
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceForging structural complexity: diastereoselective synthesis of densely substituted β-lactams with dual functional handles for enhanced core modifications†
Agustin M.
Rodriguez Treviño
 a,
Pierre
Loch-Temzelides‡
a,
Sanjay
Pandiri‡
a,
Justin K.
Kirkland
b,
Michael T.
Davenport
b,
Ulises
Aguinaga
c,
Muhammed
Yousufuddin
c,
Daniel H.
Ess
b and
László
Kürti
*a
a,
Pierre
Loch-Temzelides‡
a,
Sanjay
Pandiri‡
a,
Justin K.
Kirkland
b,
Michael T.
Davenport
b,
Ulises
Aguinaga
c,
Muhammed
Yousufuddin
c,
Daniel H.
Ess
b and
László
Kürti
*a
aDepartment of Chemistry, Rice University, Houston, Texas 77030, USA. E-mail: kurti.laszlo@rice.edu
bDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84604, USA
cDepartment of Natural Sciences, University of North Texas at Dallas, Dallas, Texas 75241, USA
First published on 8th August 2024
Abstract
The preparation of the β-lactam motif containing both C–Br and N–O bonds as functional handles remains an unmet synthetic challenge. Described herein is a novel and highly diastereoselective NBS-mediated cyclization of N-alkoxy α,β-unsaturated silyl imino ethers to furnish nearly three dozen α-bromo N-alkoxy β-lactams. The reaction gives rapid and convenient access to structurally diverse monocyclic, spirocyclic and fused β-lactams in moderate to good yields. The two functional handles were shown to be useful for the further elaboration of the β-lactam core.
Introduction
The development of efficient synthetic routes to β-lactams (2-azetidinones) has been vigorously pursued by both synthetic and medicinal chemists since Fleming's serendipitous discovery of penicillin in 1928.1 These compounds are the most widely used class of antibiotics making up to 65% of the total antibiotics market in the US.2 Monobactams or monocyclic β-lactams, a special subgroup distinguished by the lack of a fused ring to the 2-azetidinone core, have shown a broad spectrum of activity against Gram-negative bacteria. The mechanism of action of monobactams, which is similar to that of the mechanism of bicyclic β-lactam antibiotics, involves the inhibition of peptidoglycan biosynthesis in bacterial cell walls.3 Aztreonam (Fig. 1) is currently the only monobactam approved by the FDA. Due to the high efficacy of these compounds against bacterial pathogens, and with aztreonam being the first of its class of antibiotics, there is an urgent demand for the creation of efficient synthetic methods to produce monocyclic β-lactams.4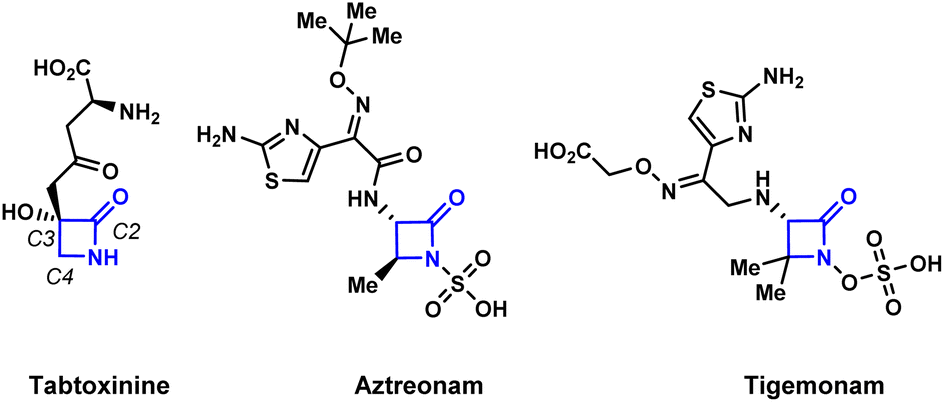 | ||
| Fig. 1 Representative examples of biologically active monocyclic β-lactams. | ||
A number of efficient synthetic methods have been developed for the preparation of the β-lactam scaffold.5 The most common disconnections (Scheme 1A) include the thermal [2 + 2] cycloadditions of either ketenes with imines6 or isocyanates with alkenes.7 Despite the existence of these methods, the synthesis of densely functionalized β-lactams remain a very difficult task. Additionally, none of these synthetic protocols allow the convenient preparation of N-alkoxy β-lactams, which are useful synthons for the further functionalization of the azetidinone ring.8 The Miller group pioneered the employment of the Mitsunobu reaction or intramolecular SN2 to effect the cyclization of the hydroxamate esters for the synthesis of N-alkoxy β-lactams (Scheme 1B).9 However, densely substituted β-lactams are difficult to prepare by these two methods due to severe steric crowding.
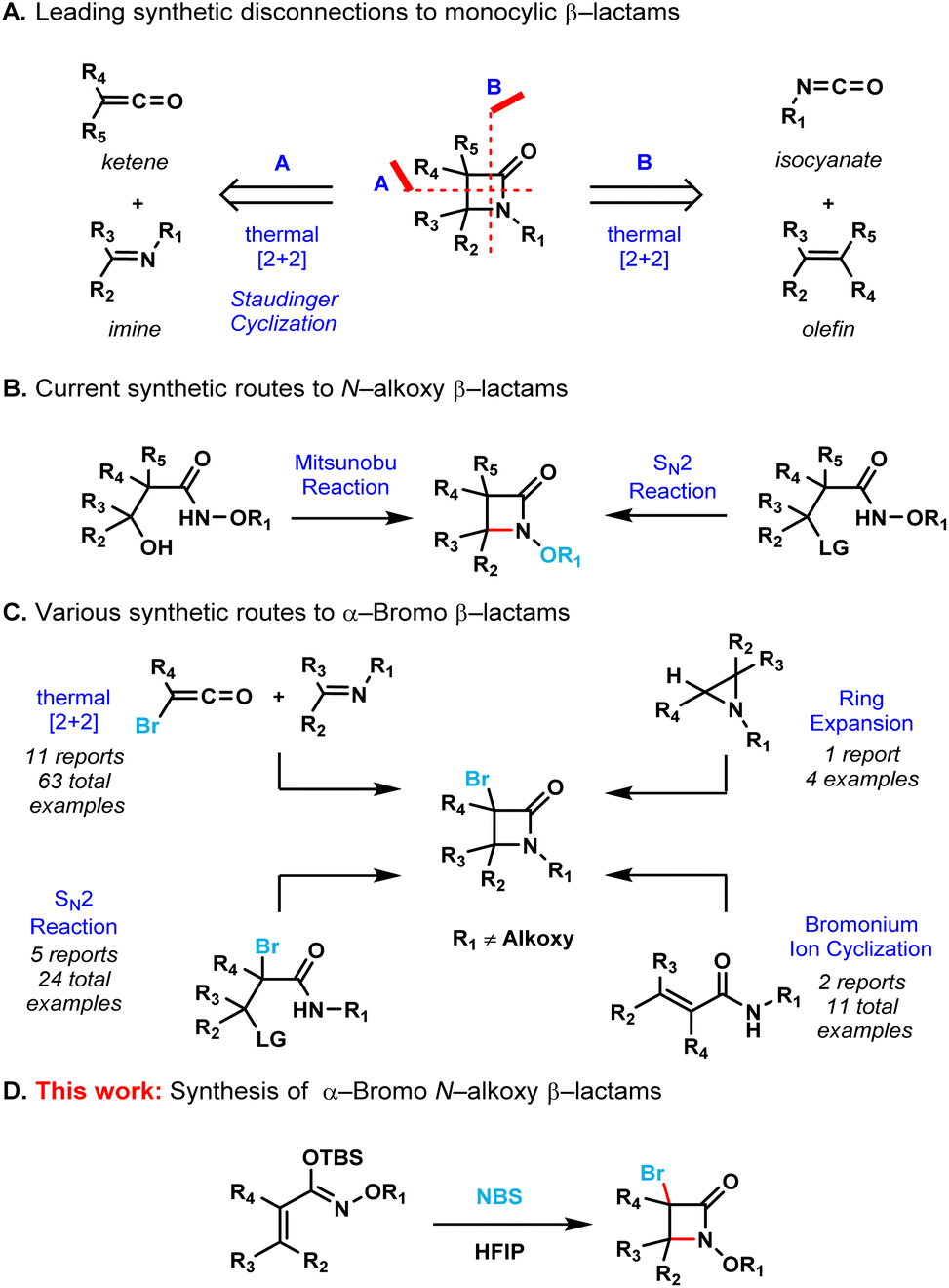 | ||
| Scheme 1 Methods for the synthesis of monocyclic β-lactams. | ||
At the same time, α-bromo β-lactams have proven to be versatile synthons for the further functionalization of the β-lactam core via cross-coupling reactions,10 metal-halogen exchange followed by trapping with activated electrophiles11 and also via nucleophilic bimolecular substitutions (SN2).12 The known synthetic protocols for the preparation α-bromo β-lactams include thermal [2 + 2] cycloadditions of imines with bromoketenes or intramolecular SN2 cyclization of amides (Scheme 1C).13 These methods are limited to substrates that already contain the bromine atom in their scaffolds. The ring-expansion of aziridines in the presence of a halogenating agent14 and the intramolecular cyclization of amides via a bromonium ion intermediate15 are clever strategies that allow the incorporation of a bromine atom into the β-lactam core. However, none of these protocols allow the expedient synthesis of β-lactams containing both C–Br and N–O bonds as functional handles.
Therefore, we decided to develop an operationally simple strategy for the synthesis of monocyclic α-bromo N-alkoxy β-lactams. The presence of both the bromine at the C3 position and the alkoxy substituent on the nitrogen allow for the further diversification of the β-lactam core to access potentially bioactive compounds quickly and efficiently.
Results and discussion
We initially attempted the preparation of these target compounds with the already known strategies for β-lactam synthesis (Scheme 1A). Applying the classical conditions of the Staudinger ketene cycloaddition reaction (i.e., [2 + 2]-cycloaddition between ketenes + imines) for the [2 + 2]-cycloaddition between ketenes and oximes failed to provide the corresponding N-alkoxy β-lactams. Another attempted [2 + 2] cycloaddition, between N-alkoxy isocyanates and olefins, similarly failed to provide the desired monocyclic N-alkoxy β-lactam core. We then shifted gears to a bromonium ion-mediated cyclization from α,β-unsaturated hydroxamate esters (Scheme 1C) as previously reported by Naskar and coworkers for the synthesis of N–H β-lactams.15 However, all attempts led to the formation of only the corresponding α,β-unsaturated ester (i.e., upon dimerization of the substrate and release of nitrogen gas as previously reported by Zhao et al. and Chattopadhyay et al., see ESI page S4†).16 With these results in hand, we proposed a bromonium ion-mediated cyclization approach starting from N-alkoxy α,β-unsaturated silyl imino ethers as substrates instead of α,β-unsaturated hydroxamate esters – this route circumvents the undesired dimerization pathway due to the absence of N–H bonds, while preserving N-nucleophilicity.We began our preliminary studies using 1.2 equivalents of N-bromosuccinimide (NBS) as the electrophilic source of bromine and acetonitrile as the solvent at 0.2 M concentration under reflux conditions (Table 1, entry 1). Gratifyingly, our proof-of-concept experiment afforded the desired 3,4-trans disubstituted monocyclic N-methoxy β-lactam 2a in 30% isolated yield. Only the trans-product was observed, which is in full agreement with the well-defined stereochemistry of the starting material (E)-silyl imino ether 1a. Our initial optimization started with solvent screening (entries 2–8) which showed that the choice of solvent was key for this transformation. When nonpolar solvents (i.e. toluene and dichloromethane) were used, the starting material remained intact even when the reaction was kept at reflux temperatures overnight. When polar solvents (i.e. tetrahydrofuran, isopropanol and methanol) were used, the starting material was fully consumed and decomposition products were observed, however, no cyclized product was detected. When trifluoroethanol was employed (entry 7) product 2a was obtained in 31% isolated yield. Finally, when hexafluoroisopropanol (HFIP) was employed, the reaction time was reduced to only 1 hour at room temperature affording product 2a in 67% isolated yield (entry 8). A result of an extensive solvent screening can be found in the ESI (page S4†). Solvent mixtures (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were also explored to decrease the amount of HFIP used in the reaction (entries 9–11). Notably, a 1:1 mixture of HFIP:DCM provided the desired product 2a with no significant change in isolated yield (70%). Hence, small-scale reactions were performed using only pure HFIP as the solvent, while large-scale reactions were conducted using solvent mixtures (entry 10). Next, we explored the effect of varying the equivalents of NBS on the outcome of the reaction (entries 12–14). Increasing the amount of NBS (1.0 → 1.5 equivalents; entry 13) led to an improvement in the isolated yield of the desired product (70 → 80%). However, further increase in the amount of NBS used (1.5 → 2.0 equivalents; entry 14) the isolated yield was slightly reduced (80 → 76%), which led us to select 1.5 equivalents of the bromine source for the optimized reaction conditions. Subsequently, we decided to explore the effect of the concentration (entries 15–17) on the reaction outcome. When the reaction was diluted (i.e., 0.2 → 0.1 M concentration), the isolated yield of the desired product remained at 80%. On the other hand, when the reaction was performed under more concentrated conditions (i.e., 0.5 M and 1.0 M), the product was obtained in reduced isolated yields (i.e., 68% and 47%, respectively). After screening eight different brominating, chlorinating and iodinating agents, we concluded that NBS was the best halogen source for the transformation (see ESI, Table 2†).
:1) were also explored to decrease the amount of HFIP used in the reaction (entries 9–11). Notably, a 1:1 mixture of HFIP:DCM provided the desired product 2a with no significant change in isolated yield (70%). Hence, small-scale reactions were performed using only pure HFIP as the solvent, while large-scale reactions were conducted using solvent mixtures (entry 10). Next, we explored the effect of varying the equivalents of NBS on the outcome of the reaction (entries 12–14). Increasing the amount of NBS (1.0 → 1.5 equivalents; entry 13) led to an improvement in the isolated yield of the desired product (70 → 80%). However, further increase in the amount of NBS used (1.5 → 2.0 equivalents; entry 14) the isolated yield was slightly reduced (80 → 76%), which led us to select 1.5 equivalents of the bromine source for the optimized reaction conditions. Subsequently, we decided to explore the effect of the concentration (entries 15–17) on the reaction outcome. When the reaction was diluted (i.e., 0.2 → 0.1 M concentration), the isolated yield of the desired product remained at 80%. On the other hand, when the reaction was performed under more concentrated conditions (i.e., 0.5 M and 1.0 M), the product was obtained in reduced isolated yields (i.e., 68% and 47%, respectively). After screening eight different brominating, chlorinating and iodinating agents, we concluded that NBS was the best halogen source for the transformation (see ESI, Table 2†).
|
|
||||
|---|---|---|---|---|
| Entrya | Solvent | Temp. (°C) | Time (h) | Yield (%) |
|
a Reactions were conducted on a 0.5 mmol scale, 0.2 M concentration and using 1.2 equivalents of NBS.
b No conversion was observed, only starting material was recovered.
c Complete conversion was observed, starting material was fully consumed.
d Solvent mixture is 1:1 in volume.
e 1.0 equivalent of NBS was used.
f 1.5 equivalents of NBS was used.
g 2.0 equivalents of NBS were used.
h 0.1 M in HFIP and 1.5 equivalents of NBS.
i 0.5 M in HFIP and 1.5 equivalents of NBS.
j 1.0 M in HFIP and 1.5 equivalents of NBS.
|
||||
| 1 | MeCN | Reflux | 16 | 30 |
| 2 | Toluene | Reflux | 16 | 0b |
| 3 | DCM | Reflux | 16 | 0b |
| 4 | IPA | r.t. | 0.5 | 0c |
| 5 | THF | r.t. | 16 | 0c |
| 6 | Methanol | r.t. | 1 | 0c |
| 7 | TFE | r.t. | 16 | 31 |
| 8 | HFIP | r.t. | 1 | 67 |
| 9d | HFIP:MeCN |
40 | 16 | 15 |
| 10d | HFIP:toluene |
r.t. | 1 | Trace |
| 11d | HFIP:DCM |
r.t. | 1 | 70 |
| 12e | HFIP | r.t. | 1 | 61 |
| 13f | HFIP | r.t. | 1 | 80 |
| 14g | HFIP | r.t. | 1 | 76 |
| 15h | HFIP | r.t. | 1 | 80 |
| 16i | HFIP | r.t. | 1 | 68 |
| 17j | HFIP | r.t. | 1 | 47 |

With the optimized reaction conditions in hand, we proceeded to explore the scope and limitations of this NBS-mediated cyclization to afford monocyclic β-lactams (Scheme 2). A diverse set of α,β-unsaturated silyl imino ethers were prepared from the corresponding hydroxamate esters (see ESI†).
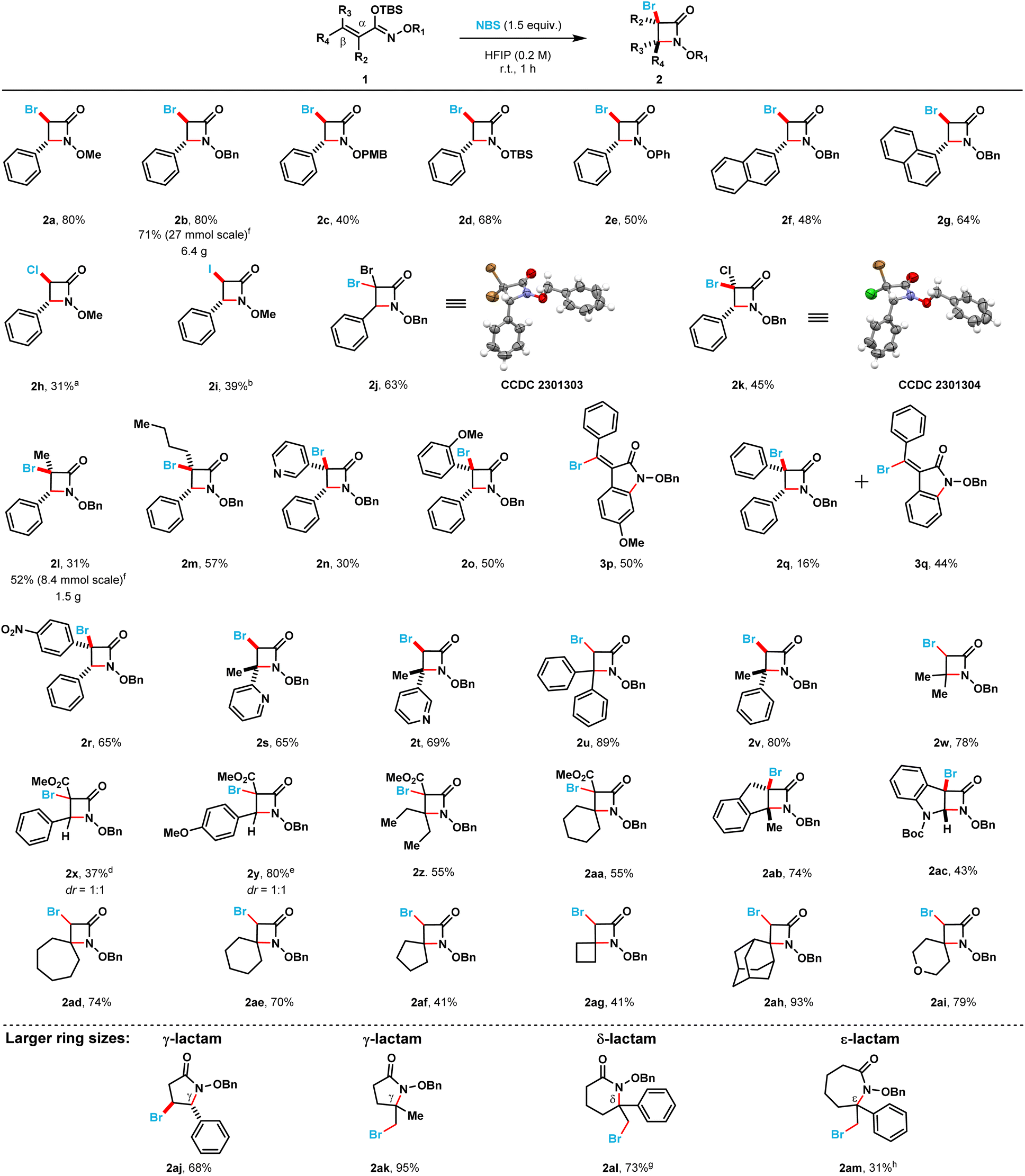 | ||
| Scheme 2 Scope of substrates for the NBS-mediated synthesis of β-lactams and a few examples of larger ring lactams. Reactions were conducted on a 0.5 mmol scale, using 1.5 equivalents of NBS at 0.2 M concentration in HFIP at room temperature for 1 h. aTCCA (1.5 equivalents) was used instead of NBS. bNIS (1.5 equivalents) was used instead of NBS. cThe reaction was heated to 40 °C for 3 h. dThe reaction was heated to reflux for 6 h. eThe reaction was stirred for 6 h. fA 1:1 mixture of DCM:HFIP was used instead of only HFIP as the solvent. gReaction was conducted on a 0.1 mmol scale. hReaction was conducted on a 0.3 mmol scale. | ||
Different N-alkoxy and N-aryloxy substituents (2a–2e) were tolerated in moderate to good yields using the optimized reaction conditions. The N-benzyloxy substituted β-lactam (2b) was obtained in 80% yield while the N-p-methoxybenzyloxy substituted β-lactam (2c) was obtained in a diminished isolated yield (40%). The N-OTBS-substituted β-lactam (2d) was also prepared in 68% isolated yield. When the phenyl ring at the C4-position was switched with 1- and 2-substituted naphthyl rings (2g and 2f) the isolated yields of the products decreased (80 → 64% and 80 → 48%, respectively). Different halogen electrophiles were evaluated (see ESI for complete screening, see page S6†). The α-chloro N-methoxy β-lactam 2h was obtained in 31% isolated yield when TCCA was employed as the chlorine source. Additionally, the α-iodo N-methoxy β-lactam 2i was obtained in 39% isolated yield when N-iodosuccinimide was used as the iodine source.
3,3,4-Trisubstituted monocyclic β-lactams (2j–2r) were obtained in moderate to poor yields (65% to 16%). 3,3-Dibromo (2j) and 3,3-bromochloro (2k) β-lactams were obtained in moderate yields 63% and 45%, respectively. Compound 2j crystallized after purification, and we were able to confirm the structure of its β-lactam core by single crystal X-ray crystallography. Compound 2k was also crystallized, and we were able to confirm the trans diastereoselectivity of this cyclization by single crystal X-ray crystallography. Alkyl substituents (compounds 2l and 2m) in the C3 position were tolerated, albeit these β-lactams were isolated in somewhat diminished yields (31% and 57%, respectively). A 3-pyridyl substituent (compound 2n) was also possible to install, however, the corresponding β-lactam was only isolated in 30% yield. When electronically dissimilar aryl substituents in the C3 position of the target β-lactams were evaluated (compounds 2o–2q), unexpected indoline-2-one side products were observed. The formation of these side products account for the decrease in the isolated yields of the corresponding β-lactams. When an o-methoxyphenyl substituent was present at the C3-position of the substrate silyl imino ether (1o), only β-lactam 2o was obtained in 50% isolated yield. However, when a p-methoxyphenyl substituent was at the C3-position instead, only the indoline-2-one product (3p) was isolated in 50% yield. When an electronically neutral phenyl group was present at the C3-position, a mixture of β-lactam (2q) and indoline-2-one (3q) products were obtained in 16% and 44% isolated yields, respectively. Finally, when an electron-deficient p-nitrophenyl substituent was introduced at the C3-position of the substrate, only the corresponding β-lactam product (2r) was obtained in 65% isolated yield.
3,4,4-Trisubstituted monocyclic β-lactams (2s–2w) were isolated in moderate to good yields (65% to 89%). C4-Heterocycle-substituted 2-pyridyl (2s) and 3-pyridyl (2t) β-lactams could also be prepared and were obtained in 65% and 69% yield, respectively. When two phenyl substituents are at the C4 position in the substrate, β-lactam 2u was obtained in an excellent isolated yield (89%). When one of the phenyl substituents is replaced by a methyl group (compound 2v) the desired β-lactam product was obtained in an 80% isolated yield. 4,4-Dimethyl β-lactam 2w was obtained in 78% yield. Fully substituted β-lactams (2x–2aa) were obtained in moderate to poor isolated yields (37% to 55%). 4,4-Diethyl β-lactam (2z) and spirocyclic β-lactam (2aa) were both obtained in 55% yield. Fused N-benzyloxy β-lactams could also be prepared in moderate to good yields. Indeno-lactam 2ab was obtained in 74% isolated yield while the dihydroindole-fused lactam 2ac was obtained in 43% isolated yield.
Additionally, the optimized reaction conditions allowed the synthesis of six spirocyclic N-benzyloxy β-lactams (2ad–2ai) in moderate to excellent yields (41% to 93%). More strained spirocyclic systems were obtained in lower isolated yields [i.e., 41% yield for both the four-membered (compound 2ag) and five-membered (compound 2af) β-lactams]. The less strained spirocyclic β-lactams were obtained in higher isolated yields [i.e., 74% for the seven-membered compound 2ad and 70% for the six-membered compound 2ae]. The 2,2-adamantyl spirocyclic β-lactam 2ah was obtained in 93%, while the heterocyclic tetrahydrofuranyl spirocyclic β-lactam 2ai was obtained in 79% isolated yield, respectively. β-Lactams 2x and 2y were prepared from the corresponding (E)-silyl imino ethers (i.e., 1x and 1y, respectively) where the phenyl ring substituent is cis to the N-alkoxy silyl imino ether substituent (Scheme 3). β-Lactams 2x and 2y were each obtained as inseparable 1:1 mixture of diastereomers in 35% and 80% isolated yields, respectively.
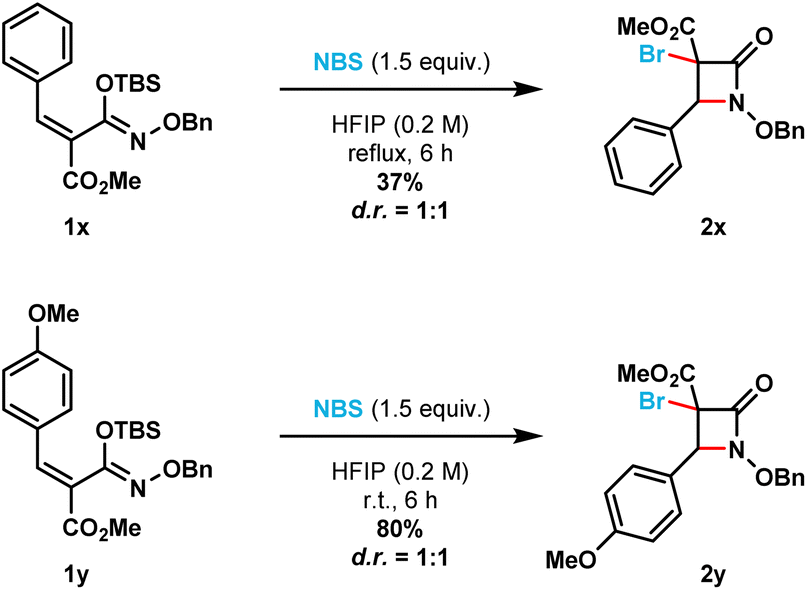 | ||
| Scheme 3 (E)-Silyl imino ethers 1x and 1y furnish a mixture of diastereomers for β-lactams 2x and 2y, respectively. | ||
The optimized reaction conditions also permitted the synthesis of 5-, 6-, and 7-membered N-benzyloxy lactams (2aj–2am) in moderate to excellent yields (31% to 95%). γ-Lactams 2aj and 2ak were both obtained in good to excellent isolated yields (68% and 95%, respectively). δ-Lactam 2al was prepared in 73% isolated yield. In contrast, the somewhat larger ε-lactam 2am was obtained in 31% isolated yield. Overall, we found that this NBS-mediated cyclization is readily scalable; the larger scale (8 to 27 mmol) afforded gram quantities of the β-lactam products 2b and 2l (6.4 g and 1.5 g, respectively) in excellent to moderate isolated yields (71% and 52%, respectively). Additionally, we found the isolation of the silyl imino ethers, via column chromatography, is not required for the cyclization to occur. The hydroxamates can be silylated under typical conditions (see ESI†), after a simple aqueous work-up with DI water and subsequent concentration, the crude silyl imino ethers can be used in the next step. NMR analysis of the crude samples indicated only the presence of silyl imino ethers and, in rare occasion, Si-based impurities that did not influence the next step. The crude silyl imino ethers can then be treated with the optimized conditions to yield the corresponding β-lactams in moderate to good yields over the 2 steps. Using this approach, β-lactam products 2b, 2l and 2v were obtained in moderate to good yields in large-scale (12 to 60 mmol) over 2 steps using a 1:1 mixture of DCM:HFIP as the reaction solvent [i.e., 49% isolated yield for compound 2b in a 60 mmol scale (9.8 g of product); 60% isolated yield for compound 2l in a 20 mmol scale (4.2 g of product); and 40% isolated yield for compound 2v in a 12 mmol scale (1.6 g of product)].
To shed light on the observed lack of diastereoselectivity in β-lactams 2x and 2y [i.e., prepared from (E)-silyl imino ethers 1x and 1y], we decided to evaluate the β-lactam cyclization step using a structurally different (Z)-silyl imino ether (1a′) under the optimized reaction conditions (in which the phenyl ring substituent is cis to the N-alkoxy silyl imino ether substituent). Thus, using a 3:1 mixture of (Z):(E) silyl imino ethers (1a′), we obtained a mixture of β-lactam diastereomers also in a 3:1 diastereomeric ratio (Scheme 4). However, to our surprise the trans-3,4-disubstituted β-lactam product (2a) was obtained as the major diastereomer, while the cis-3,4-disubstituted β-lactam product (2a′) as the minor diastereomer. The cis-relationship of the bromine and phenyl substituents in β-lactam 2a′ was confirmed using single crystal X-ray crystallography. Control experiments (see ESI, page S79†) indicated that: (1) no isomerization occurred between silyl imino ethers 1a′ and 1a upon stirring 1a′ [3:1 (Z)/(E) ratio] in HFIP in the absence of NBS at room temperature for 12 h; (2) no desilylation occurred upon stirring 1a′ in HFIP in the absence of NBS at room temperature in 1 h; and (3) no desilylation of 1a′ occurred upon stirring in HFIP using 1.5 equivalents of NH-succinimide instead of NBS at room temperature for 1 h. Therefore, we can be confident that the removal of the silyl group only occurs after the electrophilic bromination of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the imino ether.
C double bond in the imino ether.
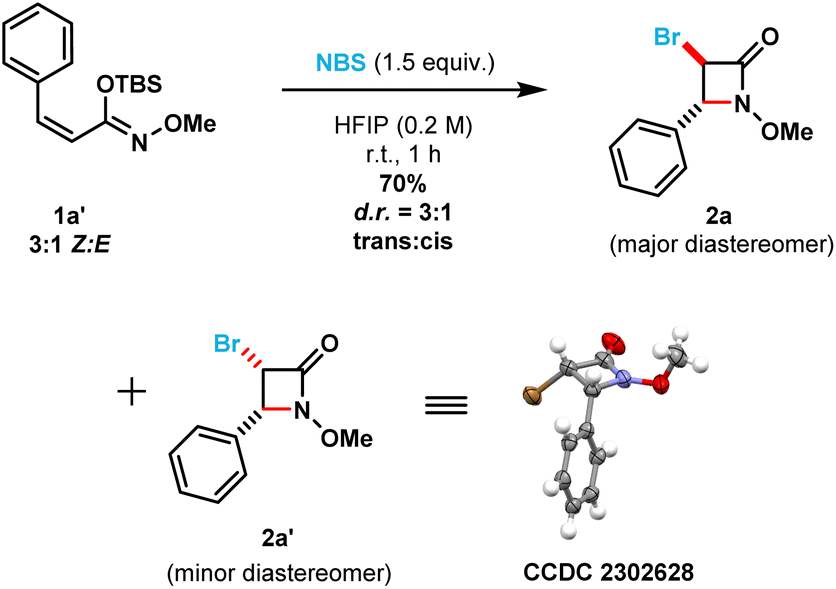 | ||
| Scheme 4 Control experiments on the diastereoselectivity of the cyclization reaction. | ||
To computationally model the mechanism and selectivity of this cyclization/β-lactam forming reaction we considered N-bromosuccinimide as a source of a cationic bromine and examined the reaction with substrates 1a, 1a′, and 1y. Based on the above control reactions, we assumed that the TBS group is not removed until after the cyclization. M06-2X17a/6-31G** (and LANL2DZ for Br)17b,c in Gaussian 16 (ref. 17d) was used to optimize all structures and frequency calculations were used to verify minima and transition states.
For bromination, a bridged bromonium type structure is generally assumed to be a possible ground state intermediate. However, for 1a (and 1a′/1y), all attempts to locate a bridged bromonium ion failed. Instead, optimized structures resulted in a benzylic carbocation intermediate, and example structure Int-Syn is shown in Fig. 2. We tried several alternative density functionals (e.g. B3LYP17e) and basis sets (e.g. def2-TZVP17f) and all gave only the benzylic carbocation structure. However, a constrained optimization suggests that the bromonium is only about 5 kcal mol−1 higher in energy. Because the barriers for cyclization are relatively small (see later discussion), it is likely that the rate limiting reaction step is formation of the benzylic carbocation. This provides a straightforward explanation for the moderate reaction yield with acetonitrile and boosted reaction yield with HFIP. This was confirmed with calculations where we examined the thermodynamics for formation of the benzylic carbocation using two explicit HFIP solvent molecules. With no explicit HFIP molecules formation of the benzylic carbocation requires 20.0 kcal mol−1. Inclusion of two HFIP molecules, one to stabilize the carbocation and one to stabilize the bromide, lowers benzylic carbocation energy to ∼4 kcal mol−1 (see ESI† for details).
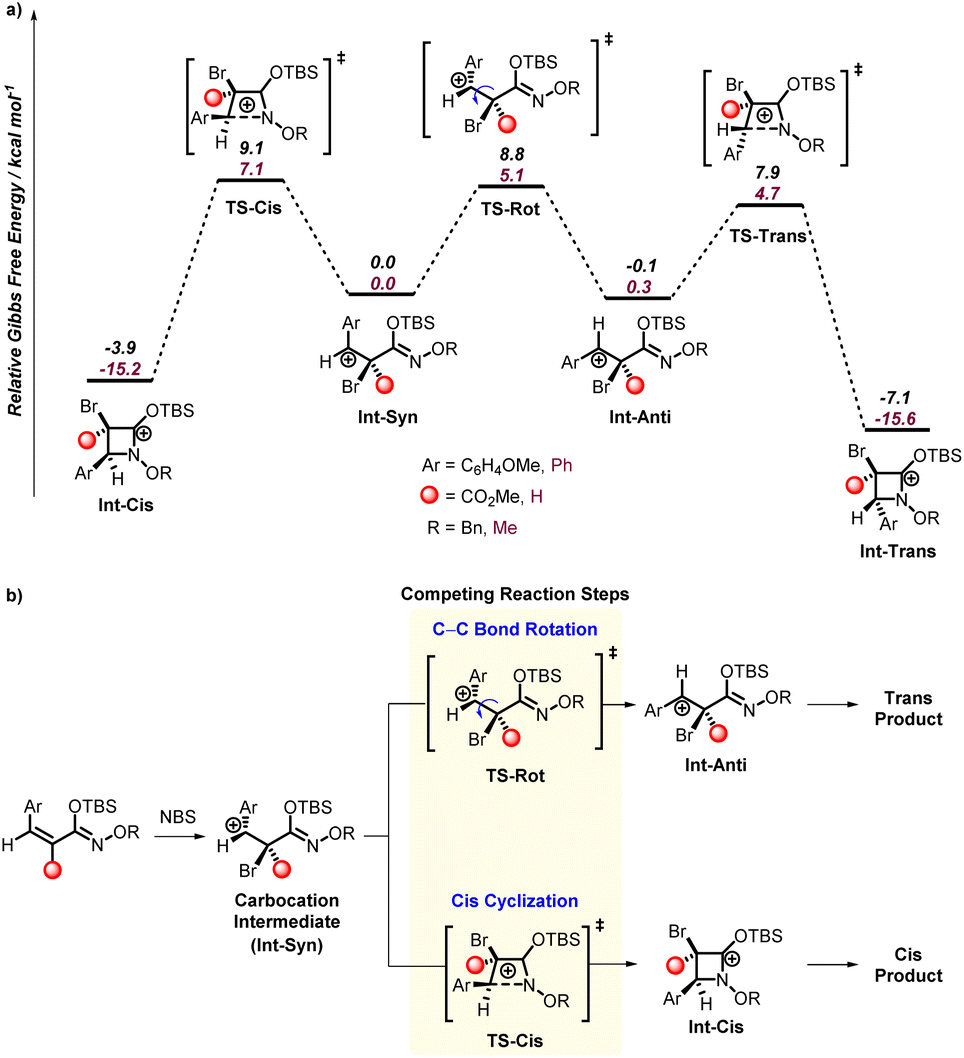 | ||
| Fig. 2 (a) Gibbs energy surface showing the comparison of the cis cyclization process with C–C bond rotation that leads to the trans cyclization intermediate. (b) Outline of cyclization selectivity model. | ||
Notwithstanding the large atomic size of bromine, the optimization of a carbocation intermediate rather than a bridged bromonium intermediate was surprising because of the exclusive trans stereoselectivity found for the cyclization of 1a. This prompted us to examine the transition states for cyclization from the benzylic carbocation intermediate. Fig. 2 shows the potential energy surface structures and the general selectivity model developed based on the intermediates and transition states. This energy surface was modelled with the SMD17g model of acetonitrile. This solvent was selected because this solvent does give conversion to products (see Table 1, entry 1) and is a parameterized solvent model available in Gaussian 16. HFIP is not available in Gaussian 16. However, test calculations with both explicit HFIP and trifluoroethanol showed very small changes to the energy surface presented in Fig. 2.
The transition state for C–N bond formation (Int-Cis and Int-Trans) is exothermic and likely not reversible once formed, and after cyclization carbonyl formation would occur. The transition state leading to cyclization with the aryl and bromide groups in a trans-relationship (TS-Trans) is only 4.7 kcal mol−1 and cyclization with these groups in a cis-relationship (TS-Cis) is 7.1 kcal mol−1. This 3.0 kcal mol−1 energy difference is consistent with nearly complete kinetic selectivity for the trans-product. DLPNO-CCSD(T) calculations (performed in ORCA)17h provide a nearly identical energy difference between these transition states and support the M06-2X energy difference.
It is useful to note that if the cyclization step is reversible, there is a 3.2 kcal mol−1 preference for the trans-cyclized intermediate (Int-Trans) for 1y. As expected, the selectivity for trans over cis cyclization is driven by the four-membered ring enforcing partial eclipsing of the bromide and aryl groups, although the aryl group can twist to relieve some of this repulsion. Consistent with our proposed benzylic carbocation mechanism, a few of the starting N-alkoxy α,β-unsaturated silyl imino ethers result in formation of a mixture of trans- and cis-products. However, it was surprising to us that these reactions generally produced a nearly equal mixture of trans- and cis-products since for 1a there is a significant kinetic and thermodynamic preference for the trans-product.
Therefore, we analyzed the cyclization from the benzylic carbocation intermediate (Int-Syn) derived from 1a′ and 1y as shown in Fig. 2. In both cases the trans-cyclization transition state (TS-Trans) is lower than the cis-cyclization transition state (TS-Cis). Also, there is a significant thermodynamic preference for the trans-cyclized intermediate (Int-Trans) compared to the cis-cyclized intermediate (Int-Cis). This prompted us to examine the possibility that cis-to-trans isomerization of the carbocation intermediates might determine the trans/cis ratio. Using a nudged elastic band method in ORCA to examine this C–C bond rotation transition states (TS-Rot) we found that they are indeed higher in energy than the trans-cyclization transition states (TS-Trans). Therefore, the model that emerged to explain the diastereoselectivity is that from these carbocation intermediates the trans/cis selectivity is likely governed by the rate of cis-cyclization (TS-Cis) versus the rate of C–C bond rotation (TS-Rot) that is followed by faster trans-cyclization (TS-Trans). For the carbocation intermediate derived from 1y the energy difference between the cis-cyclization transition state (TS-Cis) and C–C bond transition state (TS-Rot) is only 0.3 kcal mol−1, and this is qualitatively consistent with experiment. For the carbocation derived from 1a′ the difference in these transition states is 2 kcal mol−1. In both cases these small energy differences are consistent with the mixture of trans and cis products found experimentally. But again, the calculated energy difference is only expected to be qualitative. This is because transition states for bond rotations are inherently anharmonic and difficult to have highly quantitative values. Therefore, the calculated values are qualitative, but do provide a rationale for the unexpected selectivity.
This NBS-mediated cyclization method allows the straightforward synthetic access to a wide variety of α-bromo N-alkoxy β-lactams. The obtained compounds contain valuable functional handles that can be utilized for further transformations and modifications.8e,g,10c,d,11b,11c,16 To this end, we carried out a few representative transformations (Scheme 5). Debromination of compound 2b can be achieved in 79% isolated yield by treatment with tris(trimethylsilyl)silane and catalytic AIBN when heated to 100 °C (eqn (1)).13a Under this reaction conditions, compound 2l also undergoes debromination in 78% isolated yield, to provide only the cis β-lactam 4l. Treatment of the N-benzyloxy β-lactam 2a with Mo(CO)6 selectively cleaved the N–O bond while keeping bromine handle to afford N–H β-lactam 5b in 43% isolated yield (eqn (2)).18 Next, catalytic hydrogenation of 2b afforded N-hydroxy β-lactam 6b in 66% isolated yield after one hour (eqn (3)).9b The treatment of compound 2a with 2.0 equivalents of LiBH4 allowed the synthesis of N–OBn aziridine 8b in 40% yield upon reduction of the carbonyl bond and ring-opening followed by intramolecular cyclization (eqn (5)).19
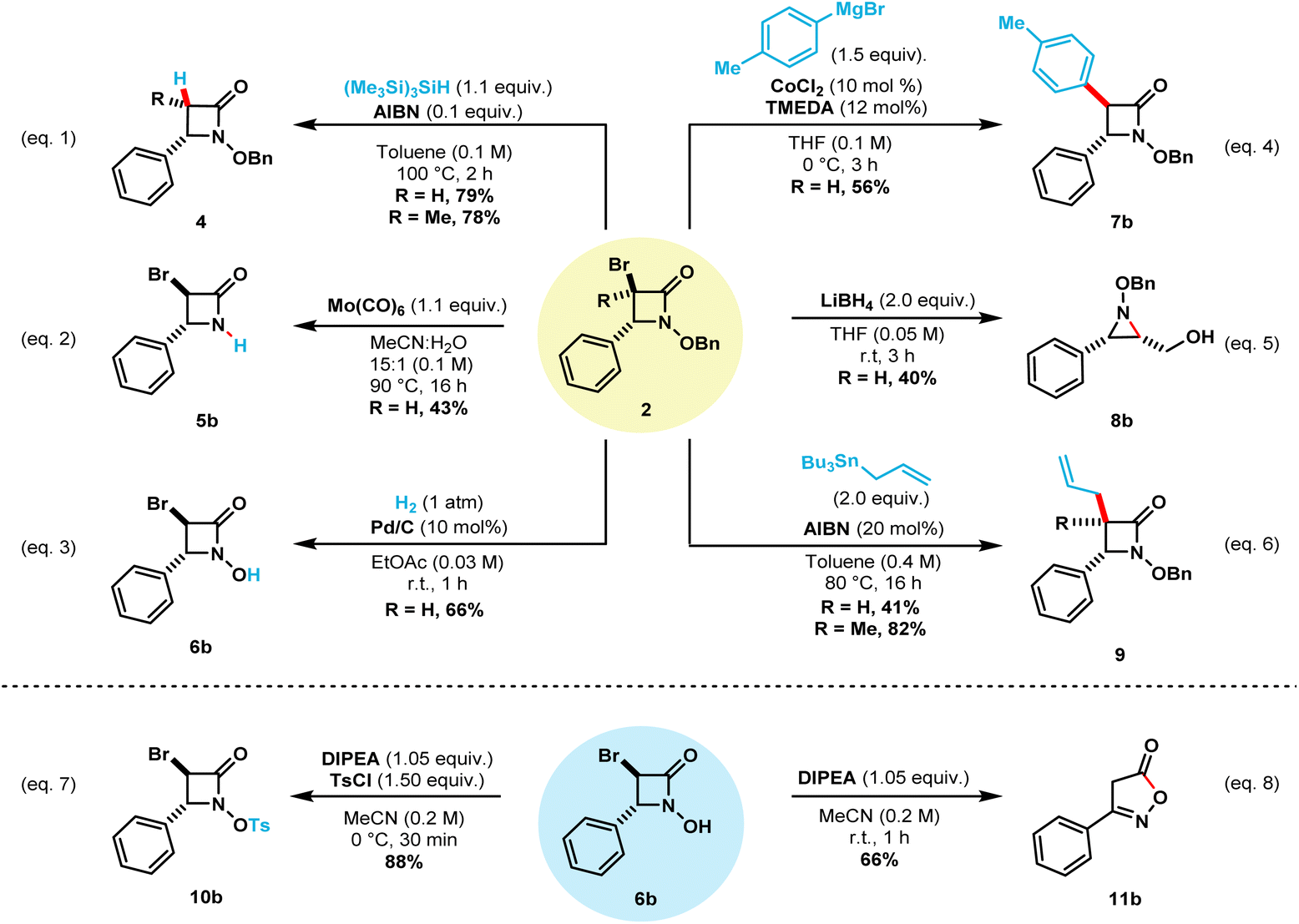 | ||
| Scheme 5 Synthetic applications of β-lactams. | ||
Keck radical allylation of compounds 2b and 2l employing allyltributylstannane afforded the corresponding allylated β-lactams 9b and 9l in 41% and 82%, respectively (eqn (6)).20 Further transformations of compound 6b are also showcased. Selective tosylation using Hunig's base in ice-cold MeCN afforded compound 10b in 88% isolated yield (eqn (7)). Finally, treatment of compound 6b with Hunig's base at room temperature afforded compound 11b in 66% yield (eqn (8)).
Conclusions
In conclusion, we report a new synthetic protocol that allows the facile preparation of α-bromo N-alkoxy β-lactams from the corresponding N-alkoxy-α,β-unsaturated silyl imino ethers using NBS. This approach permits the convenient access to a wide variety of monocyclic, fused and spirocyclic α-bromo N-alkoxy β-lactams that have never been reported before. Our novel strategy allows the synthesis of densely substituted β-lactams in both the C3 and C4 positions. The presence of both C–Br and N–O bond functional handles provides a platform for the facile modification to access highly substituted and functionalized β-lactams. In addition, we have provided a mechanistic and computational rationale for the observed diastereoselectivity of this transformation. Further studies on the potential synthetic applications of these versatile building blocks are underway.Data availability
Data supporting this manuscript are available in the associated ESI files† and via the CCDC (numbers 2301303, 2301304 and 2302628).Author contributions
L. K. conceived the idea and designed the research. A. R. performed the experiments and analyzed the data. S. P. and P. L.-T. equally contributed with experiments setup and data analysis. J. K., M. D. and D. E. performed the calculations and theoretical work. U. A. and M. Y. analyzed the crystal data by X-ray crystallography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. K. gratefully acknowledges funding from the National Science Foundation (CHE-2102462), National Institutes of Health (R35 GM-136373), Robert A. Welch Foundation (C-1764). A. R. was supported by the Mexican National Council for Science and Technology (CONACyT) PhD Scholarship Program (958417). Additionally, we thank Ms. Abbigail Ramos (visiting student from Texas A&M University College Station) for assistance with preliminary experiments. Ms. Ramos was supported by the National Science Foundation (2150216).Notes and references
- (a) A. L. Demain and R. P. Elander, Anton. Leeuw. Int. J. G., 1999, 75, 5–19 CrossRef CAS PubMed; (b) A. Fleming, Br. J. Exp. Pathol., 1929, 10, 226–236 CAS.
- B. Thakuria and K. Lahon, J. Clin. Diagn. Res., 2013, 7, 1207–1214 Search PubMed.
- For reviews on the biological activity of β-lactams: (a) L. Moreira Lima, B. Nascimiento Monteiro da Silva, G. Barbosa and E. J. Barreiro, Eur. J. Med. Chem., 2020, 208, 112829 CrossRef PubMed; (b) J. Turner, A. Muraoka, M. Badenbaugh, B. Childress, L. Pernot, M. Wiencek and Y. K. Peterson, Front. Microbiol., 2022, 13, 807955 CrossRef PubMed; (c) A. Gupta and A. K. Halve, Int. J. Pharm. Sci. Res., 2015, 6, 978–987 CAS; (d) M. I. Konaklieva, Antibiotics, 2014, 3, 128–142 CrossRef PubMed; (e) K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295–306 CrossRef CAS PubMed.
- (a) N. H. Georgopapadakou, S. A. Smith and R. B. Skyes, Antimicrob. Agents Chemother., 1982, 21, 950–956 CrossRef CAS PubMed; (b) C. Ramsey and A. P. MacGowan, J. Antimicrob. Chemother., 2016, 71, 2704–2712 CrossRef CAS PubMed.
- For reviews on the synthesis of β-lactams: (a) L. Troisi, C. Granito and E. Pindinelli, Top. Heterocycl. Chem., 2010, 22, 101–209 CAS; (b) S. Hosseyni and A. Jarrahpour, Org. Biomol. Chem., 2018, 16, 6840–6852 RSC; (c) C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930–7953 CrossRef CAS PubMed; (d) A. Saura-Sanmartin and L. Andreu-Ardil, Org. Biomol. Chem., 2023, 21, 3296–3306 RSC; (e) S. Deketelaere, T. V. Nguyen, C. V. Stevens and M. D’hooghe, ChemistryOpen, 2017, 6, 301–319 CrossRef CAS PubMed; (f) N. G. Alves, A. J. S. Alves, M. I. L. Soares and T. M. V. D. P. E. Melo, Adv. Synth. Catal., 2021, 363, 2464–2501 CrossRef CAS.
- Seminal work and selected articles: (a) H. Staudinger, Ber. Dtsch. Chem. Ges., 1907, 40, 1145–1148 CrossRef CAS; (b) H. Staudinger and H. W. Klever, Ber. Dtsch. Chem. Ges., 1907, 40, 1149–1153 CrossRef CAS; (c) F. P. Cossio, A. Arrieta and M. A. Sierra, Acc. Chem. Res., 2008, 41, 925–936 CrossRef CAS PubMed; (d) N. Fu and T. T. Tidwell, Tetrahedron, 2008, 64, 10465–10496 CrossRef CAS.
- (a) A. G. M. Barrett, M. J. Betts and A. Fenwick, J. Org. Chem., 1985, 50, 169–175 CrossRef CAS; (b) M. R. Lee, S. S. Stahl and S. H. Gellman, Org. Lett., 2008, 10, 5317–5319 CrossRef CAS PubMed; (c) W. J. Yang, B. Ling, B. W. Hu, H. L. Yin, J. Y. Mao and P. J. Walsh, Angew. Chem., Int. Ed., 2020, 59, 161–166 CrossRef CAS PubMed.
- (a) P. G. Mattingly and M. J. Miller, J. Org. Chem., 1980, 45, 410–415 CrossRef CAS; (b) T. Hirose, K. Chiba, S. Mishio, J. Nakano and H. Uno, Heterocycles, 1982, 19, 1019–1022 CrossRef CAS; (c) M. A. Krook, M. J. Miller and C. Eigenbrot, J. Chem. Soc., Chem. Commun., 1985, 1265–1266 RSC; (d) P. A. van Elburg, D. N. Reinhoudt, S. Hakerma and G. J. V. Hummel, Tetrahedron Lett., 1985, 26, 2809–2812 CrossRef CAS; (e) A. Biswas, C. Eigenbrot and M. J. Miller, Tetrahedron, 1986, 42, 6421–6428 CrossRef CAS; (f) X. B. Li, C. S. Niu and M. J. Miller, Tetrahedron Lett., 1995, 36, 1617–1620 CrossRef CAS; (g) J. R. Bellettini and M. J. Miller, J. Org. Chem., 1996, 61, 7959–7962 CrossRef CAS PubMed.
- (a) C. K. Zercher and M. J. Miller, Tetrahedron Lett., 1989, 30, 7009–7012 CrossRef CAS; (b) M. A. Williams, C. N. Hsiao and M. J. Miller, J. Org. Chem., 1991, 56, 2688–2694 CrossRef CAS; (c) C. M. Gasparski, M. Teng and M. J. Miller, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 1992, 203, 139–Orgn Search PubMed; (d) P. R. Guzzo, M. Teng and M. J. Miller, Tetrahedron, 1994, 50, 8275–8292 CrossRef CAS; (e) A. Bulychev, M. E. Obrien, I. Massova, M. Teng, T. A. Gibson, M. J. Miller and S. Mobashery, J. Am. Chem. Soc., 1995, 117, 5938–5943 CrossRef CAS; (f) S. Carosso and M. J. Miller, Bioorg. Med. Chem., 2015, 23, 6138–6147 CrossRef CAS PubMed; (g) E. K. Dolence, A. A. Minnick and M. J. Miller, J. Med. Chem., 1990, 33, 461–464 CrossRef CAS PubMed; (h) T. B. Durham and M. J. Miller, J. Org. Chem., 2003, 68, 27–34 CrossRef CAS PubMed; (i) M. Ghosh and M. J. Miller, Tetrahedron, 1996, 52, 4225–4238 CrossRef CAS; (j) P. Swaren, I. Massova, J. R. Bellettini, A. Bulycev, L. Maveryraud, L. P. Kotra, M. J. Miller, S. Mobashery and J. P. Samama, J. Am. Chem. Soc., 1999, 121, 5353–5359 CrossRef CAS; (k) A. J. Walz and M. J. Miller, Tetrahedron Lett., 2007, 48, 5103–5105 CrossRef CAS; (l) R. M. Williams, B. H. Lee, M. J. Miller and O. P. Anderson, J. Am. Chem. Soc., 1989, 111, 1073–1081 CrossRef CAS; (m) S. R. Woulfe and M. J. Miller, Tetrahedron Lett., 1984, 25, 3293–3296 CrossRef CAS.
- For examples of cross-couplings: (a) A. Hazra, J. A. Kephart, A. Velian and G. Lalic, J. Am. Chem. Soc., 2021, 143, 7903–7908 CrossRef CAS PubMed; (b) V. Koch, M. M. Lorion, E. Barde, S. Brase and J. Cossy, Org. Lett., 2019, 21, 6241–6244 CrossRef CAS PubMed; (c) M. M. Lorion, V. Koch, M. Nieger, H. Y. Chen., A. W. Lei, S. Brase and J. Cossy, Chem.–Eur. J., 2020, 26, 13163–13169 CrossRef CAS PubMed; (d) A. Tarui, S. Kondo, K. Sato, M. Omote, H. Minami, Y. Miwa and A. Ando, Tetrahedron, 2013, 69, 1559–1565 CrossRef CAS; (e) F. L. Wang, L. Liu, C. J. Yang, C. Luan, J. Yang, J. J. Chen, Q. S. Gu, Z. L. Li and X. Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202214709 CrossRef CAS PubMed.
- For examples of metal-halogen exchange: (a) F. Benfatti, G. Cardillo, S. Fabbroni, L. Gentilucci, R. Perciaccante, F. Piccinelli and A. Tolomelli, Synthesis, 2005, 61–70 CAS; (b) X. Y. Ren, C. R. Shen, G. Z. Wang, Z. L. Shi, X. X. Tian and K. W. Dong, Org. Lett., 2021, 23, 2527–2532 CrossRef CAS PubMed; (c) A. Tarui, N. Kawashima, T. Kawakita, K. Sato, M. Omote and A. Ando, J. Org. Chem., 2013, 78, 7903–7911 CrossRef CAS PubMed.
- For examples of nucleophilic substitution: (a) F. Benfatti, G. Cardillo, L. Gentilucci, R. Perciaccante, A. Tolomelli and A. Catapano, J. Org. Chem., 2006, 71, 9229–9232 CrossRef CAS PubMed; (b) G. Cardillo, S. Fabbroni, L. Gentilucci, R. Perciaccante, F. Piccinelli and A. Tolomelli, Org. Lett., 2005, 7, 533–536 CrossRef CAS PubMed; (c) H. P. Isenring and W. Hofheinz, Tetrahedron, 1983, 39, 2591–2597 CrossRef CAS.
- For selected syntheses of α-bromo β-lactams: (a) E. Bandini, G. Favi, G. Martelli, M. Panunzio and G. Piersanti, Org. Lett., 2000, 2, 1077–1079 CrossRef CAS PubMed; (b) S. Berry, S. S. Bari, B. K. Banik and A. Bhalla, Synth. Commun., 2017, 47, 2239–2246 CrossRef CAS; (c) A. K. Bose, B. N. Ghoshmazumdar and B. G. Chatterjee, J. Am. Chem. Soc., 1960, 82, 2382–2386 CrossRef CAS; (d) W. T. Brady and R. A. Owens, Tetrahedron Lett., 1976, 1553–1556 CrossRef CAS; (e) J. S. Bryans, N. E. A. Chessum, A. F. Parsons and F. Ghelfi, Tetrahedron Lett., 2001, 42, 2901–2905 CrossRef CAS; (f) S. Decamps, L. Sevaille, S. Ongeri and B. Crousse, Org. Biomol. Chem., 2014, 12, 6345–6348 RSC; (g) A. Galvan, F. N. de la Cruz, F. Cruz, M. Martinez, C. V. Gomez, Y. Alcaraz, J. M. Dominguez, F. Delgado and M. A. Vazquez, Synthesis, 2019, 51, 3625–3637 CrossRef CAS; (h) R. Joyeau, H. Molines, R. Labia and M. Wakselman, J. Med. Chem., 1988, 31, 370–374 CrossRef CAS PubMed; (i) M. S. Manhas, M. S. Khajavi, S. S. Bari and A. K. Bose, Tetrahedron Lett., 1983, 24, 2323–2326 CrossRef CAS; (j) A. Tarui, N. Kawashima, K. Sato, M. Omote, Y. Miwa, H. Minami and A. Ando, Tetrahedron Lett., 2010, 51, 2000–2003 CrossRef CAS; (k) A. Tarui, H. Nishimura, T. Ikebata, A. Tahira, K. Sato, M. Omote, H. Minam, Y. Miwa and A. Ando, Org. Lett., 2014, 16, 2080–2083 CrossRef CAS PubMed.
- H. Homsi and G. Rousseau, J. Org. Chem., 1999, 64, 81–85 CrossRef PubMed.
- D. Naskar and S. Roy, J. Chem. Soc., Perkin Trans. 1, 1999, 2435–2436 RSC.
- (a) S. Ghosh, J. Banerjee, R. Ghosh and S. K. Chattopadhyay, Synth. Commun., 2020, 50, 1353–1360 CrossRef CAS; (b) N. N. Zhang, R. Yang, D. Zhang-Negrerie, Y. F. Du and K. Zhao, J. Org. Chem., 2013, 78, 8705–8711 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (d) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed; (e) A. D. Becke, J. Chem. Phys., 1993, 98m, 5648–5652 CrossRef; (f) A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef; (g) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed; (h) F. Neese, Software update: The ORCA program system—Version 5.0, WIRES Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- D. J. Wardrop and M. S. Burge, J. Org. Chem., 2005, 70, 10271–10284 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, G. Cabrero and M. P. Ruiz, J. Org. Chem., 2007, 72, 7980–7991 CrossRef CAS PubMed.
- D. Giese, W. Damm, J. Dickhaut, F. Wetterich, S. Sun and D. P. Curran, Tetrahedron Lett., 1991, 32, 6097–6100 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental and computational results, procedures and characterization including 1H and 13C NMR spectra and X-ray crystallographic data. CCDC 2301303, 2301304 and 2302628. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01513d |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |