
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSharply expanding single-atomically dispersed Fe–N active sites through bidirectional coordination for oxygen reduction†
Huihui
Jin‡
abd,
Ruohan
Yu‡
c,
Pengxia
Ji
c,
Weihao
Zeng
c,
Zhengying
Li
*abc,
Daping
He
 *d and
Shichun
Mu
*c
*d and
Shichun
Mu
*c
aNational Engineering Laboratory for Fiber Optic Sensing Technology, Wuhan University of Technology, Wuhan 430070, China. E-mail: zhyli@whut.edu.cn
bSchool of Information Engineering, Wuhan University of Technology, Wuhan 430070, China
cState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China. E-mail: msc@whut.edu.cn
dHubei Engineering Research Center of RF-Microwave Technology and Application, School of Science, Wuhan University of Technology, Wuhan 430070, China. E-mail: hedaping@whut.edu.cn
First published on 16th April 2024
Abstract
For Fe–NC systems, high-density Fe–N sites are the basis for high-efficiency oxygen reduction reaction (ORR), and P doping can further lower the reaction energy barrier, especially in the form of metal–P bonding. However, limited to the irregular agglomeration of metal atoms at high temperatures, Fe–P bonds and high-density Fe–N cannot be guaranteed simultaneously. Here, to escape the random and violent agglomeration of Fe species during high-temperature carbonization, triphenylphosphine and 2-methylimidazole with a strong metal coordination capability are introduced together to confine Fe growth. With the aid of such bidirectional coordination, the high-density Fe–N site with Fe–P bonds is realized by in situ phosphorylation of Fe in an Fe–NC system (Fe–P–NC) at high temperatures. Impressively, the content of single-atomically dispersed Fe sites for Fe–P–NC dramatically increases from 2.8% to 65.3% compared with that of pure Fe–NC, greatly improving the ORR activity in acidic and alkaline electrolytes. The theoretical calculation results show that the generated Fe2P can simultaneously facilitate the adsorption of intermediates to Fe–N4 sites and the electron transfer, thereby reducing the reaction energy barrier and obtaining superior ORR activity.
1. Introduction
In renewable energy conversion technologies like fuel cells and metal–air batteries, the electrocatalytic oxygen reduction reaction (ORR) is a crucial process.1,2 As the best ORR catalysts, platinum (Pt)-based materials are extensively considered for such devices, but the scarcity and high prices of these materials have seriously hindered their commercialization.3,4 Therefore, it is urgent to develop cost-effective and readily available non-noble metal ORR catalysts. Among them, Fe-based catalysts, including iron carbides, iron nitrides, iron oxides and Fe–NC series catalysts, present high ORR activity.5–10 Particularly, the Fe–NC system, as one of the most promising non-noble metal catalysts, exhibits remarkable ORR activity beyond Pt/C under alkaline conditions.11–15 However, their ORR kinetics in acidic electrolytes is still inferior to that of Pt-based catalysts.To further enhance the activity of Fe–NC systems, high-density Fe–N sites should be conceived. Thus, different strategies, including increasing the specific surface area of the support, adjusting the pore structure of the support, or adding more coordination constraints,16–18 have been proposed to enrich the Fe–N sites. One of the most effective methods for increasing the dispersion of metal atoms and producing high-density Fe–N sites is the confinement strategy, which involves coordination confinement and pore confinement.19–21 Our group utilized the strong metal coordination ability of vitamin C to improve the dispersion of Fe,22 but the high oxygen content in vitamin C would be very destructive to the carbon matrix. For this reason, non-metal atoms such as N, P, S, etc. that can react with carbon to form active sites are more suitable for coordination confinement.
Hitherto, Yu et al. found that doping P atoms can simultaneously break the bond length and electroneutrality of the carbon skeleton.23 If the Fe–P structure continues to be introduced into the carbon matrix, the inactive P-doped carbon in acidic media can be activated.24 Meanwhile, through theoretical calculations, Zhao et al. proved that the Fe–P structure embedded in graphene can activate oxygen molecules and elongate the O–O bond, thereby promoting the subsequent ORR step.25 Therefore, introducing the Fe–P structure into atomically dispersed Fe–NC systems is expected to greatly boost the ORR activity of catalysts.26,27 However, most of the research is restricted to phosphatizing Fe–N–C systems under an environment that contains a phosphorus source, as typical multiple preparation steps. Unfortunately, the simultaneous presence of Fe–P and high-density Fe–N is not guaranteed at present, and the extremely dangerous exhaust gas generation cannot be prevented during heat treatment.
Herein, we propose a bidirectional coordination approach to achieve the high-density Fe–N site with the Fe–P site by a one-step construction at high temperatures. Specifically, based on the ZIF-8 preparation system, triphenylphosphine is pre-mixed with 2-methylimidazole as a ligand and then reacted with metal salts, aiming to enhance the dispersion and phase homogeneity of Fe by employing the bidirectional coordination of 2-methylimidazole and triphenylphosphorus on Fe ions. The structure characterization demonstrates that Fe–P–NC obtained by introducing triphenylphosphine forms abundant Fe–N sites and Fe–P bonds and prohibits the random aggregation of Fe in Fe–NC. As a result, Fe–P–NC exhibits higher ORR activity than Fe–NC in both acidic and alkaline electrolytes, outperforming commercial Pt catalysts in Zn–air batteries.
2. Experimental section
2.1 Catalyst synthesis
1.5 mmol of triphenylphosphine and 32 mmol of 2-methylimidazole were dissolved in 36 mL of anhydrous methanol to form a ligand solution. 0.333 mmol of FeSO4·7H2O and 4.0 mmol of Zn(NO3)2·6H2O were dissolved in 36 mL of anhydrous methanol to form a salt solution. Then, the ligand solution was added to the salt solution, and then agitated for 4 h at room temperature. Afterward, metal ions and ligands remaining in the reaction mixture were removed by several rounds of centrifugal washing, and the resulting product was dried at 60 °C for 12 h, and named as ZIF–Fe–P. Under the same solution concentration and metal salt dosage, the precursor prepared without adding triphenylphosphine was named ZIF–Fe.The dried precursors were then crushed into a fine powder and heated for 3 h at 900 °C in a tube furnace supplied with flowing N2. After naturally cooling, the catalyst derived from ZIF–Fe–P was named Fe–P–NC, and the catalyst obtained by carbonization of ZIF–Fe was named Fe–NC.
Fe–P(s)–NC (0.1 mmol triphenylphosphorus) and Fe–P(m)–NC (0.2 mmol triphenylphosphorus) were also prepared with addition of different amounts of triphenylphosphorus by the same method for comparison. Fe–P–NC was found to have the optimal ORR activity through LSV testing in 0.1 M KOH electrolyte (Fig. S1†), so the difference between Fe–NC and Fe–P–NC was mainly discussed in the follow exploration to illustrate the role of triphenylphosphorus.
2.2 Electrochemical measurements
Electrochemical tests were performed on a CHI 660E electrochemical workstation using a three-electrode working model. The three electrodes include a counter electrode (carbon rod), reference electrode (a calibrated Ag/AgCl electrode used in 0.1 M KOH, and a saturated calomel electrode (SCE) used in 0.5 M H2SO4) and working electrode (5 mm glassy carbon electrode (RDE), coated with 20 μL catalyst ink). The potential of the reference electrode can be converted to the reversible hydrogen potential using the Nernst equation. Preparation of catalyst ink: 5 mg of the prepared catalyst or 3 mg of commercial Pt/C was dispersed in a mixed solution consisting of 900 μL of isopropanol, 100 μL of deionized water and 20 μL of Nafion (5 wt%), and sonicated for 30 min to achieve a uniform ink state. All tests were performed in N2/O2 saturated electrolyte. Cyclic Voltammetry (CV) was performed at a scan rate of 50 mV s−1, Linear Sweep Voltammetry (LSV) was performed at 1600 rpm at a scan rate of 10 mV s−1, and LSV curves at different rotational speeds were measured at 800 rpm, 1200 rpm, 1600 rpm, 2000 rpm and 2400 rpm.2.3 Primary Zn–air battery fabrication
For a liquid electrolyte Zn–air battery, the anode was a 1 × 2 cm2 polished zinc foil (≥99.99%), the cathode was a 1 × 2 cm2 carbon paper loaded with catalyst (YSL-33, the area of the carbon paper loaded with catalyst is 1 × 1 cm2, and the catalyst loading is 1 mg cm−2), the electrolyte was a mixed solution composed of 6 mol per L KOH and 0.2 mol per L Zn(CH3COO)2. The performance test was carried out on the CHI 660E workstation.For the all-solid-state Zn–air battery, the anode was a polished zinc foil, the cathode was nickel foam containing catalyst with a loading of 4 mg cm−2, and the electrolyte was a polyvinyl alcohol membrane. Preparation of the polyvinyl alcohol membrane: 5 g of polyvinyl alcohol with a molecular weight of ∼145![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was placed in 50 mL of ultrapure water, stirred at room temperature for 45–50 min and then transferred to a water bath at 95 °C with continuous stirring for 1–2 h. After the solution showed an overall gel structure, it was poured into a Petri dish and shaken to form a film with a thickness of about 1 mm, cooled to room temperature and then frozen in a refrigerator for 2–4 h. After freezing, the film was dried in an oven for 1 h, and then a small amount of ultrapure water was added into the Petri dish to float the membrane for use. Preparation of catalyst ink: 20 mg of catalyst powder was dispersed in a mixture solution consisting of 100 μL of isopropanol, 100 μL of ultrapure water and 5 μL of 5 wt% Nafion and sonicated for 30 min to reach a homogeneous state.
000 was placed in 50 mL of ultrapure water, stirred at room temperature for 45–50 min and then transferred to a water bath at 95 °C with continuous stirring for 1–2 h. After the solution showed an overall gel structure, it was poured into a Petri dish and shaken to form a film with a thickness of about 1 mm, cooled to room temperature and then frozen in a refrigerator for 2–4 h. After freezing, the film was dried in an oven for 1 h, and then a small amount of ultrapure water was added into the Petri dish to float the membrane for use. Preparation of catalyst ink: 20 mg of catalyst powder was dispersed in a mixture solution consisting of 100 μL of isopropanol, 100 μL of ultrapure water and 5 μL of 5 wt% Nafion and sonicated for 30 min to reach a homogeneous state.
2.4 DFT computation details
All DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP) algorithm. The electron exchange and correlation energies were handled using the Generalized Gradient Approximation (GGA) of the Perdew–Burke–Ernzerhof scheme (PBE). The projector augmented wave (PAW) approach was used to illustrate the interaction between ions and electrons. Calculations were done using the DFT-D3 functional with PBE-PAW potentials to explore the impact of van der Waals contact on reaction energetics. The Brillouin zone was sampled using a 3 × 3 × 1 Monkhorst–Pack grid, and iterative solutions to the Kohn–Sham equations were extended in a plane-wave basis set with a kinetic energy cutoff of 450 eV. The convergence conditions were established at 10–5 eV and 0.02 eV Å−1 for the electronic self-consistent iteration and force, respectively. The Fe–N4 model was established by embedding Fe–N4 sites into a periodic 5 × 5 graphene support. The model of Fe–N4/Fe2P is to place P-doped graphene with Fe–N4 moieties in the center on an Fe2P slab, and Fe2P uses a 2 × 2 supercell with lattice parameters a = 12.44 Å, b = 12.667 Å and α = β = γ = 90°. For preventing contact, a 15 Å vacuum zone was placed between the periodically repeated slabs.3. Results and discussion
3.1 Morphology and structure characterization
The specific synthesis process of Fe–NC and Fe–P–NC is depicted in Scheme 1, which only involves a simple co-precipitation and solid-phase pyrolysis process. Here, the purpose of blending iron and zinc salts is to enable iron ions to enter the ZIF framework, and blending triphenylphosphine and 2-methylimidazole is to guarantee that Fe ions can be coordinated and anchored in time. From the scanning electron microscope (SEM) images, ZIF–Fe loses its regular cubic octahedral structure and presents a stacked state of multiple polyhedrons (Fig. S2a†), which indicates that Fe ions successfully invade the ZIF framework and break the original crystallization direction of ZIF. ZIF–Fe–P exhibits a grape-like morphology (Fig. S2b†), suggesting that triphenylphosphine coordination contributes to additional crystal orientation changes in the skeleton structure. The coordination effect of triphenylphosphorus in the precursor can also be proved through the X-ray diffraction (XRD) patterns of ZIF–Fe and ZIF–Fe–P. As observed, the diffraction peaks of ZIF–Fe in the 30–40 degrees range are not visible in ZIF–Fe–P (Fig. S3†). After high-temperature treatment, their carbonized derivatives Fe–NC and Fe–P–NC basically inherit the general morphology of ZIF–Fe and ZIF–Fe–P, respectively. Since high-temperature carbonization leads to shrinkage of the carbon structure, they both produce varying degrees of surface wrinkles (Fig. S4†). Unexpectedly, the high-resolution SEM images in Fig. S5† reveal that many conspicuous pores form on the surface of Fe–P–NC, demonstrating a looser carbon framework, compared with Fe–NC.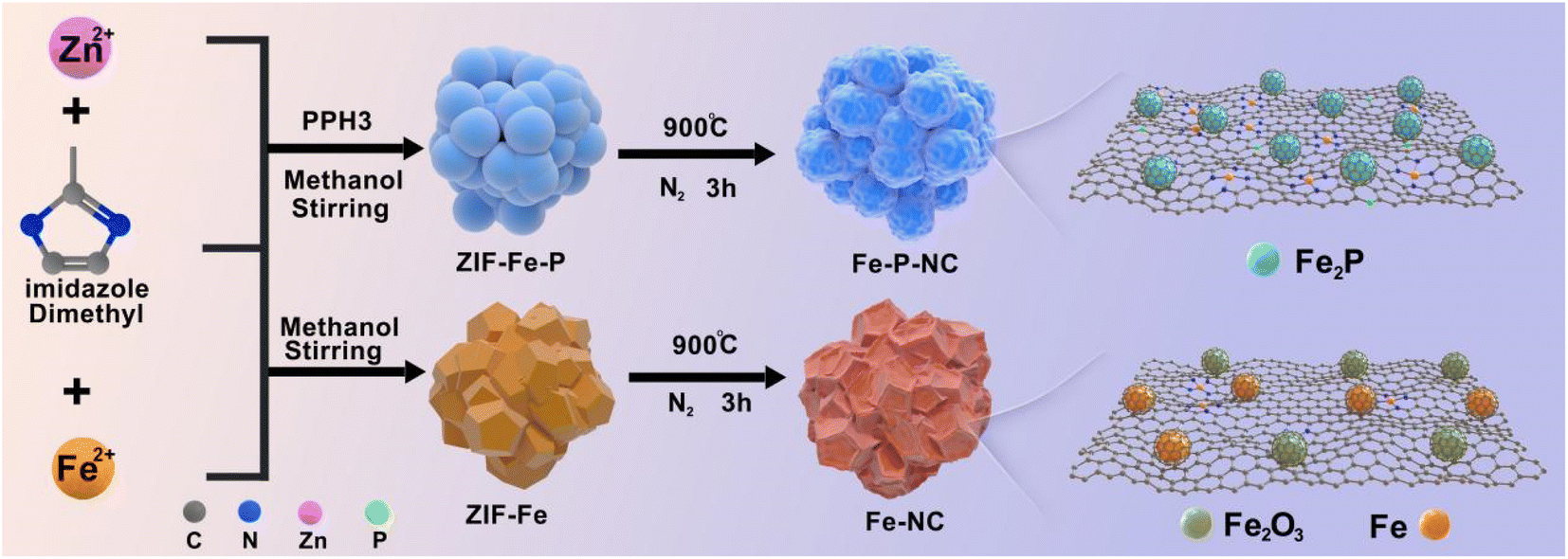 | ||
| Scheme 1 Illustration of the preparation of Fe–P–NC and Fe–NC catalysts. | ||
To confirm the pore structure of Fe–P–NC, the two samples were measured for N2 adsorption and desorption. According to Fig. 1a, both prominent hysteresis loops in the isotherms of Fe–NC and Fe–P–NC display the porous structure, as an inherent characteristic of metal–organic framework compound derivatives. Particularly, the pore size distribution plot depicts a more abundant mesoporous distribution for Fe–P–NC, in line with that exhibited in SEM images. Specifically, Fe–P–NC has a specific surface area of 367.82 m2 g−1, which is clearly larger than that of Fe–NC (298.15 m2 g−1). Additionally, as compared to Fe–NC (average pore diameter: 8.5754 nm, total pore volume: 0.6392 cm3 g−1), Fe–P–NC demonstrates a greater average pore diameter (9.0576 nm) and total pore volume (0.8329 cm3 g−1). Such a multi-mesoporous structure in the Fe–P–NC skeleton allows electrolyte penetration to accelerate transport, and also facilitates the exposure of active sites inside the carbon framework, which is beneficial to the catalytic process.28,29 Immediately, XRD was executed to ascertain the composition of Fe–P–NC and Fe–NC. Fig. 1b shows that Fe–P–NC only has some weak diffraction peaks representing Fe2P, while Fe–NC has multiple sharp diffraction peaks and is mainly composed of Fe3C and Fe2O3 particles. The difference in phase and diffraction peak intensity between Fe–P–NC and Fe–NC reflects the confinement and anchoring effect of triphenylphosphorus on Fe atoms.
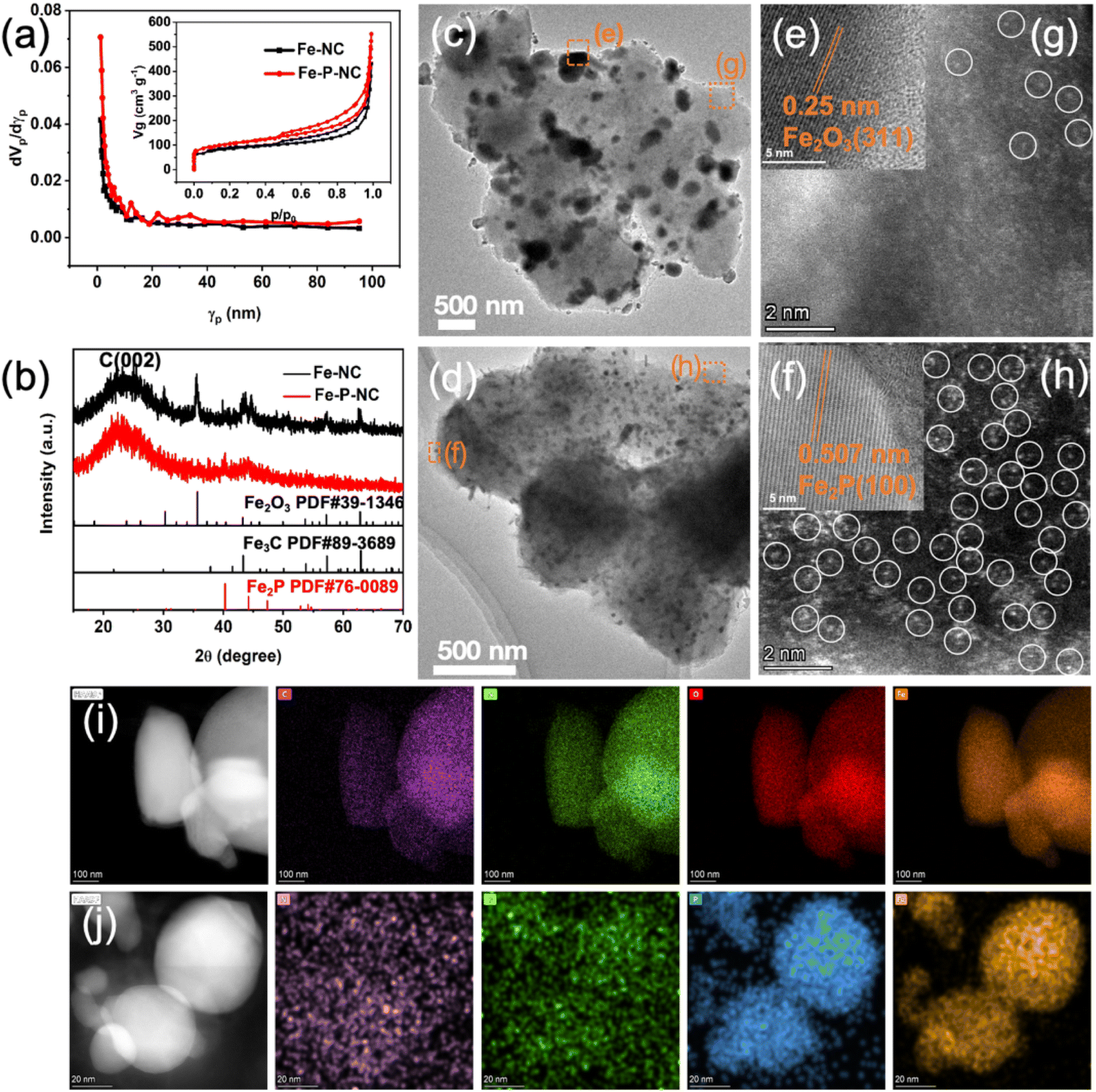 | ||
| Fig. 1 (a) Pore size distribution (inset: N2 adsorption–desorption isotherm); (b) XRD patterns; TEM images of (c) Fe–NC and (d) Fe–P–NC; HRTEM of (e) Fe–NC and (f) Fe–P–NC; HAADF STEM images of (g) Fe–NC and (h) Fe–P–NC; EDS mapping of (i) Fe–NC (purple: C, green: N, red: O, orange: Fe) and (j) Fe–P–NC (purple: N, green: O, blue: P, orange: Fe). | ||
Through transmission electron microscope (TEM) image observation (Fig. 1c and d), the particle in Fe–NC is unevenly distributed and large in size, while Fe–P–NC is uniformly dispersed with smaller-sized nanocrystals, which also explains the sharpness of the diffraction peaks expressed by the two samples in their XRD patterns. Then, high-resolution transmission electron microscope (HRTEM) observations were carried out on arbitrary particles in Fe–NC and Fe–P–NC. The measured particle interplanar spacing in Fe–NC is 0.25 nm, corresponding to the (311) crystal plane of Fe2O3 (Fig. 1e). The nanocrystal lattice spacing in Fe–P–NC is 0.507 nm, which corresponds to the (100) crystal plane of Fe2P (Fig. 1f). Aberration-corrected STEM HAADF (ac-STEM-HAADF) images reveal that traces of bright dots faintly visible at the edges of Fe–NC (Fig. 1g). Surprisingly, the surface of Fe–P–NC contains a substantial number of bright dots (Fig. 1h). Immediately, the Fe and N signals displayed in the electron energy loss spectroscopy (EELS) indicate that these bright dots in Fe–P–NC are mainly composed of monodisperse Fe–Nx (Fig. S6†). These results prove that the introduction of triphenylphosphine indeed strengthens the coordination of Fe and 2-methylimidazole and increases the dispersion and density of Fe atoms by suppressing the aggregation of Fe through bidirectional coordination. Further, the regions where particles aggregated in Fe–NC and Fe–P–NC were randomly selected for HAADF EDS characterization. Apparently, the particles in Fe–NC are concentrated with C, N, O, and Fe elements (Fig. 1i), while the particles in Fe–P–NC are only composed of Fe and P (Fig. 1j), which means the particles are complicated in Fe–NC.
To precisely understand the structure and composition differences between Fe–P–NC and Fe–NC, a variety of intricate structural characterizations were performed. In Raman spectra, Fe–P–NC has a higher ID/IG value (1.103) than Fe–NC (1.005) (Fig. 2a), indicating more defects for Fe–P–NC due to the multi-mesoporous structure and P doping. Besides, in the EPR test, Fe–P–NC has a higher G intensity (Fig. 2b), again manifesting more carbon defects in Fe–P–NC.30,31 Then, the change of each element in Fe–NC and Fe–P–NC was analyzed by X-ray photoelectron spectroscopy (XPS). In the C 1s high-resolution XPS spectrum, five different carbon peaks (C-sp2, C-sp3, C–N, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) can be distinguished.32,33 The proportion of C-sp3 in Fe–P–NC exceeds that of Fe–NC, suggesting that Fe–P–NC contains more carbon defects (Fig. S7†), which is consistent with the results of Raman and EPR analyses. Additionally, a larger C–N amount is observed in the spectrum of Fe–P–NC compared with Fe–NC, suggesting that Fe–P–NC has more active sites favorable for the ORR.34,35 The N 1s spectrum was separated into pyridinic N, Fe–N, pyrrolic N, and graphitic N.36,37 Clearly, Fe–P–NC has a greater Fe–N proportion than Fe–NC (Fig. S8†). Then, the findings of the XPS test demonstrate that Fe–P–NC has a much larger atomic content of Fe and N than Fe–NC (Table S1†). Consequently, Fe–P–NC actually has a higher concentration of Fe–N sites. The spectrum of P 2p shows that P forms Fe–P bonds with Fe in Fe–P–NC, and simultaneously incorporates into the carbon structure to form P–C and P–O bonds (Fig. S9†), increasing the active sites, which is beneficial for the ORR.38,39
O) can be distinguished.32,33 The proportion of C-sp3 in Fe–P–NC exceeds that of Fe–NC, suggesting that Fe–P–NC contains more carbon defects (Fig. S7†), which is consistent with the results of Raman and EPR analyses. Additionally, a larger C–N amount is observed in the spectrum of Fe–P–NC compared with Fe–NC, suggesting that Fe–P–NC has more active sites favorable for the ORR.34,35 The N 1s spectrum was separated into pyridinic N, Fe–N, pyrrolic N, and graphitic N.36,37 Clearly, Fe–P–NC has a greater Fe–N proportion than Fe–NC (Fig. S8†). Then, the findings of the XPS test demonstrate that Fe–P–NC has a much larger atomic content of Fe and N than Fe–NC (Table S1†). Consequently, Fe–P–NC actually has a higher concentration of Fe–N sites. The spectrum of P 2p shows that P forms Fe–P bonds with Fe in Fe–P–NC, and simultaneously incorporates into the carbon structure to form P–C and P–O bonds (Fig. S9†), increasing the active sites, which is beneficial for the ORR.38,39
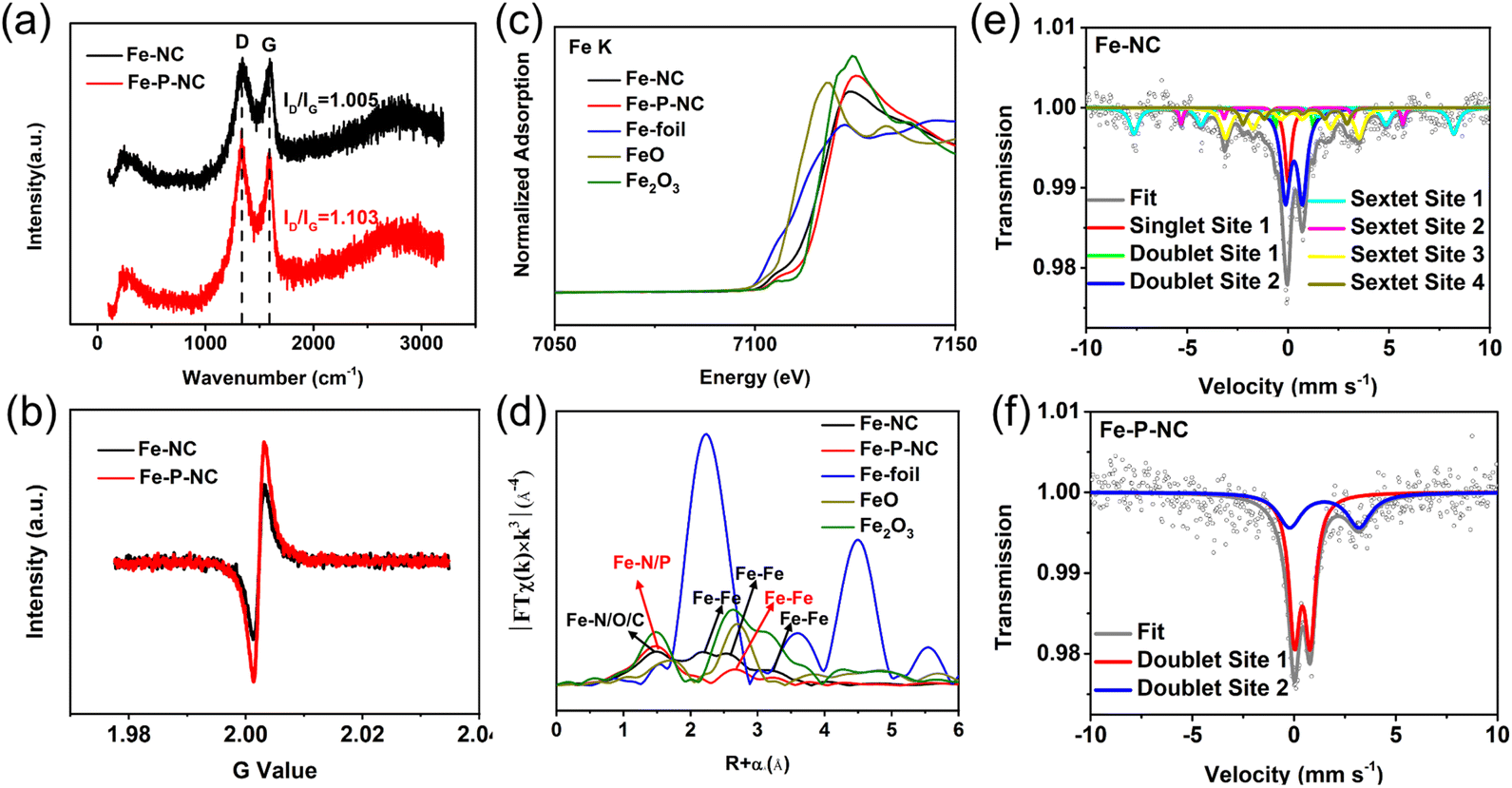 | ||
| Fig. 2 (a) Raman spectra, (b) EPR testing of carbon defects, (c) XANES spectra of the Fe-K edge, (d) FT-EXAFS spectra of the Fe-K edge, (e) 57Fe Mössbauer spectrum of Fe–NC, and (f) 57Fe Mössbauer spectrum of Fe–P–NC. | ||
Since the distribution of Fe is crucial to the catalytic activity, synchrotron radiation and Mössbauer spectroscopy were employed to accurately analyze the state of Fe in Fe–NC and Fe–P–NC. Fig. 2c shows the X-ray absorption near edge structure (XANES) spectra of the Fe-K edge, in which it is confirmed that the valence state of the Fe element in the two as-prepared samples is between those of FeO and Fe2O3, and the average Fe valence state of Fe–P–NC is higher than that of Fe–NC. The Fourier Transform (FT) extended X-ray absorption fine structure (FT-EXAFS) spectra in Fig. 2d show that Fe–NC has peaks around 1.5 Å, 2.2 Å, 2.7 Å and 3.2 Å. Specifically, the peak near 1.5 Å is the superposition state of Fe–N, Fe–O and Fe–C coordination,40,41 the peak near 2.2 Å represents the zero-valence Fe–Fe coordination and the two peaks near 2.7 Å and 3.2 Å are assigned to Fe compounds. Differently, Fe–P–NC only has a strong peak with Fe–N/P coordination around 1.5 Å, and a weak peak around 2.7 Å that belongs to Fe–Fe coordination of Fe compounds. Comparatively, Fe–NC not only contains a lot of metallic Fe–Fe bonds and metal compound Fe–Fe bonds, but its Fe–N peak is also weaker than that in Fe–P–NC, showing that the vast majority of Fe atoms in the Fe–NC system are randomly aggregated rather than being formed into single atoms. Further, through FT-EXAFS fitting, the coordination numbers of Fe–N, Fe–P, and Fe–Fe of Fe–P–NC are found to be 4.1, 0.9, and 0.8, respectively (Fig. S10 and Table S2†), confirming the co-existence of Fe–N4 single atoms and Fe2P.37,40
In the 57Fe Mössbauer spectrum, Fe–NC forms seven different splitting spectra (Fig. 2e, fitting parameters in Table S3†), which can be classified into six types of Fe, namely γ-Fe (single site 1), Fe(III)–N4 (doublet site 1), Fe2+xN (doublet site 2), Fe2O3 (sextet site 1), α-Fe (sextet site 2), and Fe3C (sextet site 3, sextet site 4).42,43 Notably, Fe–P–NC only has two different splitting spectra (Fig. 2f, fitting parameters in Table S3†), classified as Fe(III)–N4 (doublet site 1) and Fe2P (doublet site 2).37,42 Importantly, Fe(III)–N4 accounts for 65.3% of Fe sites in Fe–P–NC, but only 2.8% in Fe–NC, which is consistent with the synchrotron radiation analysis and explains why a large number of monodisperse Fe sites can be clearly observed in Fe–P–NC, while being almost invisible in Fe–NC. Overall, all of the above evidence illustrates that the Fe atoms in Fe–P–NC must be greatly dispersed through the bidirectional coordination of triphenylphosphine and 2-methylimidazole to generate a large number of Fe–N4 single-atom sites and additional Fe2P sites.
3.2 ORR and Zn–air battery performance of the Fe–P–NC system
The ORR performance of the as-prepared samples was examined in alkaline and acidic electrolytes. First, in 0.1 M KOH, Fe–NC exhibits exceptional oxygen reduction activity, as evidenced by the greater half-wave potential (E1/2: 0.878 V) and onset potential (Eonset: 0.97 V) than commercial 20% Pt/C catalysts (Eonset: 0.962 V, E1/2: 0.862 V). Distinctly, Fe–P–NC with an optimized structure and composition possesses an even higher Eonset (1.02 V) and E1/2 (0.906 V) than that of Fe–NC (Fig. 3a). Meanwhile, the calculated lowest Tafel slope also verifies the fastest reaction kinetics of Fe–P–NC (Fig. 3b). Then, the reaction path of Fe–P–NC was explored by testing its LSV curves at various rotating speeds. With the Koutecky–Levich (K–L) equation, five linear K–L plots are obtained (Fig. S11a†). The calculation result shows that Fe–P–NC has an average electron transfer number (n) of 3.97 (Fig. S11b†), which is very similar to the 4-electron transfer path. The efficient catalytic activity, fast electron transfer and a transfer path close to 4 electrons predicate that Fe–P–NC catalyzes oxygen reduction with less H2O2 generation, thus reducing the attack on Fe sites and improving the catalytic stability.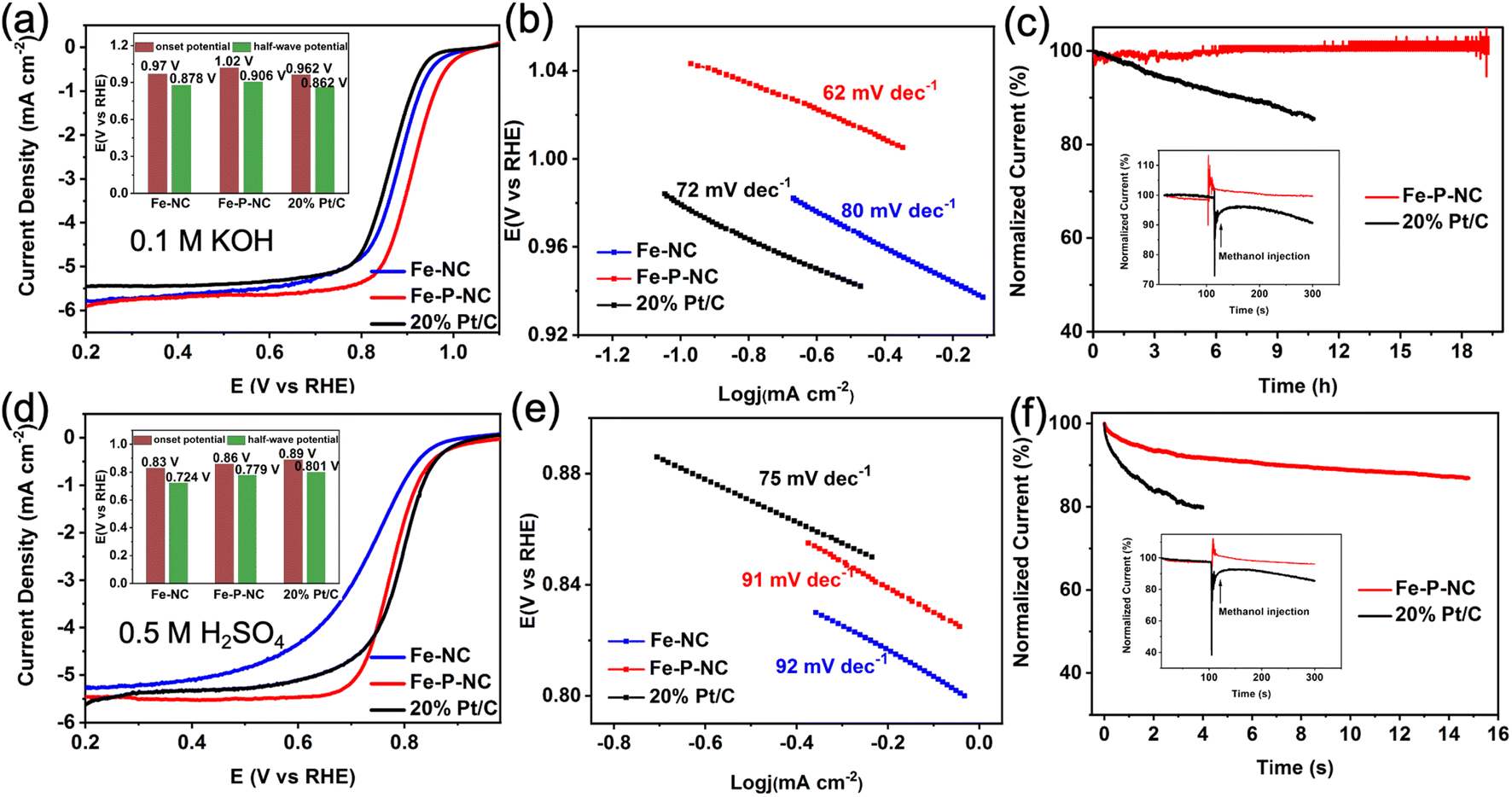 | ||
| Fig. 3 (a) LSV curves under 0.1 M KOH (inset: comparison of onset potential and half-wave potential), (b) Tafel plots, (c) chronoamperometric response test of stability (inset: methanol resistance); (d) LSV curves under 0.5 M H2SO4 (inset: comparison of onset potential and half-wave potential), (e) Tafel plots, (f) chronoamperometric response test of stability (inset: methanol resistance). | ||
Subsequently, the working stability and resistance to methanol of Fe–P–NC were probed through chronoamperometric response testing. In Fig. 3c, the normalized current of Fe–P–NC hardly changes during the 20 h potentiostatic test at 0.5 V, whereas the normalized current of 20% Pt/C drops dramatically. Moreover, in the methanol resistance test, the normalized current of Fe–P–NC fluctuates only during methanol injection and then quickly recovers the initial current density, while the normalized current of 20% Pt/C drops instantly at the moment of methanol injection, indicating an excellent methanol resistance of Fe–P–NC.
In 0.5 M H2SO4 electrolytes, Fe–P–NC maintains a catalytic ability superior to Fe–NC and comparable to 20% Pt/C, as supported by their onset potential (Fe–NC: 0.83 V, Fe–P–NC: 0.86 V, 20% Pt/C: 0.89 V), half-wave potential (Fe–NC: 0.724 V, Fe–P–NC: 0.779 V, 20% Pt/C: 0.801 V), and Tafel slope (Fe–NC: 90 mV dec−1, Fe–P–NC: 92 mV dec−1, 20% Pt/C: 75 mV dec−1) (Fig. 3d and e). Then, based on the LSV curves in Fig. S12,† the n for Fe–P–NC is calculated to be 3.81, signifying that the ORR process of Fe–P–NC in acidic media is also dominated by a 4-electron path. In the stability test, the durability and methanol resistance of Fe–P–NC are superb relative to 20% Pt/C (Fig. 3f).
In 0.1 M HClO4 electrolyte, Fe–P–NC can still obtain a half-wave potential of 0.782 V, despite being less active than commercial 20% Pt/C. And according to the half-wave potential and Tafel slope (Fig. S13 and S14†), Fe–P–NC always retains better ORR activity and reaction kinetics than Fe–NC. Crucially, in the chronoamperometric response test, Fe–P–NC shows noticeably greater stability than 20% Pt/C (Fig. S15†). The catalytic performance of Fe–P–NC in alkaline and acidic electrolytes clearly demonstrates its exceptional catalytic ability for oxygen reduction. And Fe–P–NC is likewise superior to similar materials reported in the literature, as shown by a comparison of half-wave potentials in alkaline and acidic electrolytes (Tables S4 and S5†).
To show the application potential of our prepared catalyst, Fe–P–NC and 20% Pt/C were separately assembled into Zn–air batteries as air electrodes for comparison (Fig. 4a). Fig. 4b shows that Fe–P–NC outputs a higher open-circuit voltage than 20% Pt/C. In Fig. 4c, the current density and power density of Fe–P–NC are consistently greater than those of 20% Pt/C. Moreover, Fe–P–NC also generates a higher discharge voltage (Fig. 4d). According to the mass of zinc foil consumed, the energy density of Fe–P–NC is up to 801 mA h gZn−1, larger than the 742 mA h gZn−1 of 20% Pt/C. In the galvanostatic charge–discharge test (Fig. 4e), when the catalyst was combined with RuO2 as an air electrode, the charge/discharge voltage gap of Fe–P–NC//RuO2 is substantially less than that of 20% Pt/C//RuO2. As the cycle of charging and discharging continues, the Zn–air battery fabricated with Fe–P–NC exhibits a nearly constant charge/discharge voltage gap, whereas the Zn–air battery based on 20% Pt/C exhibits a clearly increasing charge/discharge voltage gap, indicating that Fe–P–NC has significantly better stability than 20% Pt/C. The prominent performance of Fe–P–NC in Zn–air batteries can be credited to the extraordinary oxygen reduction activity and stability of Fe–P–NC compared with 20% Pt/C in an alkaline environment. Finally, we used Fe–P–NC to create an all-solid-state Zn–air battery. Fig. 4f shows photos of three all-solid-state Zn–air batteries with Fe–P–NC as the air cathode and a light bulb. The open-circuit voltage of the randomly assembled all-solid-state Zn–air battery is at least 1.38 V, revealing efficient spontaneous oxygen reduction catalytic capability of Fe–P–NC and hopeful practical application.
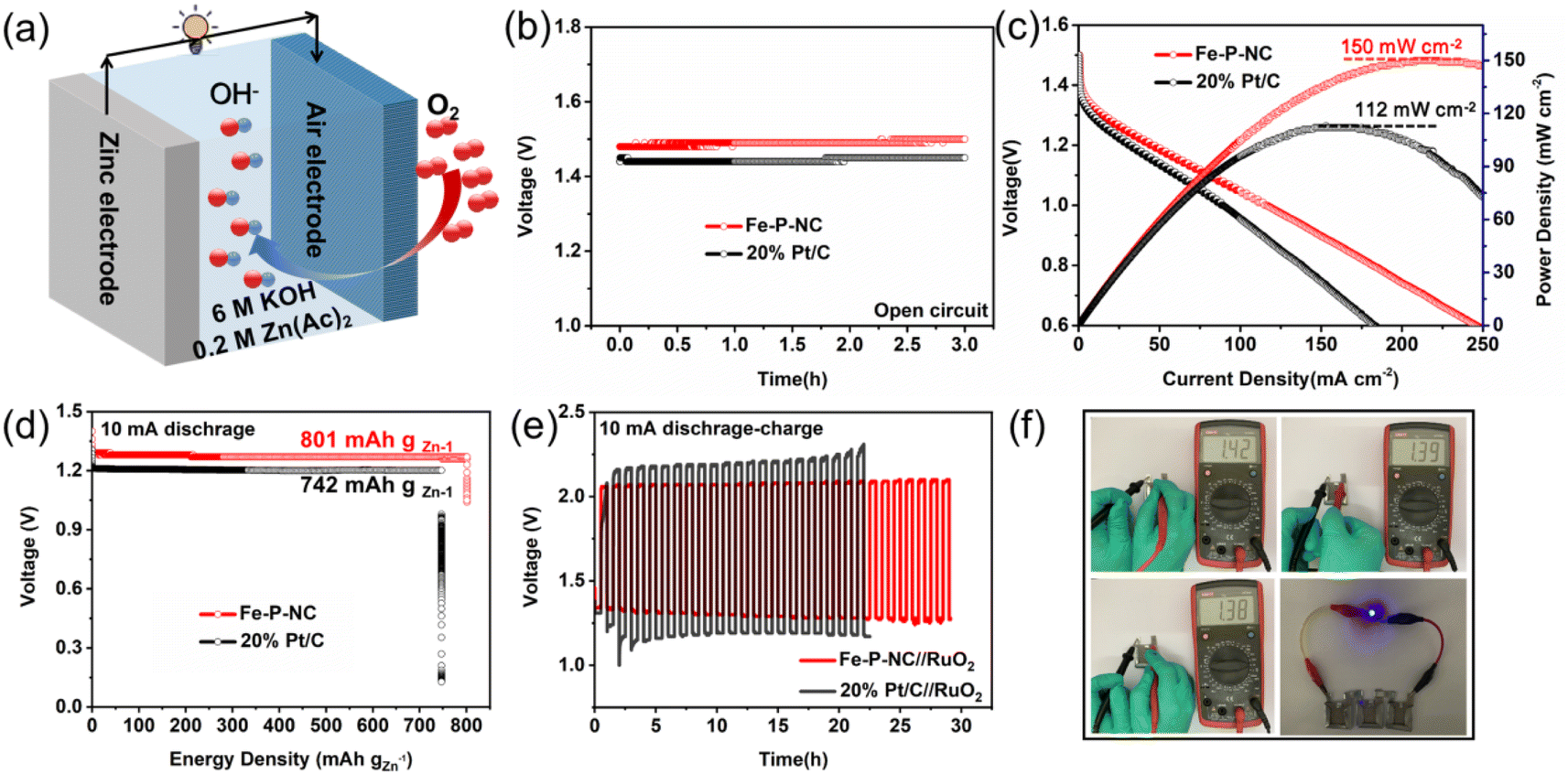 | ||
| Fig. 4 (a) Assembly diagram of a liquid electrolyte Zn–air battery, (b) open circuit potential curves, (c) polarization curves, (d) energy density curves, (e) galvanostatic charge–discharge curves, (f) the open-circuit voltage of an all-solid-state Zn–air battery assembled with Fe–P–NC and a lighted LED. | ||
3.3 Mechanism analysis of the Fe–P–NC catalyst
The previous characterizations have repeatedly proved that the Fe–P–NC catalyst contains a large number of single atom Fe–N4 sites and only a small proportion of Fe2P. Although Fe–P–NC performs better than Fe–NC in the ORR, compared with the pure Fe–N4 site, whether the presence of Fe2P indeed has a positive effect is still unknown. Thus, we conducted density functional theory (DFT) calculations to illustrate the impact of Fe2P on Fe–N4 sites. The optimized models of pure Fe–N4, pure Fe2P and the coexistence of Fe–N4 and Fe2P are shown in Fig. S16.†To confirm the main ORR active sites in Fe–P–NC, the free energy pathways of the conventional four-electron reaction process for the ORR of Fe–N4 and Fe2P at U = 1.23 V were first compared. The results show that when pure Fe2P acts as the ORR active site, the Gibbs free energy required by Fe2P (0.744 eV) in the rate-determining step (the generation of O intermediates from OOH intermediates) is significantly higher than that of Fe–N4 (0.644 eV) (Fig. S17†). This indicates that Fe–N4 serves as the predominant ORR active site in Fe–P–NC. Therefore, the reaction center was determined at the Fe–N4 site in the coexistence system of Fe2P and Fe–N4 for further exploration. Fig. 5a displays the ORR free energy pathways of Fe–N4 and Fe–N4/Fe2P. Obviously, the free energy barrier in the rate-determining step for the Fe–N4/Fe2P model is lower than that of Fe–N4, requiring only 0.58 eV. The lower reaction energy barrier determines the faster reaction process of Fe–N4/Fe2P.
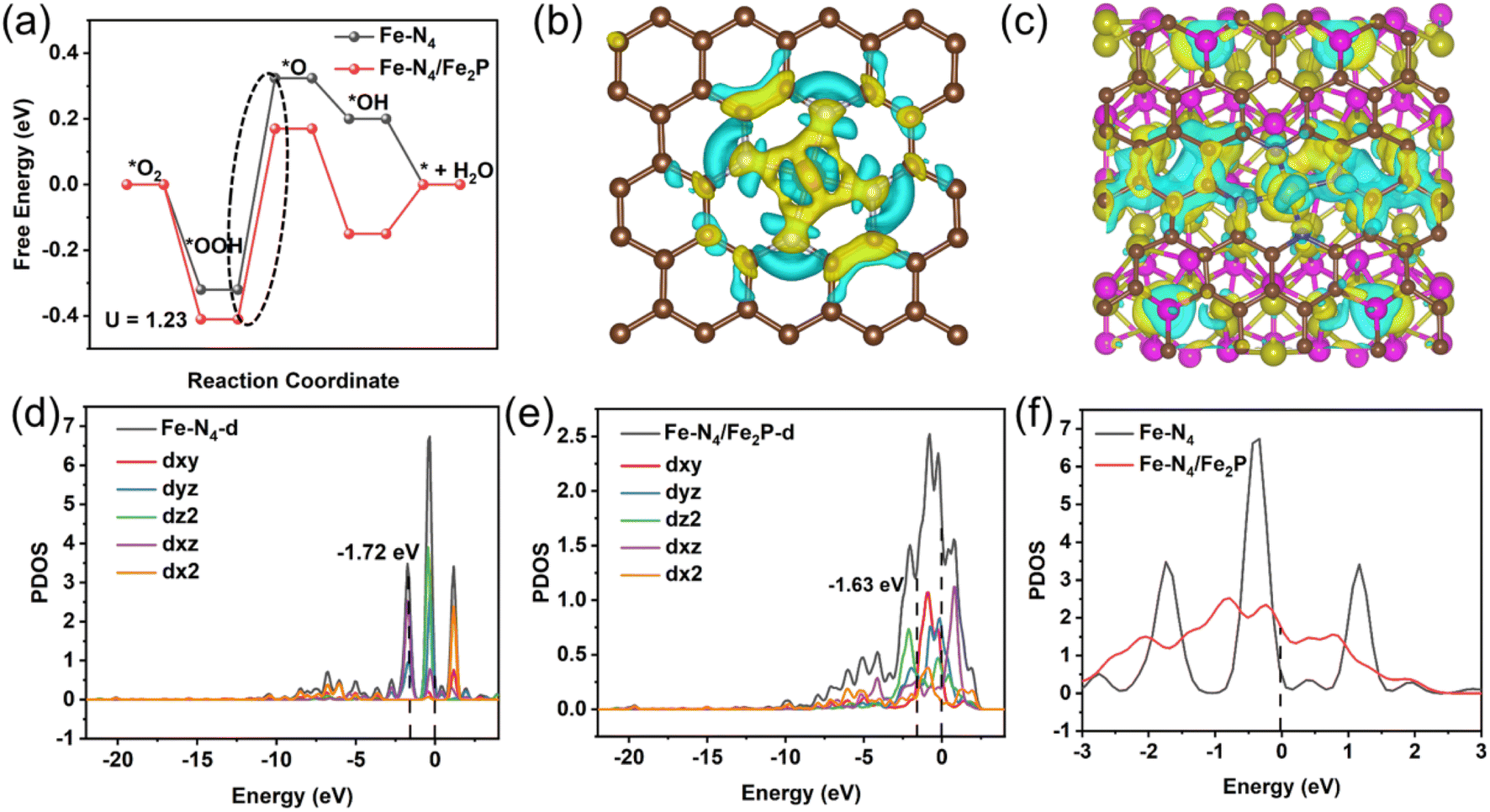 | ||
| Fig. 5 (a) ORR free energy path diagram of Fe–N4 and Fe–N4/Fe2P at U = 1.23 V, top view differential charge density of (b) Fe–N4 and (c) Fe–N4/Fe2P, partial density of states (PDOS) of d orbitals of Fe atoms in (d) Fe–N4 and (e) Fe–N4/Fe2P, and (f) comparison of the PDOS at the Fermi level. | ||
Then, how does Fe2P affect Fe–N4? The differential charge densities and Bader charges of Fe–N4 and Fe–N4/Fe2P were compared. In comparison to the charge lost by Fe–N4 (0.9955e), the Fe sites in Fe–N4/Fe2P lose less charges (0.8567e), as shown in Fig. 5b, c and S18.† This allows the adsorbed intermediates to receive more electrons from Fe sites during the reaction, making Fe–N4/Fe2P more conducive to trapping reactants or intermediates.44,45 In the density-of-states (DOS) diagram (Fig. 5d and e), the d-band center of Fe–N4/Fe2P closer to the Fermi level implies enhanced adsorption of intermediates. In addition, Fe–N4/Fe2P exhibits a substantially higher density of states at the Fermi level (Fig. 5f), indicating that the Fe site has a high concentration of valence electrons, which is favorable for rapid electron transfer.46,47 Then, we further etched the Fe–P–NC catalyst in 0.5 M H2SO4 with stirring for 12 h at room temperature, and evaluated its ORR activity after acid-etching. As indicated in Table S1,† the contents of Fe, N and P all decreased after acid-etching. According to the high-resolution XPS spectra, there is a notable decrease in the proportion of Fe–P in Fe 2p (Fig. S19†) and an increase in the proportion of Fe–N (Fig. S20†) in N 1s. This suggests that the Fe–N4 site was not greatly affected by acid-etching,48 with Fe2P suffering the most loss. Through the LSV result in Fig. S21,† Fe–P–NC exhibits a distinctly decreased half-wave potential in 0.1 M KOH electrolytes after acid-etching, which can be owing to the acid etching away part of the exposed Fe2P. Undoubtedly, such results and theoretical analysis jointly demonstrate the synergistic catalytic effect of Fe2P and Fe–N4 in the oxygen reduction process, explaining the excellent ORR performance of Fe–P–NC.
According to the aforementioned data, the triphenylphosphine-modulated Fe–P–NC catalyst has a greater potential for applications than Fe–NC. First, thanks to the strong metal coordination ability of triphenylphosphine, it strengthens the confinement of Fe atoms that are coordinated by 2-methylimidazole, thereby hindering the migration of these Fe atoms in a high-temperature environment, driving the formation of high dispersion and high-density Fe–N sites, and contributing to the high-efficiency ORR. Second, the coordination of Fe ions adsorbed on the surface of the precursor with triphenylphosphine can also promote the formation of Fe–P sites, thereby lowering the ORR reaction energy barrier and further enhancing the ORR activity of the resulting catalyst. Finally, under the attack of triphenylphosphine pyrolysis by-products, the carbon matrix acquires a richer pore structure and defect level, which can also accelerate the ORR process. In general, bidirectional coordination can effectively improve the dispersion of metal atoms, resulting in more efficient catalysis.
4. Conclusions
In summary, we successfully realize the high-density and dispersion of Fe–N sites in the carbon matrix by exploiting the bidirectional coordination of triphenylphosphine and 2-methylimidazole and introducing P atoms to form positive Fe–P bonds. Strikingly, compared with the Fe–NC catalyst, the formed Fe–P–NC catalyst is amplified from 2.8% in Fe–NC to 65.3% for single-atom dispersed Fe sites, indicating the formation of high-density monodisperse Fe–N sites in Fe–P–NC. The synergistic effect of Fe–N sites and Fe2P endows Fe–P–NC with greatly boosted oxygen reduction ability, outperforming commercial Pt catalysts in Zn–air battery performance, indicating promising application potential. This work offers workable guidelines for designing and building catalysts with high-density and highly dispersed metal atom active sites.Data availability
The authors declare that the data are available within the paper and its ESI file.†Author contributions
Huihui Jin: conceptualization, methodology, writing – original draft. Ruohan Yu: methodology, formal analysis. Weihao Zeng: methodology. Pengxia Ji: investigation. Zhengying Li, Daping He, Shichun Mu: writing-review & editing, validation. Huihui Jin and Ruohan Yu contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially sponsored by the National Natural Science Foundation of China (22102128, 22279097) and the Fundamental Research Funds for the Central Universities (WUT:2022IVA168).Notes and references
- Y. Wang, T. Zhou, S. Ruan, H. Feng, W. Bi, J. Hu, T. Chen, H. Liu, B. Yuan, N. Zhang, W. Wang, L. Zhang, W. Chu, C. Wu and Y. Xie, Directional manipulation of electron transfer by energy level engineering for efficient cathodic oxygen reduction, Nano Lett., 2022, 22, 6622–6630 CrossRef CAS PubMed.
- S. Lin, L. Xia, Y. Cao, H. Meng, L. Zhang, J. Feng, Y. Zhao and A. Wang, Electronic regulation of ZnCo dual-atomic active sites entrapped in 1D@2D hierarchical N-doped carbon for efficient synergistic catalysis of oxygen reduction in Zn-air battery, Small, 2022, 18, 2107141 CrossRef CAS PubMed.
- L. Zhang, J. Zhu, X. Li, S. Mu, F. Verpoort, J. Xue, Z. Kou and J. Wang, Nurturing the marriages of single atoms with atomic clusters and nanoparticles for better heterogeneous electrocatalysis, Interdiscip. Mater., 2022, 1, 51–87 CrossRef.
- Q. Sun, X.-H. Li, K.-X. Wang, T.-N. Yea and J.-S. Chen, Inorganic non-carbon supported Pt catalysts and synergetic effects for oxygen reduction reaction, Energy Environ. Sci., 2023, 16, 1838–1869 RSC.
- D. Yang, G. Sun, X. Wang, F. Li, Z. Gao, P. Zhang, W. Lu and M. Feng, α-Fe2O3 nanospindles as an efficient catalyst for optical and magnetic fields co-assisted Li-O2 cells, Chem. Eng. J., 2023, 474, 145712 CrossRef CAS.
- Z. Huang, F. Li, Y. Liu, S. Chen, Z. Wei and Q. Tang, The role of nitrogen sources and hydrogen adsorption on the dynamic stability of Fe–N–C catalysts in oxygen reduction reaction, Chem. Sci., 2024, 15, 1132–1142 RSC.
- C. Xu, C. Guo, J. Liu, B. Hu, J. Dai, M. Wang, R. Jin, Z. Luo, H. Li and C. Chen, Accelerating the oxygen adsorption kinetics to regulate the oxygen reduction catalysis via Fe3C nanoparticles coupled with single Fe-N4 sites, Energy Storage Mater., 2022, 51, 149–158 CrossRef.
- Z. Pei, H. Zhang, Y. Guo, D. Luan, X. Gu and X.-W. Lou, Atomically dispersed Fe sites regulated by adjacent single Co atoms anchored on N-P co-doped carbon structures for highly efficient oxygen reduction reaction, Adv. Mater., 2023, 2306047 CrossRef PubMed.
- M. Wu, X. Yang, X. Cui, N. Chen, L. Du, M. Cherif, F.-K. Chiang, Y. Wen, A. Hassanpour, F. Vidal, S. Omanovic, Y. Yang, S. Sun and G. Zhang, Engineering Fe-N4 electronic structure with adjacent Co-N2C2 and Co nanoclusters on carbon nanotubes for efficient oxygen electrocatalysis, Nano-Micro Lett., 2023, 15, 232 CrossRef CAS PubMed.
- Q. Miao, Z. Chen, X. Li, M. Liu, G. Liu, X. Yang, Z. Guo, C. Yu, Q. Xu and G. Zeng, Construction of catalytic Fe2N5P Sites in covalent organic framework-derived carbon for catalyzing the oxygen reduction reaction, ACS Catal., 2023, 13, 11127–11135 CrossRef CAS.
- D. Xue, P. Yuan, S. Jiang, Y. Wei, Y. Zhou, C. Dong, W. Yan, S. Mu and J. Zhang, Altering the spin state of Fe-NC through ligand field modulation of single-atom sites boosts the oxygen reduction reaction, Nano Energy, 2023, 105, 108020 CrossRef CAS.
- R. Wang, L. Zhang, J. Shan, Y. Yang, J. F. Lee, T.-Y. Chen, J. Mao, Y. Zhao, L. Yang, Z. Hu and T. Ling, Tuning Fe spin moment in Fe-N-C catalysts to climb the activity volcano via a local geometric distortion strategy, Adv. Sci., 2022, 9, 2203917 CrossRef CAS PubMed.
- M. Kim, K. L. Firestein, J. F. S. Fernando, X. Xu, H. Lim, D. V. Golberg, J. Na, J. Kim, H. Nara, J. Tang and Y. Yamauchi, Strategic design of Fe and N co-doped hierarchically porous carbon as superior ORR catalyst: from the perspective of nanoarchitectonics, Chem. Sci., 2022, 13, 10836–10845 RSC.
- S. Chen, X. Liang, S. Hu, X. Li, G. Zhang, S. Wang, L. Ma, C.-M. L. Wu, C. Zhi and J. A. Zapien, Inducing Fe 3d electron delocalization and spin-state transition of FeN4 species boosts oxygen reduction reaction for wearable zinc-air battery, Nano-Micro Lett., 2023, 15, 47 CrossRef CAS PubMed.
- H. Wang, M. Jerigova, J. Hou, N. V. Tarakina, S. Delacroix, N. López-Salas and V. Strauss, Modulating between 2e- and 4e- pathways in the oxygen reduction reaction with laser-synthesized iron oxide-grafted nitrogen-doped carbon, J. Mater. Chem. A, 2022, 10, 24156–24166 RSC.
- C. Jiao, Z. Xu, J. Shao, Y. Xia, J. Tseng, G. Ren, N. Zhang, P. Liu, C. Liu, G. Li, S. Chen, S. Chen and H. Wang, High-density atomic Fe-N4/C in tubular, biomass-derived, nitrogen-rich porous carbon as air-electrodes for flexible Zn-air batteries, Adv. Funct. Mater., 2023, 33, 2213897 CrossRef CAS.
- J. Li, W. Xia, J. Tang, Y. Gao, C. Jiang, Y. Jia, T. Chen, Z. Hou, R. Qi, D. Jiang, T.-R. Asahi, X. Xu, T. Wang, J. He and Y. Yamauchi, Metal-organic framework-derived graphene mesh: a robust scaffold for highly exposed Fe-N4 active sites toward an excellent oxygen reduction catalyst in acid media, J. Am. Chem. Soc., 2022, 144, 9280–9291 CrossRef CAS PubMed.
- L. Wu, R. Zhao, G. Du, H. Wang, M. Hou, W. Zhang, P. Sun and T. Chen, Hierarchically porous Fe/N/S/C nanospheres with high-content of Fe-Nx for enhanced ORR and Zn-air battery performance, Green Energy Environ., 2023, 8, 1693–1702 CrossRef CAS.
- H. Xie, B. Du, X. Huang, D. Zeng, H. Meng, H. Lin, W. Li, T. Asefa and Y. Meng, High density single Fe atoms on mesoporous N-doped carbons: noble metal-free electrocatalysts for oxygen reduction reaction in acidic and alkaline media, Small, 2023, 19, 2303214 CrossRef CAS PubMed.
- L. Qin, J. Gan, D. Niu, Y. Cao, X. Duan, X. Qin, H. Zhang, Z. Jiang, Y. Jiang, S. Dai, Y. Li and J. Shi, Interfacial-confined coordination to single-atom nanotherapeutics, Nat. Commun., 2022, 13, 91 CrossRef CAS PubMed.
- J. Zou, C. Chen, Y. Chen, Y. Zhu, Q. Cheng, L. Zou, Z. Zou and H. Yang, Facile steam-etching approach to increase the active site density of an ordered porous Fe-N-C catalyst to boost oxygen reduction reaction, ACS Catal., 2022, 12, 4517–4525 CrossRef CAS.
- C. Hu, H. Jin, B. Liu, L. Liang, Z. Wang, D. Chen, D. He and S. Mu, Propagating Fe-N4 active sites with Vitamin C to efficiently drive oxygen electrocatalysis, Nano Energy, 2021, 82, 105714 CrossRef CAS.
- K. P. Singh, E. J. Bae and J. S. Yu, Fe-P: A new class of electroactive catalyst for oxygen reduction reaction, J. Am. Chem. Soc., 2015, 137, 3165–3168 CrossRef CAS PubMed.
- F. Razmjooei, K. P. Singh, E. J. Bae and J. S. Yu, A new class of electroactive Fe- and P-functionalized graphene for oxygen reduction, J. Mater. Chem. A, 2015, 3, 11031–11039 RSC.
- L. Feng, Y. Liu and J. Zhao, Fe- and Co-P4-embedded graphenes as electrocatalysts for the oxygen reduction reaction: theoretical insights, Phys. Chem. Chem. Phys., 2017, 17, 30687–30694 RSC.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, Boosting oxygen electrocatalytic activity of Fe-N-C catalysts by phosphorus incorporation, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed.
- W. Xue, Q. Zhou, X. Cui, J. Zhang, S. Zuo, F. Mo, J. Jiang, X. Zhu and Z. Lin, Atomically dispersed FeN2P2 motif with high activity and stability for oxygen reduction reaction over the entire pH range, Angew. Chem., Int. Ed., 2023, 62, e202307504 CrossRef CAS PubMed.
- S. Lee, J. Choi, M. Kim, J. Park, M. Park and J. Cho, Material design and surface chemistry for advanced rechargeable zinc-air batteries, Chem. Sci., 2022, 13, 6159–6180 RSC.
- Z. Liu, D. Ye, X. Zhu, S. Wang, Y. Zou, L. Lan, R. Chen, Y. Yang and Q. Liao, ZIF-67-derived Co nanoparticles embedded in N-doped porous carbon composite interconnected by MWCNTs as highly efficient ORR electrocatalysts for a flexible direct formate fuel cell, Chem. Eng. J., 2022, 432, 134192 CrossRef CAS.
- P. Sha, L. Huang, J. Zhao, Z. Wu, Q. Wang, L. Li, D. Bu and S. Huang, Carbon nitrides with grafted dual-functional ligands as electron acceptors and active sites for ultra-stable photocatalytic H2O2 production, ACS Catal., 2023, 13, 10474–10486 CrossRef CAS.
- D. Ye, Q. Lan, Q. Liao, Y. Yang, R. Chen, S. Wang, Z. Liu and X. Zhu, Role of defects and oxygen-functional groups in carbon paper cathode for high-performance direct liquid fuel cells, Carbon, 2022, 192, 170–178 CrossRef CAS.
- W. Zhang, X. Guo, C. Li, J. Xue, W. Xu, Z. Niu, H. Gu, C. Redshaw and J. Lang, Ultralong nitrogen/sulfur co-doped carbon nano-hollow-sphere chains with encapsulated cobalt nanoparticles for highly efficient oxygen electrocatalysis, Carbon Energy, 2023, 5, e317 CrossRef CAS.
- Y. Zheng, P. Wang, W.-H. Huang, C.-L. Chen, Y. Jia, S. Dai, T. Li, Y. Zhao, Y. Qiu, G. I. N. Waterhouse and G. Chen, Toward More Efficient Carbon-Based Electrocatalysts for Hydrogen Peroxide Synthesis: Roles of Cobalt and Carbon Defects in Two-Electron ORR Catalysis, Nano Lett., 2023, 23, 1100–1108 CrossRef CAS PubMed.
- F. Kong, X. Cui, Y. Huang, H. Yao, Y. Chen, H. Tian, G. Meng, C. Chen, Z. Chang and J. Shi, N-doped carbon electrocatalyst: marked ORR activity in acidic media without the contribution from metal sites?, Angew. Chem., Int. Ed., 2022, 61, e202116290 CrossRef CAS PubMed.
- F. Lin, F. Lv, Q. Zhang, H. Luo, K. Wang, J. Zhou, W. Zhang, W. Zhang, D. Wang, L. Gu and S. Guo, Local coordination regulation through tuning atomic-scale cavities of Pd metallene toward efficient oxygen reduction electrocatalysis, Adv. Mater., 2022, 34, 2202084 CrossRef CAS PubMed.
- F. Gui, Q. Jin, D. Xiao, X. Xu, Q. Tan, D. Yang, B. Li, P. Ming, C. Zhang, Z. Chen, S. Siahrostami and Q. Xiao, High-performance zinc-air batteries based on bifunctional hierarchically porous nitrogen-doped carbon, Small, 2022, 18, 2105928 CrossRef CAS PubMed.
- Y. Pan, X. Ma, M. Wang, X. Yang, S. Liu, H. Chen, Z. Zhuang, Y. Zhang, W. Cheong, C. Zhang, X. Cao, R. Shen, Q. Xu, W. Zhu, Y. Liu, X. Wang, X. Zhang, W. Yan, J. Li, H. Chen, C. Chen and Y. Li, Construction of N, P co-doped carbon frames anchored with Fe single atoms and Fe2P nanoparticles as a robust coupling catalyst for electrocatalytic oxygen reduction, Adv. Mater., 2022, 34, 2203621 CrossRef CAS PubMed.
- R. Ma, X. Cui, X. Xu, Y. Wang, G. Xiang, L. Gao, Z. Lin and Y. Yang, Collaborative integration of ultrafine Fe2P nanocrystals into Fe, N, P-codoped carbon nanoshells for highly-efficient oxygen reduction, Nano Energy, 2023, 108, 108179 CrossRef CAS.
- H. Jin, H. Zhou, P. Ji, C. Zhang, J. Luo, W. Zeng, C. Hu, D. He and S. Mu, ZIF-8/LiFePO4 derived Fe-N-P Co-doped carbon nanotube encapsulated Fe2P nanoparticles for efficient oxygen reduction and Zn-air batteries, Nano Res., 2020, 13, 818–823 CrossRef CAS.
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C.-D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Simultaneously crafting single-atomic Fe sites and graphitic layer-wrapped Fe3C nanoparticles encapsulated within mesoporous carbon tubes for oxygen reduction, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS.
- Y. Zhang, Y. Jiang, G. Jiang, T. Or, R. Gao, H. Zhang, Z. Bai, N. Chen, Y. Deng and Z. Chen, Ordered mesoporous Fe2Nx electrocatalysts with regulated nitrogen vacancy for oxygen reduction reaction and Zn-air battery, Nano Energy, 2023, 115, 108672 CrossRef CAS.
- U.-I. Kramm, L. Ni and S. Wagner, 57Fe Mössbauer spectroscopy characterization of electrocatalysts, Adv. Mater., 2019, 31, 1805623 CrossRef PubMed.
- P. Chen, P. Zhang, X. Kang, L. Zheng, G. Mo, R. Wu, J. Tai and B. Han, Efficient electrocatalytic reduction of CO2 to ethane over nitrogen-doped Fe2O3, J. Am. Chem. Soc., 2022, 144, 14769–14777 CrossRef CAS PubMed.
- Z. Wang, X. Jin, R. Xu, Z. Yang, S. Ma, T. Yan, C. Zhu, J. Fang, Y. Liu, S.-J. Hwang, Z. Pan and H. Fan, Cooperation between dual metal atoms and nanoclusters enhances activity and stability for oxygen reduction and evolution, ACS Nano, 2023, 17, 8622–8633 CrossRef CAS PubMed.
- X. Liu, F. Zhao, L. Jiao, T. Fang, Z. Zhao, X. Xiao, D. Li, K. Yi, R. Wang and X. Jia, Atomically dispersed Fe/N4 and Ni/N4 Sites on separate-sides of porous carbon nanosheets with janus structure for selective oxygen electrocatalysis, Small, 2023, 19, 300289 Search PubMed.
- S. Zaman, Y. Su, C. Dong, R. Qi, L. Huang, Y. Qin, Y. Huang, F. Li, B. You, W. Guo, Q. Li, S. Ding and B. Xia, Scalable molten salt synthesis of platinum alloys planted in metal-nitrogen-graphene for efficient oxygen reduction, Angew. Chem., Int. Ed., 2022, 61, e202115835 CrossRef CAS PubMed.
- J. Zhang, W. Xu, Y. Liu, S. Hung, W. Liu, Z.-H. Lam, H. Tao, H. Yang, W. Cai, H. Xiao, H. Chen and B. Liu, In situ precise tuning of bimetallic electronic effect for boosting oxygen reduction catalysis, Nano Lett., 2021, 21, 7753–7760 CrossRef CAS PubMed.
- X. Lin, P. Peng, J. Guo, L. Xie, Y. Liu and Z. Xiang, A new steric tetra-imidazole for facile synthesis of high loading atomically dispersed FeN4 electrocatalysts, Nano Energy, 2021, 80, 105533 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01329h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |