
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotophysics of fluorescent nanoparticles based on organic dyes – challenges and design principles
Stine G.
Stenspil
 and
Bo W.
Laursen
*
and
Bo W.
Laursen
*
Nano-Science Center & Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 København Ø, Denmark. E-mail: bwl@chem.ku.dk
First published on 7th May 2024
Abstract
Fluorescent nanoparticles have become attractive for bioanalysis and imaging, due to their high brightness and photostability. Many different optical materials have been applied in fluorescent nanoparticles with a broad range of properties and characteristics. One appealing approach is the incorporation of molecular organic fluorophores in nanoparticles with the intention of transferring their known attractive solution-state properties directly to the nanoparticles. However, as molecular dyes are packed closely together in the nanoparticles their interactions most often result in fluorescence quenching and change in spectral properties making this approach challenging. In this perspective we will first discuss the origins of quenching and spectral shifts observed in dye based nanoparticles. On this background, we will then describe various designs of dye based NPs and how they address the challenges of dye–dye interactions and quenching. Our aim is to provide a general framework for understanding the supramolecular mechanisms that determine the photophysics of dye based nanoparticles. This framework of molecular photophysics and its relation to the internal structure of dye based nanoparticles can hopefully serve to assist rational design and optimization of new and improved dye based nanoparticles.
Introduction
Fluorescent nanoparticles (NPs) have proven attractive for use in bioanalysis and bioimaging.1,2 Compared to traditional molecular fluorescent labels the NPs can offer a number of advantages including highly increased brightness and increased photostability. Another key advantage of NPs is that the emissive materials are often protected from the environment making the optical properties insensitive to complex biological environments. In general, NPs also display low propensity for unspecific binding or localization in biological systems. Motivated by these potential advantages many different types of nanoparticles have been reported. In a very general manner, they can be divided into inorganic based or organic based, where inorganic NPs were more dominating in the early years. Among the inorganic nanoparticles there are many variations3,4 including the well-known quantum dots (QDs)5–8 and upconversion NPs.9,10 Fluorescent NPs where the absorption and emission stem from organic molecules and materials include NPs based on molecular dyes (neat, or embedded in a matrix material),11 conjugated polymers,12,13 and amorphous carbon materials (carbon dots).14,15In the case of QDs their spectral properties mainly result from electronic states delocalized over the entire QD and thus intimately linked to the size. By varying the size of the particle, it is possible to create emission colours spanning all wavelengths from the UV to IR.6 While this provides a direct way of engineering the optical properties of a single material, it can also be a limitation since it requires very strict preparation conditions and size control. In addition, QDs do usually not have discrete absorption bands, which limits selective excitation.6,16 Contrary to this, the optical properties of NPs based on organic dyes and conjugated polymers are largely defined by the covalent building blocks and thus independent of the size and size distribution of the NPs, while also in general being brighter than their inorganic counterparts.11 It is exactly the well-defined correlation between the covalent structure of organic molecules and their photophysical properties that is the basis for their large potential as fluorescent labels and probes.17,18 150 years of organic synthesis and study of dyes have provided a very diverse range of structures and photophysical properties, and a detailed understanding of the structure–property relations. Therefore, it is also attractive to design dyes and polymers optimized for use in fluorescent NPs, or simply use this vast back catalogue of already existing dyes and polymers and try to transfer desired photophysical properties to NPs.
Packing a large number of these organic units in one NP can bring about advantages such as very high brightness and protection from the environment. However, as molecular dyes are packed closely together in the NPs their interactions most often result in fluorescence quenching, known as aggregation caused quenching (ACQ), and change in spectral properties making this approach less straightforward and more challenging than one could have hoped. Nevertheless, the field of fluorescent organic NPs is rapidly expanding with a plethora of different design strategies and materials. All these molecular NP designs and materials face some common conditions/challenges, which needs awareness when we take the next steps towards organic fluorescent NPs with improved or more advanced photophysical properties. In this perspective, we will focus on the challenges that come up when we try to translate the photophysical properties of organic dyes to NPs. First, we will discuss various effects that influence and may change the photophysical properties of dyes when incorporated into NPs. Secondly, we will discuss examples of strategies that have been applied to mitigate, circumvent or even benefit from these effects. The intention is to provide an overview of common issues that influence the photophysical properties of dye based NPs and how these issues relate to the properties of the building blocks and the internal structure of the NPs. We will not cover other important aspects such as preparation methods, surface modification or stability in biological systems. The reader is instead directed to other reviews for a more thorough discussion of these aspects of NPs and their applications.4,11,13,19 Nanoparticles like carbon dots, that consist of amorphous to nanocrystalline cores of graphene-like structures stacked together,14,20–25 usually characterized by having unknown chromophoric systems with multiple emitter states somewhat resembling trap states in conjugated polymers, are also not within the scope of this perspective and will not be discussed further.
Challenges that influence optical properties
In the following sections, we will discuss the common effects that can influence the photophysical properties when using conjugated organic molecules and polymers as the light absorbing and emitting materials in NPs. As the field of organic dye based NPs is expanding with more and more different optical materials, matrix materials and assembly strategies it is difficult to get an overview of different challenges and their potential solutions. While some challenges are specific to certain approaches, we believe that most can be assigned to a few important general effects. We will discuss the origin of these general effects and how they manifest in the photophysical properties of NPs.Fluorescence quenching and other changes in photophysical properties when going from molecules in dilute solution to molecular materials are extremely well-studied and documented.26,27 Much can be learned from the extensive research on molecular crystals and conductive polymers for optoelectronic applications. A recent review by Gierschner et al. provides a clear and nuanced discussion of the photophysical properties of molecular solids with focus on packing effects in crystalline molecular materials.27 Even though individual chromophores used in NPs may be much different from e.g. laser or OLED molecules, the effects and challenges that are thoroughly described in this field are recurring in the field of fluorescent NPs. It can therefore prove beneficial for researchers working with dye based NPs to consult the literature in this area since the fundamental processes are very similar in many regards. In other words, there is nothing new about the photophysical effects we describe in this perspective, we just seek to give a simple general description of these effects in the specific context of organic dye based NPs. Before discussing how interactions of dyes in NPs influence the NP photophysical properties we will briefly discuss the most important descriptors of the molecular photophysical properties and their interrelations.
Photophysical descriptors of dyes
Among the most important properties of dyes is the probability (allowedness) of the optical transitions, often expressed by the transition dipole moments (μ). Experimentally the magnitude of the absorption transition moment is proportional to the molar absorption coefficient ε (integrated over the absorption band for the specific transition). For the fluorescence transition, the radiative rate kf provides a measure of the allowedness. Experimentally kf is obtained from the fluorescence quantum yield (Φf, QY) and the lifetime (τf):| kf = Φf/τf | (1) |
The Strickler–Berg relation28 predicts that ε and kf are proportional if they describe the optical transition to and from the same excited state (S1), which is most often the case for efficient fluorophores. A high quantum yield of fluorescence is obtained when the radiative rate is larger than the sum of all the competing non-radiative deactivation processes (described by knr):
| Φf = kf/(kf + knr) | (2) |
Fluorescent dyes fulfilling these criteria of intense radiative transitions and comparable low non-radiative rates are typically rigid π-systems with small structural relaxation in the excited state and thus small Stokes shifts.
When the organic fluorophores are brought close together in solid materials like molecular crystals or NPs with high dye density, efficient quenching is often observed. Often this phenomenon is just described as aggregation caused quenching (ACQ), concentration quenching, or self-quenching. These terms however cover a range of individual effects that alter the photophysical properties of fluorophores at high densities. A deeper understanding of these effects is needed to design new materials without quenched and distorted spectra. Below we discuss some of the most common issues that occur for dyes at high density in NPs.
Strong chromophore interactions
When molecular chromophores are packed closely together, the electronic transitions of a single chromophore are likely to be strongly influenced by the nearby chromophores surrounding it. In many cases, the electronic coupling between the chromophores is so strong that the excited state cannot any longer be described as only located on a single chromophore unit but is instead delocalized over two or more units. Depending on the systems and the details of the new states formed, such phenomena are described by different models like exciton coupling and excimers. We will denote all such phenomena as “strong interactions”. Strong interactions can lead to large spectral shifts in both absorption and emission spectra, just like the new delocalized excited states can display very different radiative and non-radiative rates and thus fluorescence quantum yields and lifetimes.In the context of molecular materials and dye based NPs the most common model for strong interactions is based on the molecular exciton coupling model by Kasha, which describes the new states and transitions as a result of Coulomb coupling between the transition dipole moments in neighbouring chromophores.29,30 From this model it is evident that the energy and transition probabilities of the aggregate exciton transitions strongly depend on the detailed relative arrangement of the chromophores. For a dimer, this can be illustrated by the famous cases of head-to-tail (J) and side-by-side (H) dimers, both showing new delocalized exciton states (Fig. 1). The Coulomb coupling JC depends on the distance (R) and magnitude of the monomer transition dipole moment (μ), the angle between the transition moments (θ), and the local dielectric constant (ε):
JC = μ2(1 − 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2θ)/4πεR3 cos2θ)/4πεR3 | (3) |
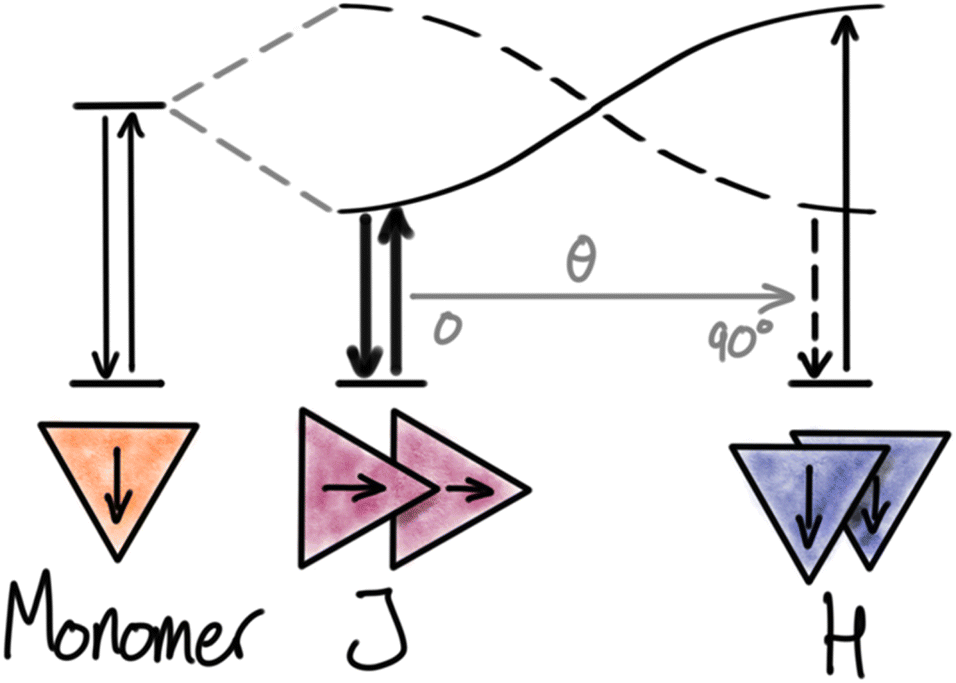 | ||
| Fig. 1 Kasha’s exciton model for dimers. Relative orientation as a function of the angle (θ) between the transition dipole moments of dyes generating J- and H-aggregates with new electronic transitions. | ||
The Coulomb coupling of the monomer transitions leads to two new excitonic transitions. The in-phase combination of the monomer transition dipole moments leads to an allowed transition, shifted by JC relative to the monomer. For the out-of-phase combination, the transition dipoles cancel out and lead to a forbidden transition, also shifted by JC relative to the monomer. In J-aggregates with head-to-tail arrangement of the transition dipoles (0° ≤ θ < 54.7°) the in-phase combination has the lowest energy, and absorption and emission spectra are found to red-shift according to the exciton coupling strength by JC relative to the monomer transition. For the side-by-side arrangement in H-aggregates (54.7° < θ ≤ 90°) the allowed in-phase combination has higher energy than the forbidden out-of-phase combination. Since the absorption is dominated by the most allowed transitions, blue-shifts are observed for H-aggregates (absorption to the S2 exciton state). On the other hand, fluorescence is originating from the lowest excited state (according to Kasha's rule) and will come from the forbidden S1 → S0 transition and appear with a large Stokes shift relative to the dominating exciton absorption band (S0 → S2). Fluorescence from H-aggregates is often weak or completely absent due to the forbidden nature of the transition, which results in low rates of emission compared to the competing non-radiative deactivation pathways. In contrast, fluorescence from J-aggregates can be very efficient due to the high radiative rate and has been actively pursued by many research groups.31 However, it is important to remember that the quantum yield is a result of the competition between the radiative rate and the non-radiative rates and cannot be predicted from the aggregate structure alone. From eqn (3) it is clear that dyes with large transition moments (μ), and thus large molar absorption coefficients, are most likely to display strong exciton coupling. As seen from eqn (3) and Fig. 1 any change in the angle between the chromophores will have large impact on the energy and transition probabilities in the aggregate. For crystals, examples of any angle can be found and in intermediate cases, both transitions can be observed (Davydov splitting).27,32 In NPs with less ordered structures a range of coexisting conformations can be expected and thus also inhomogeneous photophysical properties of the NPs.
The term excimer is usually used to describe a dimer between an excited and a ground state molecule formed dynamically in solution.33 Excimers are mainly formed by planar aromatic hydrocarbons, pyrene being the textbook example, and are characterized by a broad redshifted emission from their excited state delocalized over the π-stack dimer. In molecular solids similar emission features can be observed from some π-stack materials that either are already in a geometry favourable for excimer formation or due to structural relaxation in more dynamic systems favouring the excimer state.27,34 Since the excimer states are completely different from the monomer excited state with lower energy and different radiative and non-radiative rates, the formation of excimer states will lead to large redshifts in emission and often significant quenching.
The perspective by Bialas et al. provides a thorough discussion of different approaches for direct dye assembly to achieve specific aggregate structures with predictable properties such as excimers or J-aggregates.35 Their argument is that better understanding of the photophysical processes occurring in the solid-state will help in the development of new fluorescent solid-state materials. However, even the simplest systems require thorough analysis of aggregate photophysics and the control of aggregate structure might prove even more difficult for NPs that also need to be stable in water and biological systems. In an example by Piwonski et al.36 they carefully designed a D–π–A–π–D structure, which forms a semi-crystalline J-aggregate structure with a size of ∼3.5 nm named J-dots.
Another example by Chen et al.37 shows the encapsulation of the cyanine dye IR-140 in a hollow silica nanoparticle; the close packing results in strong interaction and J-aggregated emission in the infrared region. For the above-mentioned examples, each of the particles have their advantages. However, the formation of new emitter states by strong interactions in general leads to unpredictable photophysical properties and therefore can require a lot of trial and error before favourable photophysical properties may be achieved.
Energy migration and quenching
Energy migration is a key feature of dye based NPs. Even when the chromophores are sufficiently separated to avoid strong interactions, long range weak Coulomb interactions still mediate efficient energy transfer. This non-radiative Coulomb energy transfer is also known as resonance energy transfer (RET) or Förster resonance energy transfer (FRET).27,38,39 While energy migration between identical dyes does not itself change the photophysical properties, it often facilitates fluorescence quenching by bringing the excitation to a site where quenching occurs. In this way energy migration contributes to the phenomena of ACQ to a large extent. In a NP containing only one type of dye, homo energy transfer can occur as long as the dyes are close enough and there is a spectral overlap between absorption and emission of the chromophore. With efficient energy transfer, the excited state (the exciton) can migrate in the NP visiting many dyes during its lifetime (τ), until it eventually deactivates radiatively or non-radiatively. However, if the exciton reaches a site with a structural defect or impurity of lower energy it may get trapped there and deactivate (non-radiatively) on the trap (Fig. 2). If energy migration is efficient even a very low density of such dark trap-states may quench a large fraction or all of the NP emission.40 The most clear indication of such energy transfer mediated quenching is shortened multi-exponential fluorescence decays as the density of dye or traps in NPs is increased.41 In the absence of traps, mobile excitons (high loading of dye) will have the same mono-exponential decay and lifetime (τ) as localized excitons in a diluted system. However, in systems with energy migration and traps the lifetime of the excitons will depend on their proximity to a trap/quencher site leading to multi-exponential decays and shortened lifetimes. The efficiency of the energy transfer process itself can, in disordered/isotropic systems, be assessed by measuring the rate of anisotropy loss.42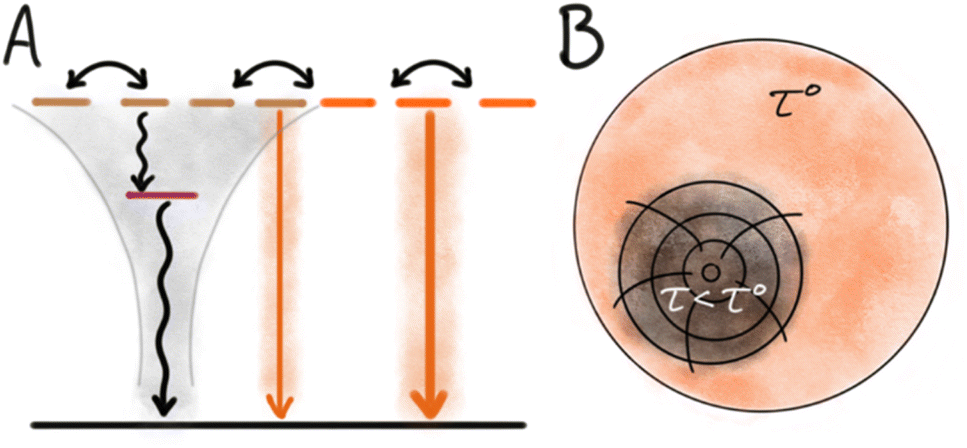 | ||
| Fig. 2 Quenching by trap states. (A) Schematic state diagram with indication of energy migration between multiple similar dyes in the NPs. Presence of a dark trap state leads to full or partial quenching of many nearby dyes. (B) Illustration of the impact of a trap state able to quench a large volume of the NPs and result in shortened multi-exponential lifetimes. | ||
The origin of dark trap states in dye based NPs is in most cases unknown and difficult to assess due to their low concentration and non-emissive nature. However, some likely origins can be considered based on structure and properties of dyes and NPs, and from knowledge of trap states in other optical materials. Traps can originate from impurities in the dye stock. Most commercial dyes have a purity >95%, the impurities could originate from synthetic by-products or degradation (e.g. photo degradation) of the dye. Such impurities can either have a low excited state energy and thus act as the trap state, or they can quench the exciton by electron transfer, in which case the trap state is a charge separated photoinduced electron transfer (PET) state.43–45 The trap state can also be a “structural defect” in the NP, like a dark H-dimer or excimer occurring in a system where the majority of dyes are organized in an emissive structure such as J-aggregates or separated by a matrix. For small NPs where the surface to volume ratio is large, surface traps may be an important contribution to quenching. Inorganic QDs illustrate this well, as they are highly crystalline materials and it is well known that the dominating sites of quenching are on the surface, and it is therefore essential to passivate these surface defects and protect the QDs from the surrounding environment.5,46 In relation to organic dye based nanoparticles it is much less clear how important surface quenching is, and it likely varies between different optical materials. However, Chen et al. found that introduction of an amphiphilic ligand, DSPE-PEG, to dye based SMILES NPs not only increased stability in water but also slightly increased the QY.47 For some dyes contact with water or dissolved oxygen is known to reduce the quantum yield and it is likely that such quenching processes occur at the NP surface and become amplified by the energy migration in NPs.48
The relative efficiency of energy migration for dyes can be estimated from their Förster distance (R0) for homo-FRET. Here R0 reports the distance between two identical dyes at which the likelihood of energy transfer is 50%. Since energy migration is a Coulomb interaction, R0 scales with the transition moments and the spectral overlap between fluorescence and absorption. This allows us to predict that dyes with strong transitions (large molar absorption coefficient, ε) and small Stokes shifts will also make up materials and NPs with the most efficient energy migration and thus larger susceptibility to quenching by trap states. For good chromophores the R0 for homo FRET can reach up to 60 Å.18,41
Efficient energy migration in NPs can however also be utilized as an advantage. Examples of this includes NP blinking by reversible formation of dark states quenching the entire NP allowing their use in super resolution microscopy40 and NPs acting as antenna systems for acceptor dyes as discussed below.
Inner filter effects
Both primary (excitation attenuation) and secondary (self-absorption) inner filter effects are common issues in optical spectroscopy of high optical density materials.49–51 In the case of NPs with high dye concentration, these effects should be kept in mind. A large effect will most likely only be an issue when working with highly absorbing dyes (high ε) in larger NPs.52 However, primary inner filter effects that can lead to deviation in quantitative measurements such as quantum yield and excitation spectra may appear even in smaller particles. For a NP with 0.6 mol L−1 concentration of a dye (Table 1) with ε ≈ 105 M−1 cm−1 this could be relevant already when sizes exceed 20 nm. In the presence of secondary inner filter effects, the blue side of the emission spectrum will be truncated due to reabsorption and as a consequence the spectrum appears red shifted.41,49 Reabsorption will also lead to stretching of the fluorescence decay in time resolved measurements resulting in apparently longer fluorescence lifetimes.53,54| wt% | C (mol L−1) | V dye (nm3) | r dye–dye (Å) |
|---|---|---|---|
| a Calculated assuming formula weight 500 g mol−1 and mass density 1 g cm−3. | |||
| 100 | 2.00 | 0.8 | 12 |
| 30.0 | 0.60 | 2.8 | 17 |
| 10.0 | 0.20 | 8.3 | 25 |
| 1.00 | 0.02 | 83 | 54 |
| 0.10 | 0.002 | 830 | 117 |
Parameters describing NP performance
Many approaches and materials are applied for dye based NPs by different research groups who also report different parameters. This can make it difficult to compare how different approaches deliver the best possible photophysical properties. Therefore, it is desirable to use some key parameters that allow for comparison between the many different NP types. Klymchenko and co-workers recently reviewed this in detail.11,52 Here we will only briefly discuss the parameters that we find most important.Brightness per volume
A commonly used parameter to evaluate the performance of an emitter is the brightness (B) defined as the product of the fluorescence quantum yield and the molar absorption coefficient (B = Φ × ε). When comparing different emitters this number usually gives a good idea of the relative emission output that can be achieved at a given excitation power. For dye based NPs the brightness can be given as B = Φ × ε × n, where n is the number of dyes in the particle. This makes sense when comparing specific NPs for imaging applications.4 However, n, and thus B, depends strongly on the NP size making the brightness values misleading in the comparison of different materials and NP constructs. Reisch and Klymchenko therefore proposed the use of brightness per volume (Bv) instead, normalised by using the nanoparticle volume VNP:11,52| Bv,NP = Φεn/VNP | (4) |
Another way to calculate BV is to normalise the brightness to the average volume occupied by each dye.
| Bv = Φε/Vdye | (5) |
Another key parameter that can differ between research groups is the description of “dye loading”, with the use of weight percentage dye compared to the total mass of the NP. Sometimes the weight used includes bulky side groups or the bulky anion instead of the actual mass of the chromophore unit. This gives inconsistent values of dye loading. Using the volume per chromophore (Vdye) will give a more fair perspective on how much of the NP actually contributes to the absorption and emission. Table 1 relates the fluorophore weight percent to the concentration in NPs and how much volume is used per fluorophore (assuming a fluorophore with formula weight 500 g mol−1, and a mass density of 1 g cm−3). At 100 wt% (the ultimate limit) the volume used per dye is below 1 nm3. Some of the brightest NP approaches reach a dye loading of around 10–30 wt% where the volume per fluorophore is increased 3–10 times compared to this ideal scenario.
Fluorescence quantum yield and lifetime
The brightness per volume gives a good indication of the overall performance of the NP. The quantum yield of fluorescence from the NP will often vary depending on the loading due to the ACQ effects discussed above. When evaluating the quantum yield of the dyes in the NP it is tempting to use the solution values for reference. This may be reasonable for rigid dyes that are not sensitive to the solvent viscosity and polarity. However, in general, a better reference will be a highly diluted NP where the dye is immobilized in the same matrix, or a dilute solid solution of the dye in a polymer thin film.41Another tool to evaluate NPs is their fluorescent lifetime. Although rarely used in this context it can prove to be a very strong tool to diagnose possible defects. If there is only one type of emitter (and no quenchers) present in the NPs a mono-exponential decay should be observed as it is the case for molecular fluorophores in dilute solid solution. If there are multiple configurations or quenchers present, the lifetime will be shortened and multi-exponential. In the paper by Kacenauskaite et al. the fluorescence decays were used as a diagnostic tool to elucidate exactly the role of quenching by energy migration to trap sites.41 In some cases the fluorescence lifetime increases when the fluorophore is encapsulated in NPs e.g. stilbenes and cyanines due to the restriction of torsional freedom, which decreases non-radiative deactivation. In particular, in these cases, the use of a solid solution (e.g. polymer thin film) as the reference for the intrinsic molecular properties becomes important. The bottom-line is the fluorescence lifetime provides important insights for understanding NP properties.
Nanoparticle design principles
In this section, we will discuss selected NP design strategies. There are many aspects to consider when working with fluorescent NPs; here we will however narrow the discussion to address only the relation between NP structure/composition and photophysical properties. Over the years many different approaches have been reported to create efficient organic based fluorescent NPs. These approaches can be divided into several classes of NP designs listed below.Nanoparticles with low loading of chromophores
A simple solution to overcome the above-mentioned challenges with ACQ and altered photophysical properties is to encapsulate low concentration of dyes in a matrix that makes up most of the NP. In such diluted dye NPs (Fig. 3A) dyes are on average far apart, randomly distributed in the matrix and thus less likely to aggregate. Energy migration is also disabled when r (dye–dye) > R0 (homo FRET). There are various types of matrices used to encapsulate dyes in this way. The matrix can be polymer based,56–60 and several examples of this type are commercially available like the polystyrene based FluoSpheres.61 Another type that was used early on with low dye loading is silica-based nanoparticles.62–65 NPs with low loading of chromophores is a reliable strategy for generating fluorescent dye based NPs. However, as soon as the loading is increased to obtain higher brightness, ACQ will become dominating. This is due to the formation of quenched aggregates or dimers as well as energy migration to these trap sites (Fig. 3B) as the average dye–dye distance becomes comparable to or smaller than R0 (Table 1). This proves to be a severe limitation for these types of NPs as the dye loading has to be kept very low and essentially most of the NP structure is the non-emissive matrix surrounding the dyes.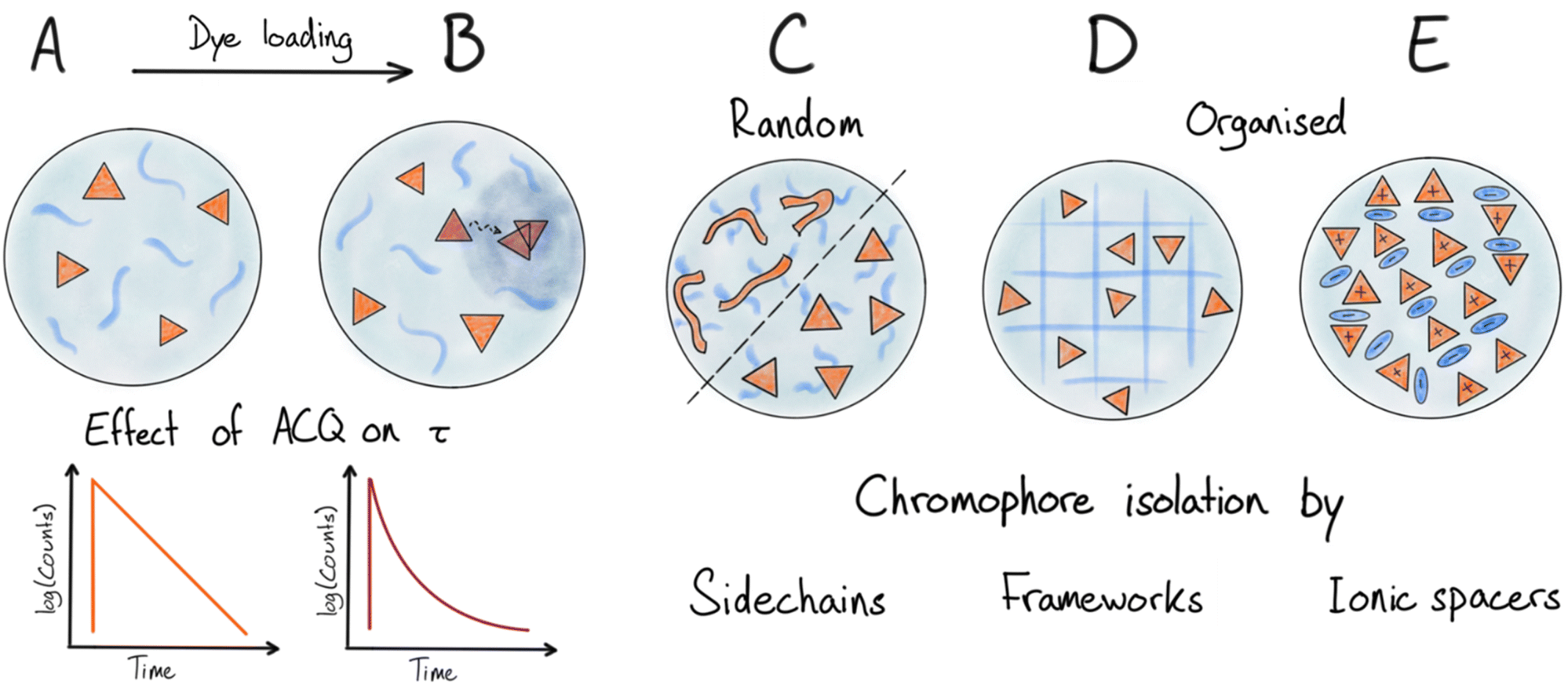 | ||
| Fig. 3 Illustrations of nanoparticle design principles. (A and B) Low to medium concentration of chromophores in matrix, and effect on fluorescence lifetime upon beginning aggregation and energy transfer mediated quenching as dye loading increases. (C), (D) and (E) Illustrate various strategies to limit chromophore interactions and mitigate ACQ at high dye densities: (C) covalently attached bulky side chains on dyes and semiconducting polymers. (D) Organization of dyes in an ordered framework/lattice. (E) Organization and separation of ionic dyes by large counterions. | ||
In the same category, we find emulsion based nanoparticles, using a water immiscible core and surfactant shell or specially designed amphiphilic dyes, which have been reported as fluorescent nanoemulsions (NEs).66,67 To ensure high efficiency of these types of emulsions the dyes have to be specifically designed to reduce both ACQ and leakage. In the work by Collot et al. different fluorophore families were used in combination with various oils to form NEs.68 The viscous oil also reduces deactivation by conformational relaxation of some of the fluorophores, thereby increasing the QY drastically compared to solution properties. Still this approach requires low concentration of the dye between 0.25 wt% to 2.5 wt% where the QY is approximately halved. Later a bulky anion tetraphenylborate (TPB) was introduced to increase the loading of cationic dyes in the oily core up to 8 wt% dye while still maintaining separation of the chromophores and keeping ACQ effects at a minimum.69
Another approach to fluorescent NEs uses the so-called fluorofluorophores that are dyes equipped with fluorinated side chains making them soluble in perfluorocarbons.70,71 In work by Sletten et al. highly fluorinated rhodamine and coumarin chromophores were used to form fluorescent perfluorocarbon emulsions with sizes of around 120 nm.70 Recently fluorinated cyanine dyes extending to the far red region were also introduced.72 NEs of these fluoro-cyanine dyes ranged in size from 190–250 nm and were found to be more stable to leaching compared to the previously reported fluorinated rhodamine. Still in all these NEs the dye loading is very low (<0.1 wt%).
Syntheses of specific amphiphiles containing chromophores to form fluorescent micelles and vesicles have also been reported,73–75 which can also be obtained by post functionalizing with fluorophores after micelle formation.76
In all of the above examples, the dyes have been randomly embedded (dissolved) in a matrix material. As the dye loading is increased formation of dimers or aggregates will take place (Fig. 3B). Even with a matrix that dissolves the dyes well ACQ is observed at relative low loadings of a few wt% or lower. This can be rationalized as a result of formation of aggregates/dimers with low emission quantum yield (e.g. H-aggregates) that not only are self-quenching but also quench nearby monomer dyes by acting as energy acceptors (traps), thereby amplifying the ACQ. As mentioned above this will be particularly true for dyes with large R0 values for homo-energy transfer where energy migration will extend the range of quenching (Fig. 2 and 3B).
Special organic materials where ACQ is limited by chromophore design
Some materials and chromophore types are more resistant to ACQ than general organic dyes, giving these materials a special advantage for the preparation of very bright NPs. Here we will discuss two such cases: polydots and the so-called AIEs.In polydot systems one can consider the covalently attached side chains as a matrix separating the dye components (the π-conjugated polymer backbone). However, contrary to systems with dyes randomly dissolved in polymer matrices, much higher effective loading is achieved since the backbones are kept from strong interaction by a much smaller fraction of the matrix (side chains) fixed in specific positions around the π-system (Fig. 3C).83,84
Because of their use in the production of OLEDs, there are already many types of semiconducting polymers available with different spectral properties and optimized for high quantum yields in the solid state. However, the development of new conjugated polymers can be time consuming and difficult. Designing new polydots with entirely new properties can therefore be a demanding process. The optical properties also slightly change during the formation of NPs compared to its solution properties. This is likely due to changes in the conjugation length and conformations of the polymer backbone, which may broaden and shift both absorption and emission.12,85,86 This can also induce quenching trap states in the structure which will influence QY and fluorescence lifetime. Piwonski et al. introduced non-planar conjugated polymers PCzBT and PCzDTBT for producing ultra-small polydots with sizes of around 3 nm.87 The quantum yield of the PCzDTBT polydots were lower than that of the corresponding polymer in THF, 0.2 and 0.5, respectively. Later they expanded the polydot spectroscopic range toward NIR emission with reasonable quantum yields.88 Further information on polydots can be found in the review by Chiu and Wu.77
General methods for suppressing ACQ
Several different strategies have been introduced to increase the loading of fluorescent dyes in general. A higher loading of dyes can be achieved by actively preventing close contact like in π–π-stacking. This is possible by introducing large or bulky groups either on the chromophore itself or on the counter ions associated with charged dyes.Doping and mix of strategies
Most of the strategies discussed above reduce ACQ by separating the chromophores (dyes) to avoid strong interactions. This is achieved using various matrices, spanning from polymers and silica acting as simple solvents with random distribution of the dyes, to highly organized structures like MOFs and SMILES. To achieve high brightness per volume a high density of chromophores is needed, which in turn requires use of an effective strategy to keep the chromophores separated. However, even when effective separation of dyes is obtained in NPs, fluorescence QYs do not reach the values expected from the chromophores in solid solution. Much of the remaining quenching that limits the performance of NPs with high chromophore density stems from the highly efficient energy migration making them sensitive to even single trap states.115 Compromises are often made where the number of traps or the energy transfer efficiency is reduced by further increasing the distance between chromophores e.g. by introducing non absorbing spacers either as an additional matrix or as “blank salts” in the case of ionic organization.41,126,127 For dye loaded polymer NPs the local density of dyes can also be modulated by the polarity of the polymer matrix to either favour segregation or isotropic distribution of dyes.115A promising approach is to use the efficient energy migration as an advantage by introducing a red shifted dopant emitter into the NP. A small fraction of dopant emitters can efficiently harvest most excitons and thus outcompete energy migration to the dark trap states (Fig. 4). This doping strategy was demonstrated for several SMILES materials showing large improvement of quantum yields and fluorescence lifetimes approaching the intrinsic molecular values and was since translated to SMILES NPs.41,128 Introduction of dopant dyes in the NP core has been used in several NP strategies to boost the fluorescence brightness,42,64,129–131 while introduction of acceptor dyes to the NP surface also has been used to develop sensing properties e.g. for DNA/RNA hybridization assays.119,120 Dopant dyes have also been used to achieve more narrow and redshifted emission of AIE NPs and polydots.44,131 In a recent example by Zhou et al. they introduced acceptor fluorophores into a AIEdot and utilize effective energy transfer while isolating the molecular chromophores to achieve more narrow emission compared to that of the AIEdot alone.130
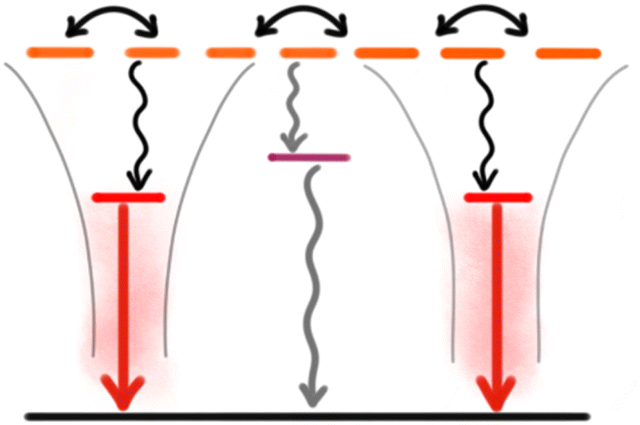 | ||
| Fig. 4 Schematic state diagram of a doped NP system with indication of energy migration between multiple similar (donor) dyes in the NP. Efficient energy transfer to a low number of isolated dopant (acceptor) dyes outcompetes quenching by the trap state. | ||
Nevertheless, doping with acceptor dyes is not a universal solution to ACQ and the brightness problem. Some dye combinations have negative impact or no effect at all on the optical properties. An example by Wu et al. shows polydots doped with 3 different molecular dyes where only 2 combinations show improved fluorescence intensity.44 This decrease could be due to quenching of the dopant dye by PET. Such PET quenching of dopant dyes can be avoided in well-organized systems like SMILES.41
Future outlook
A lot of progress in the development of organic dye based NPs has been achieved in recent years, and several materials and strategies are now available that effectively avoid strong interactions and thus allow for NPs with high chromophore density and high brightness per volume. Doping strategies additionally offer a means to further improve brightness and tuning of spectral properties. In the race for high brightness, polydots and counterion based methods are now approaching the limit set by the separating matrix and the intrinsic properties of the dyes. Still we believe that dye based NPs have much more to offer in terms of advanced photophysical properties.NPs with NIR emission are particular interesting for deep tissue imaging.132–134 NPs based on NIR dyes are however still challenging and often show low quantum yields.135,136 Typical NIR dyes like cyanines present challenges stemming from their large π-surfaces and tendency for aggregation, large oscillator strength that also favours strong coupling, their sensitivity to quenching by water and generally very fast non-radiative deactivation rates as dictated by the energy gap law.48,137 Strategies that will address these challenges and provide bright tuneable NIR-I and NIR-II NPs will be highly valuable.
New and advanced fluorescence based sensing and imaging modalities are often based on photophysical properties beyond just emission wavelength and brightness. This is the case for e.g. time-resolved techniques,58,128,138 super resolution,40 and circular polarized emission.139 Development of dye based NPs specifically for such readouts is a promising direction.
Inclusion of multiple different chromophores in NPs may provide new and advanced photophysical properties via supramolecular photophysics – examples include engineering of e.g. Stokes shifts and emission lifetimes via doping (discussed above). However, a limitation for all of the present NP strategies is the stochastic nature of these composites, and more organized structures with control of relative distances, orientations and locations of the dyes within the NP could greatly widen and improve the scope of dye based NPs.
Conclusions
This perspective discusses key aspects that must be considered when working with fluorescent organic dye based NPs. We discuss the various photophysical effects that limit brightness of fluorescent NPs; strong dye–dye interactions, and the presence of trap sites, and energy migration to these. On this background the properties and performance of various NP designs can be understood and hopefully even improved. We believe that dye based NPs have great potential stemming from the possible transfer of advanced properties from the molecular dyes into NPs when the organization and supramolecular interactions are well understood and controlled.Author contributions
SGS and BWL concieved the idea and co-wrote the perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Novo Nordisk Foundation (NNF20OC0062176) and by the Danish Council of Independent Research (DFF-0136-00122B).References
- W. R. Algar, Heroes or Villains? How Nontraditional Luminescent Materials Do and Do Not Enhance Bioanalysis and Imaging, Chem. Mater., 2020, 32(12), 4863–4883 CrossRef CAS.
- O. S. Wolfbeis, An overview of nanoparticles commonly used in fluorescent bioimaging, Chem. Soc. Rev., 2015, 44(14), 4743–4768 RSC.
- Z. Li, Q. Sun, Y. Zhu, B. Tan, Z. P. Xu and S. X. Dou, Ultra-small fluorescent inorganic nanoparticles for bioimaging, J. Mater. Chem. B, 2014, 2(19), 2793–2818 RSC.
- W. R. Algar, M. Massey, K. Rees, R. Higgins, K. D. Krause, G. H. Darwish, W. J. Peveler, Z. Xiao, H.-Y. Tsai, R. Gupta, K. Lix, M. V. Tran and H. Kim, Photoluminescent Nanoparticles for Chemical and Biological Analysis and Imaging, Chem. Rev., 2021, 121(15), 9243–9358 CrossRef CAS PubMed.
- A. P. Alivisatos, Semiconductor Clusters, Nanocrystals, and Quantum Dots, Science, 1996, 271(5251), 933 CrossRef CAS.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Quantum dots versus organic dyes as fluorescent labels, Nat. Methods, 2008, 5(9), 763–775 CrossRef CAS.
- F. P. García de Arquer, D. V. Talapin, V. I. Klimov, Y. Arakawa, M. Bayer and E. H. Sargent, Semiconductor quantum dots: Technological progress and future challenges, Science, 2021, 373(6555), eaaz8541 CrossRef.
- K. D. Wegner and N. Hildebrandt, Quantum dots: bright and versatile in vitro and in vivo fluorescence imaging biosensors, Chem. Soc. Rev., 2015, 44(14), 4792–4834 RSC.
- A. Gnach and A. Bednarkiewicz, Lanthanide-doped up-converting nanoparticles: Merits and challenges, Nano Today, 2012, 7(6), 532–563 CrossRef CAS.
- U. Resch-Genger and H. H. Gorris, Perspectives and challenges of photon-upconversion nanoparticles – Part I: routes to brighter particles and quantitative spectroscopic studies, Anal. Bioanal. Chem., 2017, 409(25), 5855–5874 CrossRef CAS.
- A. Reisch and A. S. Klymchenko, Fluorescent Polymer Nanoparticles Based on Dyes: Seeking Brighter Tools for Bioimaging, Small, 2016, 12(15), 1968–1992 CrossRef CAS PubMed.
- J. Pecher and S. Mecking, Nanoparticles of Conjugated Polymers, Chem. Rev., 2010, 110(10), 6260–6279 CrossRef CAS PubMed.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Functional nanoparticles through π-conjugated polymer self-assembly, Nat. Rev. Mater., 2021, 6(1), 7–26 CrossRef CAS.
- M. O. Alas, F. B. Alkas, A. Aktas Sukuroglu, R. Genc Alturk and D. Battal, Fluorescent carbon dots are the new quantum dots: an overview of their potential in emerging technologies and nanosafety, J. Mater. Sci., 2020, 55(31), 15074–15105 CrossRef CAS.
- S. K. Bhunia, A. Saha, A. R. Maity, S. C. Ray and N. R. Jana, Carbon Nanoparticle-based Fluorescent Bioimaging Probes, Sci. Rep., 2013, 3(1), 1473 CrossRef PubMed.
- B. N. G. Giepmans, T. J. Deerinck, B. L. Smarr, Y. Z. Jones and M. H. Ellisman, Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots, Nat. Methods, 2005, 2(10), 743–749 CrossRef CAS.
- L. D. Lavis and R. T. Raines, Bright Ideas for Chemical Biology, ACS Chem. Biol., 2008, 3(3), 142–155 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- H.-S. Peng and D. T. Chiu, Soft fluorescent nanomaterials for biological and biomedical imaging, Chem. Soc. Rev., 2015, 44(14), 4699–4722 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Carbon quantum dots and their applications, Chem. Soc. Rev., 2015, 44(1), 362–381 RSC.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Red, Green, and Blue Luminescence by Carbon Dots: Full-Color Emission Tuning and Multicolor Cellular Imaging, Angew. Chem., Int. Ed., 2015, 54(18), 5360–5363 CrossRef CAS PubMed.
- Y. Song, S. Zhu and B. Yang, Bioimaging based on fluorescent carbon dots, RSC Adv., 2014, 4(52), 27184–27200 RSC.
- J. Liu, R. Li and B. Yang, Carbon Dots: A New Type of Carbon-Based Nanomaterial with Wide Applications, ACS Cent. Sci., 2020, 6(12), 2179–2195 CrossRef CAS.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, Carbon Nanodots: Toward a Comprehensive Understanding of Their Photoluminescence, J. Am. Chem. Soc., 2014, 136(49), 17308–17316 CrossRef CAS.
- S. Tao, T. Feng, C. Zheng, S. Zhu and B. Yang, Carbonized Polymer Dots: A Brand New Perspective to Recognize Luminescent Carbon-Based Nanomaterials, J. Phys. Chem. Lett., 2019, 10(17), 5182–5188 CrossRef CAS.
- M. Pope and C. E. Swenberg, Electronic processes in organic crystals and polymers, Oxford University Press, Oxford, 2nd edn, 1999 Search PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Park, Luminescence in Crystalline Organic Materials: From Molecules to Molecular Solids, Adv. Opt. Mater., 2021, 9(13), 2002251 CrossRef CAS.
- S. J. Strickler and R. A. Berg, Relationship between Absorption Intensity and Fluorescence Lifetime of Molecules, J. Chem. Phys., 1962, 37(4), 814–822 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, The exciton model in molecular spectroscopy, Pure Appl. Chem., 1965, 11(3–4), 371–392 CAS.
- N. J. Hestand and F. C. Spano, Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer, Chem. Rev., 2018, 118(15), 7069–7163 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials, Angew. Chem., Int. Ed., 2011, 50(15), 3376–3410 CrossRef PubMed.
- A. Austin, N. J. Hestand, I. G. McKendry, C. Zhong, X. Zhu, M. J. Zdilla, F. C. Spano and J. M. Szarko, Enhanced Davydov Splitting in Crystals of a Perylene Diimide Derivative, J. Phys. Chem. Lett., 2017, 8(6), 1118–1123 CrossRef.
- B. Stevens and E. Hutton, Radiative Life-time of the Pyrene Dimer and the Possible Role of Excited Dimers in Energy Transfer Processes, Nature, 1960, 186(4730), 1045–1046 CrossRef CAS.
- R. D. Pensack, R. J. Ashmore, A. L. Paoletta and G. D. Scholes, The Nature of Excimer Formation in Crystalline Pyrene Nanoparticles, J. Phys. Chem. C, 2018, 122(36), 21004–21017 CrossRef CAS.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, Perspectives in Dye Chemistry: A Rational Approach toward Functional Materials by Understanding the Aggregate State, J. Am. Chem. Soc., 2021, 143(12), 4500–4518 CrossRef CAS.
- H. Piwoński, S. Nozue, H. Fujita, T. Michinobu and S. Habuchi, Organic J-Aggregate Nanodots with Enhanced Light Absorption and Near-Unity Fluorescence Quantum Yield, Nano Lett., 2021, 21(7), 2840–2847 CrossRef.
- W. Chen, C.-A. Cheng, E. D. Cosco, S. Ramakrishnan, J. G. P. Lingg, O. T. Bruns, J. I. Zink and E. M. Sletten, Shortwave Infrared Imaging with J-Aggregates Stabilized in Hollow Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2019, 141(32), 12475–12480 CrossRef CAS PubMed.
- T. Förster, Energy migration and fluorescence, J. Biomed. Opt., 2012, 17(1), 011002 CrossRef.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, FRET as a biomolecular research tool — understanding its potential while avoiding pitfalls, Nat. Methods, 2019, 16(9), 815–829 CrossRef CAS PubMed.
- A. Reisch, P. Didier, L. Richert, S. Oncul, Y. Arntz, Y. Mély and A. S. Klymchenko, Collective fluorescence switching of counterion-assembled dyes in polymer nanoparticles, Nat. Commun., 2014, 5(1), 4089 CrossRef CAS PubMed.
- L. Kacenauskaite, S. G. Stenspil, A. H. Olsson, A. H. Flood and B. W. Laursen, Universal Concept for Bright, Organic, Solid-State Emitters–Doping of Small-Molecule Ionic Isolation Lattices with FRET Acceptors, J. Am. Chem. Soc., 2022, 144(43), 19981–19989 CrossRef CAS PubMed.
- K. Trofymchuk, A. Reisch, P. Didier, F. Fras, P. Gilliot, Y. Mely and A. Klymchenko, Giant light-harvesting nanoantenna for single-molecule detection in ambient light, Nat. Photonics, 2017, 11(10), 657–663 CrossRef CAS.
- J.-S. Yang and T. M. Swager, Porous Shape Persistent Fluorescent Polymer Films: An Approach to TNT Sensory Materials, J. Am. Chem. Soc., 1998, 120(21), 5321–5322 CrossRef CAS.
- C. Wu, Y. Zheng, C. Szymanski and J. McNeill, Energy Transfer in a Nanoscale Multichromophoric System: Fluorescent Dye-Doped Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2008, 112(6), 1772–1781 CrossRef CAS.
- H. J. Egelhaaf, J. Gierschner and D. Oelkrug, Polarizability effects and energy transfer in quinquethiophene doped bithiophene and OPV films, Synth. Met., 2002, 127(1), 221–227 CrossRef CAS.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, (CdSe)ZnS Core−Shell Quantum Dots: Synthesis and Characterization of a Size Series of Highly Luminescent Nanocrystallites, J. Phys. Chem. B, 1997, 101(46), 9463–9475 CrossRef CAS.
- J. Chen, S. M. A. Fateminia, L. Kacenauskaite, N. Bærentsen, S. G. Stenspil, J. Bredehoeft, K. L. Martinez, A. H. Flood and B. W. Laursen, Ultra-Bright Fluorescent Organic Nanoparticles Based on Small-Molecule Ionic Isolation Lattices, Angew. Chem., Int. Ed., 2021, 60(17), 9450–9458 CrossRef CAS.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Universal quenching of common fluorescent probes by water and alcohols, Chem. Sci., 2021, 12(4), 1352–1362 RSC.
- J. B. Birks and G. T. Wright, Fluorescence Spectra of Organic Crystals, Proc. Phys. Soc., London, Sect. B, 1954, 67(9), 657 CrossRef.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, Fluorescence Quantum Yield of Aromatic Hydrocarbon Crystals, J. Phys. Chem. C, 2009, 113(7), 2961–2965 CrossRef CAS.
- S. Kumar Panigrahi and A. Kumar Mishra, Inner filter effect in fluorescence spectroscopy: As a problem and as a solution, J. Photochem. Photobiol., C, 2019, 41, 100318 CrossRef CAS.
- A. H. Ashoka, I. O. Aparin, A. Reisch and A. S. Klymchenko, Brightness of fluorescent organic nanomaterials, Chem. Soc. Rev., 2023, 52(14), 4525–4548 RSC.
- Y. Sakai, M. Kawahigashi, T. Minami, T. Inoue and S. Hirayama, Deconvolution of non-exponential emission decays arising from reabsorption of emitted light, J. Lumin., 1989, 42(6), 317–324 CrossRef CAS.
- J. S. Connolly, A. F. Janzen and E. B. Samuel, Fluorescence lifetimes of chlorophyll a: solvent, concentration and oxygen dependence, Photochem. Photobiol., 1982, 36(5), 559–563 CrossRef CAS.
- C. R. Benson, L. Kacenauskaite, K. L. VanDenburgh, W. Zhao, B. Qiao, T. Sadhukhan, M. Pink, J. Chen, S. Borgi, C.-H. Chen, B. J. Davis, Y. C. Simon, K. Raghavachari, B. W. Laursen and A. H. Flood, Plug-and-Play Optical Materials from Fluorescent Dyes and Macrocycles, Chem, 2020, 6(8), 1978–1997 CAS.
- T. Behnke, C. Würth, E.-M. Laux, K. Hoffmann and U. Resch-Genger, Simple strategies towards bright polymer particles via one-step staining procedures, Dyes Pigm., 2012, 94(2), 247–257 CrossRef CAS.
- T. Behnke, J. E. Mathejczyk, R. Brehm, C. Würth, F. Ramos Gomes, C. Dullin, J. Napp, F. Alves and U. Resch-Genger, Target-specific nanoparticles containing a broad band emissive NIR dye for the sensitive detection and characterization of tumor development, Biomaterials, 2013, 34(1), 160–170 CrossRef CAS PubMed.
- K. Hoffmann, T. Behnke, D. Drescher, J. Kneipp and U. Resch-Genger, Near-Infrared-Emitting Nanoparticles for Lifetime-Based Multiplexed Analysis and Imaging of Living Cells, ACS Nano, 2013, 7(8), 6674–6684 CrossRef CAS.
- K. Hoffmann, T. Behnke, M. Grabolle and U. Resch-Genger, Nanoparticle-encapsulated vis- and NIR-emissive fluorophores with different fluorescence decay kinetics for lifetime multiplexing, Anal. Bioanal. Chem., 2014, 406(14), 3315–3322 CrossRef CAS PubMed.
- D. Kage, L. Fischer, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, Close Spectroscopic Look at Dye-Stained Polymer Microbeads, J. Phys. Chem. C, 2018, 122(24), 12782–12791 CrossRef CAS.
- I. Johnson and M. T. Z. Spence, The Molecular Probes Handbook, Life Technologies, 11th edn, 2010 Search PubMed.
- S. Liang; C. L. John; S. Xu; J. Chen; Y. Jin; Q. Yuan; W. Tan and J. X. Zhao, Silica-Based Nanoparticles: Design and Properties, in Advanced Fluorescence Reporters in Chemistry and Biology II: Molecular Constructions, Polymers and Nanoparticles, ed. A. P. Demchenko, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp 229–251 Search PubMed.
- E.-B. Cho, D. O. Volkov and I. Sokolov, Ultrabright Fluorescent Silica Mesoporous Silica Nanoparticles: Control of Particle Size and Dye Loading, Adv. Funct. Mater., 2011, 21(16), 3129–3135 CrossRef CAS.
- D. Genovese, S. Bonacchi, R. Juris, M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Prevention of Self-Quenching in Fluorescent Silica Nanoparticles by Efficient Energy Transfer, Angew. Chem., Int. Ed., 2013, 52(23), 5965–5968 CrossRef CAS PubMed.
- S. Bonacchi, D. Genovese, R. Juris, M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Luminescent Silica Nanoparticles: Extending the Frontiers of Brightness, Angew. Chem., Int. Ed., 2011, 50(18), 4056–4066 CrossRef CAS.
- A. S. Klymchenko, F. Liu, M. Collot and N. Anton, Dye-Loaded Nanoemulsions: Biomimetic Fluorescent Nanocarriers for Bioimaging and Nanomedicine, Adv. Healthcare Mater., 2021, 10(1), 2001289 CrossRef CAS.
- A. S. Klymchenko, E. Roger, N. Anton, H. Anton, I. Shulov, J. Vermot, Y. Mely and T. F. Vandamme, Highly lipophilic fluorescent dyes in nano-emulsions: towards bright non-leaking nano-droplets, RSC Adv., 2012, 2(31), 11876–11886 RSC.
- X. Wang, N. Anton, P. Ashokkumar, H. Anton, T. K. Fam, T. Vandamme, A. S. Klymchenko and M. Collot, Optimizing the Fluorescence Properties of Nanoemulsions for Single Particle Tracking in Live Cells, ACS Appl. Mater. Interfaces, 2019, 11(14), 13079–13090 CrossRef CAS.
- V. N. Kilin, H. Anton, N. Anton, E. Steed, J. Vermot, T. F. Vandamme, Y. Mely and A. S. Klymchenko, Counterion-enhanced cyanine dye loading into lipid nano-droplets for single-particle tracking in zebrafish, Biomaterials, 2014, 35(18), 4950–4957 CrossRef CAS PubMed.
- E. M. Sletten and T. M. Swager, Fluorofluorophores: Fluorescent Fluorous Chemical Tools Spanning the Visible Spectrum, J. Am. Chem. Soc., 2014, 136(39), 13574–13577 CrossRef CAS PubMed.
- E. M. Sletten and T. M. Swager, Readily accessible multifunctional fluorous emulsions, Chem. Sci., 2016, 7(8), 5091–5097 RSC.
- I. Lim, A. Vian, H. L. van de Wouw, R. A. Day, C. Gomez, Y. Liu, A. L. Rheingold, O. Campàs and E. M. Sletten, Fluorous Soluble Cyanine Dyes for Visualizing Perfluorocarbons in Living Systems, J. Am. Chem. Soc., 2020, 142(37), 16072–16081 CrossRef CAS.
- M. Collot, J. Schild, K. T. Fam, R. Bouchaala and A. S. Klymchenko, Stealth and Bright Monomolecular Fluorescent Organic Nanoparticles Based on Folded Amphiphilic Polymer, ACS Nano, 2020, 14(10), 13924–13937 CrossRef CAS PubMed.
- I. Shulov, Y. Arntz, Y. Mély, V. G. Pivovarenko and A. S. Klymchenko, Non-coordinating anions assemble cyanine amphiphiles into ultra-small fluorescent nanoparticles, Chem. Commun., 2016, 52(51), 7962–7965 RSC.
- X. Zhang, Z. Chen and F. Würthner, Morphology Control of Fluorescent Nanoaggregates by Co-Self-Assembly of Wedge- and Dumbbell-Shaped Amphiphilic Perylene Bisimides, J. Am. Chem. Soc., 2007, 129(16), 4886–4887 CrossRef CAS.
- G. Sun, M. Y. Berezin, J. Fan, H. Lee, J. Ma, K. Zhang, K. L. Wooley and S. Achilefu, Bright fluorescent nanoparticles for developing potential optical imaging contrast agents, Nanoscale, 2010, 2(4), 548–558 RSC.
- C. Wu and D. T. Chiu, Highly Fluorescent Semiconducting Polymer Dots for Biology and Medicine, Angew. Chem., Int. Ed., 2013, 52(11), 3086–3109 CrossRef CAS.
- K. Li and B. Liu, Polymer-encapsulated organic nanoparticles for fluorescence and photoacoustic imaging, Chem. Soc. Rev., 2014, 43(18), 6570–6597 RSC.
- D. Tuncel and H. V. Demir, Conjugated polymer nanoparticles, Nanoscale, 2010, 2(4), 484–494 RSC.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, Multicolor Conjugated Polymer Dots for Biological Fluorescence Imaging, ACS Nano, 2008, 2(11), 2415–2423 CrossRef CAS PubMed.
- J. Yu, C. Wu, Z. Tian and J. McNeill, Tracking of Single Charge Carriers in a Conjugated Polymer Nanoparticle, Nano Lett., 2012, 12(3), 1300–1306 CrossRef CAS PubMed.
- L. C. Groff, X. L. Wang and J. D. McNeill, Measurement of Exciton Transport in Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2013, 117(48), 25748–25755 CrossRef CAS.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, Bioconjugation of Ultrabright Semiconducting Polymer Dots for Specific Cellular Targeting, J. Am. Chem. Soc., 2010, 132(43), 15410–15417 CrossRef CAS PubMed.
- C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Design of Highly Emissive Polymer Dot Bioconjugates for In Vivo Tumor Targeting, Angew. Chem., Int. Ed., 2011, 50(15), 3430–3434 CrossRef CAS.
- C. Szymanski, C. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, Single Molecule Nanoparticles of the Conjugated Polymer MEH−PPV, Preparation and Characterization by Near-Field Scanning Optical Microscopy, J. Phys. Chem. B, 2005, 109(18), 8543–8546 CrossRef CAS.
- J. Pecher and S. Mecking, Nanoparticles from Step-Growth Coordination Polymerization, Macromolecules, 2007, 40(22), 7733–7735 CrossRef CAS.
- H. Piwoński, T. Michinobu and S. Habuchi, Controlling photophysical properties of ultrasmall conjugated polymer nanoparticles through polymer chain packing, Nat. Commun., 2017, 8(1), 15256 CrossRef.
- H. Piwoński, W. Li, Y. Wang, T. Michinobu and S. Habuchi, Improved Fluorescence and Brightness of Near-Infrared and Shortwave Infrared Emitting Polymer Dots for Bioimaging Applications, ACS Appl. Polym. Mater., 2020, 2(2), 569–577 CrossRef.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole, Chem. Commun., 2001,(18), 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission: phenomenon, mechanism and applications, Chem. Commun., 2009,(29), 4332–4353 RSC.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: New Vistas at the Aggregate Level, Angew. Chem., Int. Ed., 2020, 59(25), 9888–9907 CrossRef CAS PubMed.
- F. Würthner, Aggregation-Induced Emission (AIE): A Historical Perspective, Angew. Chem., Int. Ed., 2020, 59(34), 14192–14196 CrossRef.
- Y. Ren, J. W. Y. Lam, Y. Dong, B. Z. Tang and K. S. Wong, Enhanced Emission Efficiency and Excited State Lifetime Due to Restricted Intramolecular Motion in Silole Aggregates, J. Phys. Chem. B, 2005, 109(3), 1135–1140 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar!, Chem. Rev., 2015, 115(21), 11718–11940 CrossRef CAS.
- S. Chen, H. Wang, Y. Hong and B. Z. Tang, Fabrication of fluorescent nanoparticles based on AIE luminogens (AIE dots) and their applications in bioimaging, Mater. Horiz., 2016, 3(4), 283–293 RSC.
- K. Li, Y. Jiang, D. Ding, X. Zhang, Y. Liu, J. Hua, S.-S. Feng and B. Liu, Folic acid-functionalized two-photon absorbing nanoparticles for targeted MCF-7 cancer cell imaging, Chem. Commun., 2011, 47(26), 7323–7325 RSC.
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. W. H. Wong, Highly Fluorescent Molecularly Insulated Perylene Diimides: Effect of Concentration on Photophysical Properties, Chem. Mater., 2017, 29(19), 8395–8403 CrossRef CAS.
- K. Trofymchuk, A. Reisch, I. Shulov, Y. Mély and A. S. Klymchenko, Tuning the color and photostability of perylene diimides inside polymer nanoparticles: towards biodegradable substitutes of quantum dots, Nanoscale, 2014, 6(21), 12934–12942 RSC.
- D. Schmidt, M. Stolte, J. Süβ, A. Liess, V. Stepanenko and F. Würthner, Protein-like Enwrapped Perylene Bisimide Chromophore as a Bright Microcrystalline Emitter Material, Angew. Chem., Int. Ed., 2019, 58(38), 13385–13389 CrossRef CAS PubMed.
- M. Stolte, T. Schembri, J. Süβ, D. Schmidt, A.-M. Krause, M. O. Vysotsky and F. Würthner, 1-Mono- and 1,7-Disubstituted Perylene Bisimide Dyes with Voluminous Groups at Bay Positions: In Search for Highly Effective Solid-State Fluorescence Materials, Chem. Mater., 2020, 32(14), 6222–6236 CrossRef CAS.
- J. Della Rocca, D. Liu and W. Lin, Nanoscale Metal–Organic Frameworks for Biomedical Imaging and Drug Delivery, Acc. Chem. Res., 2011, 44(10), 957–968 CrossRef CAS.
- A. Monguzzi, M. Ballabio, N. Yanai, N. Kimizuka, D. Fazzi, M. Campione and F. Meinardi, Highly Fluorescent Metal–Organic-Framework Nanocomposites for Photonic Applications, Nano Lett., 2018, 18(1), 528–534 CrossRef CAS.
- G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Host–Guest Antenna Materials, Angew. Chem., Int. Ed., 2003, 42(32), 3732–3758 CrossRef CAS.
- T. Doussineau, M. Smaïhi, S. Balme and J.-M. Janot, Fluorescent Hydroxyflavone–Zeolite Nanoparticles: Ship-in-a-Bottle Synthesis and Photophysical Properties, ChemPhysChem, 2006, 7(3), 583–589 CrossRef CAS PubMed.
- L. M. Grimm, S. Sinn, M. Krstić, E. D'Este, I. Sonntag, E. A. Prasetyanto, T. Kuner, W. Wenzel, L. De Cola and F. Biedermann, Fluorescent Nanozeolite Receptors for the Highly Selective and Sensitive Detection of Neurotransmitters in Water and Biofluids, Adv. Mater., 2021, 33(49), 2104614 CrossRef CAS PubMed.
- A. M. O. Azevedo, J. L. M. Santos, I. M. Warner and M. L. M. F. S. Saraiva, GUMBOS and nanoGUMBOS in chemical and biological analysis: A review, Anal. Chim. Acta, 2020, 1133, 180–198 CrossRef CAS PubMed.
- D. K. Bwambok, B. El-Zahab, S. K. Challa, M. Li, L. Chandler, G. A. Baker and I. M. Warner, Near-Infrared Fluorescent NanoGUMBOS for Biomedical Imaging, ACS Nano, 2009, 3(12), 3854–3860 CrossRef CAS PubMed.
- S. Das, D. Bwambok, B. El-Zahab, J. Monk, S. L. de Rooy, S. Challa, M. Li, F. R. Hung, G. A. Baker and I. M. Warner, Nontemplated Approach to Tuning the Spectral Properties of Cyanine-Based Fluorescent NanoGUMBOS, Langmuir, 2010, 26(15), 12867–12876 CrossRef CAS PubMed.
- A. N. Jordan, S. Das, N. Siraj, S. L. de Rooy, M. Li, B. El-Zahab, L. Chandler, G. A. Baker and I. M. Warner, Anion-controlled morphologies and spectral features of cyanine-based nanoGUMBOS – an improved photosensitizer, Nanoscale, 2012, 4(16), 5031–5038 RSC.
- P. K. S. Magut, S. Das, V. E. Fernand, J. Losso, K. McDonough, B. M. Naylor, S. Aggarwal and I. M. Warner, Tunable Cytotoxicity of Rhodamine 6G via Anion Variations, J. Am. Chem. Soc., 2013, 135(42), 15873–15879 CrossRef CAS.
- H. Yao, M. Yamashita and K. Kimura, Organic Styryl Dye Nanoparticles: Synthesis and Unique Spectroscopic Properties, Langmuir, 2009, 25(2), 1131–1137 CrossRef CAS PubMed.
- H. Yao and K. Ashiba, Highly fluorescent organic nanoparticles of thiacyanine dye: A synergetic effect of intermolecular H-aggregation and restricted intramolecular rotation, RSC Adv., 2011, 1(5), 834–838 RSC.
- T. Enseki and H. Yao, Controlled Formation of Fluorescent Organic Nanoparticles of Carbocyanine Dye via Ion-association Approach, Chem. Lett., 2012, 41(10), 1119–1121 CrossRef CAS.
- I. Shulov, S. Oncul, A. Reisch, Y. Arntz, M. Collot, Y. Mely and A. S. Klymchenko, Fluorinated counterion-enhanced emission of rhodamine aggregates: ultrabright nanoparticles for bioimaging and light-harvesting, Nanoscale, 2015, 7(43), 18198–18210 RSC.
- A. Reisch, K. Trofymchuk, A. Runser, G. Fleith, M. Rawiso and A. S. Klymchenko, Tailoring Fluorescence Brightness and Switching of Nanoparticles through Dye Organization in the Polymer Matrix, ACS Appl. Mater. Interfaces, 2017, 9(49), 43030–43042 CrossRef CAS PubMed.
- A. Reisch, A. Runser, Y. Arntz, Y. Mély and A. S. Klymchenko, Charge-Controlled Nanoprecipitation as a Modular Approach to Ultrasmall Polymer Nanocarriers: Making Bright and Stable Nanoparticles, ACS Nano, 2015, 9(5), 5104–5116 CrossRef CAS PubMed.
- N. Melnychuk and A. S. Klymchenko, DNA-Functionalized Dye-Loaded Polymeric Nanoparticles: Ultrabright FRET Platform for Amplified Detection of Nucleic Acids, J. Am. Chem. Soc., 2018, 140(34), 10856–10865 CrossRef CAS PubMed.
- C. Severi, N. Melnychuk and A. S. Klymchenko, Smartphone-assisted detection of nucleic acids by light-harvesting FRET-based nanoprobe, Biosens. Bioelectron., 2020, 168, 112515 CrossRef CAS PubMed.
- N. Melnychuk, S. Egloff, A. Runser, A. Reisch and A. S. Klymchenko, Light-Harvesting Nanoparticle Probes for FRET-Based Detection of Oligonucleotides with Single-Molecule Sensitivity, Angew. Chem., Int. Ed., 2020, 59(17), 6811–6818 CrossRef CAS.
- S. Egloff, N. Melnychuk, A. Reisch, S. Martin and A. S. Klymchenko, Enzyme-free amplified detection of cellular microRNA by light-harvesting fluorescent nanoparticle probes, Biosens. Bioelectron., 2021, 179, 113084 CrossRef CAS PubMed.
- B. Andreiuk, I. O. Aparin, A. Reisch and A. S. Klymchenko, Bulky Barbiturates as Non-Toxic Ionic Dye Insulators for Enhanced Emission in Polymeric Nanoparticles, Chem.–Eur. J., 2021, 27(50), 12877–12883 CrossRef CAS PubMed.
- Y. Haketa, K. Yamasumi and H. Maeda, π-Electronic ion pairs: building blocks for supramolecular nanoarchitectonics via iπ–iπ interactions, Chem. Soc. Rev., 2023, 52(20), 7170–7196 RSC.
- Y. Haketa and H. Maeda, Dimension-controlled ion-pairing assemblies based on π-electronic charged species, Chem. Commun., 2017, 53(20), 2894–2909 RSC.
- Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Oriented Salts: Dimension-Controlled Charge-by-Charge Assemblies from Planar Receptor–Anion Complexes, Angew. Chem., Int. Ed., 2010, 49(52), 10079–10083 CrossRef CAS PubMed.
- J. Chen, S. G. Stenspil, S. Kaziannis, L. Kacenauskaite, N. Lenngren, M. Kloz, A. H. Flood and B. W. Laursen, Quantitative Energy Transfer in Organic Nanoparticles Based on Small-Molecule Ionic Isolation Lattices for UV Light Harvesting, ACS Appl. Nano Mater., 2022, 5(10), 13887–13893 CrossRef CAS.
- D. S. Biswas, N. Melnychuk, C. Severi, P. Didier and A. S. Klymchenko, Giant Light-Harvesting in Dye-Loaded Nanoparticles Enhanced by Blank Hydrophobic Salts, Adv. Opt. Mater., 2024, 12(4), 2301671 CrossRef CAS.
- N. Adarsh and A. S. Klymchenko, Ionic aggregation-induced emission dye with bulky counterions for preparation of bright near-infrared polymeric nanoparticles, Nanoscale, 2019, 11(29), 13977–13987 RSC.
- S. G. Stenspil, J. Chen, M. B. Liisberg, A. H. Flood and B. W. Laursen, Control of the fluorescence lifetime in dye based nanoparticles, Chem. Sci., 2024, 15, 5531–5538 RSC.
- Z. Zhang, Y. Yuan, Z. Liu, H. Chen, D. Chen, X. Fang, J. Zheng, W. Qin and C. Wu, Brightness Enhancement of Near-Infrared Semiconducting Polymer Dots for in Vivo Whole-Body Cell Tracking in Deep Organs, ACS Appl. Mater. Interfaces, 2018, 10(32), 26928–26935 CrossRef CAS.
- X. Zhou, L. Zhao, K. Zhang, C. Yang, S. Li, X. Kang, G. Li, Q. Wang, H. Ji, M. Wu, J. Liu, Y. Qin and L. Wu, Ultrabright AIEdots with tunable narrow emission for multiplexed fluorescence imaging, Chem. Sci., 2023, 14, 113–120 RSC.
- X. Zhu, J.-X. Wang, L.-Y. Niu and Q.-Z. Yang, Aggregation-Induced Emission Materials with Narrowed Emission B and by Light-Harvesting Strategy: Fluorescence and Chem iluminescence Imaging, Chem. Mater., 2019, 31(9), 3573–3581 CrossRef CAS.
- S. Wang, B. Li and F. Zhang, Molecular Fluorophores for Deep-Tissue Bioimaging, ACS Cent. Sci., 2020, 6(8), 1302–1316 CrossRef CAS PubMed.
- E. D. Cosco, A. L. Spearman, S. Ramakrishnan, J. G. P. Lingg, M. Saccomano, M. Pengshung, B. A. Arús, K. C. Y. Wong, S. Glasl, V. Ntziachristos, M. Warmer, R. R. McLaughlin, O. T. Bruns and E. M. Sletten, Shortwave infrared polymethine fluorophores matched to excitation lasers enable non-invasive, multicolour in vivo imaging in real time, Nat. Chem., 2020, 12(12), 1123–1130 CrossRef CAS PubMed.
- E. Thimsen, B. Sadtler and M. Y. Berezin, Shortwave-infrared (SWIR) emitters for biological imaging: a review of challenges and opportunities, Nanophotonics, 2017, 6(5), 1043–1054 CrossRef CAS.
- Y. Yang, C. Sun, S. Wang, K. Yan, M. Zhao, B. Wu and F. Zhang, Counterion-Paired Bright Heptamethine Fluorophores with NIR-II Excitation and Emission Enable Multiplexed Biomedical Imaging, Angew. Chem., Int. Ed., 2022, e202117436 CAS.
- C. Sun, M. Zhao, X. Zhu, P. Pei and F. Zhang, One-Pot Preparation of Highly Dispersed Second Near-Infrared J-Aggregate Nanoparticles Based on FD-1080 Cyanine Dye for Bioimaging and Biosensing, CCS Chem., 2022, 4(2), 476–486 CrossRef CAS.
- R. Englman and J. Jortner, The energy gap law for radiationless transitions in large molecules, Mol. Phys., 1970, 18(2), 145–164 CrossRef CAS.
- H. S. Afsari, M. Cardoso Dos Santos, S. Lindén, T. Chen, X. Qiu, P. M. P. van Bergen en Henegouwen, T. L. Jennings, K. Susumu, I. L. Medintz, N. Hildebrandt and L. W. Miller, Time-gated FRET nanoassemblies for rapid and sensitive intra- and extracellular fluorescence imaging, Sci. Adv., 2016, 2(6), e1600265 CrossRef PubMed.
- P. Stachelek, L. MacKenzie, D. Parker and R. Pal, Circularly polarised luminescence laser scanning confocal microscopy to study live cell chiral molecular interactions, Nat. Commun., 2022, 13(1), 553 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |