
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface coating induced room-temperature phosphorescence in flexible organic single crystals†
Prodipta
Samadder
 a,
Khalid
Naim
a,
Subash Chandra
Sahoo
b and
Prakash P.
Neelakandan
*a
a,
Khalid
Naim
a,
Subash Chandra
Sahoo
b and
Prakash P.
Neelakandan
*a
aInstitute of Nano Science and Technology (INST), Knowledge City, Sector 81, Mohali 140306, India. E-mail: ppn@inst.ac.in
bDepartment of Chemistry, Panjab University, Sector 14, Chandigarh 160014, India
First published on 16th May 2024
Abstract
Materials exhibiting room temperature phosphorescence (RTP) are in high demand for signage, information encryption, sensing, and biological imaging. Due to weak spin–orbit coupling and other non-radiative processes that effectively quench the triplet excited states, RTP is sparsely observed in organic materials. Although the incorporation of a heavy atom through covalent or non-covalent modification circumvents these drawbacks, heavy-atom-containing materials are undesirable because of their deleterious side effects. Here, we designed and synthesized a new naphthalidenimine–boron complex as a coating material for the single crystals of 4,4′-dimethoxybenzophenone. The coated surface was observed to exhibit yellowish-green phosphorescence with ms lifetimes at ambient conditions through Förster resonance energy transfer (FRET). Importantly, the mechanical flexibility of the single crystals was observed to be retained after coating. The fluorescence-phosphorescence dual emission was utilised for colour-tunable optical waveguiding and anti-counterfeiting applications. As organic single crystals that can sustain mechanical deformations are emerging as the next-generation materials for electronic device fabrication, the flexible RTP organic crystals showing colour-tuneable optical waveguiding could be omnipotent in electronics.
Introduction
Phosphorescent materials are the key components of various display devices.1 Compared to inorganic phosphors, organic phosphorescent materials have the advantage of simple synthetic procedures, tuneable emission through functionalisation and higher energy efficiency.2–10 However, because of the weak spin–orbit coupling (SOC) in organic compounds, inter-system crossing from the excited singlet state to the triplet state is spin-forbidden, thereby reducing the efficiency of phosphorescence.11 Furthermore, it is well established that triplet excitons can break down through non-radiative transitions, delayed fluorescence, or triplet–triplet annihilation.12 Thus, phosphorescence from organic compounds is generally suppressed at ambient conditions and is often observed at low temperatures, thereby limiting their practical use.One commonly followed strategy to achieve phosphorescence at ambient conditions is to facilitate SOC by incorporating aromatic carbonyls or heavy atoms.13–18 Crystal engineering, doping, or supramolecular approaches are convenient compared to covalent modifications for incorporating these moieties because of the ease of preparation and the possibility of fine-tuning the structure through subtle protocol changes.19–22 These approaches further provide precise control over the positioning of the moieties, allow for tuning of material properties, and exhibit stimuli responsiveness owing to their dynamic behaviour. Among these, crystal engineering is preferable since crystals are inherently robust and show better device performance because of the ordered molecular packing.23–25 However, the hardness of single crystals curtails device fabrication and thus severely limits the use of single crystals in flexible electronic devices.
Recent advances have demonstrated that mechanically agile organic single crystals could be prepared through careful molecular engineering without compromising their physicochemical properties, and they have been successfully incorporated into flexible electronic platforms such as rollable displays, integrated optical waveguides, electronic skins, wearable devices, and stretchable sensors.26–33 The mechanical flexibility in organic single crystals arises from their unique molecular packing and structural features.34–36 Weak intermolecular forces such as van der Waals forces or hydrogen bonding allow relative motion between the molecules and facilitate flexibility. Crystals with layered or columnar structures often exhibit enhanced flexibility along specific directions and allow the materials to withstand bending, stretching, or twisting.37,38 Furthermore, surface coating is emerging as a facile strategy for improving the mechanical flexibility and physical properties of organic single crystals. Recent examples have demonstrated the suitability of this approach for modulating the flexibility, solubility, and magnetic properties of organic single crystals by coating with polymers and have led to key breakthroughs in optoelectronics.39–44
Organic single crystals that simultaneously exhibit mechanical flexibility and room temperature phosphorescence (RTP) would be priceless in developing flexible displays, sensing technologies, and biomedical imaging. Host–guest doping and insertion of halogen atoms are the commonly followed strategies for achieving RTP in single crystals.45–47 However, owing to the usage of heavy metals and halogens, these approaches result in potentially toxic and photolabile materials. Thus, heavy metal and halogen-free organic single crystals that exhibit mechanical flexibility and RTP would greatly aid the development of flexible display devices. Here, we report the modulation in the luminescence of the single crystals of 4,4′-dimethoxybenzophenone (1, Chart 1) through a surface coating approach. It was observed that the predominant blue fluorescence of the needle-like crystals if 1 was transformed to yellowish-green phosphorescence with a five-fold enhancement in lifetime at ambient conditions by surface coating with the tailor-made naphthalidenimine–boron complex 5. Importantly, the mechanical flexibility of the crystals of 1 was retained after the coating, which allowed the fabrication of colour-tuneable optical waveguides. Detailed investigations revealed that Förster resonance energy transfer (FRET) played a crucial role in the RTP process. Furthermore, a combination of RTP and flexible mechanical properties allowed the crystals to be used for anti-counterfeiting applications. Thus, the synergy between coatings and organic flexible crystals marks a significant advancement in materials research, fostering the development of next-generation, high-performance devices that will shape the future of technology.
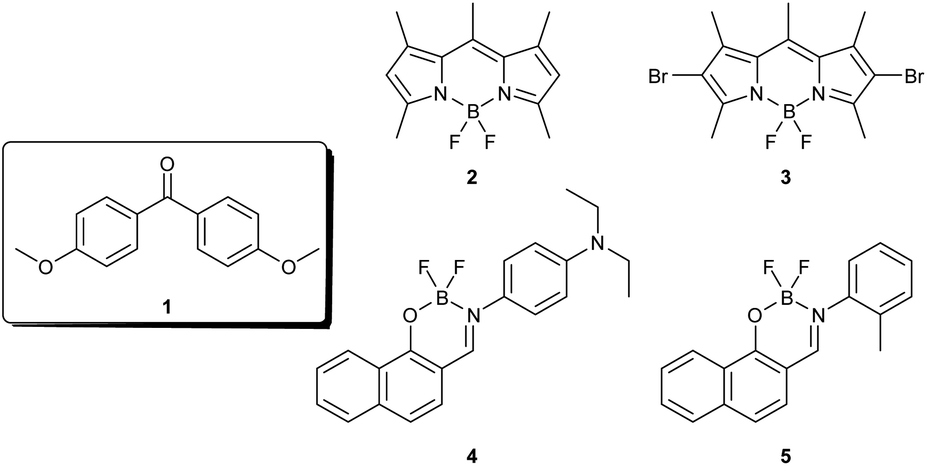 | ||
| Chart 1 Chemical structure of compounds 1–5 used in the present study. | ||
Derivatives of benzophenone are extensively explored as RTP materials because of their established photophysical properties and relatively long phosphorescence lifetimes.48–51 We selected 4,4′-dimethoxybenzophenone (1), 4-methoxybenzophenone (1′), 4-hydroybenzophenone (1′′) and 4,4′-diaminobenzophenone (1′′′) (Fig. S1†), out of which 1 and 1′′ yielded long transparent crystals. In most studies on RTP, co-crystallization is the commonly used strategy to impart phosphorescence to the crystals of benzophenone. Although the outcome of such investigations is generally positive, crystallization is a demanding process, and not all compounds are expected to yield single crystals of good quality. Moreover, obtaining co-crystals having RTP and mechanical flexibility is highly challenging. Here, we overcome these challenges by employing a surface coating protocol to impart visual RTP in flexible organic single crystals. The surface coating approach is hitherto unused for generating RTP and, importantly, is very simple and does not require any special experimental considerations. We further demonstrate that this protocol allows coating crystals completely or partially and thus could be used for tuning the emission colour.
Results and discussion
Long tape-like and needle shaped transparent crystals of 1 and 1′′, respectively, were obtained by layering hexane over a saturated solution of 1 or 1′′ in dichloromethane (DCM). The length of the crystals was in the order of a few centimetres, whereas the width was in the order of ten to two hundred microns (Fig. S2†). As shown in Fig. 1a, when subjected to an external force, the crystals of 1 exhibited elastic bending without breaking. At ambient temperature (25 ± 1 °C), we could successfully bend these crystals into a loop by applying a force to one end of the crystal with a paintbrush while fixing the other end with vacuum grease. Upon removal of the external force, the crystal instantly assumed its natural straight shape without breaking (Video S1†). The remarkable elasticity of the crystals was observable on the major faces (i.e. the (001)/(00–1) plane, Fig. S3†), and the reversible deformation could be repeated multiple times without damaging the crystal. It is noteworthy that the smooth surface of the crystals was retained in the bent state, as reflected in the SEM images (Fig. S4†). The bending strain of the crystals during the elastic deformation was calculated using the Euler–Bernoulli equation52 and was found to be 5.61 ± 0.12% for crystals having a thickness of ∼725 μm (Fig. S5†). However, under similar conditions, the crystals of 1′′ were observed to be brittle (Video S2†) and thus were not used for further experiments. | ||
| Fig. 1 (a) Images showing the sequence of elastic bending of the crystals of 1 viewed under UV light. (b) Crystal packing viewed along crystallographic b-axis showing CH–O, CH–π and π–π interactions. | ||
To gain insights into the elasticity of the crystals of 1 and to understand the correlation between the structure and the mechanical properties, we examined the crystal packing and molecular interactions in 1 (Fig. S6†). Compound 1 was observed to crystallise in the centrosymmetric triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (http://it.iucr.org/cgi-bin/gotosgtable.pl?number=2%26tabletype=S) with two crystallographically independent molecules in the asymmetric unit (Fig. 1b and Table S1†). The two independent molecules differed slightly in the orientations of the two benzene rings. The dihedral angle formed by C5–C6 and C9A-C14 rings was 56.36° and that between C20–C21 and C24–C29 planes was 52.83°. The presence of two different molecules in the asymmetric unit with two electronically different oxygens resulted in the formation of complex crystal packing held via various intermolecular interactions. On the basis of these, the elasticity exhibited by the crystals has been tentatively attributed to the intermolecular interactions such as C–H⋯O (H1B–O2 (2.635 Å), H30B–O5 (2.678 Å) and H15C–O6 (2.661 Å)) and C–H⋯π interactions. The percentage of weak intermolecular interactions in 1 was then determined through Hirshfeld surface and two-dimensional finger-print analyses (Fig. S7†).53 Analysing the Hirshfeld 2D-fingerprint plots revealed that C⋯H, O⋯H, C⋯O and H⋯H interactions governed the packing in compound 1. The contribution of each of these interactions is mentioned in Table S2.† The packing motif in the crystal states could be determined by estimating the ratio (ρ) between C⋯H and C⋯C interactions.54 The calculated value of ρ was more than 4.5, thereby suggesting a herringbone arrangement, which is a commonly observed pattern in polycyclic aromatic hydrocarbons.55
(http://it.iucr.org/cgi-bin/gotosgtable.pl?number=2%26tabletype=S) with two crystallographically independent molecules in the asymmetric unit (Fig. 1b and Table S1†). The two independent molecules differed slightly in the orientations of the two benzene rings. The dihedral angle formed by C5–C6 and C9A-C14 rings was 56.36° and that between C20–C21 and C24–C29 planes was 52.83°. The presence of two different molecules in the asymmetric unit with two electronically different oxygens resulted in the formation of complex crystal packing held via various intermolecular interactions. On the basis of these, the elasticity exhibited by the crystals has been tentatively attributed to the intermolecular interactions such as C–H⋯O (H1B–O2 (2.635 Å), H30B–O5 (2.678 Å) and H15C–O6 (2.661 Å)) and C–H⋯π interactions. The percentage of weak intermolecular interactions in 1 was then determined through Hirshfeld surface and two-dimensional finger-print analyses (Fig. S7†).53 Analysing the Hirshfeld 2D-fingerprint plots revealed that C⋯H, O⋯H, C⋯O and H⋯H interactions governed the packing in compound 1. The contribution of each of these interactions is mentioned in Table S2.† The packing motif in the crystal states could be determined by estimating the ratio (ρ) between C⋯H and C⋯C interactions.54 The calculated value of ρ was more than 4.5, thereby suggesting a herringbone arrangement, which is a commonly observed pattern in polycyclic aromatic hydrocarbons.55
It has recently been reported that the physicochemical properties of organic single crystals could be modulated by surface coatings.39 Although this approach has been of immense utility for tuning the mechanical, magnetic and optical properties of crystals, there are no reports of luminescence switching in organic single crystals through this strategy. We were thus intrigued to investigate if coating the single crystals of 1 using a suitable material would modulate its luminescence properties without interfering with their mechanical flexibility. We selected a few fluorescent dyes 2–5 (Chart 1) having complementary optical spectral features of 1 as potential surface coating agents. While compounds 2–4 are known for their excellent fluorescence properties that allowed them to be used for luminescence and photochemical applications,56,57 compound 5 was custom-made for this study. A methyl group was rationally incorporated at the ortho-position with respect to the imine bond in 5 so as to obstruct the free rotation of the phenyl group, which is anticipated to impart better luminescence features to 5 as compared to the structurally similar 4. The synthesis and characterisation of the naphthalidenimine–boron complex 5 are presented in the experimental section (Scheme S1 and Fig. S8†).
At the outset, we studied the photophysical properties of 1–5 in solution and the solid state (Fig. S9–S13†). In the solution state, compound 1 showed an emission in the UV region, whereas compounds 2–5 exhibited emission in the visible region. Among the various compounds, 5 exhibited the most intense emission in toluene with a fluorescence quantum yield of 0.63. We further studied the photophysical properties of 1–5 in the solid state. Compound 1 in the crystalline state exhibited both fluorescence and phosphorescence peaking at 439 and 442 nm, respectively with lifetimes of 2.35 ns and 0.425 ms (Fig. S14†).49 While the fluorescence of BODIPY derivatives 2 and 3 was negligible in the solid state due to aggregation-caused quenching, the naphthalidenimine-boron complexes 4 and 5 exhibited reasonably good fluorescence in the solid state with quantum yields of 0.16 and 0.19, respectively, in the powder form. In the crystalline state, the absorption and emission spectra of 5 were significantly broadened and red-shifted as compared to that in toluene (Δλ = 28 and 50 nm for absorption and emission maxima, respectively). The significant broadening and red-shift in the absorption and emission of the crystals of 5 indicate strong intralayer π–π interactions in the crystalline state. Time-resolved measurements showed an average lifetime of 23.3 ns for the crystals of 5.
Next, we studied the interaction between 1 and 2–5 in the solution and the solid state. Upon mixing a solution of 1 with that of 2–5 in toluene, no significant changes were observed in the absorption or emission features (Fig. S15–S18†), thereby indicating negligible interaction between 1 and 2–5 under these conditions. To study the interaction between 1 and 2–5 in the solid state, the crystals of 1 were coated with compounds 2–5 by drop-casting their solutions in DCM over the crystals of 1 (see Experimental section for details). Upon evaporation of the solvent at ambient conditions, a uniform layer of 2–5 was deposited on the surface of the crystals of 1. Optical microscopy and SEM analyses revealed that the surface and the crystallinity of the crystals remained intact after coating and the presence of 5 on the surface of the crystals of 1 was confirmed by EDX mapping, which showed the presence of elements such as carbon, nitrogen, oxygen, boron and fluorine on the surface of coated crystals (Fig. 2a–c and S19–S21†). It is noteworthy that by following this experimental protocol, we could coat the crystals of 1 completely or partially (Fig. 2d–g). We compared the X-ray diffraction patterns of the pristine crystals of 1 with that of the coated crystals to gain insights into the molecular level interactions in the coated crystals. It was observed that the diffraction peaks of the coated part becomes sharper and intense as compared to the uncoated part (Fig. S22 and S23†), thereby indicating that crystal packing was not altered by surface coating.
 | ||
| Fig. 2 (a–c) SEM images of the different faces of the crystals of 1 coated with 5. Digital photographs of the crystals of 1 that were (d) completely and (e, f) partially surface-coated with 5. (g) Fluorescence microscopic image of a partially surface-coated crystal. | ||
Next, we investigated the consequence of surface coating on the photophysical properties of 1–5. The absorption and emission properties of the crystals of 1 coated with 2–4 (Fig. S24 and S25†) resembled that of the individual components, thereby undermining electronic interactions between these compounds. However, the emission of the crystals of 1 coated with 5 (denoted as 5@1 hereafter) showed an emission maximum of 496 nm, which was different from the emission maxima of the individual compounds (Fig. 3a). Moreover, the difference in the emission of pristine and coated crystals was visible to the naked eye. As shown in Fig. 2d–f, the pristine crystals of 1 exhibited blue emission, whereas the sections coated with 5 exhibited green emission. Interestingly, visual observation of the coated crystals 5@1 in a UV cabinet under ambient conditions revealed an afterglow, thereby indicating phosphorescence (Fig. 3b). The RTP emanating from 5@1 was subsequently confirmed by recording the phosphorescence spectrum, which showed a maximum at 557 nm with a lifetime of 1.49 ms (Fig. 3c and Table S10†). The colour of the RTP emission was yellowish-green with Commission Internationale de l'Elcairage (CIE) colour coordinates of (0.39, 0.48) (Fig. S26†).
 | ||
| Fig. 3 (a) Emission spectra of the crystals of 1, 5, and 5@1. λex, 365 nm for 1 and 5@1, 405 nm for 5. (b) Photographs of 5@1 in a UV cabinet (λex, 365 nm) and after turning off the UV light. (c) Phosphorescence spectrum and (inset) lifetime decay of 5@1 measured at 557 nm. λex, 365 nm; delay, 0.1 ms. | ||
The absorption of the crystals of 5 is significantly overlapped by the fluorescence of the crystals of 1 (Fig. S27†). It is thus hypothesised that a Förster resonance energy transfer (FRET) from 1 to 5 would probably be the origin of RTP in the coated crystals 5@1. Upon coating the surface of the crystals of 1 with 5, it is assumed that the carbonyl group of 1 comes into close contact with the naphthalidenimine moiety, which triggers an efficient ISC leading to the formation of triplet excited states, thereby resulting in phosphorescence (Fig. S28†). The phosphorescence spectra at various excitation wavelengths were carefully examined further to corroborate the FRET mechanism (Fig. S29†). We observed that the phosphorescence intensity decreased significantly when the excitation wavelength was changed to 390 nm, which is at the edge of the absorption spectrum of 1. The phosphorescence peaks of 5@1 subsequently vanished when the excitation wavelength exceeded the absorption of 1. Further, excitation wavelengths above 390 nm that directly excited 5 resulted in fluorescence, thereby conclusively proving the role of interaction between 1 and 5 for RTP.
To validate FRET between 1 and 5, we carried out luminescence measurements of different mixtures of compounds 1 and 5. For this, solutions of 1 and 5 in DCM were mixed at varying concentrations, and thin films of the mixtures were prepared on glass slides through drop-casting. Under these conditions, 1 exhibited a phosphorescence spectrum with an emission maximum at 442 nm with an average lifetime of 0.425 ms (Fig. S30 and S31†), whereas compound 5 (in the absence of 1) did not exhibit phosphorescence. When the relative concentration of 5 in a mixture of 1 and 5 was gradually increased, the intensity and the lifetime of phosphorescence from 1 decreased proportionately. For instance, as the amount of 5 was increased to 1, 5, and 10 mol%, we observed a 90, 95, and 99% decrease in the emission intensity at 413 nm.‡ Moreover, the average lifetime of 1 at 413 nm reduced from 0.33 ms in the absence of 5 to 0.21 and 0.08 ms in the presence of 1 and 5 mol% of 5, respectively (Fig. S31a†). At 10 mol% of compound 5, the intensity decays of 1 were negligibly low to obtain meaningful data. Conversely, the phosphorescence intensity and lifetime at 600 nm were observed to increase with an increase in the relative concentration of 5 in a mixture of 1 and 5 (Fig. S31b†). At 1, 5 and 10 mol% of 5 in 1, we observed long-lived emission with lifetimes of 0.64, 0.94, and 1.01 ms at 600 nm.
Next, we studied the fluorescence excitation spectra of the mixtures of 1 and 5 at 413 and 600 nm, respectively. The fluorescence excitation spectrum of 1 at 413 nm in the absence of 5 exhibited a broad peak in the 350 to 390 nm region (Fig. S32†). When the relative concentration of 5 was increased to 1, 5, and 10 mol%, we observed that the emission intensity at 361 n decreased by 94, 96 and 98%, respectively. The excitation spectrum of 1 at 600 nm in the absence of 5 was similar to that collected at 413 nm with a broad peak in the 350 to 400 nm region. However, when the relative concentration of 5 was increased to 1, 5, and 10 mol%, we observed additional broad peaks from 420 to 460 nm. Moreover, the intensity of these peaks increased proportionately with increasing concentrations of 5. Thus, fluorescence excitation spectra suggest that the emission at 600 nm originates from the excited states that are generated by absorbing light in the 410–460 nm region. As 1 does not absorb above 400 nm and the relative concentration of 5 in the mixtures is significantly low (1, 5, or 10%), the enhanced fluorescence emission at 600 nm has been attributed to FRET from 1 to 5. We further calculated the efficiency of FRET from 1 to 5 which was calculated as 37 and 76% for 1 and 5 mol% of 5, respectively.
Under similar experimental conditions, we further recorded excitation spectra with a delay time of 0.1 ms (Fig. S33†). When the emission of 1 (in the absence of 5) was monitored at 413 nm, the observed spectra were similar with and without delays: peaks were observed till 390 nm which showed a significant decrease in intensity upon mixing with 5. However, the excitation spectra at 600 nm recorded after a delay were drastically different from those recorded without delay. As described above, the excitation spectra of mixtures of 1 and 5 at 600 nm recorded without delay showed intense peaks from 420 to 460 nm. Intriguingly, these peaks were absent in the excitation spectra when a delay time of 0.1 ms was applied and intense peaks were observed from 350 to 385 nm under these conditions (Fig. S33b†). These experiments thus suggest that the phosphorescence emission originates from 1.
It has been reported that the phosphorescence maximum of 1 varies as a function of temperature. While a predominant peak was observed at ∼440 nm at room temperature, lowering the temperature resulted in a red shift and at 77 K, the emission maximum was observed at ∼485 nm.49 The change in the emission maxima in benzophenone derivatives has been attributed to transitions occurring to different vibrational levels.58 In accordance with the literature reports and the excitation spectra with and without delays, we propose the following mechanism for the changes in the luminescence of 1 upon coating with 5. (i) Fluorescence: The emission at 498 nm of the coated crystals 5@1 originates from the singlet excited states of 5 facilitated by a FRET from 1. (ii) Room temperature phosphorescence: the emission at 557 nm of the 5@1 could be attributed to phosphorescence from 1 wherein 5 assists the formation of triplet excited states of 1.
Molecular simulations were further used to substantiate the proposed photophysical processes in the coated crystals. Time-dependent density-functional theory (TDDFT) calculations revealed an energy gap of 2.357 eV between the excited singlet and triplet states of compound 1 (Fig. S34a†). Due to the large bandgap between singlet and triplet states in compound 1, intersystem crossing is not feasible in 1 under ambient conditions. On the other hand, the energy gap between the singlet excited states of 1 and 5 was significantly lower at 0.461 eV. A smaller energy gap suggests the feasibility of energy transfer between them. Consequently, the triplet excited states of compound 1 were populated in the presence of compound 5. This suggests that the afterglow mechanism in this system relies on the T1 mediation pathway by 1. Additionally, the energy levels of the highest occupied and lowest unoccupied molecular orbitals of compound 1 was completely enclosed within the energy levels of compound 5 (Fig. S34b†). Consequently, the FRET process likely occurred in the coated crystals 5@1, where 1 serves as the energy donor and 5 as the acceptor.
Similar to 5, the absorption of the crystals of 4 was significantly overlapped by the emission of the crystals of 1 (Fig. S11†). To investigate if RTP could be achieved using 1 and 4, we carried out luminescence measurements of different mixtures of 1 and 4. When the relative concentration of 4 (in a mixture of 1 and 4) was increased, the phosphorescence intensity of 1 decreased gradually. As can be seen from Fig. S35,† as the amount of 4 was increased to 1, 5 and 10 mol%, we observed a 49, 83 and 96% decrease in the emission intensity at 413 nm. However, the extent of decrease in the case of 4 was significantly lower as compared to 5 wherein we observed 90, 95 and 99% quenching under similar conditions. Further, the calculated FRET efficiencies were 38 and 76% for a mixture of 1 with 5 mol% of 4 and 5, respectively. Thus, it is assumed that owing to the lower FRET efficiency, large amounts of 4 would be needed to impart RTP in the crystals of 1. However, during surface coating the amount of 4 deposited on the surface of 1 would be minute thus reducing the possibility of RTP. This assumption was confirmed using coated crystals 4@1 wherein we did not observe RTP (Video S3†).
Mechanical flexibility is a critical feature that defines the utility of any material for flexible optoelectronic applications. To study the effect of surface coating on the mechanical flexibility of the crystals of 1, we calculated the bending strain on the coated crystals after subjecting them to a three-point bending test. It was observed that the crystals of 1 retained their mechanical flexibility upon complete and partial coating (Fig. 4). The bending strain on the partially coated crystals was observed as 5.02 ± 0.09, 4.73 ± 0.12 and 4.27 ± 0.12% for 2–4, respectively (Fig. S36†). Similarly, the bending strain on 5@1 having a thickness of ∼725 μm was calculated as 5.20 ± 0.12 and 5.40 ± 0.13% for completely and partially coated crystals, respectively (Fig. S37†). These results indicate that the elasticity of the crystals was preserved after coating, although a marginal loss was observed upon complete coating. Next, we performed the nanomechanical analysis of the pristine crystals of 1 as well as the surface-coated crystals 5@1. Two-dimensional nanoindentation experiments carried out through the standard Oliver–Pharr method estimated the average values of E (elastic modulus – the resistance to elastic deformations), and H (hardness) of (001) face of the crystals of 1 as 2.93 ± 1.5 and 0.14 ± 0.10 GPa, respectively, indicating mild resistance to elastic deformations (Fig. S38†). In contrast, larger E and H values of 7.38 ± 2.7 and 0.22 ± 0.07 GPa, respectively, were obtained for the coated crystals 5@1, which suggested a greater sliding barrier followed by immediate release of stress, as also shown by the bending relaxation (Fig. S39†).
 | ||
| Fig. 4 Images viewed under UV light showing elastic bending of the crystals of 1 coated (a) completely and (b) partially with 5. | ||
Our photophysical investigations revealed that the emission colour of the crystals could be modulated from blue to green by surface coating. These crystals, thus, could be a step forward in the development of photonic circuits or security markings wherein switching of emission colour and lifetime is the key. We first tested the optical waveguide properties of straight and bent crystals to further explore the potential applications in this direction. As can be seen in Fig. S40,† when the straight crystals of 1 were exposed to UV light, the emission intensities at both ends were higher as compared to the bulk indicating the waveguiding ability of the crystals. After irradiating using a 365 nm pulse laser, the emission from one end of a crystal of 1 was recorded, which showed a maximum at 440 nm, which matched well with the emission of 1 in the solid state, thereby confirming active waveguiding by the crystals of 1. A similar experiment carried out using bent crystals of 1 showed that the bent crystals retained the active waveguiding properties. The spot of irradiation was then systematically varied, which revealed an inverse relation of the distance between the irradiated position and the emitting tip. The optical loss coefficient was then calculated using the fluorescence data and was found to be 1.054 ± 0.11 and 1.08 ± 0.05 dB mm−1 for straight and bent crystals of 1 (Fig. S40†), respectively, which indicated the negligible effect of mechanical deformation on optical waveguide properties.
Next, we were interested in investigating if the coated crystals 5@1 exhibited waveguide properties. When 5@1 was exposed to 365 nm light, the emission intensities at the tip of the crystals were much higher as compared to that in the bulk of the crystal, thereby indicating waveguide properties. Interestingly, we could tune the output colour by varying the spot of irradiation. For instance, when the partially coated crystal 5@1 was excited on the coated section, we observed bright green emission at the tip of the crystal (Fig. 5a(i), (ii) and S41b†). On the other hand, when the crystal was excited on the uncoated section, blue emission was observed at the tip of the crystal (Fig. 5b(iii)–(vi) and S41c†). It is important to note that the multi-colour emissions were exhibited in both the straight and bent states, as shown in Fig. 5. To corroborate the visual observations, we collected the emission spectra at one tip of the crystal (both in straight and bent states) after irradiating at different spots. When the partially coated crystal 5@1 was excited on the coated section, we observed emission at 562 and 486 nm corresponding to green emission, whereas an emission maximum at 441 nm corresponding to blue emission was observed upon irradiating the uncoated section. The emission intensity at the tip was observed to vary as a function of the distance between the tip and the irradiation spot. This data was used to calculate the optical loss coefficient for the straight and bent crystals, which were found to be 0.811 ± 0.06 and 1.23 ± 0.03 dB mm−1, respectively (Fig. S42†).
 | ||
| Fig. 5 (a, c) Digital photographs showing the different spots of irradiation of 365 nm laser on the straight and bent crystals, respectively. (b, d) Emission spectra measured at one tip of the crystal upon constantly changing the distance between the tip and the laser excitation positions on the straight and bent crystals, respectively. λex, 365 nm. | ||
Counterfeit currency, documents and electronics pose a significant threat to our daily lives. To prevent counterfeiting, various anti-counterfeiting strategies such as watermarks, holograms and barcodes are commonly resorted to. Luminescent anti-counterfeiting materials are attractive owing to their intense emission, multi-colour emission and long lifetimes. A majority of the recent examples of luminescent anti-counterfeiting materials are made of polymers, perovskites or lanthanide-based materials.59–62 The practical utility of these materials is limited because of their potential toxicity or low processability. Owing to their efficient optical waveguiding features, RTP and long lifetimes, we envisaged that the coated crystals (5@1) would be ideal for anti-counterfeiting applications. As shown in Fig. 6a, a tiny crystal of 5@1 was embedded in a paper using a commercially available bonder and the paper was further used for printing. We observed that the bright yellowish-green RTP of the crystals was retained after embedding in the paper and after printing (Fig. 6), thereby demonstrating the utility of 5@1 as a security marker. Further, the printed paper was subjected to harsh conditions like folding, exposure to water and heat to examine if the security marking would retain its luminescence during practical usage. It was observed that the RTP from the coated crystals was retained under these conditions, and importantly, the crystals could withstand the harsh real-world conditions for several cycles (Fig. 6, S43 and S44†), thereby demonstrating their effectiveness as a multi-mode security marker. Moreover, the luminescence and mechanical flexibility of the printed documents were retained for several months (Fig. S45†), thereby exemplifying the potential of the coated crystals for anti-counterfeiting applications.
 | ||
| Fig. 6 Digital photographs under different experimental conditions of a document in which the coated crystal 5@1 was embedded. The length of the crystal embedded in the document is 1 cm. | ||
Conclusions
In conclusion, we studied the luminescence and mechanical properties of centimetre-sized organic single crystals of 4,4-dimethoxybenzophenone (1) surface-coated with a custom-made naphthalidenimine–boron complex (5). The pristine crystals of 4,4-dimethoxybenzophenone showed predominantly blue fluorescence under ambient conditions and high elasticity with a strain load of ∼5.61%. By surface coating the crystals of 1 with 5, we observed that the blue fluorescence of the crystals of 1 could be transformed into bright yellowish-green phosphorescence having ms lifetimes under ambient conditions. The close contact induced by the surface coating is assumed to result in an efficient FRET process leading to room-temperature phosphorescence. Using their flexibility and RTP, the straight and bent crystals were utilised for optical waveguiding with colour-tuning. Furthermore, the combined optical and mechanical flexibility of the organic single crystals were useful for anti-counterfeiting applications wherein documents embedding the crystals could sustain real-world conditions like folding and washing. The integration of phosphorescent properties with the mechanical flexibility of organic single crystals holds great promise for applications in advanced display technologies, sensing devices, and lighting systems. This work thus lays the platform for an exciting endeavour to explore new frontiers and to develop innovative devices that can unlock the full potential of organic flexible crystals.Data availability
Crystallographic data for 1 has been deposited at CCDC under 2299361 and can be obtained from https://www.ccdc.cam.ac.uk/. All the experimental data are provided in the ESI.†Author contributions
PS, KN and PPN designed the experiments; PS and KN carried out the experiments; SCS collected, solved, and refined the single-crystal X-ray diffraction data; PS and PPN analyzed the data and cowrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Council of Scientific & Industrial Research (02(0438)/21/EMR-II) for financial support and the Indian Institute of Technology Bombay for analytical facilities.Notes and references
-
S. Huo and Y. Li, Phosphorescent Materials, American Chemical Society, 2023 Search PubMed
.
- Y. Zhang, J. Li, J. Zhao, X. Li, Z. Wang, Y. Huang, H. Zhang, Q. Liu, Y. Lei and D. Ding, Angew. Chem., Int. Ed., 2024, 63, e202313890 CrossRef CAS PubMed
- Y. Liang, P. Hu, H. Zhang, Q. Yang, H. Wei, R. Chen, J. Yu, C. Liu, Y. Wang, S. Luo, G. Shi, Z. Chi and B. Xu, Angew. Chem., Int. Ed., 2024, 63, e202318516 CrossRef CAS PubMed
- Q. Zhou, C. Yang and Y. Zhao, Chem, 2023, 9, 2446–2480 CAS
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 186 CrossRef CAS PubMed
- M. Godumala, A. V. Kumar and R. Chandrasekar, J. Mater. Chem. C, 2021, 9, 14115–14132 RSC
- A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. Santhosh Babu, Chem. Sci., 2021, 12, 4216–4236 RSC
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS
- L. Huang, C. Qian and Z. Ma, Chem.–Eur. J., 2020, 26, 11914–11930 CrossRef CAS PubMed
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed
- J. Yu, H. Ma, W. Huang, Z. Liang, K. Zhou, A. Lv, X.-G. Li and Z. He, JACS Au, 2021, 1, 1694–1699 CrossRef CAS PubMed
- L. G. Franca, P. L. Dos Santos, P. Pander, M. G. B. Cabral, R. Cristiano, T. Cazati, A. P. Monkman, H. Bock and J. Eccher, ACS Appl. Electron. Mater., 2022, 4, 3486–3494 CrossRef CAS PubMed
- Z. Zhou, X. Xie, Z. Sun, X. Wang, Z. An and W. Huang, J. Mater. Chem. C, 2023, 11, 3143–3161 RSC
- Y. Lei, W. Dai, G. Li, Y. Zhang, X. Huang, Z. Cai and Y. Dong, J. Phys. Chem. Lett., 2023, 14, 1794–1807 CrossRef CAS PubMed
- S. Datta and J. Xu, ACS Appl. Bio Mater., 2023, 6, 4572–4585 CrossRef CAS PubMed
- H. Shi, W. Yao, W. Ye, H. Ma, W. Huang and Z. An, Acc. Chem. Res., 2022, 55, 3445–3459 CrossRef CAS PubMed
- X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Small, 2022, 18, 2104073 CrossRef CAS PubMed
- P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 CAS
- S. Garain, S. N. Ansari, A. A. Kongasseri, B. Chandra Garain, S. K. Pati and S. J. George, Chem. Sci., 2022, 13, 10011–10019 RSC
- E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977–9981 CrossRef CAS PubMed
- S. Jena, J. Eyyathiyil, S. K. Behera, M. Kitahara, Y. Imai and P. Thilagar, Chem. Sci., 2022, 13, 5893–5901 RSC
- T. Su, Y.-H. Liu, Y. Chen and Y. Liu, J. Mater. Chem. C, 2022, 10, 2623–2630 RSC
- Y. Chen, Z. Chang, J. Zhang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 22424–22431 CrossRef CAS PubMed
- H. Jiang and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 1408–1428 CrossRef CAS PubMed
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed
- K. Liu, B. Ouyang, X. Guo, Y. Guo and Y. Liu, NPJ Flex. Electron., 2022, 6, 1 CrossRef CAS
- B. Fu, F. Yang, L. Sun, Q. Zhao, D. Ji, Y. Sun, X. Zhang and W. Hu, Adv. Mater., 2022, 34, 2203330 CrossRef CAS PubMed
- A. J. Thompson, A. I. C. Orué, A. J. Nair, J. R. Price, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021, 50, 11725–11740 RSC
- S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh, A. Mondal, B. B. Khatua, N. Ghosh and C. M. Reddy, Science, 2021, 373, 321–327 CrossRef CAS PubMed
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 CrossRef CAS PubMed
- M. Annadhasan, S. Basak, N. Chandrasekhar and R. Chandrasekar, Adv. Opt. Mater., 2020, 8, 2000959 CrossRef CAS
- W. Shi, Y. Guo and Y. Liu, Adv. Mater., 2020, 32, 1901493 CrossRef CAS PubMed
- Y. Wang, L. Sun, C. Wang, F. Yang, X. Ren, X. Zhang, H. Dong and W. Hu, Chem. Soc. Rev., 2019, 48, 1492–1530 RSC
- A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS PubMed
- C. M. Reddy, G. R. Krishna and S. Ghosh, CrystEngComm, 2010, 12, 2296–2314 RSC
- C. M. Reddy, R. C. Gundakaram, S. Basavoju, M. T. Kirchner, K. A. Padmanabhan and G. R. Desiraju, Chem. Commun., 2005, 3945–3947 RSC
- K. Naim, M. Singh, S. Sharma, R. V. Nair, P. Venugopalan, S. C. Sahoo and P. P. Neelakandan, Chem.–Eur. J., 2020, 26, 11979–11984 CrossRef CAS PubMed
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed
- X. Yang, M. B. Al-Handawi, L. Li, P. Naumov and H. Zhang, Chem. Sci., 2024, 15, 2684–2696 RSC
- X. Yang, L. Lan, X. Pan, Q. Di, X. Liu, L. Li, P. Naumov and H. Zhang, Nat. Commun., 2023, 14, 2287 CrossRef CAS PubMed
- X. Yang, L. Lan, X. Pan, X. Liu, Y. Song, X. Yang, Q. Dong, L. Li, P. Naumov and H. Zhang, Nat. Commun., 2022, 13, 7874 CrossRef CAS PubMed
- X. Yang, L. Lan, L. Li, X. Liu, P. Naumov and H. Zhang, Nat. Commun., 2022, 13, 2322 CrossRef CAS PubMed
- P. Marandi, K. Naim, K. Parida and P. P. Neelakandan, ACS Materials Lett., 2022, 4, 2499–2505 CrossRef CAS
- L. Lan, H. Liu, X. Yu, X. Liu and H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11283–11287 CrossRef CAS PubMed
- Y. Xia, C. Zhu, F. Cao, Y. Shen, M. Ouyang and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202217547 CrossRef CAS PubMed
- K. Huang, L. Song, K. Liu, A. Lv, M. Singh, K. Shen, J. Shen, J. Wang, H. Wang, H. Shi, H. Ma, M. Gu, G. Sun, W. Yao, Z. An and W. Huang, NPJ Flex. Electron., 2021, 5, 21 CrossRef CAS
- H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 RSC
- Y. Su, M. Wu, G. Wang, J. Li, X. Chen, X. Li, G. Wang and K. Zhang, Chem. Commun., 2023, 59, 1525–1528 RSC
- A. Huang, Y. Fan, K. Wang, Z. Wang, X. Wang, K. Chang, Y. Gao, M. Chen, Q. Li and Z. Li, Adv. Mater., 2023, 35, 2209166 CrossRef CAS PubMed
- D. Wang, Y. Xie, X. Wu, Y. Lei, Y. Zhou, Z. Cai, M. Liu, H. Wu, X. Huang and Y. Dong, J. Phys. Chem. Lett., 2021, 12, 1814–1821 CrossRef CAS PubMed
- Y. Chen, Y. Xie, H. Shen, Y. Lei, Y. Zhou, W. Dai, Z. Cai, M. Liu, X. Huang and H. Wu, Chem.–Eur. J., 2020, 26, 17376–17380 CrossRef CAS PubMed
- J.-R. Wang, M. Li, Q. Yu, Z. Zhang, B. Zhu, W. Qin and X. Mei, Chem. Commun., 2019, 55, 8532–8535 RSC
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Cryst., 2021, 54, 1006–1011 CrossRef CAS PubMed
- L. Loots and L. J. Barbour, CrystEngComm, 2012, 14, 300–304 RSC
- A. M. Philip, S. K. Manikandan, A. Shaji and M. Hariharan, Chem.–Eur. J., 2018, 24, 18089–18096 CrossRef CAS PubMed
- A. Rahman, P. P. Praveen Kumar, P. Yadav, T. Goswami, A. Shanavas, H. N. Ghosh and P. P. Neelakandan, ACS Appl. Nano Mater., 2022, 5, 6532–6542 CrossRef CAS
- S.-J. Kim, M. Choi, G. Hong and S. K. Hahn, Light Sci. Appl., 2022, 11, 314 CrossRef CAS PubMed
- T. V. Bezrodna, G. V. Klishevich, N. D. Curmei, G. M. Telbiz, N. I. Lebovka and V. I. Melnyk, J. Appl. Spectrosc., 2015, 82, 567–572 CrossRef CAS
- Z. Hu, K. Nie, X. Wang, X. Duan, R. Zhou, M. Wu, X. Ma, X. Zhang, L. Wang, L. Mei and H. Wang, Nanoscale, 2023, 15, 4893–4898 RSC
- M. Patel, R. Patel, C. Park, K. Cho, P. Kumar, C. Park and W.-G. Koh, Nano Convergence, 2023, 10, 21 CrossRef CAS PubMed
- Y. Liu, S. Liang, C. Yuan, A. Best, M. Kappl, K. Koynov, H. Butt and S. Wu, Adv. Funct. Mater., 2021, 31, 2103908 CrossRef CAS
- S. Zhao, M. Gao and J. Li, J. Lumin., 2021, 236, 118128 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, crystal data and additional figures. CCDC 2299361. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01708k |
| ‡ Experiments and analysis were focused on the peaks at 413 and 600 nm as they predominantly corresponded to the exclusive emission of 1 and 5, respectively. |
| This journal is © The Royal Society of Chemistry 2024 |