
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP-doped spherical hard carbon with high initial coulombic efficiency and enhanced capacity for sodium ion batteries†
Zheng-Guang
Liu‡
 a,
Jiahua
Zhao‡
ab,
Hao
Yao
ab,
Xiang-Xi
He
ab,
Hang
Zhang
ab,
Yun
Qiao
a,
Xing-Qiao
Wu
b,
Li
Li
*ac and
Shu-Lei
Chou
*b
a,
Jiahua
Zhao‡
ab,
Hao
Yao
ab,
Xiang-Xi
He
ab,
Hang
Zhang
ab,
Yun
Qiao
a,
Xing-Qiao
Wu
b,
Li
Li
*ac and
Shu-Lei
Chou
*b
aSchool of Environment and Chemical Engineering, Shanghai University, Shanghai 20444, P. R. China. E-mail: LiLi2020@shu.edu.cn
bInstitute for Carbon Neutralization Technology, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, Zhejiang 325035, P. R. China. E-mail: chou@wzu.edu.cn
cKey Laboratory of Advanced Energy Materials Chemistry(Ministry of Education), Nankai University, Tianjin 300071, P. R. China
First published on 11th April 2024
Abstract
Hard carbon (HC) is one of the most promising anode materials for sodium-ion batteries (SIBs) due to its cost-effectiveness and low-voltage plateau capacity. Heteroatom doping is considered as an effective strategy to improve the sodium storage capacity of HC. However, most of the previous heteroatom doping strategies are performed at a relatively low temperature, which could not be utilized to raise the low-voltage plateau capacity. Moreover, extra doping of heteroatoms could create new defects, leading to a low initial coulombic efficiency (ICE). Herein, we propose a repair strategy based on doping a trace amount of P to achieve a high capacity along with a high ICE. By employing the cross-linked interaction between glucose and phytic acid to achieve the in situ P doped spherical hard carbon, the obtained PHC-0.2 possesses a large interlayer space that facilitates Na+ storage and transportation. In addition, doping a suitable amount of P could repair some defects in carbon layers. When used as an anode material for SIBs, the PHC-0.2 exhibits an enhanced reversible capacity of 343 mA h g−1 at 20 mA g−1 with a high ICE of 92%. Full cells consisting of a PHC-0.2 anode and a Na2Fe0.5Mn0.5[Fe(CN)6] cathode exhibited an average potential of 3.1 V with an initial discharge capacity of 255 mA h g−1 and an ICE of 85%. The full cell displays excellent cycling stability with a capacity retention of 80.3% after 170 cycles. This method is simple and low-cost, which can be extended to other energy storage materials.
Introduction
The continuous consumption of fossil energy forces people to explore sustainable energy storage systems (EESs). Whilst lithium-ion batteries (LIBs), as the established technology, have been widely used in portable electronic devices and electric vehicles, the increased cost and inhomogeneous distribution of lithium resources severely impede their potential for large-scale EES applications.1,2 In contrast, sodium-ion batteries (SIBs) are considered as the most promising alternatives to LIBs, especially for large-scale energy storage due to their low cost and abundant resources.3,4 However, the development of advanced and economic electrode materials has limited the commercial application of SIBs. Anode materials, according to the reaction mechanism, can be mainly divided into intercalation, conversion and alloying types.5 As a typical intercalation-based anode, graphite is widely used in commercial LIBs, but the large ionic radius of sodium (0.102 nm) and the weak chemical bond formed between sodium and graphite lead to the poor performance of SIBs,6 and also graphite displays a pretty low specific capacity (35 mA h g−1). Hard carbon (HC) is composed of randomly-oriented curved graphene nanosheets, turbostratically stacked with rich pores; these special structural characteristics endow HC with favorable capability for Na-storage.3,7 Typically, the charge/discharge profiles of HC exhibit a slope region (>0.1 V) and a plateau region (<0.1 V), showing a reversible capacity of about 300 mA h g−1. More importantly, the unique low voltage plateau capacity can make it promising for development of high energy density electrodes for full cells. Extensive efforts have been made to promote the plateau capacity of HC materials, into which doping covalent heteroatoms (N, B, F, P or S) has been extensively explored.8,9 For example, Wu et al.10 prepared phosphorus-doped hard carbon nanofibers via electrospinning treating PAN/H3PO4 precursors, and the plateau capacity of P-doped CFs-P15 was extended from 32 to 54 mA h g−1. Also, a natural potassium-doped carbon material (HC-1100) was fabricated from coconut endocarp,11 and when used as an anode for SIBs, the plateau capacity of HC-1100 (211 mA h g−1) was higher than that of HC-1100H (169 mA h g−1) in the initial cycle. Wang et al.12 synthesized phosphorus-doped hollow carbon nanorods (P-HCNs) via a one-step grinding method followed by thermal treatment. Using the P-HCNs 0.3 electrode in SIBs, the increased plateau capacity is attributed to larger d-spacings, which are beneficial for Na+ diffusion and storage. Some other heteroatom doping strategies are listed in Table S1.† It can be found that HC is still the most state-of-the-art anode material, as well as doping covalent heteroatoms is a viable approach to improve the sodium storage capacity, whereas HC still suffers low ICE regardless of the source and preparation process. As a result, the poor ICE and unsatisfactory capacity will not be conducive to achieve high energy density anodes for SIBs. So, it is of great significance to design appropriate synthesis methods to extend the low-voltage plateau capacity along with a high ICE.PA, as shown in Scheme S1,† has attracted much attention as a P-doping source for carbon.13 It could also be used as a crosslinker and activation agent.14 The P doped carbon was achieved by hydrogen bonding between the hydroxyl groups in glucose and the phosphate groups in PA. Furthermore, the introduction of phosphates into the carbon matrix could expand the graphene layers, which facilitates the (de)insertion of Na+. When P atoms reach a certain value, they could repair some of the defects in carbon layers and promote the ICE.
Based on the above characteristics, herein, phytic acid (PA)-modified spherical hard carbon is synthesized by a hydrothermal-carbonization method and subsequent pyrolytic treatment (Scheme 1), and the optimized P-doped spherical hard carbon (PHC-0.2) delivered a total reversible capacity of 343 mA h g−1 with an enhanced plateau capacity of 227 mA h g−1 and an ultra-high ICE of 92%. The ICE of the PHC-0.2 synthesized in this work is higher than those of most of the previous heteroatom-doped carbon materials (Table S1†). Additionally, the kinetic analysis illustrates that the electrochemical reaction is dominated by the diffusion-controlled process. Moreover, coupling with Na2Fe0.5Mn0.5[Fe(CN)6] (FeMnHCF) as the cathode to assemble the corresponding full cell, the battery delivers an excellent cycling performance, implying a good application prospect of the PHC-0.2 anode for SIBs.
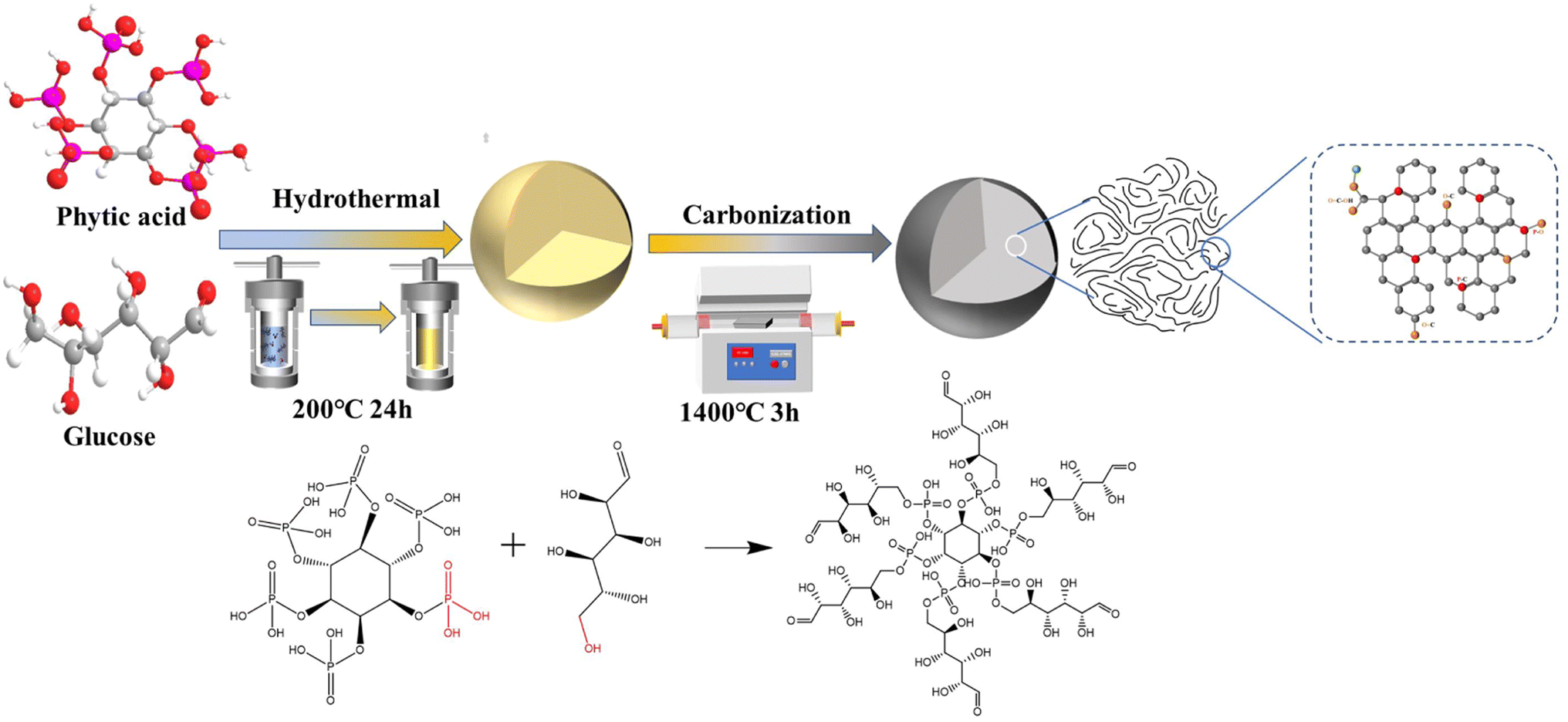 | ||
| Scheme 1 Schematic illustration showing the synthesis route of PHC-X. | ||
Results and discussion
The PHC-X was synthesized via hydrothermal carbonization of glucose and subsequent high-temperature pyrolysis. In our design, the in situ doping of P in the carbon framework was achieved via a dehydration condensation reaction between glucose and PA, in which PA not only acts as the crosslinker, but also as the heteroatom dopant and activation agent. At the same time, when the P atoms in the introduced PA reach a certain value, they can repair some of the defects in the carbon layer,12 so they may be used as a repairing agent. The structure of the obtained carbon powder is characterized by XRD and Raman spectroscopy. As shown in Fig. 1a, all specimens exhibit two typical broad diffraction peaks at ∼24° and ∼43°, which are attributed to the crystal planes of (002) and (101) in carbon materials, confirming the amorphous structure. It is noticeable that the peaks of (002) moved to smaller diffraction angles with the increase of PA, which indicates increased layer spacing. The detailed average interlayer space (d002) of PHC-X calculated by the Bragg equation is shown in Table S2.† For PHC-0.2, the d002 (2θ = 23.23°) is 0.382 nm, much larger than that of HC (0.378 nm). The larger d-spacings are beneficial for Na+ insertion into graphitic lattices. Raman spectra are shown in Fig. 1b to demonstrate the structures of PHC-X; the main peaks of Raman spectra can be fitted into four subpeaks located at ∼1200 cm−1 (D4), ∼1350 cm−1 (D1), ∼1500 cm−1 (D3), and ∼1580 cm−1 (G). Hard carbon materials mostly show common features in the 1000–1800 cm−1 range, including a defect-induced D-band and graphite crystalline G band. Generally, the degree of graphitization can be evaluated by the integral area ratio of AD1/AG.15 The AD1/AG was increased slightly from 1.006 (HC) to 1.064 (PHC-0.4) when the P-doped content was increased from 0 to 0.32 at% (Table 1), indicating that the microstructure of hard carbon tends to be more disordered to some extent. To further evaluate the specific surface area (SSA) and pore structure characteristics of PHC-X, a nitrogen adsorption–desorption isotherm test is carried out. As shown in Fig. 1c, the isotherms of all samples appear as a type-II isotherm, illustrating low porosity.16 The SSA, micropore volumes and average pore sizes are listed in Table S3.† The SSA of HC, PHC-0.1, PHC-0.2 and PHC-0.4 is 156.37, 81.26, 44.31 and 88.10 m2 g−1, respectively. It is found that the SSA of these materials first decreased and then increased, in which the SSA of PHC-0.4 is smaller than that of HC. The transverse growth SSA of PHC-0.4 may be related to the complex process, including the hydrolysis, dehydration, aromatization, crosslinking and pore-forming.14 Among these samples, PHC-0.2 possesses the lowest SSA of 44.31 m2 g−1 and a larger average pore diameter of 2.0956 nm. The decreased SSA and the increased average pore diameter may be related to the cross-linked molecular structure of the PA and glucose, which inhibited the release of small-molecule gas during high-temperature treatment, as well as some micropores form mesopores caused by P doping.17 This variation trend of SBET is critical to avoid large irreversible consumption of Na+ caused by SEI film formation in the initial cycle, realizing a high ICE.18,19Fig. 1d exhibits the corresponding pore size distributions which were calculated using the density-functional-theory (DFT) model. The micropores (1–2 nm) in HC and PHC-X are abundant, and a large number of micropores contribute to the filling of Na+,20 which was consistent with the results of the isotherm analysis. It is noted that most pores for the HC sample are found in the range of 0.5–2.2 nm, especially in the range of 1–1.5 and 1.8–2.2 nm. In contrast, there is almost no corresponding distribution in the range of 1.8–2.2 nm for PHC-X. It is also found that the total pore volume decreased from 0.086 cm3 g−1 for HC, to 0.028 cm3 g−1 for PHC-0.2. This phenomenon is attributed to that the micropores are blocked by phosphorous-containing functional groups, thus preventing N2 molecules from entering.21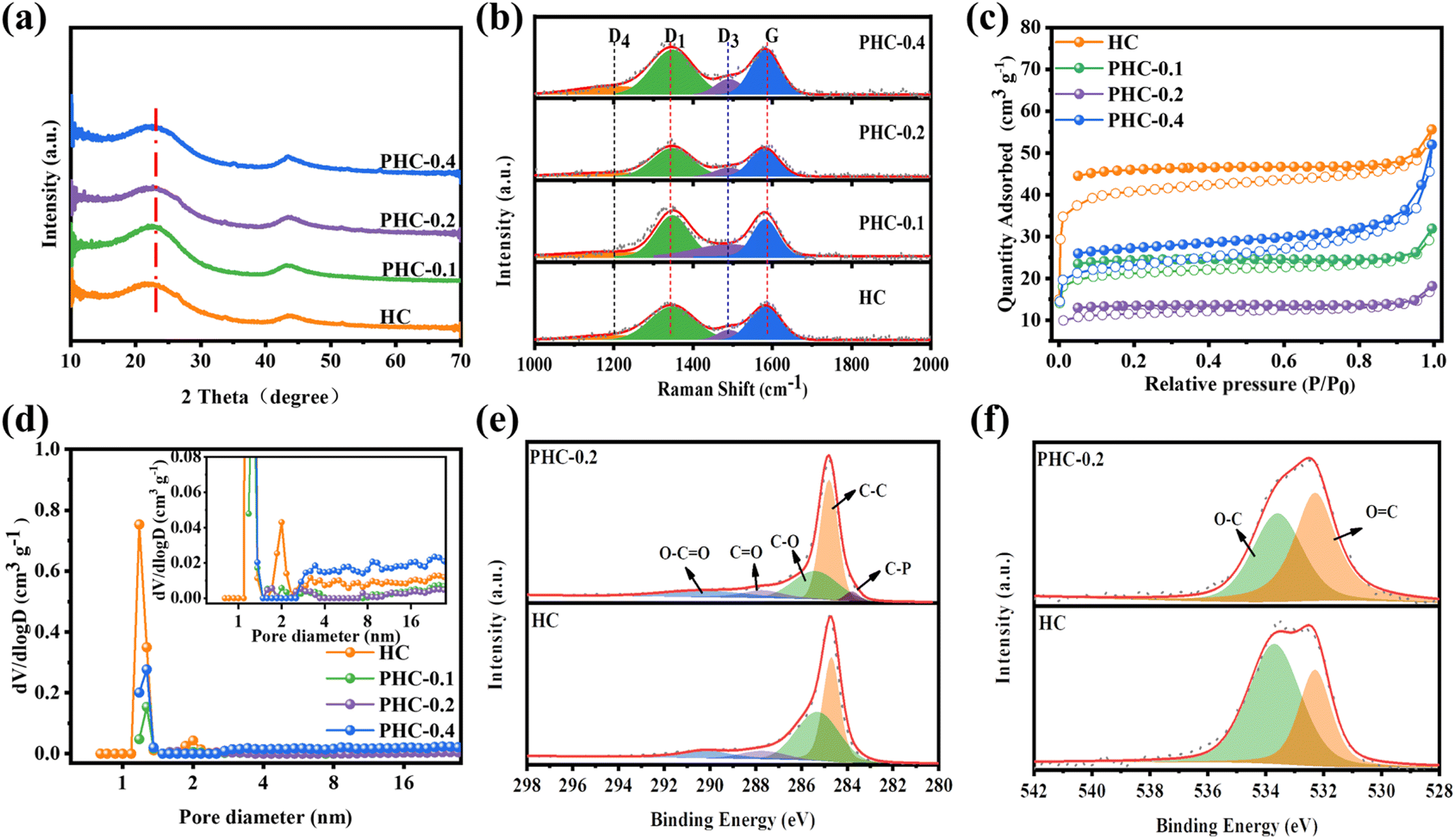 | ||
| Fig. 1 (a) XRD patterns; (b) Raman spectra; (c) N2 adsorption/desorption isotherms; (d) pore size distribution; (e) C 1s spectra; (f) O 1s spectra. | ||
| Samples | A D1/AG | S BET (m2 g−1) | Elemental compositiona | Capacityb (mA h g−1) | ICE (%) | |||
|---|---|---|---|---|---|---|---|---|
| C | O | P | 1st charge | 1st discharge | ||||
| a Obtained from XPS results. b The reversible capacity of the 1st cycle with a current density of 20 mA g−1. | ||||||||
| HC | 1.006 | 156.37 | 95.29 | 4.71 | 282.6 | 363.5 | 77.7 | |
| PHC-0.1 | 1.025 | 81.26 | 94.77 | 5.18 | 0.06 | 308.4 | 340 | 90.7 |
| PHC-0.2 | 1.050 | 44.31 | 94.29 | 5.46 | 0.26 | 343.8 | 372.7 | 92.2 |
| PHC-0.4 | 1.064 | 88.10 | 93.9 | 5.78 | 0.32 | 301.1 | 333.7 | 90.2 |
XPS was carried out to disclose the chemical composition and bonding styles of the HC-X surface. Wide scan XPS spectra (Fig. S1†) indicate that PHC-0.1, PHC-0.2 and PHC-0.4 contain the C, P and O elements, indicating that phosphorus atoms are successfully incorporated into the carbon framework through the esterification reaction between phytic acid and glucose, followed by an annealing process. The high-resolution C 1s spectrum of HC and PHC-0.2 in Fig. 1e can be deconvoluted into four peaks; the characteristic peaks at 284.6, 285.3, 287.8 and 290.2 eV belong to the C–C peak, C–O peak, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak, and O–CO peak, respectively.22,23 Additionally, compared with HC, the occurrence of C–P (283.8 eV) in PHC-0.2 can be ascribed to the effective doping of phosphorus atoms.24 The O 1s can be fitted into peaks of C–O (533.4 eV) and CO (532.3 eV), respectively.22,25 (Fig. 1f) The PHC-0.2 has more CO and less C–O than that of HC, indicating that the introduction of PA could change the content of CO, C–O bonds, and the CO functional group has the ability to react reversibly with Na+ for energy storage, which may promote the Na+ storage capacity.19,26 At the same time, the lower concentration of C–O in PHC-0.2 would result in less SEI film formation, decreasing irreversible Na+ consumption.18
O peak, and O–CO peak, respectively.22,23 Additionally, compared with HC, the occurrence of C–P (283.8 eV) in PHC-0.2 can be ascribed to the effective doping of phosphorus atoms.24 The O 1s can be fitted into peaks of C–O (533.4 eV) and CO (532.3 eV), respectively.22,25 (Fig. 1f) The PHC-0.2 has more CO and less C–O than that of HC, indicating that the introduction of PA could change the content of CO, C–O bonds, and the CO functional group has the ability to react reversibly with Na+ for energy storage, which may promote the Na+ storage capacity.19,26 At the same time, the lower concentration of C–O in PHC-0.2 would result in less SEI film formation, decreasing irreversible Na+ consumption.18
The high-resolution P 2p spectrum in Fig. S2a–c† reveals the formation of the P–C bond (133.1 eV) and P–O bond (134.6 eV) in PHC-X,8,27 and the content of the P–O in PHC-0.2 is 48.2%, which is higher than that of PHC-0.1 (25.6%) and PHC-0.4 (40.7%). These results further manifest that most of the doped phosphorus atoms can incorporate into the matrix. The higher concentration of the P–O bond in hard carbon may be beneficial to the improvement of the capacity and ICE, which is similar to that in LIBs.17 The morphologies of the HC and PHC-X were investigated by SEM and HR-TEM (Fig. 2 and S3†). The non-doped HC shows that undeveloped spherical morphologies and some microspheres grow together, forming the “cluster” of microspheres. This phenomenon is similar to previous reports.1,18,21 The introduction of PA in the precursor solution plays an important role in the regularity and dispersion of the spherical morphologies in PHC-X, and the diameter is increased. Clearly, the in situ doping of the P atom into the carbon framework was achieved as shown in Fig. 2e, which is similar to the effect of boric acid.28 The HR-TEM and SAED (Fig. 2c, d and insets) were performed to reveal the microstructure changes for the hard carbon samples, exhibiting “short-range order” and “long-range disorder” characteristics of the two samples, as well as the corresponding SAED patterns were the typical diffraction rings for the non-graphitic hard carbon structure.29 By comparing TEM images in detail, it is noteworthy the d-spacing for the PHC-0.2 sample and HC is 0.386 nm and 0.379 nm, respectively.
 | ||
| Fig. 2 SEM images of (a) HC and (b) PHC-0.2; HRTEM images and lattice distance of (c) HC and (d) HC-0.2. Inset shows the corresponding SAED and FFT patterns. (e) STEM based element mapping of PHC-0.2. | ||
As can be seen in Fig. 2d, the increase of closed nanovoids is owing to the folding of curved carbon layers and the coalescence of nanopores.30 It is difficult for the electrolyte to permeate into these closed nanovoids, thus reducing the SEI film formation and improving ICE.31 The existence of closed nanovoids and expanded d-spacing are in good agreement with the N2 adsorption–desorption isotherm test and XRD measurement. Fig. 2e shows the scanning transmission electron microscopy (STEM) and corresponding mapping images of PHC-0.2, which confirm that phosphorus atoms are effectively implanted into the carbon skeleton.
Cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) tests were used to investigate the relationship between the microstructure and electrochemical performance. Fig. 3a, b and S4† demonstrate the CV curves of PHC-X at a scan rate of 0.1 mV s−1 between 0.01 and 2 V. A reductive peak appeared at around 0.4 V in the initial CV cycle and then disappeared, which represents the formation of the solid electrolyte interface film (SEI).15 Besides, there is a pair of sharp redox peaks around 0.1 V, and the intensity is highly related to the Na+ (de)insertion process.29 The peak at 0.4 V is weaker in PHC-0.2, suggesting the decreased content of C–O bonds, as well as the enhanced intensity of the peak at 0.1 V is related to the increased CO content19 and expanded carbon spacing after P doping.32 In subsequent cycles, the curves gradually tend to overlap together, indicating a highly reversible electrochemical reaction process. The GCD profiles of various electrodes at 20 mA g−1 are composed of a sloping region and a plateau region (Fig. 3c and S4c†), which are typical characteristics of hard carbon materials.33 The sloping region corresponds to the adsorption/desorption of Na+ on the isolated graphene layers and active sites, while the plateau region is assigned to the (de)insertion of Na+ in expanded graphitic layers and pore filling,15 demonstrating the existence of three different sodium ion storage mechanisms. The initial charge/discharge capacities of HC, PHC-0.1, PHC-0.2 and PHC-0.4 are 282.6/363.5, 308.4/340, 343.8/372.7 and 301.1/333.7 mA h g−1 and detailed slope/plateau capacity values are summarized in Fig. 3d. Significantly, PHC-0.2 possesses a higher reversible capacity, corresponding to an ultrahigh ICE of 92%, which is better than those reported in most previous literature, and the ICE of hard carbon anodes in SIBs hardly exceeds 90% in ester electrolytes (Table S4†). We have also evaluated the dynamic characteristics and rate capability of PHC-X materials (Fig. 3e and f); PHC-0.2 presents the best rate performance with the reversible capacities of 332, 310, 255, 145 mA h g−1 at 20–200 mA g−1 compared with HC, PHC-0.1 and PHC-0.4. The plateau capacity decreased with the increase of current density (Fig. S5†). Fig. 3g shows the cycling performance of PHC-X at 20 mA g−1. After 100 cycles, PHC-0.2 shows an outstanding reversible capacity of 300.1 mA h g−1, and that of PHC-0.1 is 287 mA h g−1, and that of PHC-0.4 is 266 mA h g−1, all of which are better than that of HC (198 mA h g−1).
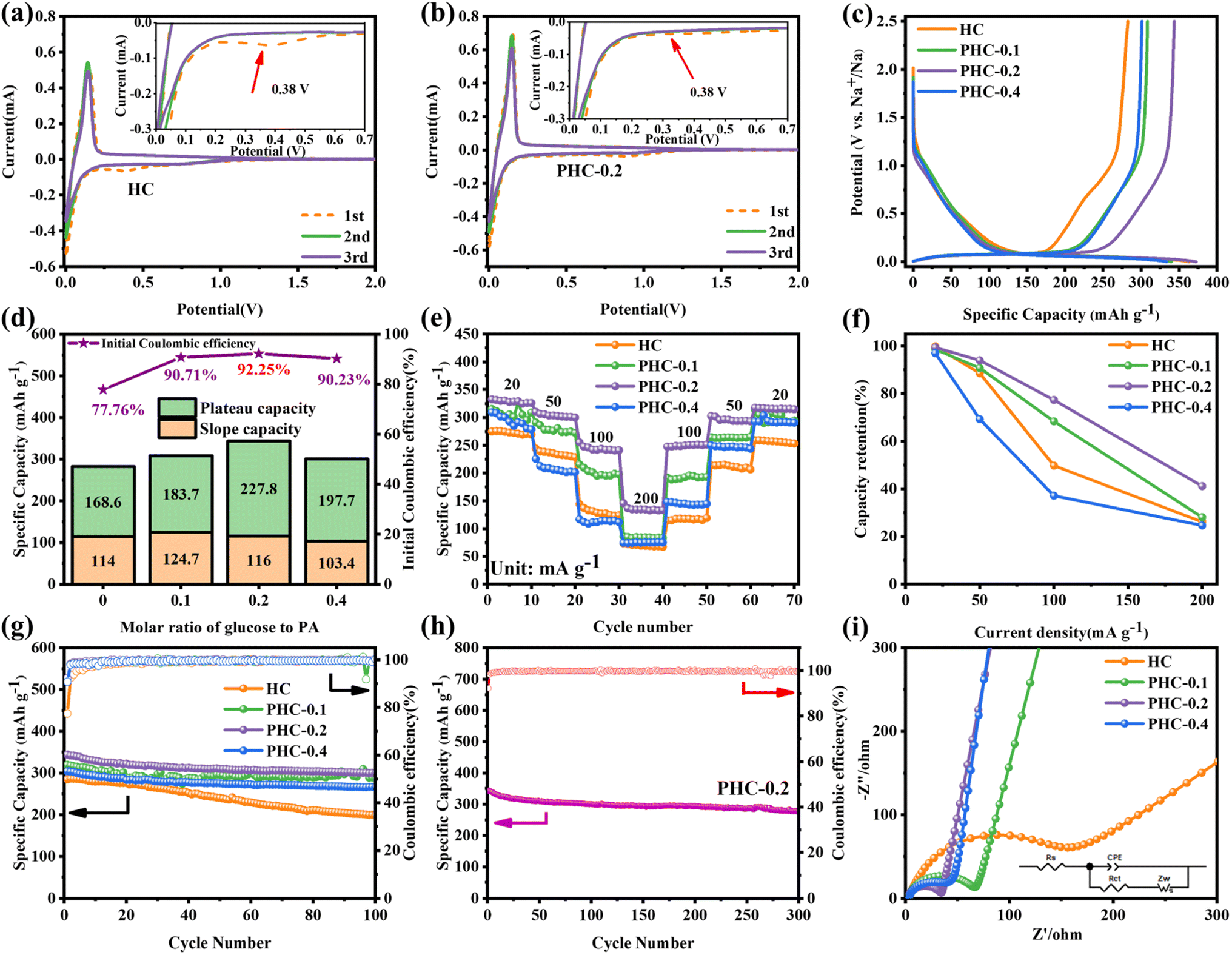 | ||
| Fig. 3 Electrochemical performance: (a and b) the CV curves of HC and PHC-0.2, (c) initial galvanostatic charge–discharge profiles of the PHC-X electrodes at 20 mA g—1, (d) plots of ICE, plateau capacity and sloping capacity in the first cycle versus PA content for PHC-X electrodes, (e and f) rate performance and capacity retention versus rate plots of PHC-X samples, (g) cycling performance of the PHC-X electrodes at 20 mA g—1, and (h) long-term cycling performance at a rate of 20 mA g−1 for the PHC-0.2 sample. (i) EIS of the samples. | ||
Moreover, the long-term cycling performance of PHC-0.2 at 20 mA g−1 is shown in Fig. 3h, which exhibits no obvious capacity decay after 300 cycles, and the capacity retention rate is 81%, demonstrating the exceptional long cycle life. Fig. 3i shows the electrochemical impedance spectra (EIS); PHC-0.2 exhibits the smallest semicircle diameter and maximum slope, implying the lowest charge transfer resistance and highest Na+ diffusion ability. To investigate the kinetics and Na+ storage behavior of the electrochemical reaction, CV tests were performed at different scan rates from 0.2 to 1.5 mV s−1. As shown in Fig. 4a, the relationship between the sweep rate (ν) and peak current (i) can be expressed as: i = aνb, where a and b are adjustable parameters, and typically, the b-value approaches 0.5 or 1, which infers a diffusion-controlled behavior or surface-induced capacitance process, respectively.34Fig. 4b shows that the anodic and cathodic peak b-value of PHC-0.2 is 0.23 and 0.34, indicating that the electrochemical reaction is mainly a diffusion-controlled process. To further quantify the ratios of capacitive contribution in total capacitance at specific scan rates, we use the formula: (V) = k1ν + k2ν1/2. As shown in Fig. 4c and S6,† the capacitance contribution reaches 36% of the total capacity at 0.2 mV s−1. As the scanning rate increases, the capacitive contribution gradually increases, indicating that the capacity mainly comes from surface sodium storage at high current density, while the slow intercalation speed cannot satisfy the fast electrochemical reaction (Fig. 4d).
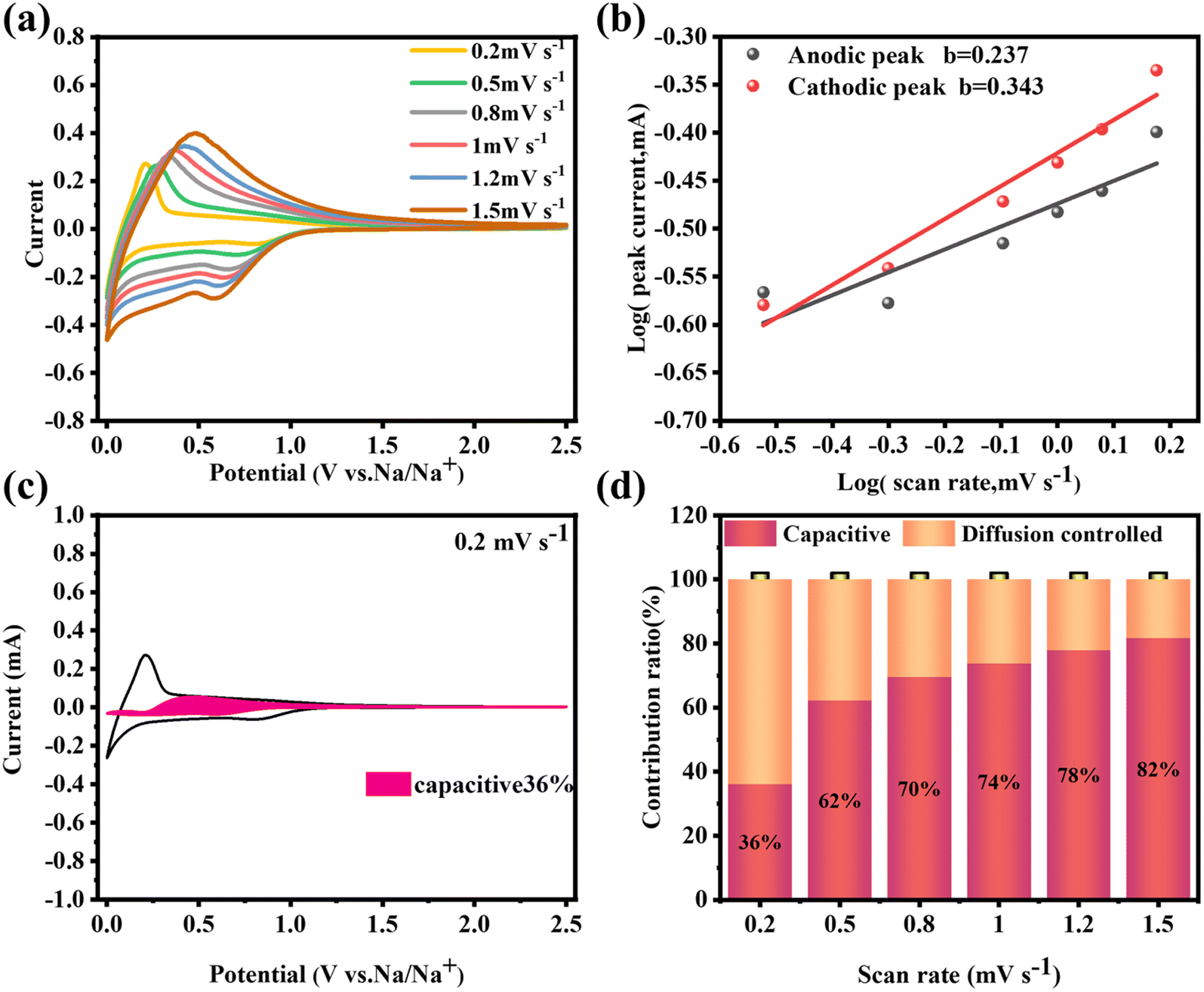 | ||
| Fig. 4 For the PHC-0.2 electrode: (a) cyclic voltammetry curves at different scan rates between 0.2 and 1.5 mV s−1, (b) the plots of log(i) versus log(ν), (c) pseudocapacitive contribution at 0.2 mV s−1, and (d) pseudocapacitive contribution at different scan rates between 0.2 and 1.5 mV s−1. | ||
To further explore the sodium storage mechanisms, GITT was used to calculate the Na+ diffusion coefficient (DNa+) in the sloping and plateau regions. According to the Fick's second law of diffusion, the DNa+ value can be calculated using the equation.35
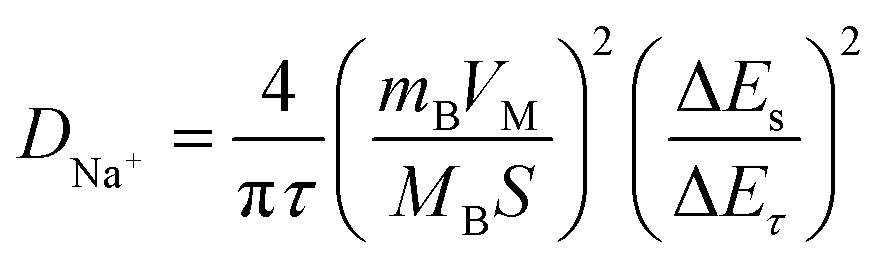
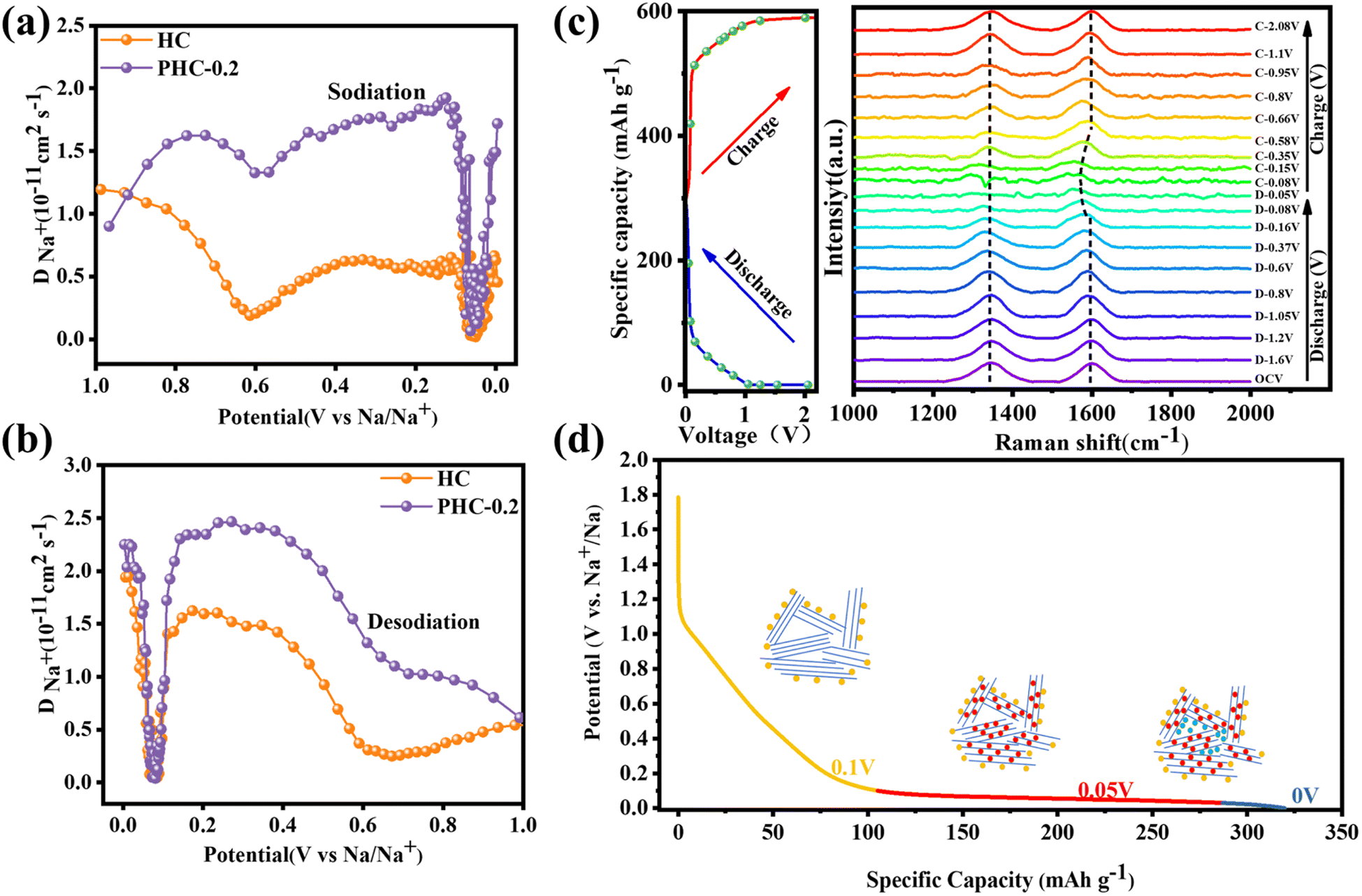 | ||
| Fig. 5 For the PHC-X anode: (a and b) Na+ diffusion coefficients during the discharge process and charge process. (c) In situ Raman spectra and the corresponding discharge–charge profiles at the first cycle of PHC-0.2 for SIBs. (d) Schematic diagram of the sodium storage stages of hard carbon. | ||
In situ Raman spectroscopy is carried out to gain further insight into the Na+ storage mechanism. As shown in Fig. 5c, D- and G-bands appear initially at 1347 cm−1 and 1597 cm−1. Upon 1st discharge to 0.16 V, no significant peak shift change can be observed, which implies that a negligible intercalation process is involved,37 but the intensity of the D band gradually decreases. As we know, the D-band reflects the presence of defects and edges of graphene layers, and the weak intensity of the D-band can be attributed to the adsorption of Na+ at defective sites, which restricts the breathing vibration of sp2 carbon rings. It is worth noting that G-band shifting to a lower wavenumber becomes higher and the signal of the G-band vanishes when discharged to 0 V, which is ascribed to the formation of graphite-intercalation-compounds.38 Additionally, the reverse increase of DNa+ and the decrease in the intensity of the D-band peak suggest the presence of pore-filling behavior. All in all, combined with GITT and in situ Raman results, the Na+ storage mechanism may belong to the “adsorption, intercalation and pore filling” model.
Application-wise, as shown in Fig. 6a, the Na2Fe0.5Mn0.5[Fe(CN)6] cathode (denoted as FeMnHCF) is coupled with anode electrodes to assemble the coin-type full SIBs and the ratio of negative and positive active material(N/P) is 1.06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Among the various kinds of cathode materials, Prussian blue (PB) and its analogues (PBAs) have been widely researched due to the three dimensional open framework, high theoretical capacity, and simple synthesis method.39 According to the XRD result in Fig. S7a,† FeMnHCF shows the monoclinic phase, with the space group P21/n, which is different from the common face centered cubic structure. Fig. S7b† displays the SEM image of FeMnHCF. The electrochemical performance of the FeMnHCF cathode has been investigated in a half coin cell which is presented in Fig. S7c and d,† and the FeMnHCF cathode provided a reversible capacity of about 135.19 mA h g−1 after 200 cycles at 50 mA g−1. Furthermore, the charge–discharge profiles of the PHC-0.2//FeMnHCF full cell at 50 mA g−1 between 2 and 3.6 V are shown in Fig. 6b. Based on the mass of the anode, the PHC-0.2//FeMnHCF exhibits an average voltage of 3.1 V, and an ICE of 85%. Moreover, a high reversible capacity of 186.7 mA h g−1 and 80.29% of the specific capacity can be maintained after 170 cycles at 50 mA g−1. As shown in Fig. 6d, the light-emitting diodes (LEDs) could be lit by an assembled PHC-0.2//FeMnHCF. These promising electrochemical performances in the full cell show that PHC-0.2 could be a promising anode material for Na-ion full cells.
:1. Among the various kinds of cathode materials, Prussian blue (PB) and its analogues (PBAs) have been widely researched due to the three dimensional open framework, high theoretical capacity, and simple synthesis method.39 According to the XRD result in Fig. S7a,† FeMnHCF shows the monoclinic phase, with the space group P21/n, which is different from the common face centered cubic structure. Fig. S7b† displays the SEM image of FeMnHCF. The electrochemical performance of the FeMnHCF cathode has been investigated in a half coin cell which is presented in Fig. S7c and d,† and the FeMnHCF cathode provided a reversible capacity of about 135.19 mA h g−1 after 200 cycles at 50 mA g−1. Furthermore, the charge–discharge profiles of the PHC-0.2//FeMnHCF full cell at 50 mA g−1 between 2 and 3.6 V are shown in Fig. 6b. Based on the mass of the anode, the PHC-0.2//FeMnHCF exhibits an average voltage of 3.1 V, and an ICE of 85%. Moreover, a high reversible capacity of 186.7 mA h g−1 and 80.29% of the specific capacity can be maintained after 170 cycles at 50 mA g−1. As shown in Fig. 6d, the light-emitting diodes (LEDs) could be lit by an assembled PHC-0.2//FeMnHCF. These promising electrochemical performances in the full cell show that PHC-0.2 could be a promising anode material for Na-ion full cells.
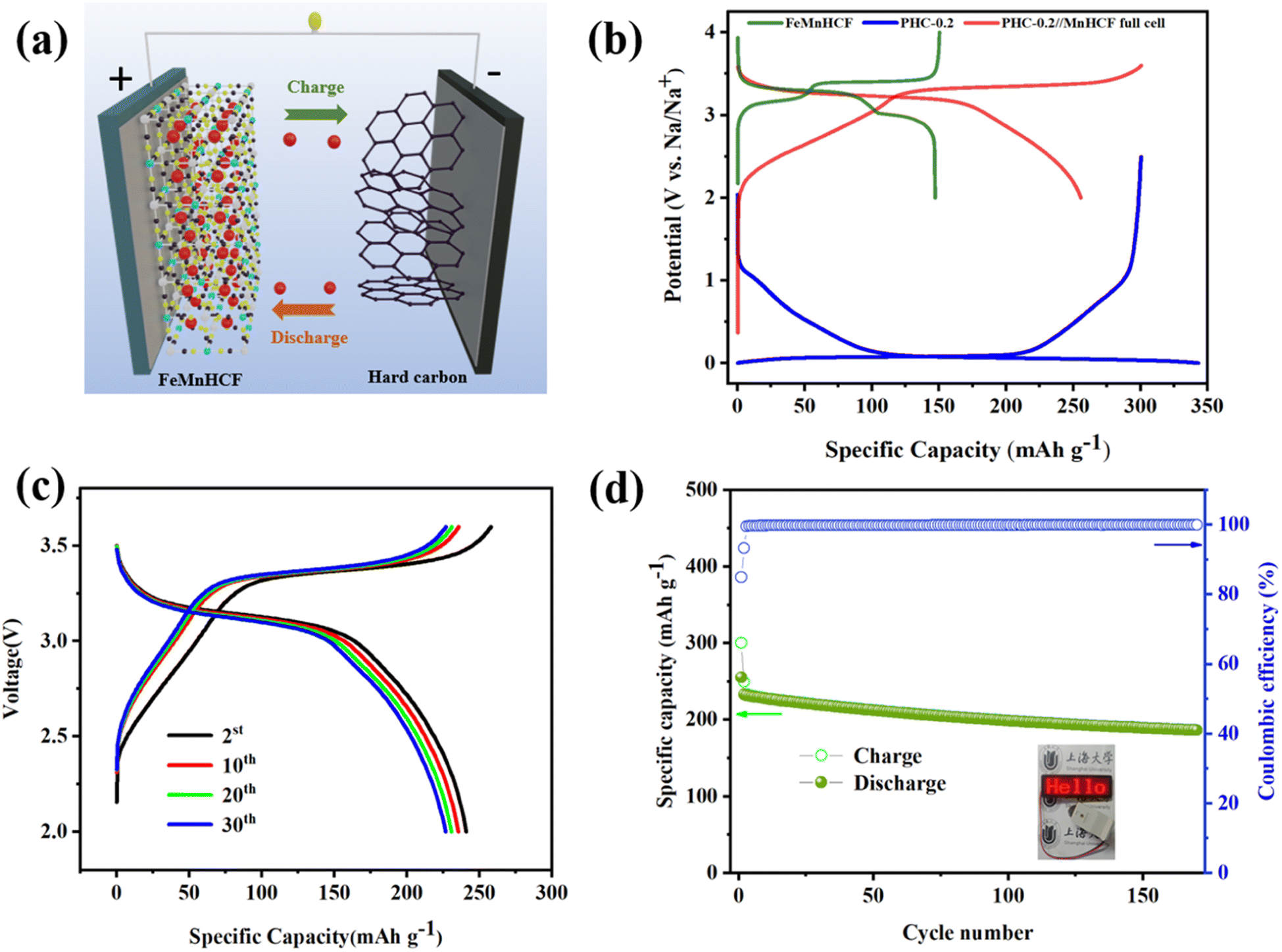 | ||
| Fig. 6 Electrochemical performances of the sodium-ion battery based on the PHC-0.2 anode and Na2Fe0.5Mn0.5[Fe(CN)6] (FeMnHCF) cathode. (a) Schematic illustration of the PHC-0.2//FeMnHCF SIB. (b) Initial three charge–discharge curves of the PHC-0.2//FeMnHCF SIB at the current density of 50 mA g−1. (c) Charge–discharge curves of the PHC-0.2//FeMnHCF SIB at various cycles. (d) Cycling performance of the PHC-0.2//FeMnHCF SIB at the current density of 50 mA g−1. Inset shows the photograph of LEDs lit by a simple PHC-0.2//FeMnHCF coin cell. | ||
Conclusions
In this work, in situ phosphorus doped hard carbon (PHC-0.2) facilely synthesized by a hydrothermal polymerization method exhibits an enhanced capacity and outstanding ICE. Introducing a suitable amount of P atoms can optimize the polydisperse spherical hard carbon, expand the interlayer spacing and repair some defects in carbon layers. Besides, GITT and in situ Raman imply that phosphorus species can elevate the diffusion coefficient of Na+ between microcrystalline interlayers to facilitate the diffusion-controlled Na+ insertion process and the sodium storage mechanism model may conform to “adsorption, intercalation and pore filling”. Lastly, the full-cell assembled with the FeMnHCF cathode exhibits a high energy density of 371 W h kg−1 based on the mass of the cathode with an operation voltage of 3.1 V, as well as an excellent cycling stability. We believe that this method may provide a new path to optimize the performance of hard carbon anodes with suitable level heteroatom doping.Data availability
The data underlying this study are available in the ESI.†Author contributions
Z. G. L. and J. H. Z. performed the material synthesis and electrochemical tests. Z. G. L., J. H. Z., and H. Y. analyzed and discussed the data. X. X. H. and H. Z. performed in situ Raman characterization. Z. G. L. and J. H. Z. wrote the manuscript with help from all of the co-authors. All authors revised the manuscript and discussed the results.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 52101265, and 52171217), and Zhejiang Provincial Natural Science Foundation of China under Grant No.ZCLY24E0201. The Innovative Research Teams of High-level Local Universities in Shanghai are gratefully acknowledged for their financial support.References
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O'Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M.-M. Titirici, Energy Environ. Sci., 2020, 13, 3469 RSC
.
- M. Ma, S. Zhang, L. Wang, Y. Yao, R. Shao, L. Shen, L. Yu, J. Dai, Y. Jiang, X. Cheng, Y. Wu, X. Wu, X. Yao, Q. Zhang and Y. Yu, Adv. Mater., 2021, 33, e2106232 CrossRef PubMed
- P. Bai, Y. He, X. Zou, X. Zhao, P. Xiong and Y. Xu, Adv. Energy Mater., 2018, 8, 1703217 CrossRef
-
(a) G. Chang, Y. Zhao, X. Gao, Z. Li and H. Zhao, Chem. Commun., 2022, 58, 7317 RSC
-
(a) X. Zhu, X. Jiang, X. Liu, L. Xiao and Y. Cao, Green Energy Environ., 2017, 2, 310 CrossRef
- Y. Liu, B. V. Merinov and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3735 CrossRef CAS PubMed
- D.-S. Bin, Y. Li, Y.-G. Sun, S.-Y. Duan, Y. Lu, J. Ma, A.-M. Cao, Y.-S. Hu and L.-J. Wan, Adv. Energy Mater., 2018, 8, 1800855 CrossRef
- C. Chen, Y. Huang, M. Lu, J. Zhang and T. Li, Carbon, 2021, 183, 415 CrossRef CAS
- K.-Y. Zhang, Y.-Q. Fu, H.-H. Liu, J.-L. Yang, M.-Y. Su, Y. Wang and X.-L. Wu, Phys. Scr., 2023, 98, 125977 CrossRef
- F. Wu, R. Dong, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 21335 CrossRef CAS PubMed
- F. Wu, L. Liu, Y. Yuan, Y. Li, Y. Bai, T. Li, J. Lu and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 27030 CrossRef CAS PubMed
- X. Wang, M. Hou, Z. Shi, X. Liu, I. Mizota, H. Lou, B. Wang and X. Hou, ACS Appl. Mater. Interfaces, 2021, 13, 12059 CrossRef CAS PubMed
-
(a) Y. Li, K. Zhang, Z. Chen, Y. Wang, L. Wang, F. Liang and Y. Yao, Nano, 2020, 15, 2050003 CrossRef CAS
- Z. Yang, Y. Gao, Z. Zhao, Y. Wang, Y. Wu and X. Wang, J. Power Sources, 2020, 474, 228500 CrossRef CAS
- X.-X. He, J.-H. Zhao, W.-H. Lai, R. Li, Z. Yang, C.-M. Xu, Y. Dai, Y. Gao, X.-H. Liu, L. Li, G. Xu, Y. Qiao, S.-L. Chou and M. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 44358 CrossRef CAS PubMed
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef
- H. Tao, S. Du, F. Zhang, L. Xiong, Y. Zhang, H. Ma and X. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 34245 CrossRef CAS PubMed
- L. Yang, M. Hu, Q. Lv, H. Zhang, W. Yang and R. Lv, Carbon, 2020, 163, 288 CrossRef CAS
- Y. Liao, F. Luo, T. Lyu, M. Chen, C. Liu, D. Xu, P. Chen, Q. Liu, Z. Wang, S. Li, Y. Ye, D. Wang, C. Miao, Z. Liu, D. Wang and Z. Zheng, Diamond Relat. Mater., 2022, 129, 109392 CrossRef CAS
- X. Lin, Y. Liu, H. Tan and B. Zhang, Carbon, 2020, 157, 316 CrossRef CAS
- Y. Li, Z. Wang, L. Li, S. Peng, L. Zhang, M. Srinivasan and S. Ramakrishna, Carbon, 2016, 99, 556 CrossRef CAS
- R.-R. Li, X.-X. He, Z. Yang, X.-H. Liu, Y. Qiao, L. Xu, L. Li and S.-L. Chou, Mater. Chem. Front., 2021, 5, 7595 RSC
- C. Chen, Y. Huang, Y. Zhu, Z. Zhang, Z. Guang, Z. Meng and P. Liu, ACS Sustain. Chem. Eng., 2020, 8, 1497 CrossRef CAS
-
(a) F. Wu, R. Dong, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 21335 CrossRef CAS PubMed
- T. Xu, X. Qiu, X. Zhang and Y. Xia, Chem. Eng. J., 2023, 452, 139514 CrossRef CAS
-
(a) C. Shen, C. Wang, T. Jin, X. Zhang, L. Jiao and K. Xie, Nanoscale, 2022, 14, 8959 RSC
-
(a) W. Xu, C. Lv, Y. Zou, J. Ren, X. She, Y. Zhu, Y. Zhang, S. Chen, X. Yang, T. Zhan, J. Sun and D. Yang, J. Power Sources, 2019, 442, 227184 CrossRef CAS
- D. Wu, F. Sun, Z. Qu, H. Wang, Z. Lou, B. Wu and G. Zhao, J. Mater. Chem. A, 2022, 10, 17225 RSC
- K. Yu, X. Wang, H. Yang, Y. Bai and C. Wu, J. Energy Chem., 2021, 55, 499 CrossRef CAS
- Y. Li, Y. Lu, Q. Meng, A. C. S. Jensen, Q. Zhang, Q. Zhang, Y. Tong, Y. Qi, L. Gu, M.-M. Titirici and Y.-S. Hu, Adv. Energy Mater., 2019, 9, 1902852 CrossRef CAS
- K. Wang, F. Sun, H. Wang, D. Wu, Y. Chao, J. Gao and G. Zhao, Adv. Funct. Mater., 2022, 32, 2203725 CrossRef CAS
- C. Chen, Y. Huang, Z. Meng, M. Lu, Z. Xu, P. Liu and T. Li, Carbon, 2020, 170, 225 CrossRef CAS
- Y. Li, Y. Yuan, Y. Bai, Y. Liu, Z. Wang, L. Li, F. Wu, K. Amine, C. Wu and J. Lu, Adv. Energy Mater., 2018, 8, 1702781 CrossRef
- S. Qiu, L. F. Xiao, M. L. Sushko, K. S. Han, Y. Y. Shao, M. Y. Yan, X. M. Liang, L. Q. Mai, J. W. Feng, Y. L. Cao, X. P. Ai, H. X. Yang and J. Liu, Adv. Energy Mater., 2017, 7, 1700403 CrossRef
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569 CrossRef CAS
- C. Bommier, T. W. Surta, M. Dolgos and X. L. Ji, Nano Lett., 2015, 15, 5888 CrossRef CAS PubMed
- H. Tan, R. Zhou and B. Zhang, J. Power Sources, 2021, 506, 230179 CrossRef CAS
- J. Li, L. Yu, Y. Li, G. Wang, L. Zhao, B. Peng, S. Zeng, L. Shi and G. Zhang, Nanoscale, 2021, 13, 692 RSC
- J.-Q. Huang, R. Du, H. Zhang, Y. Liu, J. Chen, Y.-J. Liu, L. Li, J. Peng, Y. Qiao and S.-L. Chou, Chem. Commun., 2023, 59, 9320 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01395f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |