
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introducing AFS ([Al(SO3F)3]x) – a thermally stable, readily available, and catalytically active solid Lewis superacid†
Johanna
Schlögl
a,
Ole
Goldammer
a,
Julia
Bader
a,
Franziska
Emmerling
 b and
Sebastian
Riedel
*a
b and
Sebastian
Riedel
*a
aFachbereich Biologie, Chemie, Pharmazie, Institut für Chemie und Biochemie – Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34/36, 14195 Berlin, Germany. E-mail: schjo@zedat.fu-berlin.de; s.riedel@fu-berlin.de
bDepartment Materials Chemistry, Federal Institute for Material Research and Testing, Richard-Willstätter-Straße 11, 12489 Berlin, Germany
First published on 2nd May 2024
Abstract
Common Lewis superacids often suffer from low thermal stability or complicated synthetic protocols, requiring multi-step procedures and expensive starting materials. This prevents their large-scale application. Herein, the easy and comparably cheap synthesis of high-purity aluminium tris(fluorosulfate) ([Al(SO3F)3]x, AFS) is presented. All starting materials are commercially available and no work-up is required. The superacidity of this thermally stable, polymeric Lewis acid is demonstrated using both theoretical and experimental methods. Furthermore, its synthetic and catalytic applicability, e.g. in bond heterolysis reactions and C–F bond activations, is shown.
Introduction
Trivalent aluminium species are archetypical Lewis acids and their applications in synthetic chemistry are manifold.1,2 Solid Lewis acids, like high-surface aluminium fluoride, HS-AlF3, or aluminium chlorofluoride (ACF, AlClxF3x, with x = 0.3–0.05), serve as powerful catalysts in a variety of industrial transformations, e.g. C–H activation, C–F activation, (de)fluorination, or Friedel–Crafts type conversions.2–4 In fundamental research, the generation of highly reactive cations, e.g. [P9]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 or [C(C6F5)3]+,6 has been realized using molecular Lewis acids like Al[OC(CF3)3]37,8 or [Al(OTeF5)3]2.9,10 In the last two decades, the scope of aluminium-based Lewis acids has significantly broadened with the emergence of so-called Lewis superacids.7–14 These molecular Lewis acids outperform SbF5 in terms of acidity and handling. Furthermore, efforts to introduce superacidity to solid Lewis acids have been made, e.g. by treatment of partially dehydroxylated silica with Al[OC(CF3)3]315 or by anion-doping of ACF using [Al(OTeF5)3]2.16 However, although the potential of these new Lewis superacids is well-acknowledged in fundamental research, their application in industry is all but popular. This is because they are either not easily accessible in bulk quantities, as they require multistep synthetic procedures involving rather expensive starting materials, or they exhibit low thermal stability.
5 or [C(C6F5)3]+,6 has been realized using molecular Lewis acids like Al[OC(CF3)3]37,8 or [Al(OTeF5)3]2.9,10 In the last two decades, the scope of aluminium-based Lewis acids has significantly broadened with the emergence of so-called Lewis superacids.7–14 These molecular Lewis acids outperform SbF5 in terms of acidity and handling. Furthermore, efforts to introduce superacidity to solid Lewis acids have been made, e.g. by treatment of partially dehydroxylated silica with Al[OC(CF3)3]315 or by anion-doping of ACF using [Al(OTeF5)3]2.16 However, although the potential of these new Lewis superacids is well-acknowledged in fundamental research, their application in industry is all but popular. This is because they are either not easily accessible in bulk quantities, as they require multistep synthetic procedures involving rather expensive starting materials, or they exhibit low thermal stability.
