (red circle). Each group contains four molecules, with the number of linearly fused benzenes (N) ranging from 0 to 3. In both groups, the calculated wavelengths corresponding to S1, T1, and Stokes shifts increase as N increases, and the wavelengths in the meso-alkynyl group are always larger than those in the meso-phenyl group with the same N value. The plots of the S1 and T1 wavelengths as a function of N exhibit strong linear correlations in both groups, with the slopes of the fitting lines for T1 being larger than those for S1. The meso-alkynyl group with a larger N yields greater calculated Stokes shifts, which is consistent with the remarkably large experimental Stokes shift of Pt-TNP (314 nm) resulting from the meso-β double regulation.
Based on the optimized structure of Pt-TNP and Zn-TNP, we evaluated the ring currents and induced magnetic shielding with a nucleus-independent chemical shift52 (NICS), anisotropy of the induced current density53 (ACID), and the gauge-including magnetically induced current54,55 (GIMIC) calculations (Table S7 and Fig. S76–S78†). The NICS(0) values are negative in both the fused naphthene and porphyrin core for both compounds. The diatropic ring currents of Zn-TNP and Pt-TNP continuously flow throughout the whole structure, showing the global aromaticity of these two compounds. Additionally, localized diatropic ring currents are observed on the naphthalene moieties and the inner 16-bond porphyrin core.
Conclusions
In summary, we successfully synthesized porphyrin complexes with remarkable red-shifted NIR absorption and emission (ranging from 800 to 1200 nm) by tailoring the molecular energy levels through meso- and β-substituents. The electronic structures and optical properties of target meso-alkynyl naphthoporphyrin complexes Zn-TNP and Pt-TNP were fully elucidated via spectroscopic measurements and theoretical calculations. The solid structures of the complexes were analyzed using single-crystal X-ray diffraction, revealing saddling or mixed saddling–ruffling distortions. Zn-TNP and Pt-TNP displayed Q bands at 816 nm and 792 nm, respectively. Zn-TNP exhibited a fluorescence band at a wavelength of 847 nm. Pt-TNP exhibited room-temperature NIR-II phosphorescence at 1106 nm with a huge Stokes shift, and its ability to produce singlet oxygen was directly shown by the emission at 1270 nm. Pt-TNP also demonstrated excellent NIR photothermal conversion capacity. The dual functionality of Pt-TNP in singlet oxygen and heat generation makes it a promising candidate for combined photothermal/photodynamic therapy. While aromatic-ring-fusion is a well-established approach for reducing the HOMO–LUMO gap by HOMO-lifting, the additional functionalization of alkynyl groups in meso positions causes more than 100 nm of shifting in absorption/emission bands by LUMO-reduction when compared to their aryl-substituted counterparts. The DFT calculations provided an in-depth explanation of the correlation between the decrease in S1/T1 energy levels and the incorporation of meso-β substituents, as well as enhanced ISC activity in the Pt complex. The meso-β dual regulation of porphyrins effectively reduces the energy gap and modulates the excited states, offering a promising strategy for developing novel stable NIR materials.Data availability
All experimental details including synthetic procedures, spectroscopic data, and theoretical calculation results, are available in the ESI.†Author contributions
C. Q. carried out the synthesis, characterization, manuscript writing and theoretical calculations. X. G., Y. S., H. G. and F. C. contributed to the synthesis, characterization, and data analysis. C. Q., H. G. and Y. Z. performed the single-crystal X-ray diffraction measurements and analysis. F. W. and Z. S. designed and supervised the project, and revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 22271140, 22071103, and 22371118). The theoretical calculations are performed using the supercomputing resources at the High-Performance Computing Center of Nanjing University.Notes and references
- F. Ding, Y. Zhan, X. Lu and Y. Sun, Recent advances in near-infrared II fluorophores for multifunctional biomedical imaging, Chem. Sci., 2018, 9, 4370–4380 RSC.
- S. He, J. Song, J. Qu and Z. Cheng, Crucial breakthrough of second near-infrared biological window fluorophores: design and synthesis toward multimodal imaging and theranostics, Chem. Soc. Rev., 2018, 47, 4258–4278 RSC.
- G. Hong, A. L. Antaris and H. Dai, Near-infrared fluorophores for biomedical imaging, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- L. Li, X. Han, M. Wang, C. Li, T. Jia and X. Zhao, Recent advances in the development of near-infrared organic photothermal agents, Chem. Eng. J., 2021, 417, 128844 CrossRef CAS.
- V.-N. Nguyen, Z. Zhao, B. Z. Tang and J. Yoon, Organic photosensitizers for antimicrobial phototherapy, Chem. Soc. Rev., 2022, 51, 3324–3340 RSC.
- G. Anguera, W.-Y. Cha, M. D. Moore, J. Lee, S. Guo, V. M. Lynch, D. Kim and J. L. Sessler, Hexadecaphyrin-(1.0.0.0.1.1.0.1.1.0.0.0.1.1.0.1): A Dual Site Ligand That Supports Thermal Conformational Changes, J. Am. Chem. Soc., 2018, 140, 4028–4034 CrossRef CAS PubMed.
- Y. Wang, H. Kai, M. Ishida, S. Gokulnath, S. Mori, T. Murayama, A. Muranaka, M. Uchiyama, Y. Yasutake, S. Fukatsu, Y. Notsuka, Y. Yamaoka, M. Hanafusa, M. Yoshizawa, G. Kim, D. Kim and H. Furuta, Synthesis of a Black Dye with Absorption Capabilities Across the Visible-to-Near-Infrared Region: A MO-Mixing Approach via Heterometal Coordination of Expanded Porphyrinoid, J. Am. Chem. Soc., 2020, 142, 6807–6813 CrossRef CAS PubMed.
- Y. Wang, K. Ogasahara, D. Tomihama, R. Mysliborski, M. Ishida, Y. Hong, Y. Notsuka, Y. Yamaoka, T. Murayama, A. Muranaka, M. Uchiyama, S. Mori, Y. Yasutake, S. Fukatsu, D. Kim and H. Furuta, Near-Infrared-III-Absorbing and -Emitting Dyes: Energy-Gap Engineering of Expanded Porphyrinoids via Metallation, Angew. Chem., Int. Ed., 2020, 59, 16161–16166 CrossRef CAS PubMed.
- T. Higashino, Y. Kurumisawa, S. Nimura, H. Iiyama and H. Imahori, Enhanced Donor–π–Acceptor Character of a Porphyrin Dye Incorporating Naphthobisthiadiazole for Efficient Near-Infrared Light Absorption, Eur. J. Org Chem., 2018, 2018, 2537–2547 CrossRef CAS.
- H. Mori, T. Tanaka and A. Osuka, Fused porphyrinoids as promising near-infrared absorbing dyes, J. Mater. Chem. C, 2013, 1, 2500–2519 RSC.
- L. Edwards, M. Gouterman and C. B. Rose, Synthesis and vapor spectrum of zinc tetrabenzporphine, J. Am. Chem. Soc., 1976, 98, 7638–7641 CrossRef CAS PubMed.
- S. Ito, T. Murashima and N. Ono, A new synthesis of pyrroles fused with polycyclic skeletons, J. Chem. Soc., Perkin Trans. 1, 1997, 3161–3166 RSC.
- C. M. B. Carvalho, T. J. Brocksom and K. T. de Oliveira, Tetrabenzoporphyrins: synthetic developments and applications, Chem. Soc. Rev., 2013, 42, 3302–3317 RSC.
- S. Ito, N. Ochi, H. Uno, T. Murashima and N. Ono, A new synthesis of [2,3]naphthoporphyrins, Chem. Commun., 2000, 893–894 RSC.
- O. S. Finikova, A. V. Cheprakov and S. A. Vinogradov, Synthesis and Luminescence of Soluble Meso-Unsubstituted Tetrabenzo- and Tetranaphtho[2,3]porphyrins, J. Org. Chem., 2005, 70, 9562–9572 CrossRef CAS PubMed.
- T. Khoury and M. J. Crossley, A strategy for the stepwise ring annulation of all four pyrrolic rings of a porphyrin, Chem. Commun., 2007, 4851–4853 RSC.
- M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchenkov and A. V. Cheprakov, Tetraaryltetraanthra[2,3]porphyrins: Synthesis, Structure, and Optical Properties, J. Org. Chem., 2012, 77, 11119–11131 CrossRef CAS PubMed.
- V. Yakutkin, S. Aleshchenkov, S. Chernov, T. Miteva, G. Nelles, A. Cheprakov and S. Baluschev, Towards the IR Limit of the Triplet–Triplet Annihilation-Supported Up-Conversion: Tetraanthraporphyrin, Chem.–Eur. J., 2008, 14, 9846–9850 CrossRef CAS PubMed.
- F. Wu, Y. Sun, H. Gao, X. Zhi, Y. Zhao and Z. Shen, Boosting near-infrared photothermal/photoacoustic conversion performance of anthracene-fused porphyrin via paramagnetic ion coordination strategy, Sci. China: Chem., 2023, 66, 164–173 CrossRef CAS.
- Y. Fan, Z. Zeng, H. Shu, M. Zhou, L. Xu, Y. Rao, T. Gu, X. Liang, W. Zhu and J. Song, Two- and three-dimensional β,β′-N-heterocycle fused porphyrins: concise construction, singlet oxygen production and electro-catalytic hydrogen evolution reaction, Org. Chem. Front., 2021, 8, 6080–6088 RSC.
- J. R. Sommer, A. H. Shelton, A. Parthasarathy, I. Ghiviriga, J. R. Reynolds and K. S. Schanze, Photophysical Properties of Near-Infrared Phosphorescent π-Extended Platinum Porphyrins, Chem. Mater., 2011, 23, 5296–5304 CrossRef CAS.
- H. Yamada, D. Kuzuhara, T. Takahashi, Y. Shimizu, K. Uota, T. Okujima, H. Uno and N. Ono, Synthesis and Characterization of Tetraanthroporphyrins, Org. Lett., 2008, 10, 2947–2950 CrossRef CAS PubMed.
- N. Kobayashi, T. Furuyama and K. Satoh, Rationally Designed Phthalocyanines Having Their Main Absorption Band beyond 1000 nm, J. Am. Chem. Soc., 2011, 133, 19642–19645 CrossRef CAS PubMed.
- T. Furuyama, K. Satoh, T. Kushiya and N. Kobayashi, Design, Synthesis, and Properties of Phthalocyanine Complexes with Main-Group Elements Showing Main Absorption and Fluorescence beyond 1000 nm, J. Am. Chem. Soc., 2014, 136, 765–776 CrossRef CAS PubMed.
- T. Hoshi and N. Kobayashi, Spectroscopic and structural properties of phthalocyanines deduced from their frontier molecular orbitals (MOs) and MO calculations, Coord. Chem. Rev., 2017, 345, 31–41 CrossRef CAS.
- H. L. Anderson, Meso-alkynyl porphyrins, Tetrahedron Lett., 1992, 33, 1101–1104 CrossRef CAS.
- T. Tanaka and A. Osuka, Conjugated porphyrin arrays: synthesis, properties and applications for functional materials, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- M. D. Peeks, T. D. W. Claridge and H. L. Anderson, Aromatic and antiaromatic ring currents in a molecular nanoring, Nature, 2017, 541, 200–203 CrossRef CAS PubMed.
- A. Summerfield, M. Baldoni, D. V. Kondratuk, H. L. Anderson, S. Whitelam, J. P. Garrahan, E. Besley and P. H. Beton, Ordering, flexibility and frustration in arrays of porphyrin nanorings, Nat. Commun., 2019, 10, 2932 CrossRef PubMed.
- H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Organic molecule-based photothermal agents: an expanding photothermal therapy universe, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- Y. Ishii, S. Ito, Y. Saito, D. Uno and T. Oba, Synthesis of [2,3]naphthoporphyrins using 4,9-dihydro-4,9-ethano-2H-benz[f]isoindole as a benz[f]isoindole equivalent, Tetrahedron, 2015, 71, 8892–8898 CrossRef CAS.
- G. A. Brito, F. Della-Felice, G. Luo, A. S. Burns, R. A. Pilli, S. D. Rychnovsky and M. J. Krische, Catalytic Enantioselective Allylations of Acetylenic Aldehydes via 2-Propanol-Mediated Reductive Coupling, Org. Lett., 2018, 20, 4144–4147 CrossRef CAS PubMed.
- Y. Sun, F. Wu, H. Gao, C. Qu, K. Wang, Y. Zhao and Z. Shen, Ruffle-distorted meso-silylethynyl-substituted naphthoporphyrin showing near-infrared Q band beyond 800 nm, J. Porphyrins Phthalocyanines, 2023, 27, 114–120 CrossRef CAS.
- S. Ito, T. Murashima, N. Ono and H. Uno, A new synthesis of benzoporphyrins using 4,7-dihydro-4,7-ethano-2H-isoindole as a synthon of isoindole, Chem. Commun., 1998, 1661–1662 RSC.
- J. E. Rogers, K. A. Nguyen, D. C. Hufnagle, D. G. McLean, W. Su, K. M. Gossett, A. R. Burke, S. A. Vinogradov, R. Pachter and P. A. Fleitz, Observation and Interpretation of Annulated Porphyrins: Studies on the Photophysical Properties of Meso-Tetraphenylmetalloporphyrins, J. Phys. Chem. A, 2003, 107, 11331–11339 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Klod and E. Kleinpeter, Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes-application in conformational and configurational analysis, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- H. M. Marques and I. Cukrowski, Molecular mechanics parameters for the modelling of four-coordinate Zn(II) porphyrins, Phys. Chem. Chem. Phys., 2003, 5, 5499–5506 RSC.
- A. Hazell, Structure of (5,10,15,20-tetraphenyl-21H,23H-porphinato)platinum(II), C44H28N4Pt, Acta Crystallogr., Sect. C: Struct. Chem., 1984, 40, 751–753 CrossRef.
- T. M. Krygowski, Crystallographic studies of inter-and intramolecular interactions reflected in aromatic character of pi-electron systems, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- M. K. Cyrañski, T. M. Krygowski, M. Wisiorowski, N. J. R. van Eikema Hommes and P. v. R. Schleyer, Global and Local Aromaticity in Porphyrins: An Analysis Based on Molecular Geometries and Nucleus-Independent Chemical Shifts, Angew. Chem., Int. Ed., 1998, 37, 177–180 CrossRef.
- W. Jentzen, X.-Z. Song and J. A. Shelnutt, Structural Characterization of Synthetic and Protein-Bound Porphyrins in Terms of the Lowest-Frequency Normal Coordinates of the Macrocycle, J. Phys. Chem. B, 1997, 101, 1684–1699 CrossRef CAS.
- C. J. Kingsbury and M. O. Senge, The shape of porphyrins, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- J. Mack, M. J. Stillman and N. Kobayashi, Application of MCD spectroscopy to porphyrinoids, Coord. Chem. Rev., 2007, 251, 429–453 CrossRef CAS.
- J. Mack, Y. Asano, N. Kobayashi and M. J. Stillman, Application of MCD Spectroscopy and TD-DFT to a Highly Non-Planar Porphyrinoid Ring System. New Insights on Red-Shifted Porphyrinoid Spectral Bands, J. Am. Chem. Soc., 2005, 127, 17697–17711 CrossRef CAS PubMed.
- W. Zhang, S. Chen, P. Sun, S. Ye, Q. Fan, J. Song, P. Zeng, J. Qu and W.-Y. Wong, NIR-II J-Aggregated Pt(II)-Porphyrin-Based Phosphorescent Probe for Tumor-Hypoxia Imaging, Adv. Healthcare Mater., 2022, 11, 2200467 CrossRef CAS PubMed.
- S. Kumar, Y. K. Maurya, T. Lis and M. Stępień, Synthesis of a donor–acceptor heterodimer via trifunctional completive self-sorting, Nat. Commun., 2022, 13, 3204 CrossRef PubMed.
- B. Golec, J. Buczyńska, K. Nawara, A. Gorski and J. Waluk, Photodegradation of free base and zinc porphyrins in the presence and absence of oxygen, Photochem. Photobiol. Sci., 2023, 22, 2725–2734 CrossRef CAS PubMed.
- Z. Zhou, X. Zhou, Q. Liu, X. Zhang and H. Liu, Fixation of Zinc(II) Ion to Dioxygen in a Highly Deformed Porphyrin: Implications for the Oxygen Carrier Mechanism of Distorted Heme, Org. Lett., 2015, 17, 4078–4081 CrossRef CAS PubMed.
- F. Rusydi, M. Kemal Agusta, A. Gandaryus Saputro and H. Kasai, A First Principles Study on Zinc–Porphyrin Interaction with O2 in Zinc–Porphyrin(Oxygen) Complex, J. Phys. Soc. Jpn., 2012, 81, 124301 CrossRef.
- J. Yang, Y. Zhang, X. Wu, W. Dai, D. Chen, J. Shi, B. Tong, Q. Peng, H. Xie, Z. Cai, Y. Dong and X. Zhang, Rational design of pyrrole derivatives with aggregation-induced phosphorescence characteristics for time-resolved and two-photon luminescence imaging, Nat. Commun., 2021, 12, 4883 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, The gauge including magnetically induced current method, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- H. Fliegl, J. Jusélius and D. Sundholm, Gauge-Origin Independent Calculations of the Anisotropy of the Magnetically Induced Current Densities, J. Phys. Chem. A, 2016, 120, 5658–5664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, characterization, additional spectroscopic data, theoretical analysis, X-ray crystallographic data, and simulation of magnetically induced ring currents. CCDC 2325275 and 2265412. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01806k |
| This journal is © The Royal Society of Chemistry 2024 |