
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmploying racemization strategies to simultaneously enhance the quantum yield, lifetime, and water stability of room-temperature phosphorescent materials†
Zenggang
Lin
a,
Peng
Zhang
ab,
Fuqiang
Song
a,
Yuzhu
Yang
a,
Xuan
Miao
a and
Weisheng
Liu
 *a
*a
aKey Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: liuws@lzu.edu.cn
bCollege of Chemistry and Biology Engineering, Hechi University, Yizhou 546300, P. R. China
First published on 2nd May 2024
Abstract
Room temperature phosphorescence (RTP) materials are increasingly recognized for their superior luminescent properties, which are pivotal in applications such as anti-counterfeiting, information storage, and optoelectronics. Despite this, the sensitivity of most RTP systems to humidity presents a significant challenge in achieving durable RTP performance in aqueous environments. This study proposes a strategy to enhance organic room-temperature phosphorescence through racemization. By incorporating external racemates of various chiral phosphors—NDBD-Ph, NDBD-Ph-Ph, NDBD-CH3, and NDBD-O-CH3—into a polyacrylonitrile (PAN) matrix, we significantly enhance the RTP properties (quantum yield, lifetime, and afterglow-time) of the resultant films. This enhancement can be attributed to the increased density of racemic molecules in the matrix and the increased spin–orbit coupling (SOC), facilitating the development of a long-lasting polymer RTP system in water. Notably, the racemic rac-NDBD-Ph@PAN film exhibits a persistent bright turquoise afterglow, even after immersion in water for a month. Furthermore, for the first time, we achieved an enhanced green to cyan RTP response to pH variations under both acidic and alkaline conditions (pH = 2–12), with the maximum phosphorescence emission intensity increasing up to threefold. The remarkable water stability, reversible response characteristics, and enhanced phosphorescence properties of this system offer promising potential for dynamic information encryption in aqueous environments.
Introduction
Recent interest in long-lived RTP has revitalized research into organic luminophores,1–3 leading to innovative applications in next-generation optoelectronics,4 anti-counterfeiting,5,6 sensors,7 and information storage.8,9 These advancements capitalize on the extended lifespan and spin multiplicity of triplet excitons in organic molecules.10,11 Organic RTP, particularly through the strategic incorporation of dopants, has seen significant progress in enhancing RTP capabilities. Despite these advances, fully understanding the complex photophysics of multiphase solid-state systems remains a formidable challenge.12,13 The transition from the lowest excited singlet state (S1) to the triplet state (T) and from the triplet excited state (T1) to the ground state (S0) involves intersystem crossing (ISC), a process inherently inhibited by spin restrictions. Moreover, the excited states are susceptible to deactivation through non-radiative vibrational modes and quenching by molecular oxygen, further complicating the achievement of stable, long-lived RTP in aqueous environments at room temperature.14–17 Notably, while the primary focus of doped RTP materials has been on encryption and anti-counterfeiting, emerging applications in sensing highlight the unique benefits of organic RTP doping.18–20Current literature offers limited insight into the relationship between molecular solid forms and RTP properties. Research indicates that the luminescence of organic molecules is highly susceptible to variations in molecular orientation and packing density. This underscores the importance of examining molecular packing patterns to augment RTP, particularly for chiral molecules with analogous structures and photophysical characteristics.21,22 According to Wallach's rule, racemic single crystals exhibit higher density compared to chiral molecules.23 This study aims to substantiate this hypothesis by embedding luminescent molecules within a polymer matrix through electrostatic attraction and additional intermolecular forces in a racemic configuration of axial chiral molecules. The objective is to determine if a denser racemic cross-stacking arrangement can enhance RTP.
Polymer-based RTP materials exhibit advantages over crystalline counterparts in terms of ease of preparation, flexibility, and transparency.24–26 However, limitations exist, notably the abbreviated lifespan of polymer RTP systems due to rapid and significant non-radiative decay of triplet excitons. RTP films employing polyvinyl alcohol (PVA) as a substrate demonstrate heightened sensitivity to air and humidity. This sensitivity is particularly pronounced due to the susceptibility of hydrogen or ionic bonds between the phosphor and the polymer matrix to decomposition in air or water, leading to phosphorescence quenching. This underscores a notable drawback: such polymer RTP materials are ill-suited for applications in aqueous environments. Consequently, the development of long-lived RTP materials stable in water is imperative for polymer systems. Achieving room temperature phosphorescence materials with extended lifespans necessitates long-life chromophores and a rigid polymer microenvironment.27,28 Typically, in polymer-based RTP materials, bright and durable emissions are derived from discrete chromophores. Identifying monomeric molecules with optimal RTP behavior is crucial for generating ultra-long organic room-temperature phosphorescence (UORTP).29 The chiral binaphthyl structure, characterized by an alkyl chain linking two naphthalene rings in a nearly parallel orientation, is notable. The torsional angle of the alkyl chain results in incomplete spatial overlap and a deflection angle between the naphthalene rings. This configuration facilitates a charge transfer channel through the intermediate naphthalene ring stack structure, enabling resonance transfer between the HOMO and LUMO electron clouds, rendering it an ideal framework for superior organic long-persistence materials.30,31 In systems comprising electron-rich and electron-deficient molecules, the formation of strong electrostatic interactions is facilitated by opposing electrostatic potential (ESP) regions. Aromatic carbonyl and hydroxyl groups in organic molecules enhance charge transfer, promoting ISC from singlet to triplet states. The rigid environment within the polymer system curtails molecular vibration and mitigates non-radiative deactivation of triplet excitons, thereby augmenting phosphorescence efficacy.32–34
In this study, we aim to harness electrostatic interactions with robust environmental resilience to develop long-lived RTP materials that exhibit stable phosphorescence under various conditions, including aqueous environments. Guided by this design principle, we introduce a universal strategy for enhancing photoluminescence in organophosphorus materials. This strategy involves doping films of racemic materials featuring axial chiral characteristics (Fig. 1b). Polyacrylonitrile (PAN) is selected as the polymer matrix due to its abundant cyanide groups. These groups not only facilitate the generation of triplet excitons during the ISC process but also form a rigid polymer network via electrostatic interactions opposite to the electrostatic potential of the binaphthyl derivatives. This network effectively immobilizes chromophore molecules, minimizing non-radiative decay and enabling UORTP under environmental conditions. The amorphous chiral polymer films detailed herein are synthesized through a doping strategy coupled with a constraint effect (Fig. 1a and b), demonstrating a pronounced racemic RTP enhancement. The peak phosphorescence emission intensity surpasses that of the pure enantiomer configuration by over twofold, with a phosphorescence lifetime reaching 1357 ms. Due to its potent electrostatic attraction, the racemate RTP system exhibits remarkable stability under aqueous conditions. Moreover, this study pioneers an enhanced in situ reversible pH-responsive organic afterglow system, triggered by environmental pH shifts altering molecular stacking patterns, thereby facilitating its application in pH sensing (Fig. 1c). This research offers a novel approach to augment RTP emission and achieve highly stable RTP systems capable of stimulus-responsive luminescence in aqueous environments.
 | ||
| Fig. 1 Schematic diagram of the photoluminescence phenomenon of doped thin films. (a) Chemical structures of NDBD-Ph, NDBD-Ph-Ph, NDBD-CH3, and NDBD-O-CH3. (b) Preparation process of doped films. (c) Luminescence images of doped films R-NDBD-Ph@PAN and rac-NDBD-Ph@PAN after UV-254 nm irradiation at room temperature, in an aqueous environment and after pH sensing, respectively. | ||
Results and discussion
To validate the proposed concepts, we synthesized a series of axial chiral enantiomers: R-NDBD-Ph@PAN, S-NDBD-Ph@PAN, and rac-NDBD-Ph@PAN, through the polymerization of PAN with axial chiral binaphthyl derivatives NDBD-Ph via drop casting (Fig. 2a). The doped thin films were fabricated by dispensing a homogeneous mixture of organic phosphor, PAN, and dimethylformamide (DMF) onto quartz substrates followed by annealing at 110 °C for 1 hour to eliminate DMF residues. Thermal annealing further enhances the matrix rigidity and the intermolecular interactions between the organic phosphor and the substrate, thereby improving the RTP properties of the films. Notably, all doped films exhibited a visible green afterglow under ambient conditions. Post UV irradiation removal, a pronounced green super-long afterglow lasting 5–12 seconds was observed in all samples (Fig. 2g). Remarkably, the racemic doped films exhibited superior luminous properties, with phosphorescence intensity approximately 2.3 times greater than that of the chiral enantiomer films (Fig. 2c). The estimated luminescence lifetimes, derived from fitting the fluorescence and phosphorescence decay curves, span nanoseconds to seconds for fluorescence and phosphorescence, respectively (Fig. 2e, f and Table S1†), attributed to the strong electrostatic interactions between electron donors and acceptors, confirming the persistent RTP characteristics. Among the systems, rac-NDBD-Ph@PAN demonstrated the longest phosphorescence lifetime (1357 ms@543 nm), likely owing to Wallach's rule. Furthermore, upon switching the excitation source from 254 nm to 365 nm, the long-lived green emission persisted, attributable to the axial molecular structure of the binaphthyl derivatives. The transient emission spectra of the doped films revealed main emission bands between 350 and 475 nm, with a shoulder peak at 290 nm, aligning with the blue fluorescence observed under UV light (Fig. 2b). After a 2 ms delay, rac-NDBD-Ph@PAN showed a dual emission peak between 465 and 700 nm, indicative of RTP (Fig. 2c). Similarly, R-NDBD-Ph@PAN and S-NDBD-Ph@PAN also exhibited dual RTP emission peaks, with lifetimes of 1068 ms and 1159 ms at 543 nm, respectively. The CIE coordinates of the phosphorescence spectra corresponded closely with the observed afterglow colors (Fig. 2d). These results demonstrate the successful achievement of long-lived RTP by doping binaphthyl derivatives in the PAN matrix. XRD analysis of the films showed no significant difference, indicating their amorphous nature (Fig. S1, ESI†). We tentatively hypothesize that the stronger RTP emission properties produced by the racemate-doped films are due to the tighter molecular packing of rac-NDBD-Ph molecules in the PAN matrix. SEM characterization of the films revealed large clusters in the rac-NDBD-Ph@PAN films (Fig. S2, ESI†). In addition, Fig. S3† shows the fluorescence emission spectra and ultraviolet visible absorption spectra of R-NDBD-Ph@PAN, S-NDBD-Ph@PAN, and rac-NDBD-Ph@PAN in solution state, respectively. We can see that when the three films are in a solution state, the fluorescence emission and ultraviolet absorption of rac-NDBD-Ph@PAN have some redshift compared to R-NDBD-Ph@PAN and S-NDBD-Ph@PAN. This may be due to the more compact arrangement of molecules in the film of rac-NDBD-Ph@PAN, which further proves our speculation. | ||
| Fig. 2 Fundamental photophysical properties of doped films based on NDBD-Ph. (a) Chemical structures of polymer PAN and organic phosphor NDBD-Ph. (b) Prompt and (c) delayed PL spectra of doped films (λEX = 300 nm, delay: 2 ms). (d) The CIE 1931 coordinates of doped films. (e) Time-resolved fluorescence decay curves at 392 nm (λEX = 300 nm). (f) Time-resolved phosphorescent decay curves at 543 nm (λEX = 300 nm). (g) Luminescence photographs of doped films under UV-254 nm irradiation and after removal of UV-254 nm irradiation with different duration times. The concentration of rac/R/S-NDBD-Ph for rac/R/S-NDBD-Ph@PAN is 2.5 wt%. | ||
We systematically explored the influence of the monomer molar ratio on the phosphorescence properties of polymers (Fig. S4, ESI†). Polymer films were synthesized with varying molar feed ratios ranging from 0.1% to 5%. As the molar ratio of rac-NDBD-Ph@PAN increased from 0.1% to 2.5%, the phosphorescence lifetime at 543 nm extended from 189 ms to 1357 ms. This trend suggests that both the rigidity of the polymer network and the exciton quenching process were substantially enhanced with an increase in PAN content. The integration of numerous cyanide groups into the polymer matrix restricts molecular motion through augmented intermolecular interactions between the chromophore and polymer chains. Concurrently, the suppression of triplet exciton quenching was observed, attributed to the broader dispersion of monomers within the polymer membranes. Conversely, at a 5% molar feed ratio of rac-NDBD-Ph@PAN, the lifetime of the resulting polymer at 543 nm slightly decreased from 1357 ms to 1056 ms. Furthermore, SEM images (Fig. S2c and d, ESI†) reveal that a more uniform cluster formation occurs in the rac-NDBD-Ph@PAN thin film at a doping ratio of 2.5%. These findings underscore the critical role of the optimal monomer molar ratio in maximizing the ultra-long phosphorescence lifetime of the targeted polymer under environmental conditions.
To validate the RTP characteristics of the doped films, phosphorescence emission and excitation spectra of the rac-NDBD-Ph@PAN films were compared with those of pure PAN films (Fig. S5a, ESI†). The emission bands of the doped films distinctly differ from those of the pure PAN film, indicating that the doped films' persistent RTP is not derived from the intrinsic triplet emission of the PAN polymer matrix. Subsequently, the phosphorescence emission spectra of the doped films were compared with those of the organic phosphor in both solution and solid states to ascertain the source of the doped films' RTP properties. As illustrated in Fig. S5b,† the phosphorescence emission bands of the doped rac-NDBD-Ph@PAN film and the organic phosphor in the solid state partially overlap. However, a significant overlap exists between the phosphorescence emission band of rac-NDBD-Ph solid powder in solution and the doped film's emission, suggesting that the doped film's persistent RTP can be attributed to the discrete molecular phosphorescence of the organic phosphor. Moreover, increasing the doping concentration from 0.5% to 5% did not significantly alter the UV absorption spectrum of rac-NDBD-Ph@PAN, implying efficient dispersion of the phosphor rac-NDBD-Ph within the PAN matrix (Fig. S6, ESI†). Hence, the RTP properties of the rac-NDBD-Ph@PAN doping film are linked to the discrete molecular phosphorescence of the rac-NDBD-Ph compound.
Next, we explored the phosphorescence enhancement effect in racemic organic phosphors. Fig. S7† presents the phosphorescence emission spectra of rac-NDBD-Ph and R/S-NDBD-Ph in their single crystal forms. Rac-NDBD-Ph exhibited the most pronounced phosphorescence emission at 579 nm, accompanied by a significant shoulder peak between 535 and 560 nm, whereas R/S-NDBD-Ph displayed a singular phosphorescence emission peak in the range of 535–600 nm. The peak phosphorescence emission intensity of the racemates was sixfold higher than that of the single crystals with absolute configurations. Furthermore, the maximum RTP emission peak of rac-NDBD-Ph showed a slight red shift compared to R/S-NDBD-Ph, likely due to denser stacking and enhanced intramolecular interactions. Notably, chiral R-NDBD-Ph and S-NDBD-Ph exhibited subtle differences in the spectral shape of their circularly polarized luminescence (CPL) signals, as demonstrated in Fig. S8,† with |gem| = 1.72 × 10−3, nearly mirroring each other. Fig. S9† displays the UV absorption spectra of rac-NDBD-Ph, R-NDBD-Ph, and S-NDBD-Ph in both dichloromethane solution and single crystal states. The UV absorption of the single crystal state shows a pronounced redshift compared to that of the solution state, additionally, the UV absorption of rac-NDBD-Ph in solution exhibited a slight redshift compared to R/S-NDBD-Ph, possibly attributed to closer molecular packing in the crystal structure.
Fig. 3 depicts the crystal structures of rac-NDBD-Ph and R-NDBD-Ph, detailed in Table S3, ESI.† Both belong to the monoclinic system yet reside in different space groups, P2_1n for rac-NDBD-Ph and C2y for R-NDBD-Ph, respectively. While rac-NDBD-Ph and its chiral enantiomers exhibit similar layered stacking, their molecular arrangements diverge. In the rac-NDBD-Ph's crystal, mirror-symmetric molecules R-NDBD-Ph and S-NDBD-Ph are symmetrically positioned on opposite sides at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio. Conversely, in R-NDBD-Ph crystals, identical chiral molecules pair up, forming rectangular solids within pure enantiomer's unit cells, leading to a layered structure akin to rac-NDBD-Ph (Fig. S10, ESI†). The filling patterns of racemic and chiral analogues differ slightly, significantly influencing molecular assembly within the crystals. NDBD-Ph structures adopt layered configurations with extensive intermolecular interactions, such as C–H⋯π, and both intra- and intermolecular hydrogen bonds (indicated by dotted lines in Fig. 3). This arrangement encourages the system to achieve maximal order, culminating in crystal formation.35,36 In contrast, rac-NDBD-Ph's arrangement is denser, characterized by closer intramolecular (2.022 Å, 2.156 Å) and intermolecular (1.887 Å) hydrogen bonds, alongside more C–H⋯π interactions (3.644 Å, 3.342 Å) between R- and S-configurations. The chiral R-NDBD-Ph, however, exhibits longer intramolecular (2.093 Å, 2.325 Å, 2.141 Å) and intermolecular (1.937 Å) hydrogen bonds and weaker C–H⋯π (2.882 Å) interactions. The denser molecular packing is posited to favor phosphorescence emission enhancement. Through analyzing the crystal structures of these samples, we deduce that C–H⋯π interactions positively influence phosphorescence intensity, while intra- and intermolecular hydrogen bonds strongly mitigate the non-radiative deactivation pathways of the triplet state, akin to matrix stacking mechanisms.37
:1 stoichiometric ratio. Conversely, in R-NDBD-Ph crystals, identical chiral molecules pair up, forming rectangular solids within pure enantiomer's unit cells, leading to a layered structure akin to rac-NDBD-Ph (Fig. S10, ESI†). The filling patterns of racemic and chiral analogues differ slightly, significantly influencing molecular assembly within the crystals. NDBD-Ph structures adopt layered configurations with extensive intermolecular interactions, such as C–H⋯π, and both intra- and intermolecular hydrogen bonds (indicated by dotted lines in Fig. 3). This arrangement encourages the system to achieve maximal order, culminating in crystal formation.35,36 In contrast, rac-NDBD-Ph's arrangement is denser, characterized by closer intramolecular (2.022 Å, 2.156 Å) and intermolecular (1.887 Å) hydrogen bonds, alongside more C–H⋯π interactions (3.644 Å, 3.342 Å) between R- and S-configurations. The chiral R-NDBD-Ph, however, exhibits longer intramolecular (2.093 Å, 2.325 Å, 2.141 Å) and intermolecular (1.937 Å) hydrogen bonds and weaker C–H⋯π (2.882 Å) interactions. The denser molecular packing is posited to favor phosphorescence emission enhancement. Through analyzing the crystal structures of these samples, we deduce that C–H⋯π interactions positively influence phosphorescence intensity, while intra- and intermolecular hydrogen bonds strongly mitigate the non-radiative deactivation pathways of the triplet state, akin to matrix stacking mechanisms.37
 | ||
| Fig. 3 Structural information of NDBD-Ph in crystals. Intermolecular interactions of a single molecule with adjacent ones of (a) rac-NDBD-Ph and (b) R-NDBD-Ph. | ||
To elucidate the potential mechanism behind RTP emission, polymethyl methacrylate (PMMA) and polystyrene (PS) were selected as matrices for comparative analysis due to their glass Tg being similar to that of the PAN matrix.38 Intriguingly, the doped film rac-NDBD-Ph@PS exhibited orange phosphorescence under ambient conditions (Fig. S11a, ESI†), whereas rac-NDBD-Ph@PMMA displayed only faint cyan phosphorescence under the same conditions (Fig. S11c, ESI†). Unlike PMMA or PS, PAN not only possesses inherent rigidity but also exhibits strong electron absorption capacity, attributed to its abundant cyanide groups. This characteristic likely facilitates strong electrostatic attraction interactions with organic phosphors. Conversely, the phenyl groups in PS and organic phosphors might engage in strong π–π stacking interactions. We hypothesize that initiating RTP properties in organic phosphors within doped thin films requires not merely the matrix's curing effect but also substantial interactions between the polymer matrix and organic phosphors.
To test this hypothesis, we utilized density functional theory to compare the SOC, energy levels, HOMO, LUMO, and ESP distribution between rac-NDBD-Ph and R-NDBD-Ph. The binding energy between these organic phosphors and the PAN matrix qualitatively illustrates the electrostatic interactions between the organic phosphors and the PAN matrix. Given the similar electronic configurations and crystal structures of the chiral molecules R-NDBD-Ph and S-NDBD-Ph, R-NDBD-Ph was selected for calculations. NDBD-Ph, containing Br atoms and other heavy atoms such as N and O, enhances SOC, which slightly increases after racemization (Fig. 4a). Fig. 4b illustrates that the ESP distribution on the surface of the PAN matrix encompasses both positive and negative charges. Additionally, rac-NDBD-Ph and R-NDBD-Ph exhibit positive ESP values along their backbones, facilitating strong electrostatic interactions with the PAN matrix. The backbone of rac-NDBD-Ph shows a higher positive charge, indicating stronger non-covalent interactions between rac-NDBD-Ph and the PAN matrix. The frequency analysis results for rac-NDBD-Ph@PAN and R-NDBD-Ph@PAN showed no imaginary frequencies, indicating that both sets of complexes are at the minimum point structure. The strong electrostatic interactions between the organic phosphors and the PAN matrix can also be clearly seen in the ESP surfaces of the two sets of compounds (Fig. S13, ESI†). The binding of rac-NDBD-Ph to the PAN matrix tends to be planar, resulting in stronger non-covalent interactions in the complex rac-NDBD-Ph@PAN. Notably, the optimal binding mode between the PAN matrix and the organic phosphor is defined by the combination energy (Fig. 4c); the results show that the cyano group on the PAN backbone points to the planar backbone of the organic phosphor, and the binding energies for rac-NDBD-Ph@PAN and R-NDBD-Ph@PAN doped films are −33.83 and −29.78 kcal mol−1, respectively, with the larger values observed when rac-NDBD-Ph is incorporated into the PAN matrix. This suggests that stronger electrostatic interaction occurs between rac-NDBD-Ph and the PAN matrix, leading to enhanced phosphorescence intensity and extended phosphorescence lifetime compared to the other films.
 | ||
| Fig. 4 Mechanism of long-lived RTP of polymeric systems. (a) Calculated SOC constant, calculated energy diagram and SOC (ξ) of small molecule rac-NDBD-Ph and R-NDBD-Ph. (b) Molecular structures and calculated ESP distribution of polymer PAN and two synthetic phosphors. (c) Calculated combining energy between organic phosphor-doped PAN matrix films. (d) Illustration of RTP emission based on the mechanism of electrostatic interaction. | ||
Based on these findings, we propose a mechanism for ultra-long RTP observed in amorphous polymers (Fig. 4d). Initially, under UV lamp irradiation, the axial chiral units efficiently generate excited states within the polymer via exciton coupling of adjacent chromophores. Subsequently, the abundance of cyanide groups within the polymer matrix facilitates the formation of triplet excitons by bridging singlet and triplet excited states. Concurrently, the polymer's rigid microenvironment restricts molecular motion, stabilizing the triplet excitons and enabling sustained organic phosphorescence under ambient conditions. To further validate the universality of our approach for achieving long-lived RTP at room temperature, we also explored the incorporation of organic phosphors with various terminal groups, including (R/S/rac)-NDBD-Ph-Ph, (R/S/rac)-NDBD-CH3, and (R/S/rac)-NDBD-O-CH3, into PAN (Fig. S14, ESI†) and PS matrices (Fig. S15, ESI†). As anticipated, the introduction of axial chiral structures into the polymer matrix resulted in polymers exhibiting prolonged phosphorescence lifetimes. Notably, the introduction of external racemates significantly amplified the phosphorescence emission intensity of the polymers.
Exploring the ultra-long phosphorescence emission of rac-NDBD-Ph@PAN, we investigated the luminous properties of these doped thin films in water. Typically, the presence of hydrophobic alkyl chains in polymers under high humidity conditions reduces water diffusion rates, enhancing the polymer's water resistance. As depicted in Fig. 5g, rac-NDBD-Ph@PAN exposed to water for one month still exhibited visible cyan RTP to the naked eye. Fig. S16† shows a slight decrease in the UV absorption intensity of the film after water immersion. To delve deeper into this unique luminous behavior, we examined the photophysical properties of the thin films immersed in water over a month (Fig. 5a). Interestingly, the overall intensity of the phosphor emission spectrum slightly diminished from its initial state, while the ratio of the RTP emission intensity (I514 nm/I543 nm) increased marginally. The afterglow color of the rac-NDBD-Ph@PAN film gradually shifted from green to cyan. Further investigating the water's impact on phosphorescence properties, we assessed the phosphorescence emission intensity of rac-NDBD-Ph@PAN thin films under dry and humid conditions over more than eight cycles (Fig. 5b). Comparing the emission intensities in these two states revealed minimal changes in maximum emission intensity, demonstrating the rac-NDBD-Ph@PAN thin films' remarkable stability and their rare turquoise afterglow emission in water within organic polymer systems. Surprisingly, control group films including rac-NDBD-Ph-PH@PAN, rac-NDBD-CH3@PAN, and rac-NDBD-O-CH3@PAN also exhibited high luminous stability in water (Fig. S17, ESI†). Conversely, after immersing the absolute configuration film (R/S)-NDBD-Ph@PAN in water for 30 minutes, only weak phosphorescence was emitted from the film's edge (Fig. S18, ESI†). This indicates that the stable, long-lived RTP of these doped films in water is not solely due to PAN waterproof properties but also suggests that racemization enhances the film's water stability.
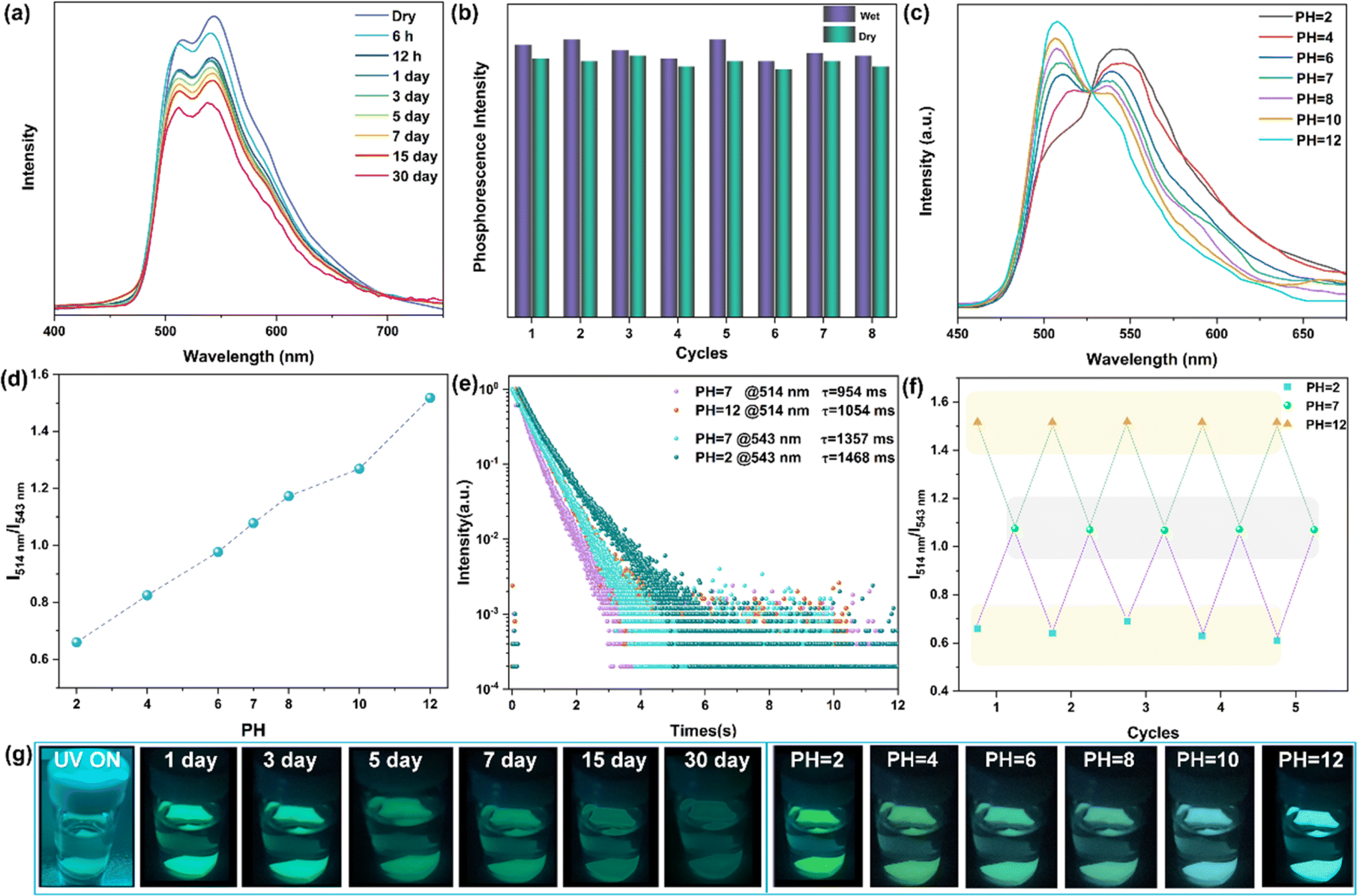 | ||
| Fig. 5 Photophysical properties of the rac-NDBD-Ph@PAN film in an aqueous environment. (a) Record the phosphorescence spectrum at regular intervals (λEX = 300 nm, delay: 2 ms). (b) Phosphorescence intensity evolution of the rac-NDBD-Ph@PAN film at 543 nm in two states (dry and wet) for eight cycles (λEX = 300 nm). (c) Phosphorescence spectra of the rac-NDBD-Ph@PAN film at different pH values (λEX = 300 nm). (d) Ratio of I514 nm/I543 nm phosphorescence intensity of the rac-NDBD-Ph@PAN film at different pH values (λEX = 300 nm). (e) Comparison of time-resolved phosphorescence decay curves at 543 nm at pH 2 versus 514 nm at pH 12 (λEX = 300 nm). (f) Cycling experiments on the I514 nm/I543 nm phosphorescence intensity ratio of rac-NDBD-Ph@PAN films at different pH values (λEX = 300 nm). (g) Photographs of afterglow after immersion in water for different times and pH values in aqueous solutions. | ||
Given the high luminous stability of rac-NDBD-Ph@PAN thin films in water, we investigated their RTP response to varying pH values. The delayed emission spectra revealed a distinct relationship between the intensity of two RTP emission bands and pH levels. As illustrated in Fig. 5c, in an aqueous solution at pH 6, the phosphorescence emission at 543 nm gradually increased, while emission at 514 nm decreased. As acidity heightened, the 514 nm RTP intensity further diminished, whereas the 543 nm RTP intensity rose, shifting the luminous color from cyan to green (Fig. 5g, right), and markedly enhancing the film's overall emission performance (Fig. S19, ESI†). Conversely, with increasing pH values, the 543 nm RTP intensity decreased and the 514 nm intensity increased, enabling acid–base–dependent continuous luminous color transition from pH 2 to pH 12. The emission properties of the films were significantly improved. Additionally, the ratio of RTP emission intensity (I514 nm/I543 nm) assessed the repeatability of the acid–base response (Fig. 5f). The rac-NDBD-Ph@PAN polymer films, subjected to cycles between pH 2 and pH 12 solutions, demonstrated consistent phosphorescence color changes with the acid-base response and reversible behavior over multiple cycles. The I514 nm/I543 nm ratios remained stable across identical pH levels (Fig. 5d). These results indicate that the doped polymer films possess robust reversible acid-base response characteristics, regaining their initial emission intensity and color after immersion in deionized water. This reproducibility is crucial for practical sensing applications. Phosphorescence lifetime attenuation curves under alkaline (pH 12) and acidic (pH 2) conditions showed a slower decay trend (Fig. 5e), RTP lifetimes to 1054 ms (514 nm) and 1468 ms (543 nm), respectively. This achievement marks a novel enhancement of phosphorescence emission under acid–base conditions, a rarity given molecular vibration and oxygen sensitivity, facilitating continuous emission over extended periods for the first time.39,40
In an attempt to elucidate the mechanism behind enhanced pH phosphorescence sensing, we sought to obtain the single crystal structures of the organic phosphor NDBD-Ph under varying conditions. Despite efforts, single crystal structures were successfully obtained only for the absolute configuration compound R-NDBD-Ph under acidic and alkaline conditions, denoted as R-NDBD-Ph-acid and R-NDBD-Ph-base, respectively. As depicted in Fig. S20 and Table S4,† R-NDBD-Ph-base exhibits a greater number and stronger intramolecular (2.032, 2.051, 2.061, 2.724 Å) and intermolecular (1.903 Å) hydrogen bonds compared to R-NDBD-Ph. Moreover, the dihedral angles of R-NDBD-Ph-acid and R-NDBD-Ph-base are 74.051° and 79.614°, respectively, exceeding that of R-NDBD-Ph (72.812°). The increased torsional angle, indicative of a larger non-coplanar structure, likely facilitates enhanced spin–orbit coupling (SOC), thereby promoting a more efficient ISC process. Density functional theory was employed to compare the SOC, energy levels, binding energy, and electrostatic potential (ESP) distribution between R-NDBD-Ph-acid, R-NDBD-Ph-base, and R-NDBD-Ph, along with their binding energy with the PAN matrix. As shown in Fig. S21,† the SOC constant and positive ESP values on the backbone of R-NDBD-Ph-acid and R-NDBD-Ph-base are marginally higher than those of R-NDBD-Ph, suggesting stronger non-covalent interactions between R-NDBD-Ph-acid/R-NDBD-Ph-base and the PAN matrix. The calculated binding energies are −31.17 kcal mol−1 and -33.21 kcal mol−1 for R-NDBD-Ph-acid and R-NDBD-Ph-base, respectively. This indicates enhanced electrostatic interactions between R-NDBD-Ph and the PAN matrix post acidic or alkaline treatment, leading to increased phosphorescence intensity and extended lifetime. These findings also imply that variations in intramolecular stacking configurations can influence the luminous properties of the system.
RTP materials exhibiting stable, high-efficiency, and long-lived phosphorescence emission are highly desirable for applications, with the simplicity of thin film fabrication enhancing their versatility. RTP systems that are large-scale, flexible, transparent, and easily fabricated are ideally suited for information storage and sensing applications. Leveraging the durable turquoise phosphorescence emission and pH-responsive RTP characteristics of rac-NDBD-Ph@PAN in water, we explored their potential in information security. Initially, using a rac-NDBD-Ph-doped PAN solution as ink, we wrote encrypted messages on commodities. After irradiation with UV-254 nm under ambient conditions the “LZU” green phosphorescence was visibly sustained for an extended period (Fig. 6a); spraying a small amount of water on the pattern shifted the encrypted information to a cyan afterglow, and subsequent heat treatment of the encrypted message shows the initial green afterglow. As shown in Fig. 6b, spraying an acidic or alkaline solution on the encrypted message changes the luminescent color from green to bright green or bright cyan. This multilevel information storage and the reversible encryption-decryption process underscore the potential for advanced anti-counterfeiting applications. Furthermore, the development of recyclable pH test papers underscores their significant practical utility.
 | ||
| Fig. 6 Application of long-lived RTP systems for multilevel information dynamic encryption. (a) Photomicrographs of luminescence based on rac-NDBD-Ph films under 254 nm UV light irradiation with the UV light turned off, sprayed with water and dried for anti-counterfeiting applications. (b) Anti-counterfeit applications of pH test strips prepared from rac-NDBD-Ph films. | ||
Conclusion
In this study, we devised a novel approach to significantly enhance the long-life RTP of polymer systems by incorporating racemates of chiral molecules NDBD-Ph, NDBD-Ph-Ph, NDBD-CH3, and NDBD-O-CH3 into a PAN matrix. The RTP intensity of the rac-NDBD-Ph@PAN thin films markedly increased, with a lifetime extending to 1357 ms, substantially surpassing that of R-NDBD-Ph@PAN (1068 ms) and S-NDBD-Ph@PAN (1159 ms). Further analysis, including single crystal X-ray diffraction and theoretical calculations (covering the spin–orbit coupling (SOC), energy levels, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), electrostatic potential (ESP) distribution, and binding energy analysis of the doped thin films), revealed that electrostatic interactions between the PAN matrix and rac-NDBD-Ph, along with tighter molecular packing, contribute to the improved RTP longevity. This study also demonstrates that optimizing the molecular stacking mode enhances the binding energy within the polymer system. Remarkably, a series of racemate polymer films maintained stable turquoise phosphorescence even after a month of water immersion, indicating exceptional water resistance and broadening the potential applications for racemically doped films. For the first time, we obtained polymer thin films rac-NDBD-Ph@PAN with superior pH-responsive properties in water. The unique RTP characteristics, electrostatic interactions, and formation of dense stacking configurations of racemates introduce an innovative strategy for achieving high-performance polymer RTP systems through non-covalent bond interactions, even in aqueous environments. This research advances the development of durable RTP systems in water, paving the way for sophisticated applications in multi-level information dynamic encryption, display, and sensing.Data availability
All experimental data are available from the corresponding author upon reasonable request.Author contributions
Zenggang Lin: conceptualization, validation, and writing (preparation of the original draft and reviewing). Peng Zhang: investigation and validation. Fuqiang Song, Yuzhu Yang, and Xuan Miao: software use and investigation. Weisheng Liu: supervision, conceptualization, visualization, project administration, and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21871122 and 21431002) and the Fundamental Research Funds for the Central Universities (Grant No. lzujbky-2021-kb17).References
- Y. Y. Zhao, J. H. Yang, C. Liang, Z. J. Wang, Y. F. Zhang, G. C. Li, J. M. Qu, X. Wang, Y. H. Zhang, P. Sun, J. B. Shi, B. Tong, H. Y. Xie, Z. X. Cai and Y. P. Dong, Angew. Chem., Int. Ed., 2024, 63, e202317431 CrossRef CAS PubMed.
- P. S. Cao, Y. Y. Wang, H. Y. Zheng and P. Wu, Aggregate, 2023, e468 Search PubMed.
- G. W. Xiao, Y. J. Ma, Z. H. Qi, X. Y. Fang, T. H. Chen and D. P. Yan, Chem. Sci., 2024, 15, 3625 RSC.
- B. Zhou, Z. Qi and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202208735 CrossRef CAS PubMed.
- C. Xing, B. Zhou, D. P. Yan and W. H. Fang, Adv. Sci., 2024, 2310262 CrossRef PubMed.
- C. Xing, Z. H. Qi, B. Zhou, D. P. Yan and W. H. Fang, Angew. Chem., Int. Ed., 2024, e202402634 Search PubMed.
- Z. Wu, K. Bergmann and Z. M. Hudson, Angew. Chem., 2024, e202319089 CAS.
- M. Gmelch, T. Achenbach, A. Tomkeviciene and S. Reineke, Adv. Sci., 2021, 8, 2102104 CrossRef CAS PubMed.
- C. Xing, B. Zhou, D. P. Yan and W. H. Fang, CCS Chem., 2023, 5, 2866–2876 CrossRef.
- B. Zhou, Z. H. Qi, M. Q. Dai, C. Xing and D. P. Yan, Angew. Chem., Int. Ed., 2023, 62, e202309913 CrossRef CAS PubMed.
- X. Y. Fang, Y. Q. Tang, Y. J. Ma, G. W. Xiao, P. Y. Li and D. Y. Yan, Sci. China Mater., 2023, 66, 664–671 CrossRef CAS.
- X. Y. Dou, Xu. Wang, X. L. Xie, J. Zhang, Y. Li and B. Tang, Adv. Funct. Mater., 2024, 2314069 CrossRef.
- R. Q. Zhao, C. Wang, K. Huang, L. Li, W. R. Fan, Q. X. Zhu, H. H. Ma, X. W. Wang, Z. H. Wang and W. Huang, J. Am. Chem. Soc., 2023, 145, 26532–26539 CrossRef CAS PubMed.
- H. Su, K. Hu, W. H. Huang, T. Wang, X. L. Zhang, B. Chen, H. Miao, X. P. Zhang and G. Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218712 CrossRef CAS PubMed.
- X. F. Wang, H. Y. Xiao, P. Z. Chen, Q. Z. Yang, B. Chen, C. H. Tung, Y. Z. Chen and L. Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- Q. Q. Xia, X. H. Wang, J. L. Yu, Z. Y. Chen, X. Y. Lou, X. M. Liu, M. X. Wu and Y. W. Yang, Aggregate, 2023, 4, e370 CrossRef CAS.
- X. F. Wang, W. J. Guo, H. Y. Xiao, Q. Z. Yang, B. Chen, Y. Z. Chen, C. H. Tung and L. Z. Wu, Adv. Funct. Mater., 2020, 30, 1907282 CrossRef CAS.
- L. K. Huang, B. Chen, X. P. Zhang, C. O. Trindle, F. Liao, Y. C. Wang, H. Miao, Y. Luo and G. Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16046 CrossRef CAS PubMed.
- T. H. Song, H. L. Liu, J. Ren and Z. W. Wang, Adv. Opt. Mater., 2024, 12, 2301215 CrossRef CAS.
- X. Y. Wu, C. H. Ma, J. C. Liu, Y. S. Liu, S. Luo, M. C. Xu, P. Wu, W. Li and S. X. Liu, ACS Sustainable Chem. Eng., 2019, 7, 18801–18809 CrossRef CAS.
- W. W. Xie, W. B. Huang, J. T. Li, Z. K. He, G. X. Huang, B. S. Li and B. Z. Tang, Nat. Commun., 2023, 14, 8098 CrossRef CAS PubMed.
- Y. F. Zhang, J. S. Li, J. L. Zhao, X. F. Li, Z. M. Wang, Y. C. Huang, H. K. Zhang, Q. Liu, Y. X. Lei and D. Ding, Angew. Chem., Int. Ed., 2024, 63, e20231389 Search PubMed.
- K. H. Ernst, Chimia, 2018, 72, 399–403 CrossRef CAS PubMed.
- P. F. She, J. Y. Duan, F. Y. Li, Y. X. Zhou, Y. Y. Qin, J. Wei, S. J. Liu, Y. Ma and Q. Zhao, Sci. China Mater., 2023, 66, 4749–4755 CrossRef CAS.
- X. H. Lin, J. Wang, B. B. Ding, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 3459 CrossRef CAS PubMed.
- Y. F. Zhang, Q. K. Sun, L. T. Yue, Y. G. Wang, S. W. Cui, H. C. Zhang, S. F. Xue and W. J. Yang, Adv. Sci., 2022, 9, 2103402 CrossRef CAS PubMed.
- L. Gao, J. Y. Huang, L. J. Qu, X. H. Chen, Y. Zhu, C. Li, Q. C. Tian, Y. L. Zhao and C. L. Yang, Nat. Commun., 2023, 14, 7252 CrossRef CAS PubMed.
- N. Gan, X. Zou, M. Y. Dong, Y. Z. Wang, X. Wang, A. Q. Lv, Z. C. Song, Y. Y. Zhang, W. Q. Gong, Z. Zhao, Z. Y. Wang, Z. X. Zhou, H. L. Ma, X. W. Liu, Q. S. Chen, H. F. Shi, H. H. Yang, L. Gu, Z. F. An and W. Huang, Nat. Commun., 2022, 13, 3995 CrossRef CAS PubMed.
- L. W. Ma, Y. W. Liu, H. Tian and X. Ma, JACS Au, 2023, 3, 1835–1842 CrossRef CAS PubMed.
- L. Gu, W. P. Ye, X. Liang, A. Q. Lv, H. L. Ma, M. J. Singh, W. Y. Jia, Z. C. Shen, Y. Guo, Y. R. Gao, H. Z. Chen, D. D. Wang, Y. L. Wu, J. W. Liu, H. Wang, Y. X. Zheng, Z. F. An, W. Huang and Y. L. Zhao, J. Am. Chem. Soc., 2021, 143, 18527–18535 CrossRef CAS PubMed.
- R. H. Liu, B. B. Ding, D. Z. Liu and X. Ma, Chem. Eng. J., 2021, 421, 129732 CrossRef CAS.
- C. Wang, L. J. Qu, X. H. Chen, Q. Zhou, Y. Yang, Y. Zheng, X. Zheng, L. Gao, J. Q. Hao, L. Y. Zhu, B. X. Pi and C. L. Yang, Adv. Mater., 2022, 34, 2204415 CrossRef CAS PubMed.
- K. Chen, Y. L. Wang, B. Chu, Z. S. Yan, H. R. Li, H. K. Zhang, S. L. Hu, Y. Z. Yang, B. Liu and X. H. Zhang, J. Mater. Chem. C, 2022, 10, 16420–16429 RSC.
- X. Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem, 2023, 7, 854 CrossRef CAS PubMed.
- J. D. Dunitz, Chem. Commun., 2003, 5, 545–548 RSC.
- M. A. F. Afzal, A. Sonpal, M. Haghighatlari, A. J. Schultz and J. Hachmann, Chem. Sci., 2019, 10, 8374–8383 RSC.
- G. J. Niday and P. G. Seybold, Anal. Chem., 1978, 50, 1577–1578 CrossRef CAS.
- H. Z. Wu, D. L. Wang, Z. Zhao, D. Wang, Y. Xiong and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2101656 CrossRef CAS.
- Q. Li, M. Zhou, Q. Yang, Q. Wu, J. Shi, A. Gong and M. Yang, Chem. Mater., 2016, 28, 8221–8227 CrossRef CAS.
- J. Joseph and A. A. Anappara, Phys. Chem. Chem. Phys., 2017, 19, 15137–15144 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2336502–2336505. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01719f |
| This journal is © The Royal Society of Chemistry 2024 |