In this context, the fluorosulfate group (–SO3F) presents an interesting ligand, as it can be introduced using comparably cheap, commercially available starting materials and it imposes great thermal stability on its compounds due to their tendency to polymerize. Compared to ACF, the bulkiness of the –SO3F group introduces a distortion to the three-dimensional network that could lead to enhanced Lewis acidity. Of the related trifluormethanesulfonate group (–SO3CF3, OTf) the Lewis acid Al(OTf3)3 is already known and well-established as a catalyst in a variety of organic transformations.17 However, quantum-chemical calculations render it to be non-superacidic.11
Aluminium tris(fluorosulfate) (Al(SO3F)3, AFS) was introduced already in 1983 by Verma and Singh.18 Preliminary reports on aluminium fluorosulfates included the partially substituted AlCl(SO3F)2 and the acetonitrile adduct Al(SO3F)3·3CH3CN, for none of which a characterization was provided.19 Verma and Singh published a synthetic route starting from amalgamated aluminium and HOC(O)CF3 and subsequent conversion of the obtained Al[OC(O)CF3]3 with 3 equivalents of HSO3F. However, all our attempts to reproduce this reaction have always resulted in an incomplete substitution (Fig. S1†). Only by introducing a large excess of HSO3F a full substitution could be achieved. However, removing the excess acid afterwards is a tedious task, which makes the whole procedure impractical, apart from its multi-step nature and the need for an amalgam (Fig. S2†). Thus, an alternative to Al[OC(O)CF3]3 as a starting material and a more practical route to the published synthesis needs to be developed.
Herein, we report on the preparation and isolation of AFS through an easy, straightforward process using AlMe3 and HSO3F, two commercially available and comparably cheap starting materials. Furthermore, the new synthetic protocol avoids the use of mercury for the first step, the activation of aluminium. The synthetic procedure requires no work-up and can be performed on a multigram scale. The polymeric nature and thermal stability of AFS is demonstrated and its superacidity is proven by applying both theoretical and experimental methods. Finally, its synthetic and catalytic applicability, e.g. in bond heterolysis reactions and C–F bond activations, is shown.
Results and discussion
Synthesis and characterization of AFS
The addition of 3 equivalents of HSO3F to a frozen solution of AlMe3 in 1,2,3-trifluorobenzene and subsequent warming of the mixture to room temperature leads to the evolution of methane and the formation of AFS (1) (Scheme 1). The latter can be isolated as a colourless powder in 96% yield after removal of all volatiles under reduced pressure and drying overnight. | ||
| Scheme 1 Synthesis of AFS (1). | ||
The choice of solvent is crucial for the success of the synthesis of 1. While SO2Cl2 reacts with the starting materials, SO2ClF, a popular solvent in superacid chemistry, does not allow the warming of the reaction mixture to room temperature.20 Thus it results in an incomplete conversion yielding a temperature sensitive, in some cases explosive reaction product. Using n-pentane as a solvent leads to the formation of a bright yellow reaction mixture and the subsequent formation of a slurry, suggesting polymerization as a side reaction. Finally, standard aromatic solvents such as toluene or pyridine undergo electrophilic aromatic substitution (Fig. S4†). Hence, deactivated arenes need to be used as solvents. 1,2-Difluorobenzene seems to form adducts with the Lewis acid, as can be seen in the corresponding IR spectrum (Fig. S5†), so we opted for the even more strongly deactivated 1,2,3-trifluorobenzene.
1 is a colourless powder that can be stored for at least one year under an inert atmosphere at room temperature, as opposed to the reaction product obtained by Verma and Singh, which was only stable for a few days.18 TGA/DSC measurements reveal that the thermal decomposition of 1 occurs only at temperatures above 140 °C (Fig. S6†). 1 is sparingly soluble in acetonitrile and insoluble in HSO3F and most of the common inorganic and organic solvents (see ESI† for more information). This indicates a high degree of polymerization and therefore the formulation as [Al(SO3F)3]x seems more appropriate than the formula based on composition. The polymeric structure is a common trait of metal fluorosulfates, due to the polydentate nature of the –SO3F ligand, and together with the high reactivity, this has often prevented their solid-state characterization by single-crystal X-ray diffraction.21,22 Only two molecular structures in the solid state are known of metal tris(fluorosulfates), Sb(SO3F)323 and Au(SO3F)3.24 Indeed, powder XRD studies of 1 suggest an amorphous nature (Fig. S7†).
The proposed polymeric structure of 1 is further supported by spectroscopic investigations. The IR spectrum shows a strongly blue-shifted S–F stretching band, indicating a covalent coordination of the –SO3F ligand as opposed to an ionic one (Fig. 1a).21 Though the spectrum contains six vibrational modes, suggesting a tridentate bridging –SO3F ligand,21 its band positions fit those of related polymeric Ga(SO3F)325 and In(SO3F)3,26 which were assigned to be a bidentate bridging coordination mode. This is in agreement with the findings published by Verma and Singh.18 Taking into account our own investigations, a satisfactory assignment of the denticity of the –SO3F ligand is not possible, but a polymeric nature of 1 can be assumed.
 | ||
| Fig. 1 Spectroscopic characterization of 1: (a) IR spectrum (ATR, 25 °C). (b) 27Al and 19F MAS NMR spectra (νrot = 21 kHz, 25 °C). | ||
The 27Al magic angle spinning (MAS) NMR spectrum of 1 shows two overlapping signals at −17 and −23 ppm that can be assigned to an octahedral coordination sphere around the aluminium (Fig. 1b).27 Due to the presence of strongly distorted [AlO6] moieties the signals are significantly broadened and indicate the presence of at least two different coordination polyhedra within the bulk. The 19F MAS NMR spectrum contains a broad singlet in the typical fluorosulfate region at 36 ppm (Fig. 1b).
To further study the polymerization of 1, the gas-phase structures of the monomer, dimer, and trimer were calculated at the B3LYP-D3(BJ)/def2-TZVPP level (Fig. 2).28–30 A comparison of the respective Gibbs free energies reveals a free energy gain per monomer addition of roughly 56 kJ mol−1, suggesting that the polymerization of 1 is thermodynamically favoured.
 | ||
| Fig. 2 Calculated gas phase structures of [Al(SO3F)3]n (n = 1, 2, 3). Some fluorosulfate ligands have been omitted for clarity and only their coordinated O-atoms are shown. (B3LYP-D3(BJ)/def2-TZVPP). | ||
The theoretical Lewis acidity of Al(SO3F)3
To estimate the Lewis acidity of 1, monomeric Al(SO3F)3 was chosen as a simplified model and its gas-phase fluoride ion affinity (FIA) was computed at the BP86-D3(BJ)/def-SVP28–30 level using the isodesmic reaction with trimethylsilyl fluoride as anchor point.31The obtained FIA value of 538 kJ mol−1 is higher than the benchmark Lewis acid SbF5 with 489 kJ mol−1 and comparable to other aluminium-based Lewis superacids, thus theoretically rendering monomeric Al(SO3F)3 to be superacidic (Fig. 3).
 | ||
| Fig. 3 Calculated FIAs of selected Lewis acids. | ||
The experimental Lewis acidity of AFS
To evaluate the Lewis acidity of 1 in the condensed state, three different methods were applied: (I) investigating the blueshift of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration of CD3CN upon adduct formation with AFS, (II) the Gutmann–Beckett method, and (III) a competition experiment with [PPh4][SbF6].
N stretching vibration of CD3CN upon adduct formation with AFS, (II) the Gutmann–Beckett method, and (III) a competition experiment with [PPh4][SbF6].
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) N stretching vibration of CD3CN upon adduct formation with AFS.
The wavenumber of the CN stretching mode of CH3CN is a sensitive measure of Lewis acidity and its blueshift upon coordination to a Lewis acidic centre is frequently used for the evaluation of both solid and molecular Lewis acids.11 Fermi coupling between ν(CN) and ν(CC) + δs(CH3) complicates the exact determination of Δν(CN) as it results in additional modes of medium intensity. Hence, the adduct with deuterated acetonitrile is normally prepared, as here no additional Fermi resonances appear. Upon coordination of CD3CN to 1, the CN stretching vibration of the adduct AFS·CD3CN (2) is blue-shifted by 78 cm−1 compared to free CD3CN (2336 cm−1, 2258 cm−1, Fig. S8†). This shift is higher than the one of SbF5·CD3CN (65 cm−1) and lies within the range of other aluminium-based Lewis superacids, such as Al[OC(C6F5)3]3 (79 cm−1) or [Al(OTeF5)3]2 (70 cm−1).9,14,32 Furthermore, it is higher than those of other aluminium-based solid Lewis acids, such as ACF (68 cm−1) or ACF teflate (73 cm−1).16,33
N stretching vibration of CD3CN upon adduct formation with AFS.
The wavenumber of the CN stretching mode of CH3CN is a sensitive measure of Lewis acidity and its blueshift upon coordination to a Lewis acidic centre is frequently used for the evaluation of both solid and molecular Lewis acids.11 Fermi coupling between ν(CN) and ν(CC) + δs(CH3) complicates the exact determination of Δν(CN) as it results in additional modes of medium intensity. Hence, the adduct with deuterated acetonitrile is normally prepared, as here no additional Fermi resonances appear. Upon coordination of CD3CN to 1, the CN stretching vibration of the adduct AFS·CD3CN (2) is blue-shifted by 78 cm−1 compared to free CD3CN (2336 cm−1, 2258 cm−1, Fig. S8†). This shift is higher than the one of SbF5·CD3CN (65 cm−1) and lies within the range of other aluminium-based Lewis superacids, such as Al[OC(C6F5)3]3 (79 cm−1) or [Al(OTeF5)3]2 (70 cm−1).9,14,32 Furthermore, it is higher than those of other aluminium-based solid Lewis acids, such as ACF (68 cm−1) or ACF teflate (73 cm−1).16,33
 | ||
| Fig. 4 31P{1H} NMR spectrum of the reaction between Et3PO and 1 to determine the Lewis acidity of 1 by the Gutmann–Beckett method. Two species are initially formed: (a) the unstable “classical” Gutmann–Beckett complex (3a) and (b) the chemisorbed Et3PO unit within the polymer (3b). The 31P{1H} NMR spectrum of Et3PO is shown for comparison. | ||
The reactivity of AFS
 | ||
| Scheme 2 Synthesis of the trityl cation via chloride abstraction of Ph3CCl by 1. | ||
The perfluorinated analog of the trityl cation, [PhF3C]+, was recently synthesized in our group through halide abstraction using the Lewis superacid [Al(OTeF5)3]2.6 This inspired us to test the effectiveness of 1 in a likewise reaction. Compared to its non-fluorinated analog, which is known as a versatile hydride and methanide abstraction agent, [PhF3C]+ is expected to show an even higher reactivity.35 The addition of PhF3CCl to a suspension of 1 in SO2 at −80 °C and subsequent warming of the reaction mixture to room temperature yields an intense red-violet suspension. The 19F NMR spectrum reveals four signals, that can be assigned to PhF3COSO2F (6), with the fluorosulfate ligand bound to the central carbon (Fig. S19†). A similar outcome was observed by Dutton et al., who isolated the respective PhF3COSO2CF3 upon conversion of PhF3CCl with stoichiometric amounts of HSO3CF3.35 The formation of 6 can be explained with the generation of the perfluorotrityl cation and [AFS–Cl]− (5) and the subsequent decomposition to 6 (Scheme 3). In contrast to what we found in the case of 4, [AFS–Cl]− is not stable in the presence of the strongly electrophilic [PhF3C]+, which is likely why the abstraction of one –SO3F ligand occurs, followed by the attack at the central carbon atom. As a side reaction, the attack can also happen at the para-position of one perfluorinated aryl ring resulting in the formation of ketone 7 (Scheme 3, Fig. S20†). This reaction pathway can be followed by the appearance of five additional signals in the 19F NMR spectrum over time (Fig. S21†) and was also observed by Dutton et al.35 Intriguingly, the reaction mixture remains red-violet in color, though 6 should be colorless (Table S1†) and 7 is light-tan.35 Moreover, a similar red-violet color has been described multiple times with respect to the formation of [PhF3C]+ and UV-Vis spectroscopy reveals an absorption maximum at λ = 500 nm, which is also the reported value for [PhF3C]+ (Fig. S22†).6,36 Nevertheless, all efforts to detect signals corresponding to the cation via low-temperature 19F NMR spectroscopy failed. These observations infer that the –SO3F group in 6 is only loosely bound to the central carbon atom, owing to the highly delocalized charge and its polydentate nature, and that 6 is in an equilibrium with 8. However, this reaction is too fast to be discernible on the NMR timescale, yet detectable through UV-Vis spectroscopy.
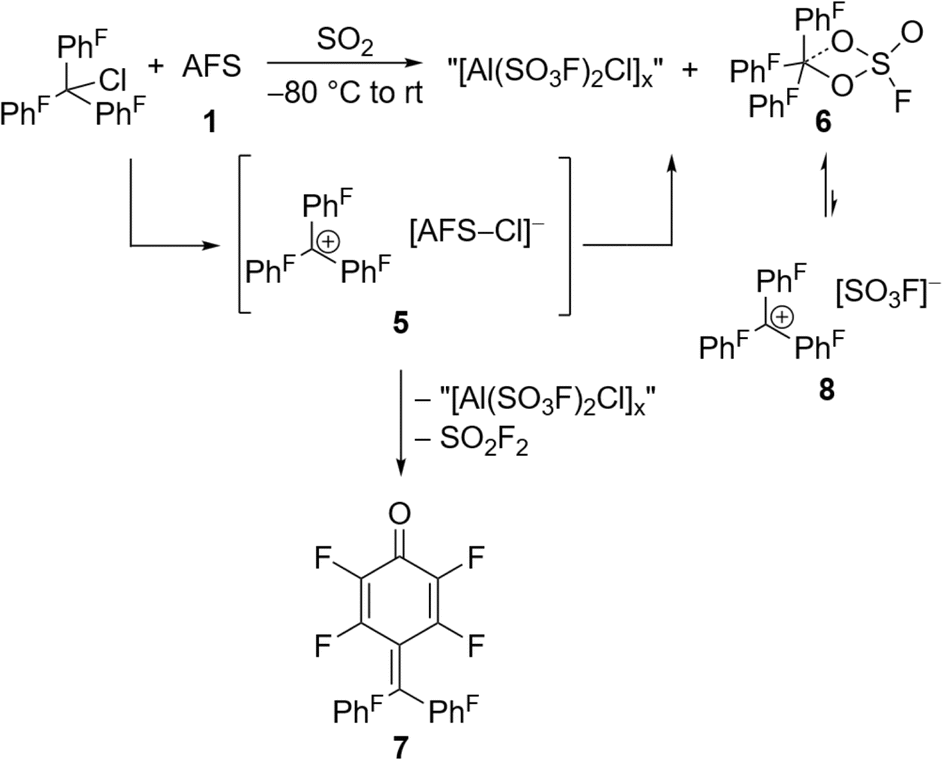 | ||
| Scheme 3 In situ generation of the [PhF3C]+ cation via halide abstraction of perfluoro trityl chloride and further reactions leading to fluorosulfate 6 and ketone 7. | ||
In an effort to investigate the hydride abstraction ability of the reaction mixture, Ph3CH was added resulting in an immediate color change from red-violet to yellow. This color change is consistent with the formation of the trityl cation [Ph3C]+, which is further supported by 1H NMR (Fig. S23†).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C10H6, CH2), where an oxide-fluoride exchange yielded [PEt3F]+.40 Electrophilic phosphonium cations (ECPs) similar to [PEt3F]+ have been introduced by Stephan et al. as versatile main group catalysts, e.g. in the hydroarylation of olefins,41 isomerization and polymerizations of olefins or hydrosilylation of olefins or alkynes.38,42
C10H6, CH2), where an oxide-fluoride exchange yielded [PEt3F]+.40 Electrophilic phosphonium cations (ECPs) similar to [PEt3F]+ have been introduced by Stephan et al. as versatile main group catalysts, e.g. in the hydroarylation of olefins,41 isomerization and polymerizations of olefins or hydrosilylation of olefins or alkynes.38,42
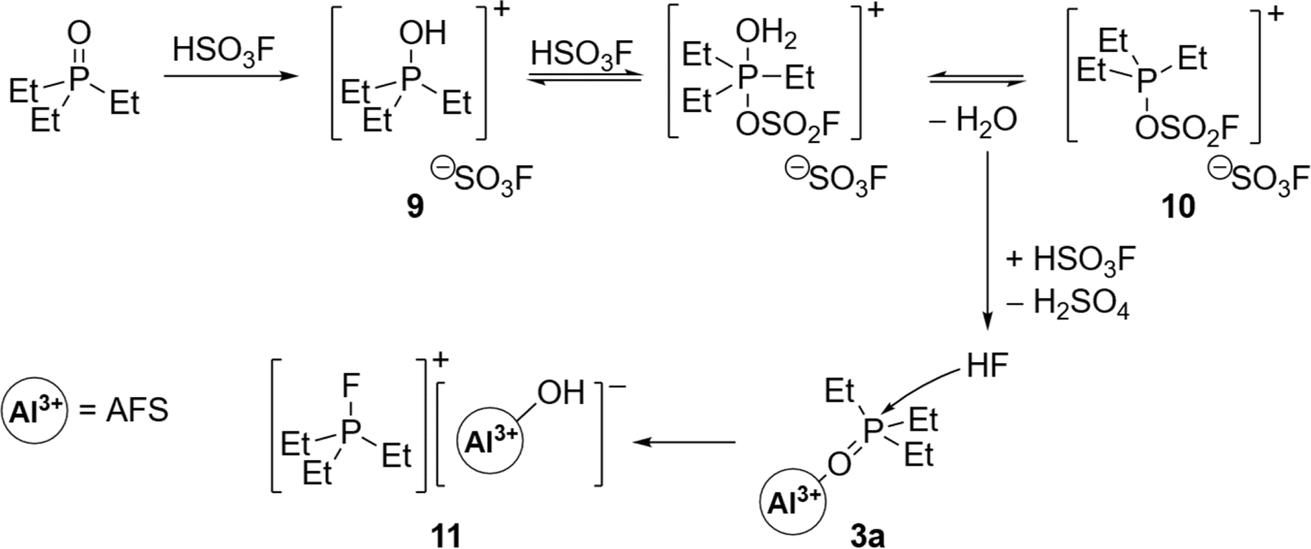 | ||
| Scheme 4 Proposed mechanism for the formation of fluorophosphonium salt 11. Residual HSO3F in the presence of Et3PO leads to the in situ formation of HF and subsequent attack of the Gutmann–Beckett complex 3a. | ||
 | ||
| Fig. 5 Catalytic C–F bond activation of fluoroalkanes in the presence of AFS and Et3SiH (RH, alkyl; R′ = alkyl, aryl). (a) Dehydrofluorination (DHF), (b) hydrodefluorination (HDF). | ||
| Substrate | t [h] | Conv.b [%] | C–F activation type | Main productc |
|---|---|---|---|---|
| a 30 mg [46 mol% (100% active sites)] of the catalyst in a J Young NMR tube using CD2Cl2 as solvent (see ESI for more information). b Conversions were determined through 19F NMR spectroscopy and are based on the converted fluorinated substrate into the corresponding products using CFCl3 as internal standard. c In addition to Et3SiF. d In this case only 10 mg [15 mol% (100% active sites)] of the catalyst were used. | ||||
| 1-Fluoropentane | 2 | >99 | DHF | (E/Z)-2-Pentene |
| 1-Fluoroheptane | 2 | 61 | DHF | cis/trans-2-Heptene |
| 24 | >99 | |||
| 2-Fluoropentane | 1 | >99 | DHF | (E/Z)-2-Pentene |
| 1-Fluoroadamantaned | 1 | >99 | HDF | Adamantane |
| Fluorocyclohexane | 1 | >99 | DHF/HDF | Cyclohexene, cyclohexane |
| 2,2-Difluorobutane | 2 | 59 | DHF/HDF | (E)-2-Fluoro-2-butene, (E/Z)-2-butene |
| 24 | 80 | |||
| (CF3)C6H5 | 24 | 2 | — | — |
At room temperature, 1-fluoropentane, 2-fluoropentane, and fluorocyclohexane were transformed quantitatively within 1–2 h through dehydrofluorination into the corresponding olefins, whereas 24 h were needed for 1-fluoroheptane. In the case of 1-fluoropentane and 1-fluoroheptane subsequent isomerization always yielded the respective 2-olefin. This high activity of AFS is remarkable, as for comparison, dehydrofluorination using ACF as catalyst and Et3GeH as hydride source only occurs at elevated temperatures.4 However, the conversion is not as fast as observed for ACF teflate catalysing the same reactions at room temperature.16 In the case of fluorocyclohexane, also hydrodefluorination occurred, leading to the formation of cyclohexane aside from cyclohexene. 1-Fluoroadamantane was consumed within one hour undergoing hydrodefluorination into adamantane. 2,2-Difluorobutane was mostly consumed within 24 h and was transformed almost selectively into (E)-2-fluoro-2-butene through dehydrofluorination. The latter partly underwent hydrodefluorination into (E/Z)-2-butene. Finally, in the case of trifluorotoluene almost no conversion could be observed, even after 24 h.
Conclusion
In this work, we introduced a more convenient synthesis for high-purity AFS, which avoids the use of an aluminium amalgam. Our one-step synthesis relies on AlMe3 and HSO3F as commercially available and comparably cheap starting materials, and has the additional advantage that no work-up is required. The superacidity of this polymeric, thermally stable aluminium Lewis acid was demonstrated both computationally and experimentally and its applicability in typical Lewis acid transformations was shown. In bond heterolysis reactions it was not only possible to stabilize the trityl cation [Ph3C]+, but also to generate its perfluorinated analogue [PhF3C]+. Moreover, AFS was able to deoxygenate Et3PO in the presence of HF to afford the corresponding fluorophosphonium cation [PEt3F]+. Finally, the high catalytic activity of AFS was successfully tested in dehydrofluorination and hydrodefluorination reactions at room temperature using Et3SiH as the hydride source. AFS arises as a new thermally stable, polymeric Lewis acid which is easy to manage, comparably cheap to prepare and which showcases superacidic character. With that, AFS builds a bridge between molecular Lewis superacids, which are either non-easily accessible or thermally unstable, and solid Lewis acids, which are normally not as acidic. In conclusion, we hope to pave the way for a Lewis superacid that is suitable for large-scale applications.Data availability
Additional details regarding experimental methods and experimental data are given in the ESI.†Author contributions
J. S. and O. G. performed synthetic work and collected vibrational spectroscopy data. J. S. and J. B. collected LT-NMR data. F. E. collected powder-XRD data. J. S. wrote the manuscript. All authors discussed and commented on the manuscript. J. S., J. B. and S. R. revised the manuscript. J. S. and S. R. conceptualized and coordinated the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 387284271 – SFB 1349. The assistance provided by the Core Facility BioSupraMol, supported by the DFG, is also acknowledged. Computing time was made available by the High-Performance Computing Center at the ZEDAT, Freie Universität Berlin. We thank the AG Braun at HU Berlin for providing 2-fluoropentane and 2,2-difluorobutane. J. S. acknowledges the Verband der Chemischen Industrie (VCI) for providing PhD funding (Kekulé Fellowship).Notes and references
-
(a) G. A. Olah, J. Kaspi and J. Bukala, J. Org. Chem., 1977, 42, 4187 CrossRef CAS
; (b) A. Corma and H. García, Chem. Rev., 2002, 102, 3837 CrossRef CAS PubMed
- T. Krahl and E. Kemnitz, Catal. Sci. Technol., 2017, 7, 773 RSC
-
(a) T. Krahl, R. Stösser, E. Kemnitz, G. Scholz, M. Feist, G. Silly and J.-Y. Buzaré, Inorg. Chem., 2003, 42, 6474 CrossRef CAS PubMed
- G. Meißner, D. Dirican, C. Jäger, T. Braun and E. Kemnitz, Catal. Sci. Technol., 2017, 7, 3348 RSC
- J. Frötschel-Rittmeyer, M. Holthausen, C. Friedmann, D. Röhner, I. Krossing and J. J. Weigand, Sci. Adv., 2022, 8, eabq8613 CrossRef PubMed
- K. F. Hoffmann, D. Battke, P. Golz, S. M. Rupf, M. Malischewski and S. Riedel, Angew. Chem., Int. Ed., 2022, 61, e202203777 CrossRef CAS PubMed
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659 CrossRef PubMed
- A. Kraft, N. Trapp, D. Himmel, H. Böhrer, P. Schlüter, H. Scherer and I. Krossing, Chem.–Eur. J., 2012, 18, 9371 CrossRef CAS PubMed
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2017, 56, 8263 CrossRef CAS PubMed
- K. F. Hoffmann, A. Wiesner, S. S. Steinhauer and S. Riedel, Chem.–Eur. J., 2022, 28, e202201958 CrossRef CAS PubMed
- L. Greb, Chem.–Eur. J., 2018, 24, 17881 CrossRef CAS PubMed
- J. Chen and E. Y.-X. Chen, Dalton Trans., 2016, 45, 6105 RSC
- J. F. Kögel, A. Y. Timoshkin, A. Schröder, E. Lork and J. Beckmann, Chem. Sci., 2018, 9, 8178 RSC
-
(a) J. F. Kögel, D. A. Sorokin, A. Khvorost, M. Scott, K. Harms, D. Himmel, I. Krossing and J. Sundermeyer, Chem. Sci., 2018, 9, 245 RSC
- K. K. Samudrala, W. Huynh, R. W. Dorn, A. J. Rossini and M. P. Conley, Angew. Chem., Int. Ed., 2022, 61, e202205745 CrossRef CAS PubMed
- M. Bui, K. F. Hoffmann, T. Braun, S. Riedel, C. Heinekamp, K. Scheurell, G. Scholz, T. M. Stawski and F. Emmerling, ChemCatChem, 2023, 15, e202300350 CrossRef CAS
-
(a) G. A. Olah, O. Farooq, S. Morteza, F. Farnia and J. A. Olah, J. Am. Chem. Soc., 1988, 110, 2560–2565 CrossRef CAS
- S. Singh and R. D. Verma, Polyhedron, 1983, 2, 1209 CrossRef CAS
-
(a) E. Hayek, J. Puschmann and A. Czaloun, Monatsh. Chem., 1954, 85, 359 CrossRef CAS
-
G. A. Olah, G. K. Surya Prakash, R. Molnr and J. Sommer, Superacid Chemistry, JohnWiley & Sons,Inc,, Hoboken, NJ, USA, 2009 Search PubMed
- G. A. Lawrance, Chem. Rev., 1986, 86, 17 CrossRef CAS
- T. Michałowski, P. J. Malinowski, W. Grochala and J. Fluor, Chem, 2016, 189, 102 Search PubMed
- D. Zhang, S. J. Rettig, J. Trotter and F. Aubke, Inorg. Chem., 1995, 34, 3153 CrossRef CAS
- H. Willner, S. J. Rettig, J. Trotter and F. Aubke, Can. J. Chem., 1991, 69, 391 CrossRef CAS
- A. Storr, P. A. Yeats and F. Aubke, Can. J. Chem., 1972, 50, 452 CrossRef CAS
- R. C. Paul, R. D. Sharma, S. Singh and R. D. Verma, Inorg. Nucl. Chem. Lett., 1981, 43, 1919 CrossRef CAS
-
K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials. 27Al NMR, Elsevier Science Ltd., Oxford, 2002, vol. 6 Search PubMed
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
-
(a) H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489 RSC
- B. v. Ahsen, B. Bley, S. Proemmel, R. Wartchow, H. Willner and F. Aubke, Z. Anorg. Allg. Chem., 1998, 624, 1225 CrossRef
- T. Krahl, E. Kemnitz and J. Fluor, Chem, 2006, 127, 663 CAS
-
(a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235 CrossRef CAS
- E. G. Delany, S. Kaur, S. Cummings, K. Basse, D. J. D. Wilson and J. L. Dutton, Chem.–Eur. J., 2019, 25, 5298 CrossRef CAS PubMed
- G. A. Olah and M. B. Comisarow, J. Am. Chem. Soc., 1967, 89, 1027 CrossRef CAS
-
(a) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed
- E. Pires and J. M. Fraile, Phys. Chem. Chem. Phys., 2020, 22, 24351 RSC
-
(a) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 6538 CrossRef CAS PubMed
- M. Pérez, T. Mahdi, L. J. Hounjet and D. W. Stephan, Chem. Commun., 2015, 51, 11301 RSC
- M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01753f |
| This journal is © The Royal Society of Chemistry 2024 |