
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical recycling of polymeric materials
Weizhe
Zhang
,
Lars
Killian
and
Arnaud
Thevenon
 *
*
Organic Chemistry and Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht University, Universiteitsweg 99, Utrecht, The Netherlands. E-mail: a.a.thevenon-kozub@uu.nl
First published on 21st May 2024
Abstract
Polymeric materials play a pivotal role in our modern world, offering a diverse range of applications. However, they have been designed with end-properties in mind over recyclability, leading to a crisis in their waste management. The recent emergence of electrochemical recycling methodologies for polymeric materials provides new perspectives on closing their life cycle, and to a larger extent, the plastic loop by transforming plastic waste into monomers, building blocks, or new polymers. In this context, we summarize electrochemical strategies developed for the recovery of building blocks, the functionalization of polymer chains as well as paired electrolysis and discuss how they can make an impact on plastic recycling, especially compared to traditional thermochemical approaches. Additionally, we explore potential directions that could revolutionize research in electrochemical plastic recycling, addressing associated challenges.
 Weizhe Zhang | Weizhe Zhang is a postdoctoral fellow at the group of Organic Chemistry and Catalysis (OCC), Utrecht University, the Netherlands. He received his BSc (2014) and MSc (2017) from Beihang University, China. He received his PhD (2023) from Leiden University, the Netherlands. His research focuses on materials science of electrochemical systems and applications of novel chemical conversion technologies. |
 Lars Killian | Lars Killian graduated with an MSc in chemistry from Utrecht University in 2021. In the same year, he started his PhD research at the same university under the supervision of Arnaud Thevenon, working on the development of novel ligands with large, extended π-systems. |
 Arnaud Thevenon | Arnaud Thevenon received his PhD (2018) from the University of Oxford. He was a Marie Skłodowska-Curie postdoctoral researcher at Caltech (2018–2021) and the University of Konstanz (2021) before joining Utrecht University as an Assistant Professor in 2021. His research interest includes the development of homogeneous thermo/electrocatalysts for small molecules, biomass, and waste (plastic) valorization as well as the development of novel polymers that are intrinsically circular by design. |
1 Introduction
Plastics are ubiquitous and essential to modern society. They are relatively inexpensive to manufacture and display many notable advantages over other materials such as light weight and processability, as well as tunable thermal, physical, and mechanical properties. By combining polymeric materials with diverse additives, compatibilizers, among other components, the material properties are tailored to make countless plastic products in industrial sectors ranging from packaging, agriculture, construction and automotive, to medicine and household. Since the beginning of the plastic era, over 8 billion tons of plastics have been produced. Most of these have been designed with specific end-user properties in mind, as opposed to their end-of-life management. Consequently, more than 60% of the plastic produced has ended up in landfills or is lost in the environment. This represents both a waste of potential resources as well as an ecological disaster.1In Europe, approximately 65 Mt of plastic waste is collected every year.2 Half of it is shipped to non-EU countries and the other half is used for energy recovery (21%), and landfilled (13%), while only 16% is recycled. One of the reasons for this low percentage in recycling is that waste streams consist of mixtures of various plastics of unknown compositions and are potentially contaminated by organic (e.g., food remains) or inorganic (e.g., lead in polyvinylchloride (PVC)) fractions.3 Other reasons include the limited recycling technology, improper disposal procedures and lack of incentive policies. There are currently four types of strategies implemented for plastic recycling: reusing (primary), mechanical (secondary), chemical (tertiary) and energy recovery.4 Primary reusing strategies require uncontaminated and pristine-like quality feeds of single-use waste plastics representing a very small portion of the plastic waste stream.5 Mechanical recycling can give a second life to plastic waste by reprocessing it. However, recycled plastics typically show inferior mechanical/thermal properties compared to pristine feeds, due to the deterioration of the polymer backbone (i.e., lower molecular weight from chain scission) and are therefore limited to few recycling cycles.6 Energy recovery, the process of burning plastic waste to produce energy in the form of heat, electricity, or fuel, has the advantage that it works effectively for any complicated mixtures of plastics, while it is at the cost of releasing greenhouse gases and/or toxic byproducts.7 Moreoever, current technologies are not able to recover incineration products, such as monomers or chemical building blocks, for reuse.
Chemical recycling offers an exciting opportunity to recover valuable products from polymeric materials, such as monomers, chemical building blocks, or new polymers from single plastic waste streams that are not compatible or cannot be recycled (any more) by the primary or secondary strategies.5,8,9 In chemical recycling, polymers can be classified into two categories: polymers with chemically cleavable units (e.g., polyester, polyamide, polycarbonate, polyurethane) and chemically inert polymers (e.g., polyolefins, PVC, polystyrene). In contrast to promising examples of industrial processes for the depolymerization of polyesters (e.g., methanolysis process from Eastman, Volcat process from IBM, or catalytic PET process from Ioniqa),10 no industrial process exists for chemically inert polymers despite representing the biggest volume of plastic waste generated yearly.1 In other words, chemical recycling is still not commonly implemented on an industrial scale as it is energy-intensive, notably due to the various chemical treatments involved throughout the recycling process (e.g., chemical reactions, extractions, and purifications).5 The challenges in implementing chemical recycling are motivating the search for novel mild processes for the catalytic conversion of polymers directly to monomers, building blocks and new polymers.
The renaissance of (organic-)electrochemistry since the 2000s is contributing to fantastic breakthroughs in the development of novel methodologies to functionalize small molecules with outstanding precision (i.e., chemo/regioselectivity) under mild conditions.11 We envision that these recent advancements will greatly inspire the emerging field of electrochemical recycling of polymeric materials and, to a greater extent, of plastics, to push the limits of traditional chemical recycling techniques (Fig. 1). For instance, electrochemistry offers direct control over the potential energy of electrons via applied voltages. This real-time tuning of catalyst activity and selectivity could be particularly advantageous for selectively recycling multilayered polymeric materials or even dealing with mixed plastic waste (Fig. 1a). Since electrochemistry uses traceless reagents (electrons) derived increasingly from renewable sources, it circumvents the use of potentially harmful and stoichiometric chemical reductants and oxidants which generates additional waste.12 Moreover, it allows for separated half-reactions, providing more opportunities than conventional thermochemical processes, for example through paired catalysis where two value-added products would be generated from a single (electro-)chemical reaction (Fig. 1d).13
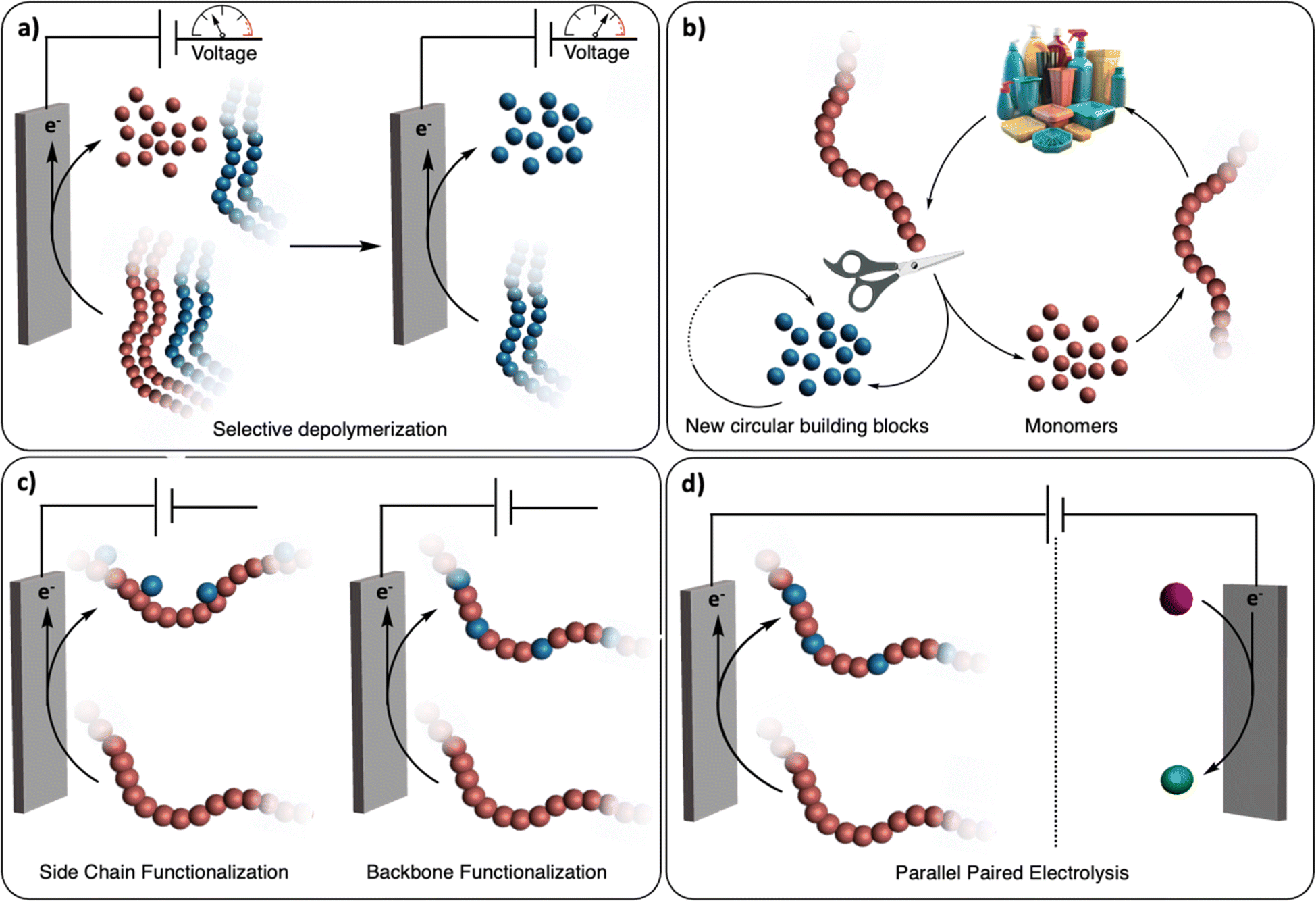 | ||
| Fig. 1 Electrochemical recycling of polymeric materials. (a) Electrochemical depolymerization of multilayered polymeric materials. Selective depolymerization of the first polymer component (red polymer) is achieved at a given electrochemical potential. The second polymer component (blue polymer) is then depolymerized at a higher potential. (b) General scheme for closed-loop polymer recycling. Polymers are either selectively depolymerized into monomers (red spheres) or other building blocks (blue spheres). In the former case, monomers can be repolymerized back to (the same) polymer and in the latter case, the building blocks can be used for other applications. (c) Post-polymerization functionalization. Functional groups are electrochemically added to the side chain or the backbone of the polymer. (d) Parallel paired electrolysis. The electrochemical oxidative functionalization of the polymer is performed simultaneously with the reduction of a small molecule (purple sphere) to another value-added product (green sphere). | ||
Several recent reviews have examined the field of electrochemical recycling of polymeric materials, notably the review by Petersen et al.14 which offers a comprehensive review, principally focusing on biopolymers, until 2021.12,15,16 Given the rapid progress in electrochemically-driven recycling of synthetic polymers in recent years, we offer perspectives on the future trajectory of this field, incorporating discussions on the latest achievements. By contextualizing other studies on thermochemical and electrochemical functionalization of small molecules as well as functionalization/depolymerization of synthetic and biopolymers, we aim to provide the community with a source of inspiration for the development of new approaches. Depolymerization into building blocks, post-polymerization functionalization, and paired electrocatalysis are discussed based on the distinct requirements for their use in polymer and plastic recycling. These approaches cater to diverse needs, including complete cleavage into segments, repurposing plastic waste, and construction of a comprehensive electrochemical device (Fig. 1). Beyond conceptualizing and classifying these ideas of electrochemical recycling of polymers, we also discuss the challenges linked with the development of such novel technologies. From there, we speculate on new research directions that could spur the advancement of (electro-)chemical recycling technologies of polymeric materials and plastics.
2 Electrochemical recovery of building blocks
Chemical recycling to building blocks (CRBB) refers to approaches in which polymers are cleaved into small molecules such as their monomers (Fig. 1b). CRBB is an appealing and promising recycling strategy as valuable chemicals can be obtained from end-of-life polymers and plastics. Moreover, if the building blocks are recycled back to monomers, the original starting materials can be recovered, purified, and subsequently repolymerized into new polymers with identical properties to their virgin counterparts. This closed-loop approach ideally creates a circular polymer economy, wherein polymeric materials are continuously recycled and reused, reducing the need for new raw materials.9 For polymer backbones containing chemically cleavable bonds (e.g., C–O bonds such as in esters and acetals), CRBB is particularly effective.Herein, we will first discuss the recent progress in the development of electrochemical methods to cleave C–O bonds in the backbone of polymers for polyethylene terephthalate (PET), polyoxymethylene and biopolymers. We will then briefly present an example of S–S bond scission before discussing the more challenging scission of C–C bonds in polyolefins. To the best of our knowledge, electrochemical CRBB methods have not been developed for breaking C–N bonds to date; we therefore provide our perspective on new potential research directions based on our previous discussion on electrochemical C–O and C–C bond cleavage.
2.1. Electrochemical C–O bond cleavage
Polyesters and polycarbonates represent a major class of polymeric materials with versatile physical properties making them ubiquitous to a large range of applications from construction materials to medical applications.17 Among the polyester family, PET is the third most produced and discarded plastic worldwide.1 It is a semi-crystalline polymer and its thermochemical depolymerization has been widely studied.18 Thermochemical CRBB approaches are based on solvolysis methods (hydrolysis, glycolysis, alcoholysis), which enable the recovery of ethylene glycol (EG) and terephthalic acid (TPA), the two monomers of PET (Fig. 2a). Solvolysis methods require high temperature/pressure as well as extreme pH conditions to allow for high conversion.18 Furthermore, they can generate a substantial amount of (inorganic) waste during the neutralization step required, for instance, if the solvolysis is performed under basic conditions. Despite these limitations, solvolysis is used at large scale by several companies (e.g., Eastman, IBM, Ioniqa, DuPont, Shell Polyester, Zimmer, and Goodyear).10,19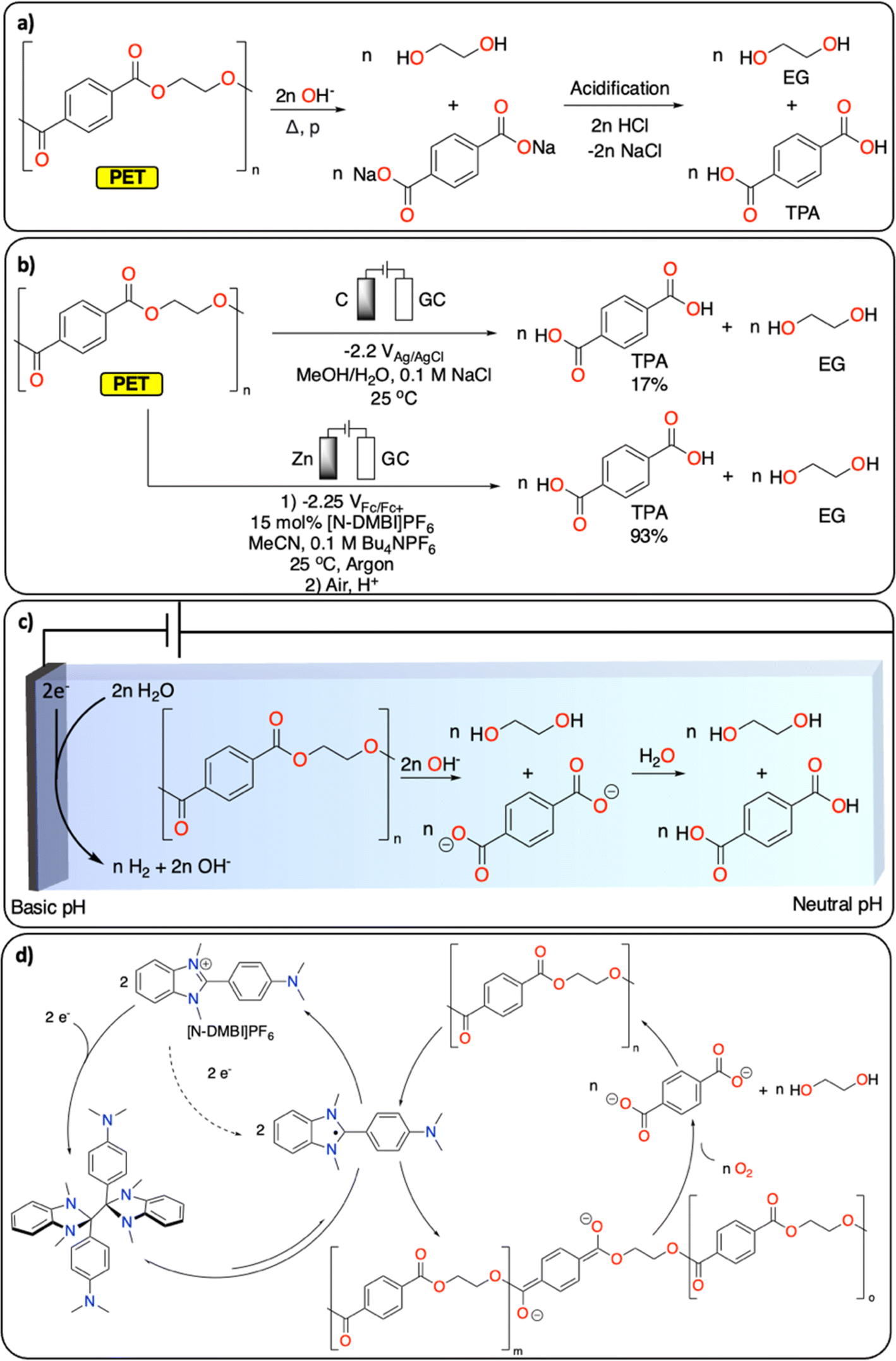 | ||
| Fig. 2 Depolymerization of PET to TPA and EG. (a) Hydrolysis under basic conditions; (b) electrochemical approaches reported;20,21 (c) electrochemically generated pH gradient;21 (d) redox-mediated reductive depolymerization.20 | ||
Electrochemically induced depolymerization is a great potential alternative that enables the recovery of EG and TPA under mild conditions (Fig. 2b). For instance, Luca et al. developed an electrochemical depolymerization strategy for PET that avoids the use of highly corrosive caustic solutions (Fig. 2c).21 By generating a local basic environment in a water/MeOH-based electrolyte, they were able to recover up to 17% of TPA within one hour at room temperature. These encouraging results motivate further development of electrochemical solvolysis methodologies. However, electrochemistry allows to go beyond simple solvolysis, notably by reductive catalytic depolymerization reactions, which are not possible using traditional thermochemical approaches. For instance, Luca et al. also demonstrated the electrochemical depolymerization of insoluble semi-crystalline PET particles using benzimidazolium cations ([N-DMBi]PF6]) as redox mediator to generate ester anion radicals, leading to almost quantitative depolymerization of PET into TPA and EG upon exposure of the reaction mixture to air after electrocatalysis (Fig. 2d).20
Once the depolymerization is completed, the recovery of TPA and EG is essential for subsequential repolymerization and chemical conversions. In contrast to the straightforward isolation of TPA (via precipitation from aqueous solution by changing the pH), the recovery of EG from aqueous solutions is more challenging. EG's high boiling point (197 °C) and excellent water solubility make the distillation energy intensive. Alternatively, electrochemical methods enable the direct transformation of EG into various value-added products. For instance, EG can selectively be oxidized to formate22 or glycolic acid (GA)23 (with the concomitant hydrogen evolution reaction), making the overall PET CRBB process more cost-effective (cf. part 3).22
More generally, polyesters and polycarbonates are characterized by a large electrochemical window making them attractive as electrolytes for battery and electrosynthesis applications, yet this same characteristic may restrict their potential for electrochemical depolymerization reactions.24 Consequently, other examples of electrochemical recycling of polyesters and polycarbonates beside PET remain scarce. Promisingly, Voloshchuk et al. demonstrated that bisphenol A can be recovered from polycarbonate via electrochemical reductive depolymerization in DMF, in the presence of Bu4NClO4, on a platinum electrode.25 Separately, Mindemark et al. reported that these polymers can decompose under high oxidative and reductive potentials on carbon and lithium electrodes, resulting in a mixture of oligomers, hydrocarbons (e.g., C3H8), CO2 and CO.26,27 We envision that further development of redox mediators and efficient electrocatalysts could broaden the scope of reductive/oxidative depolymerization. This development has a great potential to enhance control over selectivity toward monomers or other building blocks and lower their onset potentials.
Polyether represents the last class of polymers containing C–O groups. The depolymerization of polyoxymethylene (POM) is a promising example of CRBB performed on highly crystalline polymers (Fig. 3). A total yield of over 50% consisting of various building blocks (mostly formaldehyde, oxydimethanol and 1,3,5-trioxane) could be recovered at ambient temperature, after 2.5 h in an undivided cell using graphite working and counter electrodes. The authors further highlight the dual role of the solvent (hexafluoropropanol, HFIP) in breaking down some of the crystalline parts of the polymer to expose more of the acetal linkages, and in liberating protons during its anodic oxidation to trigger POM depolymerization. This work demonstrates a new chemical tool for recycling insoluble and highly crystalline polymers under mild conditions; something that is especially challenging using thermochemical approaches.28
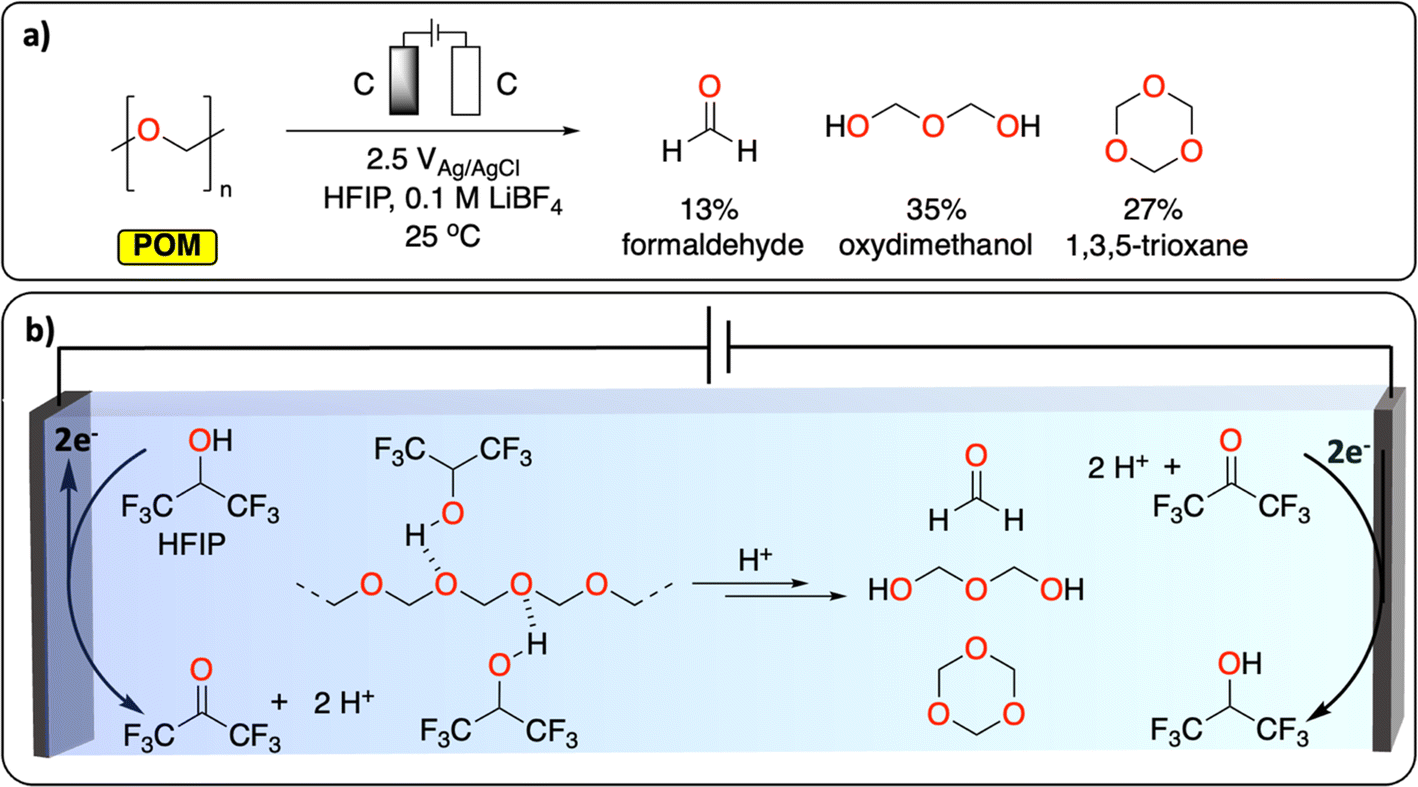 | ||
| Fig. 3 Electrochemical depolymerization of POM. (a) Reaction conditions reported to generate formaldehyde, formic acid and 1,3,5-trioxane via electrochemical depolymerization of POM. (b) The dual role of HFIP in breaking the crystallinity of POM and in generating H+ during its oxidation to trigger POM depolymerization.28 | ||
In comparison with synthetic polymers, the electrochemical depolymerization of biopolymers to recover building blocks (e.g., glucose, furfural, 5-HMF, levulinic acid, monomers) is relatively far in development, with many studies published over recent years (Fig. 4).29–31 Most electrochemical depolymerization methods essentially follow traditional thermochemical approaches, with the added benefit of low reaction temperatures and pressures.14 The most common biopolymers studied are cellulose, lignin and chitin. Cellulose and chitin are both connected via β(1 → 4) acetal linkages, but differ in the structure of the monomeric sugars. While cellulose and chitin are semi-crystalline polymers, lignin, due to the heterogeneity of its composition, is amorphous with more variation in linkages and different monomers.
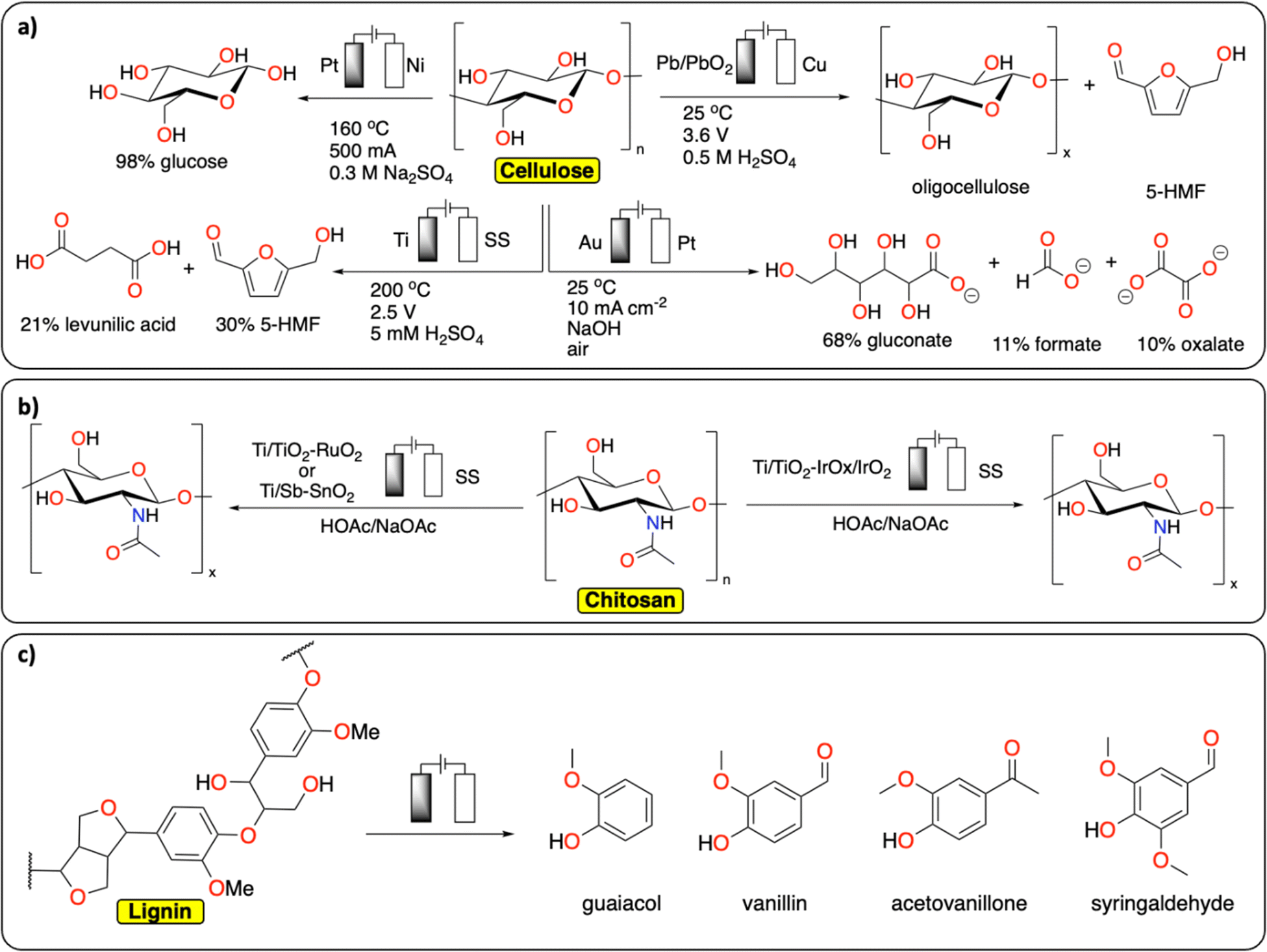 | ||
| Fig. 4 Electrochemical depolymerization of biopolymers. (a) Cellulose;32–34,36 (b) chitosan;39,40 (c) lignin and the corresponding products obtained. For lignin, only a general scheme is provided, we refer the readers to these reviews for a more detailed description of the existing methodologies.43–49 | ||
A number of electrochemical approaches have been developed to selectively cleave the β(1 → 4) linkages of cellulose to isolate glucose as a final product (Fig. 4a). One of the early reports details the electrochemical generation of an acidic solution in the anode chamber to hydrolyze cellulose at high temperatures in an electrochemical batch reactor.32 A maximum yield of 98% glucose could be obtained by applying 500 mA current on a 2.5 cm square Pt anode, at 160 °C. Besides glucose, 30% 5-HMF and 21% levulinic acid were recovered from cellulose in a different study, using a batch reactor with 5 mM H2SO4 electrolyte and a cell voltage of 2.5 V, at 200 °C.33 Several other approaches were investigated at room temperature. In these strategies, radicals formed at the anode facilitate the depolymerization reactions. Li et al. reported on a Pb/PbO2 anode that generates hydroxyl radicals in a sulfuric acid solution to depolymerize cotton into a mixture of oligomeric sugars.34 It is proposed the hydroxyl radicals formed at the anode oxidize the cellulose substrate. Similarly, Li et al. used Fenton-type chemistry to depolymerize cellulose using graphite/PTFE electrodes modified by 2-ethylanthraquinone.35 The total yield of soluble sugars and other products reached a maximum of 15.8%. In a different approach using alkaline conditions in conjunction with a gold-based electrode, Zhao et al. were able to obtain gluconate with a yield of 67.8%, together with lower yields of formate and oxalate.36 The ability to control the concentration of acidic species or radicals at the electrode surface has been further discussed in a publication by the group of Koper.37 In this study, the electrochemical hydrolysis and decomposition of cellobiose – a dimer of glucose – has been compared to the equivalent thermochemical reactions. In contrast to thermochemical methods, electrochemical conversion of cellulose benefits from milder reaction conditions and better control of radical-assisted oxidation.
Another common biopolymer is chitosan, which constitutes partially deacetylated chitin (Fig. 4b). As with cellulose, most electrochemical methods are aimed at partial depolymerization by selectively targeting the linkages between the monomeric glucosamine units. One of the first reports on chitosan depolymerization details the use of a Ti/TiO2–RuO2 anode in an aqueous acetic acid/sodium acetate solution.38 Using this method, the authors successfully decrease the degree of polymerization without notable deacetylation. In a similar study, a Ti/Sb–SnO2 electrode was used with increased performance compared to the study mentioned before.39 Other electrode materials have also been used successfully to decrease the degree of polymerization in chitosan, such as an iridium based electrode, Ti/TiO2–IrOx/IrO2, used by Hu et al.40 Another interesting finding from Zeng et al. could inspire new ideas in the field of plastic electrochemical recycling.41 They reported an approach consisting of inducing partial depolymerization of chitosan directly on solid particles. They found that by using a pulsed electric field treatment of 25 kV cm−1, the crystalline phase of the polymer was significantly damaged inducing the degradation of high molecular weight chitosan. Relating back to plastic recycling, the development of pulsed-electrochemical methods could be of high interest for breaking the crystalline phase of polymers.
Lignin constitutes a key component of lignocellulosic biomass and its valorization has gathered substantial interest over recent years.42 In extension to this, it is also one of the most studied polymers in terms of electrochemical reactivity (Fig. 4c). Due to the heterogeneity of its composition, many different products can be formed from its depolymerization and subsequent reactions. These different products consist of a wide range of low molecular weight compounds, mostly different phenolics. There are both oxidative and reductive methods known for the electrochemical conversion of lignin, which mostly follow thermochemical approaches.14 The current state of the art and limitations in electrochemical lignin conversion have been extensively reviewed in recent publications.43–49 Deng et al. notably mention four main challenges that need to be overcome before electrochemical processes for lignin conversion can become viable.44 These are unfavourable side-reactions (e.g., water splitting), low selectivity, overoxidation (e.g., decarboxylation and generation of CO2) and limited electrolyte options. Besides lignin, the electrochemical conversion of lignin model compounds has also been studied extensively in recent years.50 Because of the wide variety of products that are obtained in these reactions, the main developments in this field concern ways of steering selectivity and isolating products.
2.2. Electrochemical S–S bond cleavage
S–S bonds are principally found in vulcanized polymers in the tire industry. During chemical vulcanization, the double bonds in polyisoprene or poly(styrene–butadiene) react with sulfur to form cross-linked materials with several cyclic and mono-, di-, and polysulfide linkages.51,52 Nowadays in Europe nearly all used tires are recycled to recover energy or materials.53 However, one of the unsolved challenges in the chemical recycling of vulcanized polymers is to selectively cleave the C–S (273 kJ mol−1) and/or S–S (227 kJ mol−1) bonds over C–C bonds (347 kJ mol−1) for recovering of original polymers.52 The known methods [thermomechanical, thermochemical, physical (microwave, ultrasound) and biological] lead to partial oxidation, depolymerization or other degradation products which alter the properties of the devulcanized polymer. While electrochemical methods have not been reported yet on the devulcanization of polymer, a recent study from Daasbjerg et al. demonstrates a successful electrochemical-depolymerization of poly(dithiothreitol) derivatives via S–S bond cleavage using anthraquinone as a redox mediator (Fig. 5).54 These results could inspire further development of methodologies to selectively break C–S/S–S bonds to chemically recycle vulcanized polymers.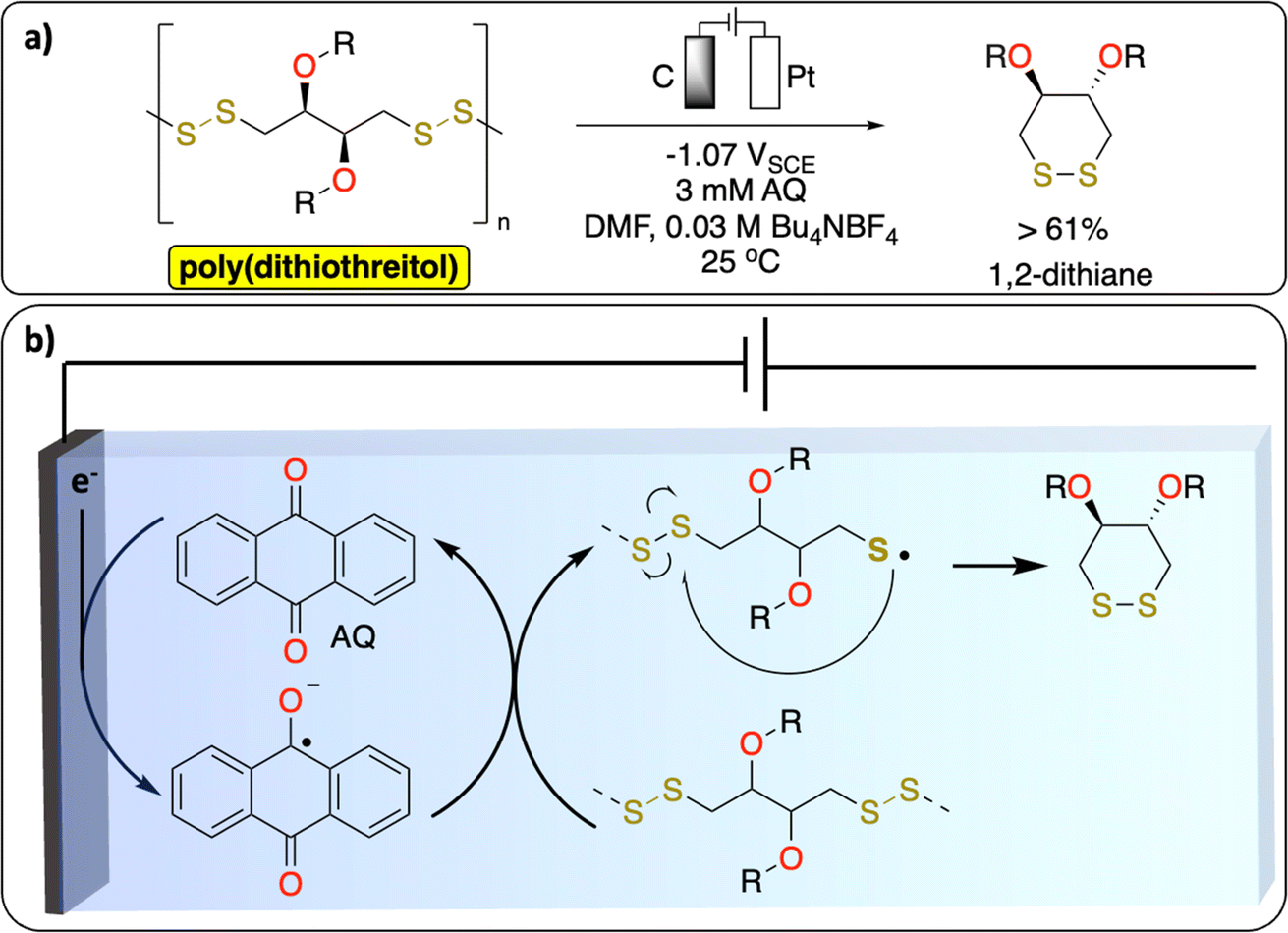 | ||
| Fig. 5 Redox-mediated electrochemical depolymerization of poly(dithiothreitol). (a) Scheme of the reaction. (b) Proposed mechanism.54 | ||
2.3. Electrochemical C–C bond cleavage
Polyolefins, including low-density polyethylene (LDPE), high-density polyethylene (HDPE), polypropylene (PP), polyvinylchloride (PVC) and polystyrene (PS), account for more than 60% of global plastic production and waste.55 They offer a wide range of properties (e.g., lightweight, tunable mechanical strength, flexibility, chemical inertness) making them uniquely versatile materials. While the production of polyolefins is expected to quadruple by 2050, global recycling remains excessively low (5% for LDPE, 10% for HDPE, and <1% for PP).5 Current chemical methods for polyolefin recycling are mainly based on thermal degradation (e.g., pyrolysis, gasification) that requires high temperatures, making the process energy-intensive.56 We refer the reader to the following reviews for a more detailed account of recent advances in the catalytic thermochemical recycling of polyolefins.57–60The electrochemical cleavage of polar C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds has been studied extensively on small molecules.61 However, there is still a large knowledge gap in approaches to breaking nonpolar C–C bonds such as those in polyolefins. Efficient electrochemical methods for the direct depolymerization of polyolefins back to monomers or building blocks have therefore not been reported to date. Nevertheless, recently Reisner et al. reported a tandem chemical-electrocatalytic process to recover a mixture of ethylene and propylene from polyethylene (Fig. 6). Both products were obtained from a Kolbe oxidation of succinic acid and glutaric acid generated from the chemical oxidation of polyethylene in 6% HNO3 at 180 °C.62 With this strategy, the authors managed to obtain a polyethylene-to-hydrocarbon (ethylene and propylene) yield of 7.6%. One of the main challenges in the further development of electrochemical oxidation methods to cleave nonpolar C–C bonds is to prevent overoxidation which ultimately leads to the decomposition of the polymer to CO2. For instance, Drogui et al. report an electrochemical method to decompose polystyrene microparticles to CO2. While this is not desirable from a recycling point of view, this approach has great potential to purify contaminated water streams containing micro and nanoparticles of plastics.63
C bonds has been studied extensively on small molecules.61 However, there is still a large knowledge gap in approaches to breaking nonpolar C–C bonds such as those in polyolefins. Efficient electrochemical methods for the direct depolymerization of polyolefins back to monomers or building blocks have therefore not been reported to date. Nevertheless, recently Reisner et al. reported a tandem chemical-electrocatalytic process to recover a mixture of ethylene and propylene from polyethylene (Fig. 6). Both products were obtained from a Kolbe oxidation of succinic acid and glutaric acid generated from the chemical oxidation of polyethylene in 6% HNO3 at 180 °C.62 With this strategy, the authors managed to obtain a polyethylene-to-hydrocarbon (ethylene and propylene) yield of 7.6%. One of the main challenges in the further development of electrochemical oxidation methods to cleave nonpolar C–C bonds is to prevent overoxidation which ultimately leads to the decomposition of the polymer to CO2. For instance, Drogui et al. report an electrochemical method to decompose polystyrene microparticles to CO2. While this is not desirable from a recycling point of view, this approach has great potential to purify contaminated water streams containing micro and nanoparticles of plastics.63
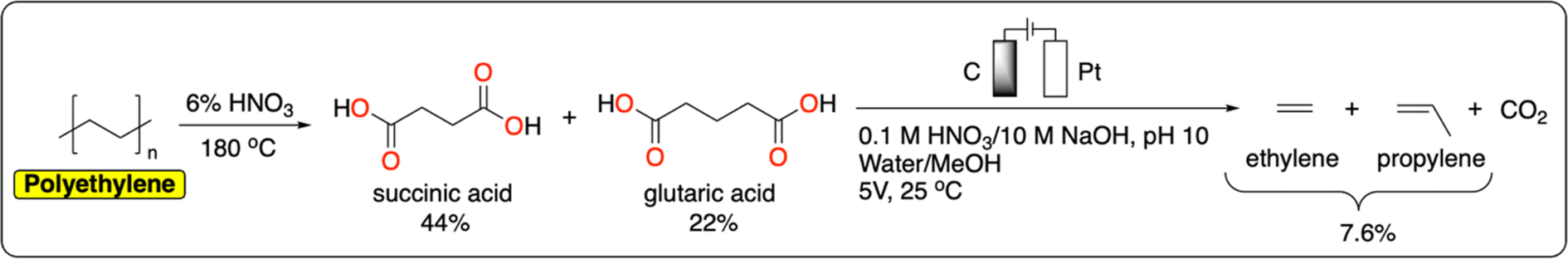 | ||
| Fig. 6 Integrated tandem chemical/electrochemical depolymerization of polyethylene to ethylene and propylene.62 | ||
2.4. Electrochemical C–N bond cleavage
Following polyolefins, polymeric materials containing C–N groups constitute the second main class of polymers.1 This category includes polyurethanes, polyamides, and polyamines (Fig. 7a), which are extensively used as thermoplastic or thermosetting materials.17 They find a wide range of applications from rigid/flexible foams to coatings, fibers, adhesives, and elastomers.64,65 In Europe in 2017, the end-of-life management of this class of polymeric materials principally consisted of landfilling (0.9 Mt per year), incinerating (0.6 Mt per year) and recycling (mechanical and chemical, 0.5 Mt per year).66 The wide range of chemical structures, molecular weights, degrees of crystallinity, crosslinking densities, and hard-to-soft segment ratios make the chemical recycling of polymeric materials containing C–N groups particularly challenging.65 Similar to PET, the principal chemical recycling technique is solvolysis to (partially) recover monomers from polyurethanes and polyamides.64,67 For example, BASF, Maincoop and Polytecna, among others, have implemented this technique at the industrial scale to recover the polyol fractions from flexible polyurethane foams.67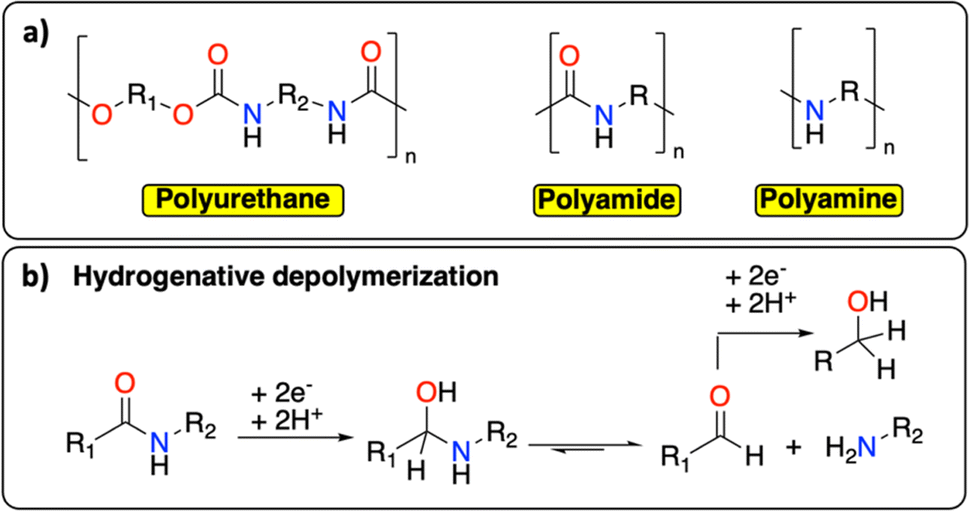 | ||
| Fig. 7 C–N containing polymers. (a) Schematic structure of polyurethane, polyamide and polyamine; (b) example of hydrogenative depolymerization of polyamide back to amine, aldehyde, or alcohol. | ||
Recently, Kumar and Milstein developed a versatile thermochemical hydrogenative depolymerization method where polyamides could partially be converted into the corresponding amino-alcohol with good conversion and selectivity.68 The proposed mechanism involves the hydrogenation of the amide bond leading to C–N bond dissociation and further hydrogenation of the formed aldehyde to the amino-alcohol (Fig. 7b). In the 1970s, Sabol et al. reported a similar reductive electrochemical approach on small aliphatic amides using an undivided electrochemical cell and two Pt electrodes.69 Through careful control of the reaction conditions, they could selectively isolate either the alcohol or the aldehyde product. We envision that a revisit of this methodology could open new doors for the (electro-)chemical recycling of C–N containing polymers.
3 Electrochemical post-polymerization functionalization
In post-polymerization functionalization (PPF), polymers undergo conversion to new polymers by editing the polymer backbone with new functional groups (Fig. 1c).60 Adjusting the degree of functionalization is a powerful means to fine-tune the thermal/mechanical/rheological properties of the resulting polymer without altering the bulk structure. These new polymers with tailored properties are especially attractive for specific/niche applications, that cannot or are difficult to be prepared through traditional “bottom-up” approaches. Alternatively, maintaining a low degree of functionalization enables the retention of the same properties as the parent polymers. Such an editing strategy is particularly appealing to introduce weak linkages into polymer backbones that are especially challenging to be chemically recycled such as polyolefins. PPF provides a way to add an end-of-life management tool into persistent polymers that have not been designed with this functionality.While the first examples of electrochemical PPF methods have only recently been reported, thermochemical PPF methodologies have been developed for decades.70 These have been applied to a wide range of polymers such as polyesters, polyurethanes, polyethers and polyenes,70–73 as well as to the more challenging polyolefins.60 Most of these methods involve a change in the oxidation state of the polymer backbone. We therefore see great potential for electrochemical approaches, which could benefit from a more effective and controllable oxidation process (see Section 5). In this section, we summarize the main advances in electrochemical approaches for PPF. Similar to the first part, the discussions are organized by the type of chemical bond that is oxidized or reduced. When reported, we also briefly discuss the changes in the thermo/mechanical properties of the resulting polymer.
3.1. Electrochemical-oxidation of C–OH bonds
TEMPO (2,2,6,6-tetramethyl-1-piperidinyl-oxy) mediated oxidation of alcohol functionalities is the most reported strategy for the functionalization of cellulose (Fig. 8a).74 Isogai et al. reported on the TEMPO-mediated electrochemical oxidation of polysaccharides, including cellulose fiber. Interestingly, using this method the authors show that the cellulose fiber underwent little depolymerization – maintaining the original surface structure – while increasing aldehyde and carboxylic acid contents (Fig. 8b).75 A similar TEMPO-mediated oxidation of carbohydrates was reported to selectively oxidize the primary alcohols to carboxylic acids.76 More recently, Ivaska et al. investigated the mechanism of electrocatalytic oxidation of cellulose on gold electrodes.77 The main products were oxidized cellulose derivatives with increased carboxylic acid content and hybrid materials containing cellulose and gold nanoparticles. The group of Sugano further investigated the gold-catalyzed oxidation of hemicelluloses with similar results.78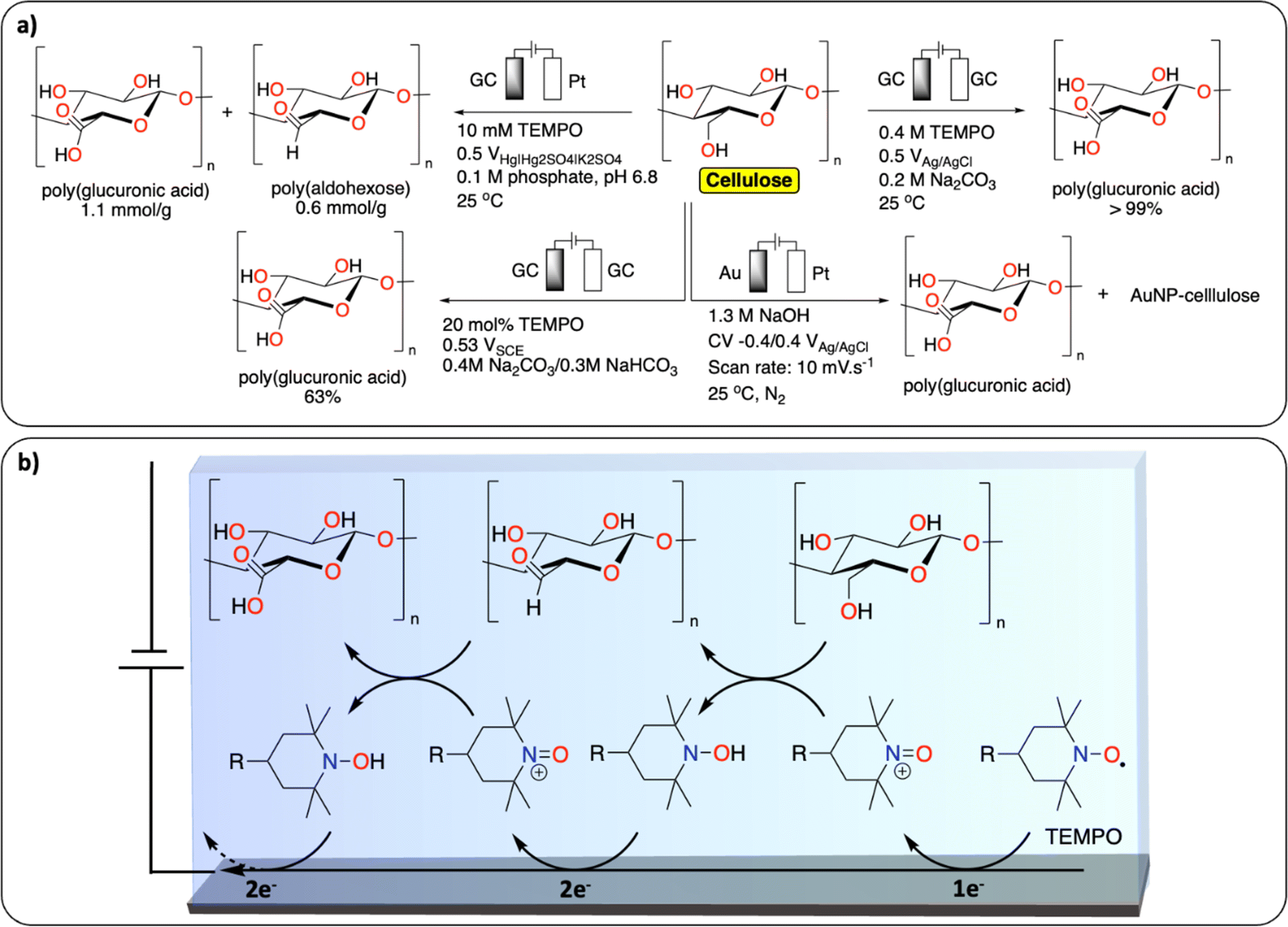 | ||
| Fig. 8 Electrochemical PPF of cellulose. (a) Scheme of reaction conditions reported for the electrochemical PPF of cellulose;74–76,78 (b) example of proposed mechanism of TEMPO mediated electrochemical oxidation of cellulose. | ||
3.2. Electrochemical oxidation of C–X bonds (X = Cl, F)
In Europe, approximately 27% (0.8 Mt) of waste PVC has been recycled and introduced back onto the market in 2022.79 Most of it has been mechanically recycled to generate materials with similar properties to pristine PVC. The challenge resides in properly sorting PVC waste to avoid cross-contamination of recycled PVC with the various toxic stabilizers (e.g., cadmium, lead) and plasticizers (e.g., phthalates) used to enhance the thermal stability or tune the properties of PVC for specific applications.80,81 Alternative recycling methods include the thermal and oxidative dechlorination of PVC to recover HCl, Cl2 and hydrocarbons.81Various electrochemical approaches have been developed for the reductive dehalogenation and oxidative halogenation of small molecules.11,82,83 On polymers, Batsalva et al. reported the reductive partial dehydrochlorination of PVC in DMF, on Pt electrode (Fig. 9a). They reported the formation of a cross-linked materials containing unsaturated groups that deposited on the cathode upon bulk electrolysis at −2 V vs. SCE.84 More recently, McNeil et al. reported the reductive partial dechlorination of PVC with concomitant oxidative chlorination of electron-rich arenes on graphite electrodes at room temperature in DMF electrolyte (Fig. 9b).85 Interestingly, di(2-ethylhexyl)phthalate, a common plasticizer additive used in PVC, served as a redox mediator to facilitate the reductive dechlorination reaction (Fig. 9d). While limited C–C bond scission was observed, the characterization of the resulting polymer reveals the presence of a complex mixture of functional groups including alkenyl, aromatic, ester/ether, alkyl and unreacted chloride groups. Consequently, the authors observed a substantial decrease in the glass transition temperature (Tg, from 83 °C to 59 °C) and a higher degree of crystallinity in the resulting polymer. This proof of concept not only paves the way for paired-catalysis plastic recycling (the concept of paired-electrolysis is developed in Section 4) but also demonstrates that additives contained in plastics can even be beneficial for plastic electrochemical recycling. Besides reductive approaches, the oxidative dechlorination of PVC was initially studied by Ushida et al., on Pt electrodes by electrogenerated superoxide anions, in THF/DMF mixed solvent (Fig. 9a).86 While dechlorination was achieved, substantial C–C bond cleavage was also observed together with the formation of hydroxy groups, conjugated carbonyl groups and polyene structures. In another study, the partial electrochemical degradation of PVC particles was investigated using hydrogen peroxide, which was generated from the oxygen reduction reaction on a TiO2/graphite cathode in an acidic aqueous solution at 100 °C. This method ultimately led to C–C bond cleavage, generating a mixture of dehalogenated polymers containing ketones and alcohol groups, oligomers and small molecules (formic acid, acetic acid and propionic acid).87
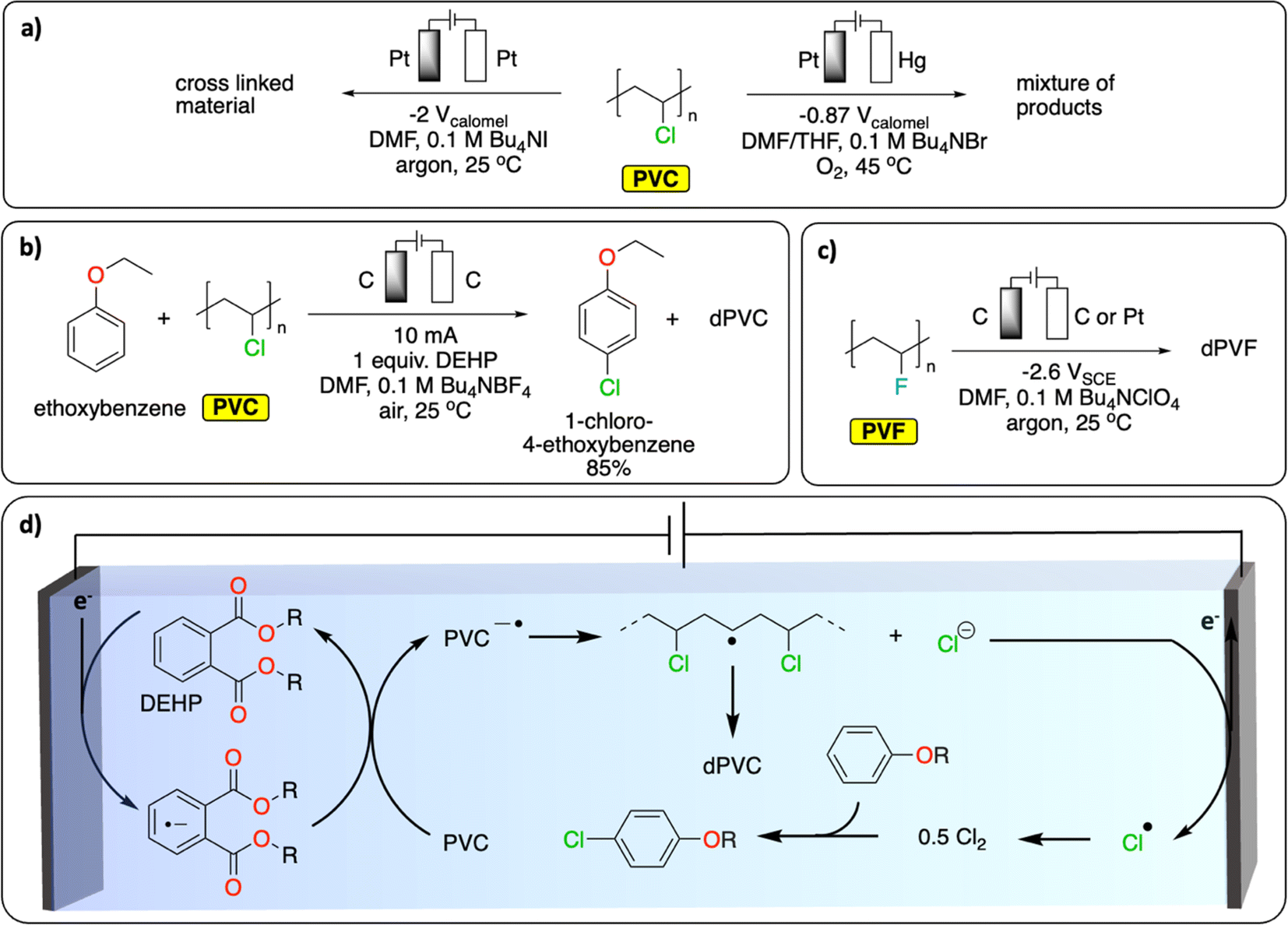 | ||
| Fig. 9 Electrochemical dehalogenation. (a) Electrochemical degradation of PVC; (b) tandem electrochemical dechlorination of PVC with concomitant chlorination of an electron-rich arene;85 (c) electrochemical defluorination of PVF;89 (d) schematic representation of the DEHP-mediated reductive dichlorination of PVC coupled with chlorination of an electron-rich arene. The abbreviations dPVC and dPVF refer to (partial) dechlorinated PVC and (partial) defluorinated PVF, respectively.85 | ||
Similar to PVC, fluorinated polymers are mostly recycled mechanically.88 Chemical recycling principally concerns pyrolytic depolymerization to recover a mixture of fluorinated small molecules including tetrafluoroethylene. Apart from the issue of low selectivity, the process also requires high temperatures (>650 °C) and therefore a high energy input. Fluorinated polymers are well-known for their inertness to (electro-)chemical treatment. However, one report on polyvinyl fluoride (PVF) and polyvinylidene fluoride (PVDF) suggests that reductive defluorination is possible in DMF electrolyte on a glassy carbon electrode to form polyene-type materials (Fig. 9c).89
3.3. Electrochemical oxidation of C–H bonds
PPF of polyolefins provides an elegant alternative way of generating functionalized polyolefins, traditionally produced from the challenging olefin/polar monomer (e.g., acrylates, amine-containing functional groups) copolymerization reaction.90–92 Over the years, various thermochemical approaches have been reported for Csp3–H functionalization of polyolefins (especially PE but also polyisobutylene, PP and PS), through oxidation, radical-based, carbene/nitrene, transition-metal catalyzed and dehydrogenation approaches.60,73,93Despite precedent on small molecules, reports on electrochemical methods for Csp3–H functionalization of polyolefins remain scarce.94,95 A recent study from the group of Botte demonstrates an electrochemical method to introduce ketones, alcohol functionalities and unsaturation in LDPE (Fig. 10a).96 By applying a low, oscillating cell potential of ±1 V vs. SHE on a Cu electrode, suspended LDPE in an acidic electrolyte containing CuSO4 was partially electrochemically oxidized, as observed by a significant increase of CO, C–O and CC signals in the post-catalysis IR spectrum of the resulting polymer.97 However, the approach also led to Csp3–Csp3 bond scission and the formation of smaller molecules such as dodecanoic acid.96 In another study, Ackermann et al. reported the electrochemical azidation of Csp3–H bonds in polystyrene (Fig. 10b).97 The Csp3–H azidation using a salen-type manganese complex afforded modified polystyrene materials with minimum C–C chain scission (up to 77% mass retention) and 1.6 mol% azidation using a graphite electrode. The high regioselectivity for the benzylic Csp3–H was achieved by careful design of the ligand motif to prevent side-reactions, including C–C bond breaking, Fig. 10d. The thermo/mechanical properties of the resulting azidated polymer show negligible changes compared to pristine PS. Electrochemical azidation reactions were also successfully performed on polynorbornene in an undivided cell (graphite anode/Zn cathode), in the presence of MnBr2 and trimethylsilyl azide in DCM (Fig. 10c). Interestingly, in a mixture of polynorbornene and PS, this method selectively cleaved Csp2–Csp2 bond of polynorbornene over PS.98
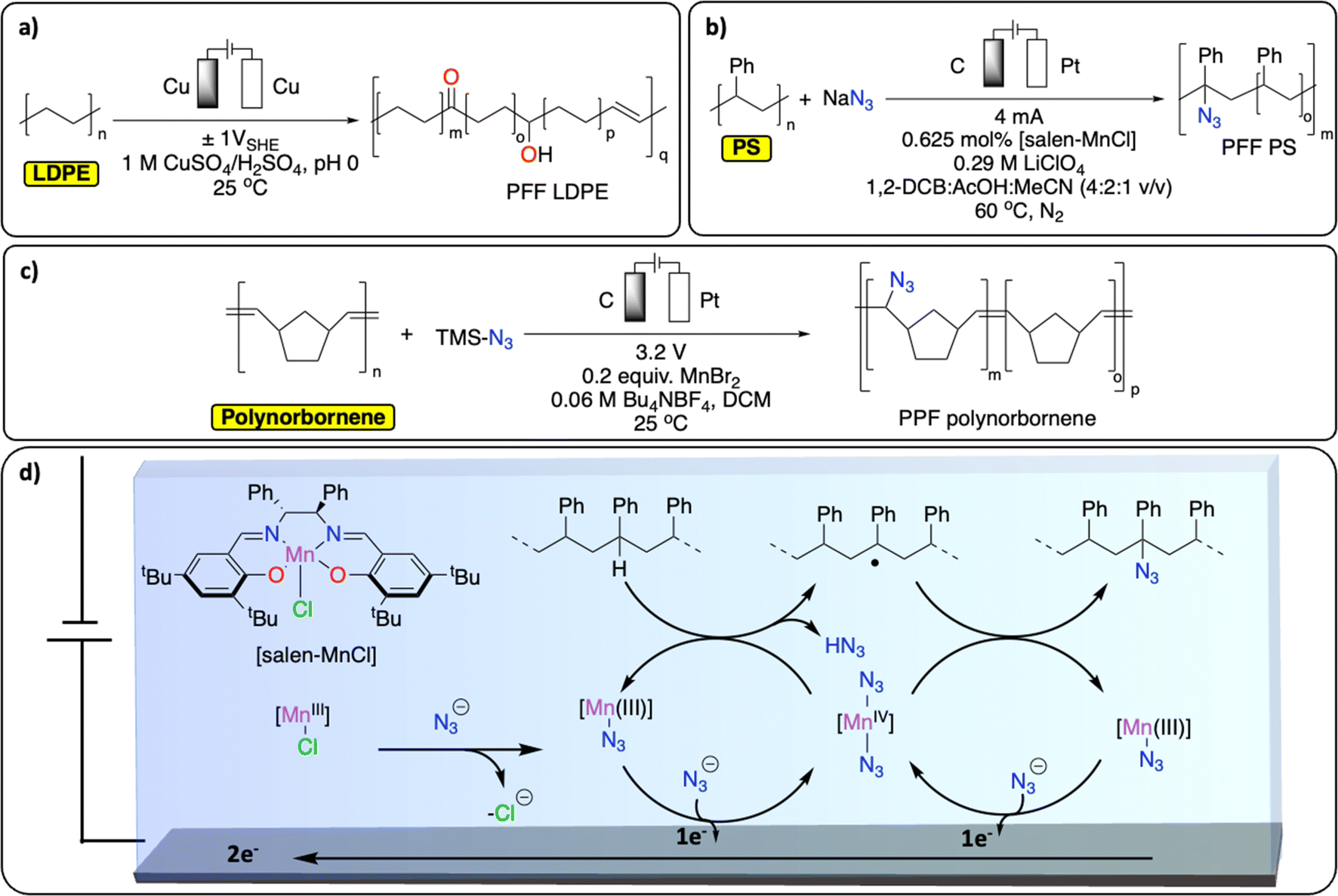 | ||
| Fig. 10 Electrochemical PPF strategies for C–H bond functionalization. (a) Electrochemical oxidation of LDPE;96 (b) electrochemical azidation of PS;97 (c) electrochemical azidation of polynorbornene;98 (d) schematic representation of the Mn-mediated oxidative azidation of PS. | ||
3.4. Electrochemical functionalization of aromatic groups
Polymers containing an aromatic ring in each repeating unit offer an additional handle for PPF. In particular, the introduction of functional groups onto PS, polysulfones, polyesters or polycarbonates is targeted to transform the polymers' properties for applications as compatibilizers, ion-exchange resins, or catalysts.99 In thermochemistry, these functionalization reactions are mostly performed via traditional electrophilic aromatic substitution (e.g., Friedel–Crafts) although more exotic approaches have also been studied.99A recent study from Sarlah et al. demonstrates the electrochemical reductive dearomatization of polystyrene (Fig. 11).100 While the reaction was not possible using traditional Birch or Benkeser conditions, the authors adapted a previously reported method from Baran et al. on PS.101 A graphite cathode and a sacrificial Mg anode were used in a THF-based electrolyte to completely dearomatize commercial PS. Dimethylurea and hexamethylphosphoramide were used as additives to prevent the passivation of the cathode by lithium plating. Analysis of the resulting polymer showed no chain scission, with minor changes to the bulk thermoproperties of the material (Tg = 95–103 °C). Finally, it was demonstrated that the reduced polymer could serve as a platform for further functionalization by epoxidation of the formed cyclohexene rings (Fig. 11).100 This not only provides new functionalities for PS, it also gives a handle to tailor its Tg by controlling the degree of epoxidation (Tg = 128–162 °C). Next to this pioneering example, the functionalization of polymers containing aromatic groups has great potential for the electrochemical recycling of plastics. As previously discussed, electrochemical electrophilic aromatic substitution has been widely studied on small molecules. Once applied to aromatic polymers, this new strategy could be applied to prepare a wide variety of new materials.
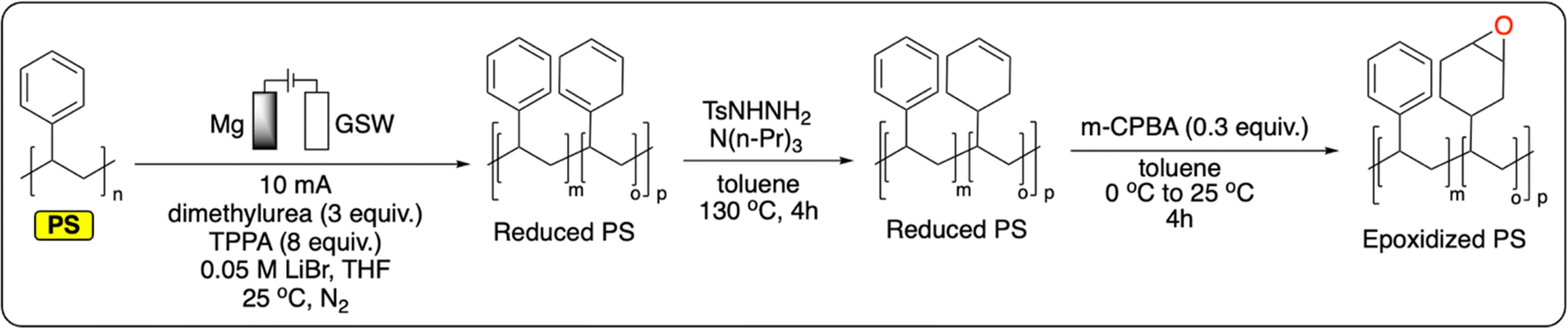 | ||
| Fig. 11 Electrochemical reduction of PS followed by chemical reduction and epoxidation.100 | ||
4 Paired electrolysis
Paired electrolysis allows both half-reactions to generate valuable products in a complete electrochemical system, as opposed to the common focus on one of the half-reactions, where the other half-reaction only plays an auxiliary role. In an ideal process, all the electrons from the oxidized starting material at the anode (used as fuel) would be transferred to the reduced starting material at the cathode to achieve a combined electrochemical yield. The faradaic efficiency (FE) is then calculated by:where ncathode and nanode are the moles of the desired products formed both at the cathode and the anode, respectively, Q is the charge passed, Zcathode and Zanode are the numbers of electron transferred to generate the product from starting materials at the cathode and anode, respectively, and F is the Faraday constant (96
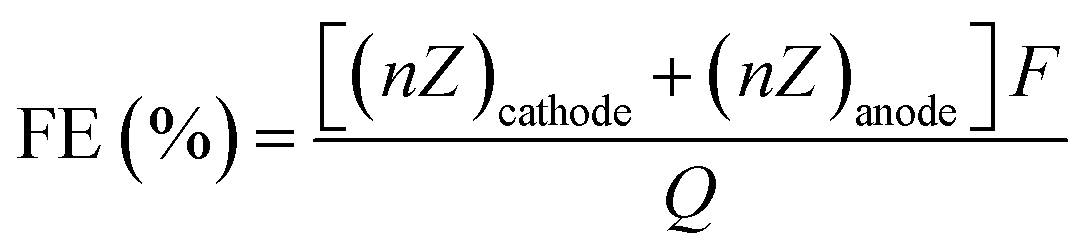
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1). In paired electrolysis, the FE can reach 200% in the case where all the electrons harvested at the anode are delivered at the cathode to selectively form the two desired products. BASF has developed the first commercial paired electrocatalysis process for the electrochemical oxidation of 4-tert-butylbenzene to 4-tert-butylbenzaldehyde with concomitant electrochemical reduction of phthalic acid dimethylester to phthalide with a FE as high as 180%.102
485 C mol−1). In paired electrolysis, the FE can reach 200% in the case where all the electrons harvested at the anode are delivered at the cathode to selectively form the two desired products. BASF has developed the first commercial paired electrocatalysis process for the electrochemical oxidation of 4-tert-butylbenzene to 4-tert-butylbenzaldehyde with concomitant electrochemical reduction of phthalic acid dimethylester to phthalide with a FE as high as 180%.102
Paired electrolysis can be categorized into four different approaches based on the interplay between two half-reactions: parallel, convergent, divergent, and linear (Fig. 12).102 In parallel paired electrolysis, distinct cathodic and anodic reactions occur simultaneously. In convergent paired electrolysis, the final products are formed through the reaction of intermediates generated from both the anode and cathode. In divergent paired electrolysis, the same substrate is converted to different products at the anode and cathode. Finally, in linear paired electrolysis, the substrate is converted to an intermediate at one electrode and converted to the final product at the other electrode. The vast possibilities of paired electrocatalysis open up new possibilities for the electrochemical recycling of plastics. Herein, we summarize the first examples of parallel and convergent paired electrolysis applied to plastic recycling. To the best of our knowledge, no plastic recycling strategies have been reported using the divergent and linear paired electrolysis approaches to date, leaving space for further development of novel dual catalysis methodologies.
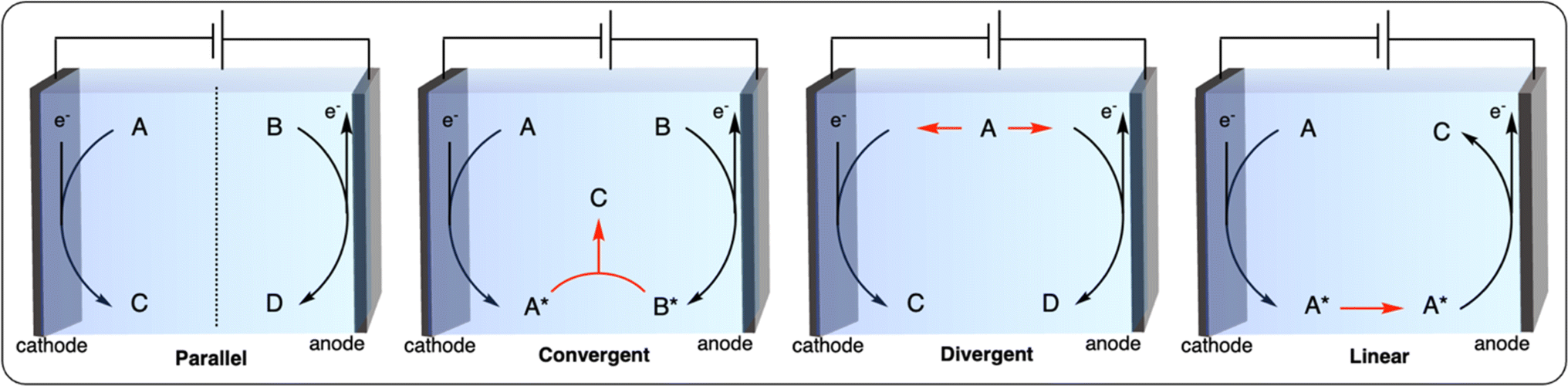 | ||
| Fig. 12 Schematic representation of the most common paired-electrolysis reactions. | ||
By carefully choosing the anodic and cathodic reactions, parallel paired electrolysis systems allow for the reduction of energy input of the electrochemical cell, in addition to the generation of two value-added products. In this context, the reduction of small molecules (e.g., H2O, CO2) has been investigated in parallel paired electrolysis with oxidation of organic molecules derived from biomass (e.g., HMF), as well as biopolymers.103–105 Chu et al. coupled the oxidation of a mixture of sugars derived from cellulose with CO2 reduction in a sunlight-driven electrochemical cell (Fig. 13a), producing formate at both electrodes in excellent faradaic efficiencies (>85%).106 By replacing conventional water oxidation at the anode with biomass conversion, the cell voltage was lowered by 32%. In a similar fashion, Li et al. reported on chitin oxidation to acetate with over 90% yield combined with hydrogen generation at the cathode, lowering the overall energy consumption with 15% compared to a system using conventional water oxidation (Fig. 13b).107
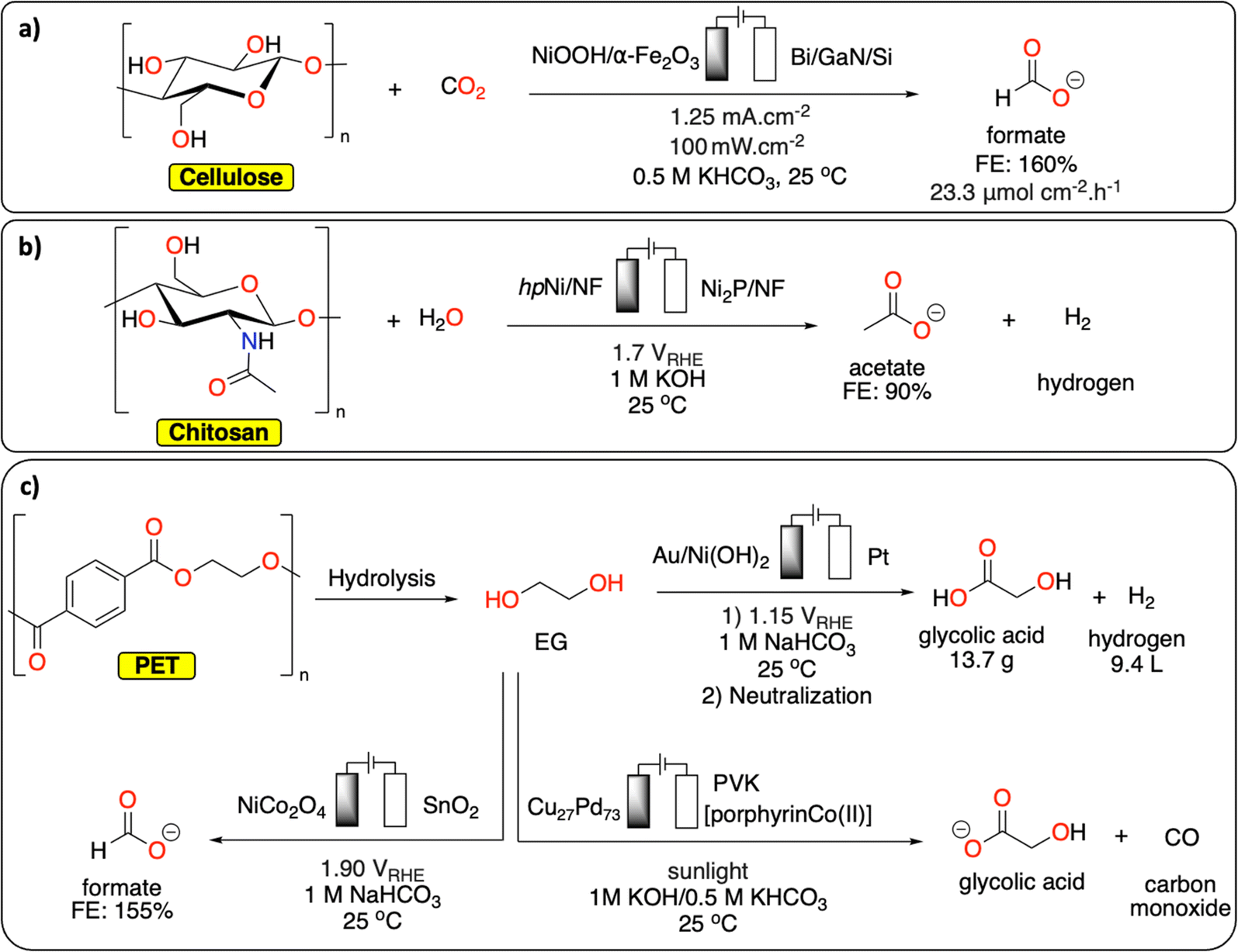 | ||
| Fig. 13 Parallel paired electrolysis polymer recycling methodologies. (a) Electrochemical conversion of cellulose/CO2 to formate;106 (b) electrochemical chitosan to acetate conversion with concomitant hydrogen production;107 (c) electrochemical conversion of EG/CO2 to various products.16,108–110 | ||
Although reports on direct polymer conversion with paired electrolysis are relatively scarce compared to other electrochemical topics, there are several promising examples using building blocks derived from polymer waste. For example, parallel paired electrocatalysis has successfully been used for EG recycling from PET waste. In a recent study from Ma et al., EG produced from the alkaline depolymerization of PET was electrochemically oxidized to formic acid on a NiCo2O4 anode (Fig. 13c). The reaction was combined with the CO2 electrochemical reduction reaction on Sn electrode to also generate formic acid. Under optimized conditions, the parallel paired electrolysis generated formic acid with faradaic efficiency up to 155% at a cell voltage of 1.90 V.108 In another example from Duan et al., EG from PET was electrochemically converted to glycolic acid on Au/Ni(OH)2 electrode with high selectivity (91%) and current density (326.2 mA cm−2) at 1.15 V vs. RHE while hydrogen was generated at the cathode (Fig. 13c).16 As a proof-of-concept, it was further demonstrated that by using a membrane-free flow electrolyzer, 13.7 g of glycolic acid and 9.4 L of H2 could be generated revealing the high potential of co-production of valuable chemicals and fuel from waste. Reisner et al. reported a photoelectrochemical approach to transform CO2 (from various feedstocks) into CO, syngas and formate while oxidizing EG from PET into glycolate using a perovskite-based photocathode containing an immobilized molecular Co-phthalocyanine catalyst and a Cu/Pd alloy anode (Fig. 13c).109,110 This study paves the way toward the development of integrated carbon capture and sunlight-driven utilization system for both CO2 and polymeric/plastic waste.
In convergent paired electrocatalysis, two different substrates undergo oxidation or reduction to afford intermediates/products that react among themselves to generate the final product. We previously described the work of McNeil et al. on the PPF of PVC which was paired with the chlorination of electron-rich arenes (Fig. 9d). This strategy could be seen as a convergent paired electrolysis where the HCl produced from the reduction of PVC reacts with the one-electron oxidized arene to form the final chlorinated arene. While this report constitutes the only example of convergent paired electrocatalysis applied to plastic recycling, it paves the way for future development (further discussed in Section 5) of using waste polymeric materials or plastics as reagents to form novel (polymeric) products.
5 Future directions and strategies
Polymer electrochemistry is an emerging field of electrochemistry providing complementary opportunities to traditional thermochemical approaches to notably tackle challenges in the recycling of polymeric materials. While considerable progress has been made in the electrochemical recycling of certain classes of polymers such as PET and biopolymers, a large gap in knowledge remains when it comes to the broader range of commodity and engineering polymeric materials and plastics. We therefore envision that the field will significantly grow in the coming years. Herein, we provide an outlook on future research directions in the field and highlight some of the most important challenges that need to be overcome.5.1. Electrochemical activation of terminated polymer chains
A polymerization reaction consists of three essential steps: initiation, propagation, and termination. Most exergonic polymerization reactions have a critical temperature (Tc) at which the propagation stops. Above Tc, depolymerization becomes thermodynamically favored and monomers can eventually be recovered from polymer chains that are not terminated. In plastic waste, polymer chains are terminated. One of the challenges is to find efficient methodologies to reactivate these dormant polymer chains, so that depolymerization may be possible at conditions above Tc. In thermochemistry, this approach has been successfully developed for the depolymerization of commodity polymers synthesized via controlled radical (ATRP, RAFT, ITP), ionic, metathesis and ring opening-type polymerization.111,112 For instance, the group of Gramlich reported the radical depolymerization of Reversible Addition-Fragmentation Chain-Transfer RAFT-synthesized polymethylmethacrylate to recover methacrylate.113 Electrochemistry could provide a novel way to activate dormant polymer chains to induce depropagation and recover monomers with no concomitant waste formation. For instance, electrochemically mediated atom transfer radical polymerization has been studied extensively.114 It would be interesting to use similar strategies at conditions above Tc to recover monomers (Fig. 14a). It could also open new ways to recover monomers from polyolefins. A dual catalysis approach could be envisioned where electrochemically generated active polyolefin chains would be depolymerized via β-alkyl elimination using a thermocatalyst.115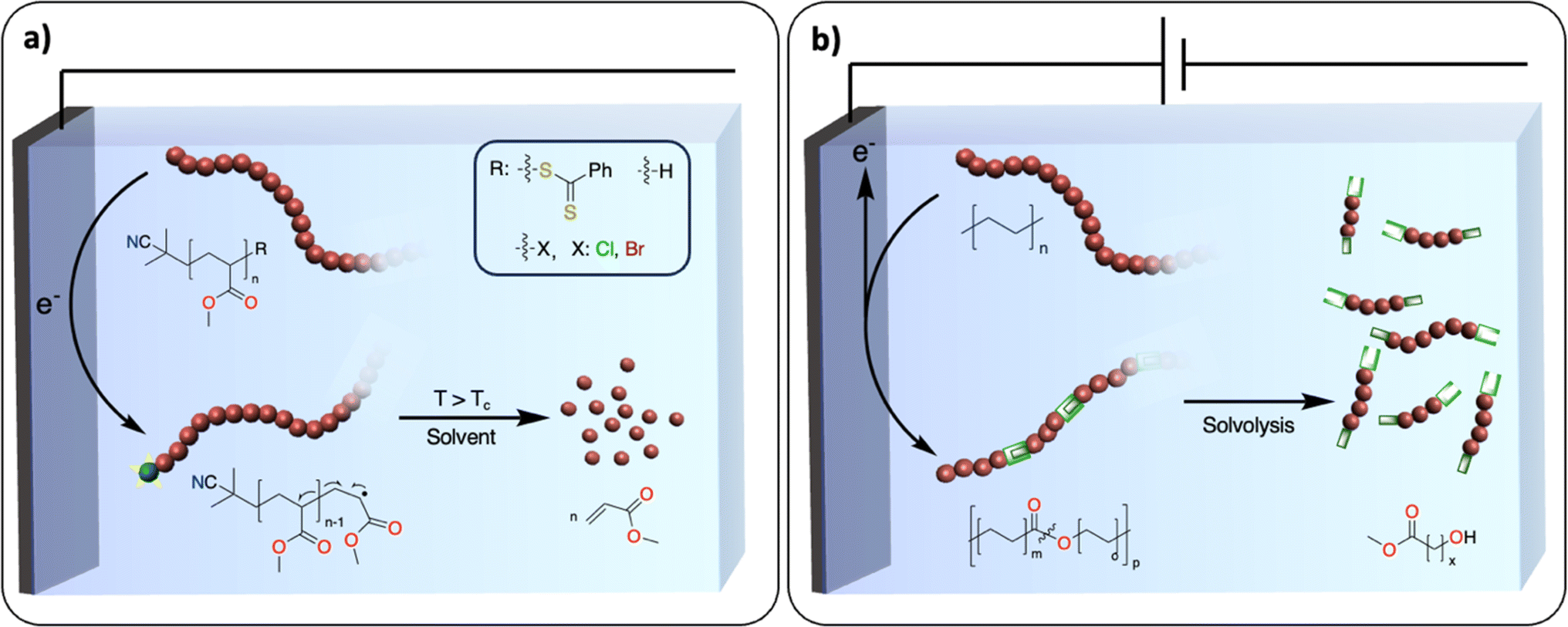 | ||
| Fig. 14 Two examples of future challenges for electrochemical recycling of polymeric materials. (a) Electrochemical activation of terminated polymer chains. The blue ball with the yellow star represents the activated chain-end of the polymer; (b) introducing weak linkages within the polymer backbone. The double green boxes represent the cleavable unit. | ||
5.2. Introducing weak linkages within the polymer backbone
With the inspiration from recent works on the redesign of polymers for circularity, the introduction of weak linkages within the polymer backbone represents an appealing strategy for closing the loop of the plastic economy.116,117 Taking the example of polyolefins, the polymer backbones solely consist of carbon–carbon repeating units, presenting no easily accessible handles for degradation/recycling. Recent work, pioneered by the group of Mecking, has demonstrated that synthesizing novel polymers consisting of isolated ester or ketone linkages embedded within an aliphatic C–C backbone affords recyclable/biodegradable polyolefin-like polymers.117–119 The insertion of weak linkages into backbones could also be used to convert polyolefin waste into recyclable counterparts while keeping the original properties. For instance, thermochemical oxidation of the polyolefin backbone enables the introduction of various functional groups such as alcohols, ketones, esters and oximes,56,57,60,120 which could serve as chemical handles for depolymerization reactions.121The electrochemical oxidation of aliphatic C–H bonds on small molecules – notably on cyclohexane – to alcohols or ketones has been thoroughly investigated.122 There is also an example of successful electrochemically driven Baeyer–Villiger rearrangement to generate esters from ketones on cyclohexane-based substrates.123 In comparison with thermochemical approaches, electrocatalytic transformations favor reactions involving fewer proton and electron transfers, which may prevent the formation of overoxidized products, C–C bond scission and therefore CO2 release.122 Moreover, electrochemical oxidation generates active oxidants controllably and locally, which prevents the excess use of chemical oxidants needed in thermochemical reactions. Despite these promising studies, a selective method for the oxidation of polyolefins to polyesters (Fig. 14b) without substantial C–C bond breaking is still lacking.
5.3. Polymer-to-polymer electrochemical conversion
The polymer-to-polymer (electrochemical) conversion enables the synthesis of polymers with new properties from post-consumer plastics to target different types of applications. Using such a repurposing recycling strategy, plastic waste can be valorized by creating materials with higher economic value. For instance, waste polyethylene was successfully transformed into water-soluble antifungal polymers by thermochemical aerobic oxidation.124 In Section 3, we discussed the few reported examples of polymer-to-polymer electrochemical conversion demonstrating the potential of electrochemistry in performing such reactions. Herein, we want to go further into the possibilities that electrochemistry offers. Starting from PVC, the formation of a C-centered radical upon electrochemical dechlorination could be used as a platform to generate new polymers by reaction with various reagents (e.g., CO2, Michael acceptors, alkynes, arenes), also with the possibility to use different paired electrolysis strategies. Similarly, the electrochemical functionalization of aromatic groups such as PS, PET, polycarbonate and polyurethane opens up new perspectives for the synthesis of novel polymers with totally different properties. However, it is important to mention that transforming a polymer into a new polymer implies that a novel strategy for its end-of-life management must be developed in parallel, otherwise, the approach merely delays the problem of waste plastic accumulation.5.4. Challenges
In this section, we highlight some of the biggest challenges in the electrochemical recycling of polymeric materials and plastics to provide potential directions for future research. Although the discussion on these challenges may not be comprehensive, we hope to stimulate the community to develop new strategies and solutions to tackle these issues.Firstly, conventional NMR spectroscopy techniques employed for solution state material characterization often fail to accurately map and depict the degree of post-polymerization functionalization, particularly in case of a low degree of functionalization. Isotopic labelling such as 13C, 18O, 15N, and/or fluor/phosphor probes offers additional avenues for characterization but may pose challenges to introduce within the polymer backbone or require the use of costly chemicals.129,130 Less conventional NMR spectroscopy techniques such as solid state and Dynamic Nuclear Polymerization (DNP) NMR spectroscopy which are increasingly recognized as powerful and versatile tools for the characterization of polymeric materials, are still underutilized in the field but could provide valuable complementary insights to traditional solution state NMR spectroscopy techniques.138,139
Secondly, optical microscopy can offer essential information about the degree of functionalization of modified polymers, although this method requires additional fluorescent decoration.131 For instance, the groups of Chen and Coates have reported a powerful tool to monitor the precise monomer sequencing at a single polymer chain level on various synthetic polymers using a coupled reaction approach toward super-resolution imaging.140 Developing a reminiscent versatile method to characterize PPF polymers would be of high interest to this field of research.
Thirdly, the characterization of polymers on GPC/SEC with multiple detectors is a routine technique in polymer chemistry to extract a plethora information on polymers (e.g., Mn, Mw, PDI, viscosity, branching, cross-linking). Specifically, GPC/SEC-IR is a powerful tool to analyze the degree of functionalization as a function of elution time. However, the development of novel detectors for a quantitative measure when the degree of functionalization is below 5%, range typically achieved in most PPF on synthetic polymers, is needed.
Finally, atomic force microscope coupled with IR (AFM-IR) provides a means to characterize both physical properties (e.g., surface stiffness, morphology) and chemical compositions of polymers, offering useful information on the degree and spatial distribution of functionalization on 2D polymeric films.132
Author contributions
All authors wrote, revised, and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
AT thanks the department of Chemistry of Utrecht University and the Dutch Research Council (NWO) for funding via Veni.212.039 and OCENW.XS21.4.035.References
- R. Geyer, J. R. Jambeck and K. Lavender Law, Sci. Adv., 2017, 3, 958 CrossRef.
- Plastic waste and recycling in the EU, https://www.europarl.europa.eu/news/en/headlines/society/20181212STO21610/plastic-waste-and-recycling-in-the-eu-facts-and-figures, accessed September 26, 2023.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS.
- N. Singh, D. Hui, R. Singh, I. P. S. Ahuja, L. Feo and F. Fraternali, Composites, Part B, 2017, 115, 409–422 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem, 2017, 1, 1–11 CrossRef.
- A. Jansson, K. Möller and T. Gevert, Polym. Degrad. Stab., 2003, 82, 37–46 CrossRef CAS.
- R. Verma, K. S. Vinoda, M. Papireddy and A. N. S. Gowda, Procedia Environ. Sci., 2016, 35, 701–708 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- T. W. Walker, N. Frelka, Z. Shen, A. K. Chew, J. Banick, S. Grey, M. S. Kim, J. A. Dumesic, V. Lehn and G. W. Huber, Sci. Adv., 2020, 6, eaba7599 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- C. Karuppalah, N. Vasiljevic and Z. Chen, Electrochem. Soc. Interface, 2021, 30, 55 CrossRef.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- H. A. Petersen, T. H. T. Myren, S. J. O'Sullivan and O. R. Luca, Mater. Adv., 2021, 2, 1113–1138 RSC.
- M. Jiang, X. Wang, W. Xi, H. Zhou, P. Yang, J. Yao, X. Jiang and D. Wu, Chem. Eng. J., 2023, 461, 141962 CrossRef CAS.
- Y. Y. Haohong Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef PubMed.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124(5), 2617–2650 CrossRef CAS PubMed.
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- A. Barredo, A. Asueta, I. Amundarain, J. Leivar, R. Miguel-Fernández, S. Arnaiz, E. Epelde, R. López-Fonseca and J. I. Gutiérrez-Ortiz, J. Environ. Chem. Eng., 2023, 11, 109823 CrossRef CAS.
- P. H. Pham, S. Barlow, S. R. Marder and O. R. Luca, Chem Catal., 2023, 3, 100675 CrossRef CAS.
- T. H. T. Myren, T. A. Stinson, Z. J. Mast, C. G. Huntzinger and O. R. Luca, Molecules, 2020, 25, 2742 CrossRef CAS PubMed.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. C. M. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300094 CrossRef CAS PubMed.
- J. Zhang, J. Yang, T. Dong, M. Zhang, J. Chai, S. Dong, T. Wu, X. Zhou and G. Cui, Small, 2018, 14, 1800821 CrossRef PubMed.
- A. A. Pud, S. P. Rogal’skii, G. S. Shapoval and V. G. Voloshchuk, Theor. Exp. Chem., 1998, 34, 292–296 CrossRef CAS.
- C. Sångeland, B. Sun, D. Brandell, E. J. Berg and J. Mindemark, Batteries Supercaps, 2021, 4, 785–790 CrossRef.
- L. Xing, W. Li, C. Wang, F. Gu, M. Xu, C. Tan and J. Yi, J. Phys. Chem. B, 2009, 113, 16596–16602 CrossRef CAS.
- Y. Zhou, J. Rodríguez-López and J. S. Moore, Nat. Commun., 2023, 14, 4847 CrossRef CAS PubMed.
- C. Coutanceau, N. Neha and T. Rafaïdeen, Curr. Opin. Electrochem., 2023, 38, 101210 CrossRef CAS.
- Z. I. Lai, L. Q. Lee and H. Li, Micromachines, 2021, 12, 1405 CrossRef PubMed.
- Y. Holade, N. Tuleushova, S. Tingry, K. Servat, T. W. Napporn, H. Guesmi, D. Cornu and K. B. Kokoh, Catal. Sci. Technol., 2020, 10, 3071–3112 RSC.
- J. C. Yu, M. M. Baizer and K. Nobe, J. Electrochem. Soc., 1988, 135, 83 CrossRef CAS.
- O. Akin and A. Yuksel, Cellulose, 2017, 24, 4729–4741 CrossRef CAS.
- D. Meng, G. Li, Z. Liu and F. Yang, Polym. Degrad. Stab., 2011, 96, 1173–1178 CrossRef CAS.
- Z.-X. Wang, G. Li, F. Yang, Y.-L. Chen and P. Gao, Carbohydr. Polym., 2011, 86, 1807–1813 CrossRef CAS.
- H. Xiao, M. Wu and G. Zhao, Catalysts, 2016, 6, 5 CrossRef.
- Y. Kwon, S. E. F. Kleijn, K. J. P. Schouten and M. T. M. Koper, ChemSusChem, 2012, 5, 1935–1943 CrossRef CAS PubMed.
- Q. Cai, Z. Gu, Y. Chen, W. Han, T. Fu, H. Song and F. Li, Carbohydr. Polym., 2010, 79, 783–785 CrossRef CAS.
- Z. Gu, Q. Cai, Y. Liu and F. Li, J. Polym. Environ., 2013, 21, 479–486 CrossRef CAS.
- P. C. Deng, G. Wang, J. Z. Hu, Y. W. Tian, C. Weng and X. Gao, Mater. Sci. Forum, 2016, 847, 281–286 Search PubMed.
- W. Luo, Z. Han, X. Zeng, S. Yu and J. F. Kennedy, IFSET, 2010, 11, 587–591 CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- G. Chatel and R. D. Rogers, ACS Sustainable Chem. Eng., 2014, 2, 322–339 CrossRef CAS.
- X. Du, H. Zhang, K. P. Sullivan, P. Gogoi and Y. Deng, ChemSusChem, 2020, 13, 4318–4343 CrossRef CAS PubMed.
- R. Ayub and A. Raheel, Int. J. Mol. Sci., 2022, 23, 3767 CrossRef CAS PubMed.
- D. Gao, D. Ouyang and X. Zhao, Green Chem., 2022, 24, 8585–8605 RSC.
- R. S. Weber and K. K. Ramasamy, ACS Omega, 2020, 5, 27735–27740 CrossRef CAS PubMed.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Green Chem., 2020, 22, 7233–7264 RSC.
- P. Li, H. Zhou, Y. Tao, J. Ren, C. Wu and W. Wu, Electroanalysis, 2022, 34, 1529–1539 CrossRef CAS.
- M. Garedew, C. H. Lam, L. Petitjean, S. Huang, B. Song, F. Lin, J. E. Jackson, C. M. Saffron and P. T. Anastas, Green Chem., 2021, 23, 2868–2899 RSC.
- A. Dorigato, D. Rigotti and G. Fredi, Adv. Ind. Eng. Polym. Res., 2023, 6, 288–309 CAS.
- R. Saputra, R. Walvekar, M. Khalid, N. M. Mubarak and M. Sillanpää, Chemosphere, 2021, 265, 129033 CrossRef CAS.
- M. Sienkiewicz, J. Kucinska-Lipka, H. Janik and A. Balas, Waste Manage., 2012, 32, 1742–1751 CrossRef CAS PubMed.
- M. Hansen-Felby, S. U. Pedersen and K. Daasbjerg, Molecules, 2022, 27, 6292 CrossRef CAS.
- Y. Miao, Y. Zhao, G. I. N. Waterhouse, R. Shi, L.-Z. Wu and T. Zhang, Nat. Commun., 2023, 14, 4242 CrossRef CAS.
- D. Jubinville, E. Esmizadeh, S. Saikrishnan, C. Tzoganakis and T. Mekonnen, SM&T, 2020, 25, e00188 Search PubMed.
- X.-Y. Wang, Y. Gao and Y. Tang, Prog. Polym. Sci., 2023, 143, 101713 CrossRef CAS.
- K. Faust, P. Denifl and M. Hapke, ChemCatChem, 2023, 15, e202300310 CrossRef CAS.
- L. Zou, R. Xu, H. Wang, Z. Wang, Y. Sun and M. Li, Natl. Sci. Rev., 2023, 10, nwad207 CrossRef CAS PubMed.
- N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC.
- S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- M. Kiendrebeogo, M. R. Karimi Estahbanati, A. Khosravanipour Mostafazadeh, P. Drogui and R. D. Tyagi, Environ. Pollut., 2021, 269, 116168 CrossRef CAS.
- V. Hirschberg and D. Rodrigue, J. Polym. Sci., 2023, 61, 1937–1958 CrossRef CAS.
- G. Rossignolo, G. Malucelli and A. Lorenzetti, Green Chem., 2024, 26, 1132–1152 RSC.
- J. Datta, P. Kopczyńska, D. Simón and J. F. Rodríguez, J. Polym. Environ., 2018, 26, 166–174 CrossRef CAS.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Waste Manage., 2018, 76, 147–171 CrossRef.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- R. A. Benkeser, H. Watanabe, S. J. Mels and M. A. Sabol, J. Org. Chem., 1970, 35, 1210–1211 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- X. Chen and T. Michinobu, Macromol. Chem. Phys., 2022, 223, 2100370 CrossRef CAS.
- T. J. Farmer, J. W. Comerford, A. Pellis and T. Robert, Polym. Int., 2018, 67, 775–789 CrossRef CAS.
- Y. Zhao, D. Li and X. Jiang, Eur. J. Org Chem., 2023, 26, e202300664 CrossRef CAS.
- P. Parpot, K. Servat, A. P. Bettencourt, H. Huser and K. B. Kokoh, Cellulose, 2010, 17, 815–824 CrossRef CAS.
- T. Isogai, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1593–1599 CrossRef CAS PubMed.
- K. Schnatbaum and H. J. Schäfer, Synthesis, 1999, 864–872 CrossRef CAS.
- Y. Sugano, R.-M. Latonen, M. Akieh-Pirkanniemi, J. Bobacka and A. Ivaska, ChemSusChem, 2014, 7, 2240–2247 CrossRef CAS.
- Y. Sugano, T. Saloranta, J. Bobacka and A. Ivaska, Phys. Chem. Chem. Phys., 2015, 17, 11609–11614 RSC.
- Z. Ait-Touchente, M. Khellaf, G. Raffin, N. Lebaz and A. Elaissari, Polym. Adv. Technol., 2023, 35(1), e6228 CrossRef.
- K. Lewandowski and K. Skórczewska, Polymers, 2022, 14, 3035 CrossRef CAS PubMed.
- X. Jiang, B. Zhu and M. Zhu, Green Chem., 2023, 25, 6971–7025 RSC.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507–514 CrossRef CAS.
- G. S. Shapoval, A. P. Tomilov, A. A. Pud and K. V. Batsalova, Polym. Sci. U.S.S.R., 1987, 29, 1564–1572 CrossRef.
- D. E. Fagnani, D. Kim, S. I. Camarero, J. F. Alfaro and A. J. McNeil, Nat. Chem., 2023, 15, 222–229 CrossRef CAS PubMed.
- Z. Osawa and Y. Uchida, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 2259–2266 CrossRef CAS.
- F. Miao, Y. Liu, M. Gao, X. Yu, P. Xiao, M. Wang, S. Wang and X. Wang, J. Hazard. Mater., 2020, 399, 123023 CrossRef CAS.
- B. Améduri and H. Hori, Chem. Soc. Rev., 2023, 52, 4208–4247 RSC.
- A. A. Pud, O. E. Mikulina and G. S. Shapoval, J. Macromol. Sci., Part A: Pure Appl.Chem., 1995, 32, 621–628 CrossRef.
- L. Guo, W. Liu and C. Chen, Mater. Chem. Front., 2017, 1, 2487–2494 RSC.
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1494 CrossRef CAS PubMed.
- H. Mu, G. Zhou, X. Hu and Z. Jian, Coord. Chem. Rev., 2021, 435, 213802 CrossRef CAS.
- K. Faust, P. Denifl and M. Hapke, ChemCatChem, 2023, 15(13), e202300310 CrossRef CAS.
- Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11191–11774 CrossRef PubMed.
- C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- R. Rahimzadeh, J. Ortega-Ramos, Z. Haque and G. G. Botte, ChemElectroChem, 2023, 10, e202300021 CrossRef CAS.
- I. Maksso, R. C. Samanta, Y. Zhan, K. Zhang, S. Warratz and L. Ackermann, Chem. Sci., 2023, 14, 8109–8118 RSC.
- A. D. Fried, B. J. Wilson, N. J. Galan and J. N. Brantley, J. Am. Chem. Soc., 2022, 144, 8885–8891 CrossRef CAS PubMed.
- J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed.
- Z. Siddiqi and D. Sarlah, J. Am. Chem. Soc., 2021, 143, 21264–21269 CrossRef CAS PubMed.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- N. Sbei, T. Hardwick and N. Ahmed, ACS Sustain. Chem. Eng., 2021, 9, 6148–6169 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- Y. Pan, H. Zhang, B. Zhang, F. Gong, J. Feng, H. Huang, S. Vanka, R. Fan, Q. Cao, M. Shen, Z. Li, Z. Zou, R. Xiao and S. Chu, Nat. Commun., 2023, 14, 1013 CrossRef CAS PubMed.
- H. Zhao, D. Lu, J. Wang, W. Tu, D. Wu, S. W. Koh, P. Gao, Z. J. Xu, S. Deng, Y. Zhou, B. You and H. Li, Nat. Commun., 2021, 12, 2008 CrossRef CAS PubMed.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- S. Kar, M. Rahaman, V. Andrei, S. Bhattacharjee, S. Roy and E. Reisner, Joule, 2023, 7, 1496–1514 CrossRef CAS.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Nat. Synth., 2023, 2, 182–192 CrossRef.
- T. M. McGuire, A. C. Deacy, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 18444–18449 CrossRef CAS PubMed.
- G. R. Jones, H. S. Wang, K. Parkatzidis, R. Whitfield, N. P. Truong and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 9898–9915 CrossRef CAS PubMed.
- M. J. Flanders and W. M. Gramlich, Polym. Chem., 2018, 9, 2328–2335 RSC.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- Y. Gu, Y. Lin, H. Jiang, B. Li, C. Wu, J. Liu, J. Liu, Y. Wang, D. Cui and T. Tang, Macromolecules, 2022, 55, 7732–7739 CrossRef CAS.
- M. Eck, S. T. Schwab, T. F. Nelson, K. Wurst, S. Iberl, D. Schleheck, C. Link, G. Battagliarin and S. Mecking, Angew. Chem., Int. Ed., 2023, 62, e202213438 CrossRef CAS PubMed.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- S. T. Schwab, M. Baur, T. F. Nelson and S. Mecking, Chem. Rev., 2024, 124, 2327–2351 CrossRef CAS PubMed.
- M. Eck and S. Mecking, Acc. Chem. Res., 2024, 57(6), 971–980 CrossRef CAS PubMed.
- M. Baur, N. K. Mast, J. P. Brahm, R. Habé, T. O. Morgen and S. Mecking, Angew. Chem., Int. Ed., 2023, 62, e202310990 CrossRef CAS PubMed.
- M. R. Barone and A. M. Jones, Org. Biomol. Chem., 2017, 15, 10010–10015 RSC.
- N. H. Attanayake and M. Tang, Curr. Opin. Electrochem., 2021, 30, 100791 CrossRef CAS.
- A. Herman, J.-L. Mathias and R. Neumann, ACS Catal., 2022, 12, 4149–4155 CrossRef CAS.
- C. W. S. Yeung, M. H. Periayah, J. Y. Q. Teo, E. T. L. Goh, P. L. Chee, W. W. Loh, X. J. Loh, R. Lakshminarayanan and J. Y. C. Lim, Macromolecules, 2023, 56, 815–823 CrossRef CAS.
- S. Rejman, I. Vollmer, M. J. Werny, E. T. C. Vogt, F. Meirer and B. M. Weckhuysen, Chem. Sci., 2023, 14, 10068–10080 RSC.
- K. Hatakeyama-Sato, K. Sadakuni, K. Kitagawa and K. Oyaizu, Sci. Rep., 2023, 13, 5711 CrossRef CAS PubMed.
- Z. Han, H. Jiang, A. Xue, G. Ni, Y. Sun, Y. Tang, P. Wan and Y. Chen, J. Electroanal. Chem., 2022, 923, 116814 CrossRef CAS.
- T. K. F. Dier, D. Rauber, D. Durneata, R. Hempelmann and D. A. Volmer, Sci. Rep., 2017, 7, 5041 CrossRef PubMed.
- D. Schmitt, C. Regenbrecht, M. Hartmer, F. Stecker and S. R. Waldvogel, Beilstein J. Org. Chem., 2015, 11, 473–480 CrossRef CAS PubMed.
- S. Stiefel, A. Schmitz, J. Peters, D. D. Marino and M. Wessling, Green Chem., 2016, 18, 4999–5007 RSC.
- M. K. Eriksen, K. Pivnenko, M. E. Olsson and T. F. Astrup, Waste Manage., 2018, 79, 595–606 CrossRef CAS PubMed.
- Y. Li, L. Wen and W. Guo, Chem. Soc. Rev., 2023, 52, 1168–1188 RSC.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- W. Mo, X.-Q. Tan and W.-J. Ong, ACS Sustainable Chem. Eng., 2023, 11, 5782–5799 CrossRef CAS.
- S. K. Nabil, S. McCoy and M. G. Kibria, Green Chem., 2021, 23, 867–880 RSC.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS.
- T. Bligaard, R. M. Bullock, C. T. Campbell, J. G. Chen, B. C. Gates, R. J. Gorte, C. W. Jones, W. D. Jones, J. R. Kitchin and S. L. Scott, ACS Catal., 2016, 6, 2590–2602 CrossRef CAS.
- A. G. M. Rankin, J. Trébosc, F. Pourpoint, J.-P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2019, 101, 116–143 CrossRef CAS PubMed.
- E. Verde-Sesto, N. Goujon, H. Sardon, P. Ruiz, T. V. Huynh, F. Elizalde, D. Mecerreyes, M. Forsyth and L. A. O'Dell, Macromolecules, 2018, 51, 8046–8053 CrossRef CAS.
- R. Ye, X. Sun, X. Mao, F. S. Alfonso, S. Baral, C. Liu, G. W. Coates and P. Chen, Nat. Chem., 2024, 16, 210–217 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |