
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClinically used drug arsenic trioxide targets XIAP and overcomes apoptosis resistance in an organoid-based preclinical cancer model†
Liwa
Shi‡
ab,
Jing
Lu‡
abc,
Xin
Xia‡
ab,
Xue
Liu
ab,
Hongyan
Li
d,
Xinghua
Li
e,
Jun
Zhu
 ab,
Xiaofeng
Li
*c,
Hongzhe
Sun
*d and
Xinming
Yang
*ab
ab,
Xiaofeng
Li
*c,
Hongzhe
Sun
*d and
Xinming
Yang
*ab
aGuangdong Provincial Engineering Research Center of Molecular Imaging, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, China. E-mail: yangxm57@mail.sysu.edu.cn
bGuangdong-Hong Kong-Macao University Joint Laboratory of Interventional Medicine, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai 519000, China
cDepartment of Gastroenterology, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, China
dDepartment of Chemistry, CAS-HKU Joint Laboratory of Metallomics on Health and Environment, The University of Hong Kong, Hong Kong SAR, China. E-mail: hsun@hku.hk
eDepartment of Infectious Diseases, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, China. E-mail: lixiaofeng@mail.sysu.edu.cn
First published on 3rd May 2024
Abstract
Drug resistance in tumor cells remains a persistent clinical challenge in the pursuit of effective anticancer therapy. XIAP, a member of the inhibitor of apoptosis protein (IAP) family, suppresses apoptosis via its Baculovirus IAP Repeat (BIR) domains and is responsible for drug resistance in various human cancers. Therefore, XIAP has attracted significant attention as a potential therapeutic target. However, no XIAP inhibitor is available for clinical use to date. In this study, we surprisingly observed that arsenic trioxide (ATO) induced a rapid depletion of XIAP in different cancer cells. Mechanistic studies revealed that arsenic attacked the cysteine residues of BIR domains and directly bound to XIAP, resulting in the release of zinc ions from this protein. Arsenic-XIAP binding suppressed the normal anti-apoptosis functions of BIR domains, and led to the ubiquitination-dependent degradation of XIAP. Importantly, we further demonstrate that arsenic sensitized a variety of apoptosis-resistant cancer cells, including patient-derived colon cancer organoids, to the chemotherapy drug using cisplatin as a showcase. These findings suggest that targeting XIAP with ATO offers an attractive strategy for combating apoptosis-resistant cancers in clinical practice.
Introduction
The drug resistance of cancer cells to apoptosis is a major problem for efficient anticancer therapy. The versatile inhibitor of apoptosis proteins (IAPs) play a critical role in the regulation of a cell's decision to live or die in response to anticancer agents in cancer cells.1 Eight IAP family members have been identified in humans, including XIAP, cIAP1, cIAP2, ML-IAP, NAIP, ILP2, Survivin and Bruce that are characterized by the presence of the conserved Baculoviral IAP Repeat (BIR) domain with a zinc finger (ZF) structure.2,3 The best-known function of IAPs, just as their names imply, is anti-apoptosis via caspase inhibition.4 Furthermore, IAPs are also recognized as multifunctional proteins that are involved in various physiological processes, such as receptor signaling, metal ion homeostasis and ubiquitination to guide protein degradation.5–9 Dysregulated expression of IAPs is believed to have important consequences with respect to resistance of tumor cells to apoptosis.10 Therefore, IAP family members, especially XIAP, which is the most intensively studied member of this family, have been heavily pursued as potential therapeutic targets.11–13XIAP, the most potent caspase inhibitor in the IAP family, is a promising target for cancer drug development.12,14,15 Up-regulation of XIAP has been observed in a variety of human cancers16 and is responsible for resistance to chemotherapy-induced cell death.17 Indeed, siRNA or antisense oligonucleotides that down regulate XIAP could enhance sensitivity of various malignant cell lines to chemotherapy.18,19 Currently, there are two main strategies to develop XIAP inhibitors: (a) antisense oligonucleotides to down-regulate the XIAP level, which is advantageous as they target the whole XIAP protein; (b) small molecular antagonist mimicking Smac peptide to block the caspase inhibition ability of BIR domains, which offers a more rapid suppression potential for their targets.3 Although some of these inhibitors have been evaluated in clinical trials or in preclinical development, they are still not clinically available, either due to the limited efficacy or safety issues.3,8 Further effort is still urgently needed to develop XIAP antagonists.
Arsenic and its derivatives have been used in traditional Chinese medicine for centuries.20 In modern times, arsenic-based drugs have attracted great interest due to their promising broad efficacy in inducing apoptosis across various types of cancers.21–23 To date, arsenic trioxide (As2O3, ATO) combined with all-trans retinoic acid, has been clinically used to treat acute promyelocytic leukemia (APL) by promoting the degradation of the PML-RARα fusion protein and demonstrated a remarkable 5 year survival rate of ca. 90%.20,24–26 Moreover, arsenic has shown promising effects for chronic myelocytic leukemia,27 and is under clinical study for a variety of hematologic malignancies and solid tumors.22,28,29 The broad anticancer activity of arsenic has attracted significant attention, and great efforts have been made to explore the underlying mechanisms for arsenic in different contexts.28–31 However, the precise mechanisms underlying the diverse mode of action of arsenic to induce apoptosis are still not fully understood.28,29,32
Previous drug screening and assessment have faced challenges due to the high heterogeneity of cancer cells. Conventional models, such as cancer cell line-based models, have notable limitations in accurately representing the clinical response observed in patients. In contrast, cancer patient-derived organoids preserve the phenotypic and genetic diversity of the original tumor from patients. Therefore, patient-derived organoids offer a more precise testing platform to establish correlations between organoid responses and patient responses to cancer treatments. They have emerged as a powerful tool for drug testing, providing enhanced accuracy and reliability.33
In this study, we surprisingly observed an arsenic-induced rapid depletion of XIAP in different cancer cells. We found that arsenic targeted the cysteine residues of BIR domains and replaced zinc ions from XIAP, which induced a significant conformational change of the protein. Arsenic showed a dual-action inhibitory effect on XIAP by suppressing the normal anti-apoptosis functions of the BIR domains, and inducing the ubiquitination-mediated degradation of the whole protein. Furthermore, using cisplatin as a showcase, we showed that arsenic sensitized a variety of apoptosis-resistant cancer cells, as well as patient-derived colon cancer organoids, to the chemotherapy drug. These findings suggest that targeting XIAP by ATO offers an attractive strategy to combat apoptosis-resistant cancers.
Results
Arsenic induces XIAP depletion in cancer cells
We collected tumor tissue samples from patients diagnosed with colon cancer and evaluated XIAP levels using RT-qPCR, and found that approximately 60% of the patients' tumors (6 out of 10 patients) exhibited higher expression levels of XIAP (Fig. S1†). We also studied the expression patterns of XIAP in different tumors by referring to the recently published OncoDB database,34 which revealed that there are high expression levels of XIAP in a variety of human cancers (Fig. S2†). The highly effective apoptosis-inducing activity of ATO for different cancers has been well documented; however, this broad anticancer mode of action is still not fully understood.28–31 In view of the master role of XIAP in the resistance to apoptosis,16,17 we wondered whether ATO potentially affects XIAP functionality. To address this issue, we first incubated AGS gastric adenocarcinoma cells with a gradient concentration of ATO, and the protein level of endogenous XIAP was examined by western blot analysis. We found that arsenic induced the depletion of XIAP in AGS cells in a dose-dependent manner (Fig. 1A and S3†). Notably, a significantly reduced level of XIAP was observed even with the treatment of 1–2 μM arsenic (Fig. 1A and S3†) concentrations that could be safely achieved in patients according to a previous clinical study.35 We further incubated 2 μM arsenic with AGS and examined the protein level of endogenous XIAP at the indicated time point, and found a time-dependent reduction of XIAP (Fig. 1B and S3†). The treatment with arsenic induced an obvious decrease in the level of XIAP at 4 h and almost complete depletion of endogenous XIAP in AGS after 8 h (Fig. 1B and S3†).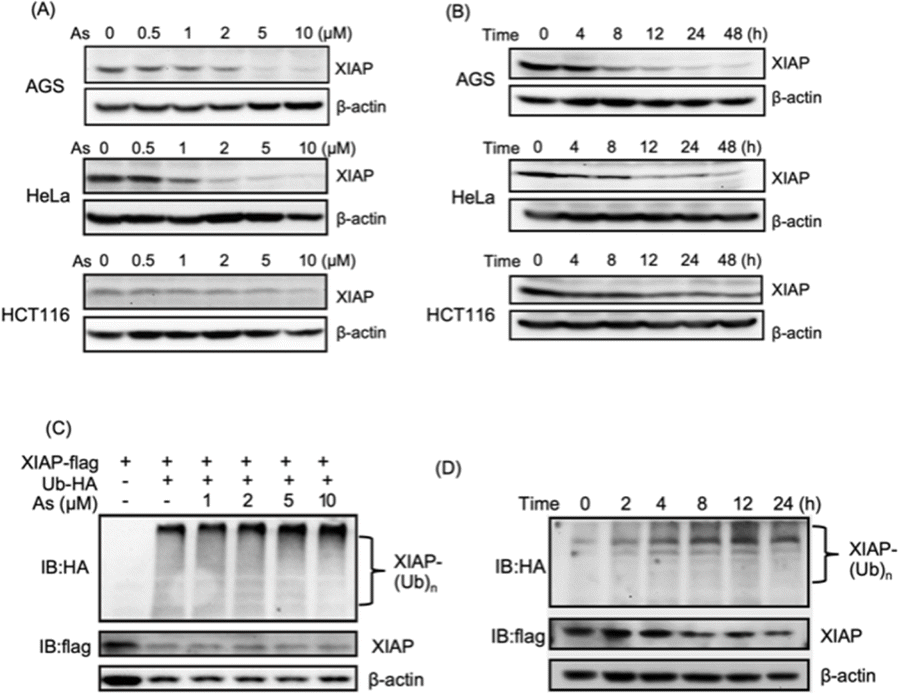 | ||
| Fig. 1 Arsenic trioxide induces depletion of XIAP in different cancer cells. (A) Arsenic induces depletion of XIAP protein in a dose-dependent manner. Gradient arsenic trioxide was added into the cell cultured medium for 48 h. (B) Arsenic induces depletion of XIAP protein in a time-dependent manner. In the case of HCT116, it is noteworthy that approximately 30% of XIAP expression was still observable after 12 h of arsenic treatment. This observation could potentially be attributed to the inherent resistance of the HCT116 cell line to arsenic. XIAP levels were revealed by immunoblotting with anti-XIAP antibody. (C) Arsenic induces ubiquitination of XIAP in a dose-dependent manner. (D) Arsenic induces ubiquitination of XIAP in a time-dependent manner. | ||
To exclude cell type specificity, we also performed similar experiments using various cancer cell lines, including the cervical carcinoma cell line HeLa and colorectal carcinoma cell line HCT116. Western blot analysis showed that arsenic treatment led to both dose- and time-dependent reduction in the level of XIAP in all tested cancer cell lines (Fig. 1A, B and S3†).
We further investigated whether the arsenic-induced XIAP depletion depends on ubiquitination modification. HEK293t cells expressing HA-labeled ubiquitin and Flag-labeled XIAP were treated with arsenic for 16 h, and the ubiquitination levels of XIAP were examined by western blot analysis. As shown in Fig. 1C, arsenic treatment resulted in XIAP ubiquitination in a dose-dependent manner (Fig. 1C and S3†). In the time-dependent experiment, 2 μM arsenic resulted in the apparent ubiquitination of XIAP as early as 2 h, and the ubiquitin conjugation to XIAP was further enhanced with the incubation time, correlated with the reduced level of XIAP in cells (Fig. 1D and S3†), implying that arsenic sufficiently promotes XIAP in the ubiquitin-proteasome system.
Arsenic binds to BIR domains of XIAP directly
We next investigated the underlying mechanism of arsenic-induced XIAP depletion. We first examined whether arsenic binds to XIAP in cells by cellular thermal shift assay (CETSA).36 The treatment of cells with arsenic resulted in the melting points of XIAP shifting from ca. 47 °C to ca. 45 °C with ΔTm = 2 °C, indicating that the binding of arsenic destabilized XIAP (Fig. 2A), and XIAP is a potential target of arsenic in cells. Similar to full-length XIAP, BIR1–BIR2–BIR3 protein was less thermally stable upon arsenic treatment with a remarkable ΔTm of ca. 20 °C (Fig. 2B). The solo BIR domains of XIAP i.e., BIR1, BIR2 and BIR3 also showed less thermal stability upon arsenic binding (Fig. S5†), confirming that arsenic interacts with the BIR domains of XIAP in cells. To further confirm that the low thermal stability of XIAP protein is a result of direct arsenic binding, we prepared recombinant apo-BIR domain proteins of XIAP and blocked the free Cys residues with the N-ethylmaleimide (NEM) agent. We found that arsenic could not induce a melting point change in BIR proteins without free Cys (Fig. S5†).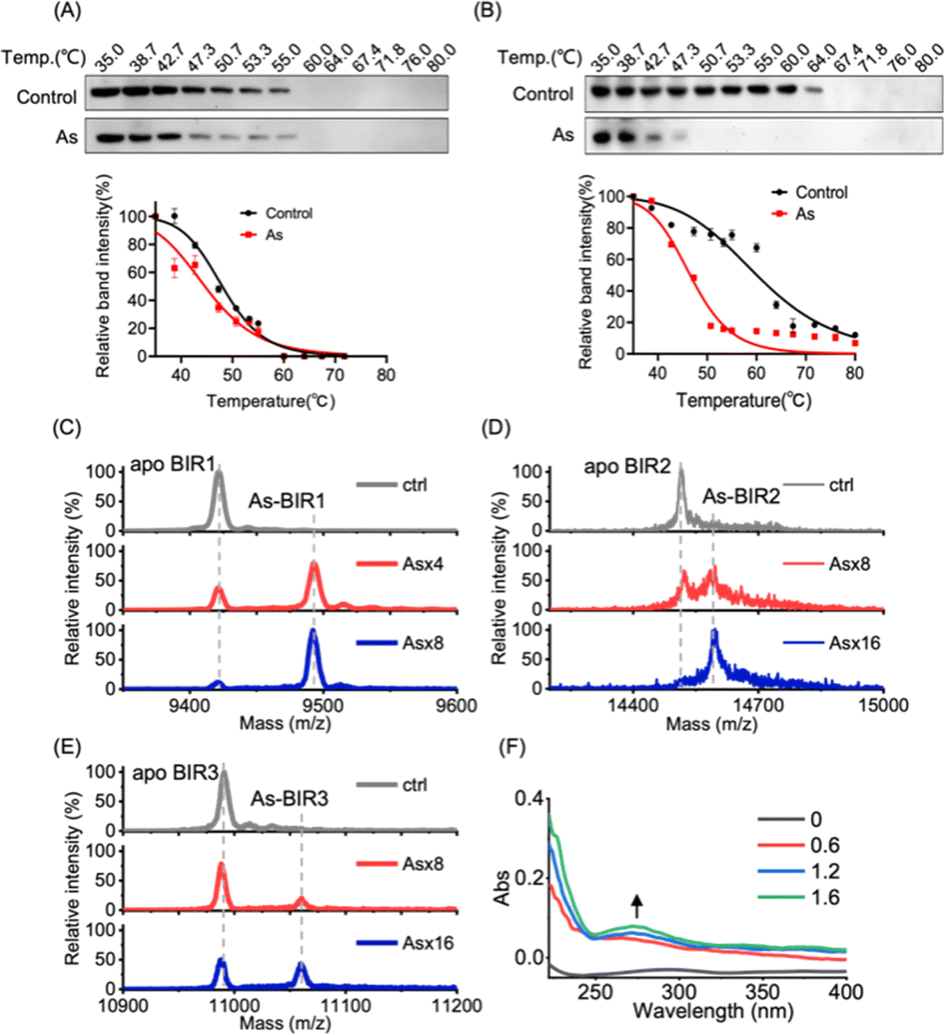 | ||
Fig. 2 Arsenic binds to BIR domains of XIAP directly. (A) Representative CETSA blot for arsenic–XIAP binding in cell. HEK293t cells expressing XIAP–Flag were pretreated with arsenic for 4 h following indicated heat shocks. Soluble Flag-fused XIAP full-length protein in the sample supernatant was revealed by immunoblotting with anti-Flag antibody. The melting temperature shift (ΔTm) between treated and control samples was measured based on CETSA melt curves. (B) Representative CETSA blot for arsenic–BIR1–BIR2–BIR3 binding in cells. HEK293t cells expressing BIR1–BIR2–BIR3–Flag were pretreated with arsenic for 4 h following indicated heat shocks. Soluble Flag-fused XIAP truncated protein (BIR1–BIR2–BIR3) in the sample supernatant was revealed by immunoblotting with anti-Flag antibody. (C–E) MALDI-TOF mass spectrometry for monitoring the interaction between arsenic and BIR domains of XIAP. Apo-form BIR1 (20–99), BIR2 (120–240) and BIR3 (241–356) were pre-incubated with or without the gradient arsenic trioxide. The molar ratios of binding between As3+ and BIR domains were determined to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. (F) UV absorption spectra of apo-BIR3 upon addition of gradient arsenic as indicated. :1. (F) UV absorption spectra of apo-BIR3 upon addition of gradient arsenic as indicated. | ||
To further study the potential arsenic–BIR interaction in vitro, we studied the arsenic binding by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF-MS). Incubation of gradient amounts of ATO with apo-BIR domain proteins resulted in the decreased intensities of the peaks at m/z ratios of 9421.4, 14515.0 and 10990.7 assignable to apo-BIR1, apo-BIR2 and apo-BIR3, accompanied by the increase in the peaks at m/z ratios of 9492.7, 14596.0 and 11059.5 (calcd m/z of 9496.4, 14590.0 and 11065.7) respectively; the increase in the m/z ratios ca. 75 (atomic mass of arsenic) indicates that BIR1, BIR2 and BIR3 bound to one arsenic ion (As3+) respectively (Fig. 2C–E).
It is known that arsenic preferentially binds to the sulfhydryl group of cysteine residues29,32 and all three BIR domains of XIAP contain a 3Cys–His motif in the ZF structures. We therefore hypothesize that arsenic ions (As3+) may target the cysteine residues in the BIR domains of XIAP. In the optical absorption experiment, titration of arsenic solution into the apo-form BIR3 protein resulted in the appearance of a new absorption peak in the range of 250 to 340 nm (Fig. 2F), which results from the ligand-to-metal charge transfer (LMCT).20 In contrast, no absorption peak could be observed when arsenic was titrated into BIR3 protein with the blocking of Cys by NEM (Fig. S6†).
Arsenic induces zinc release from BIR domains and conformational changes in XIAP
Our UV absorption data suggest that arsenic attacked the sulfhydryl group of cysteine residues in BIR domains (Fig. 2F), which are critical for Zn2+ binding in zinc finger proteins,37 and we therefore hypothesized that arsenic may replace Zn2+ from BIR domains. The complexation of the chromophoric chelator 4-(2-pyridylazo)resorcinol (PAR) with Zn2+ results in the visible absorption spectra at ca. 480 nm, which can be used to assess Zn2+ content. We thus prepared Zn-bound BIR1, BIR2 and BIR3 in HEPES buffer and monitored the potential Zn2+ release from BIR domains upon arsenic treatment by PAR Zn-chelating assay. Surprisingly, no Zn2+ release could be observed upon addition of ATO into the Zn-BIR protein solution. Considering that there is a high level of GSH in cells38 and GSH has been reported to enhance the anti-cancer activity of arsenic,39 we performed the arsenic competition experiment in PAR Zn-chelating assay in the presence of 5 mM GSH to mimic physiological conditions. Interestingly, we observed the appearance of new peaks at ca. 480 nm, assignable to the Zn(PAR)2 complex in all BIR proteins, suggesting that arsenic induces Zn2+ release from BIR domains in the presence of GSH (Fig. 3A). Thus, ATO could target BIR domains and induce Zn2+ release from XIAP under near physiological conditions.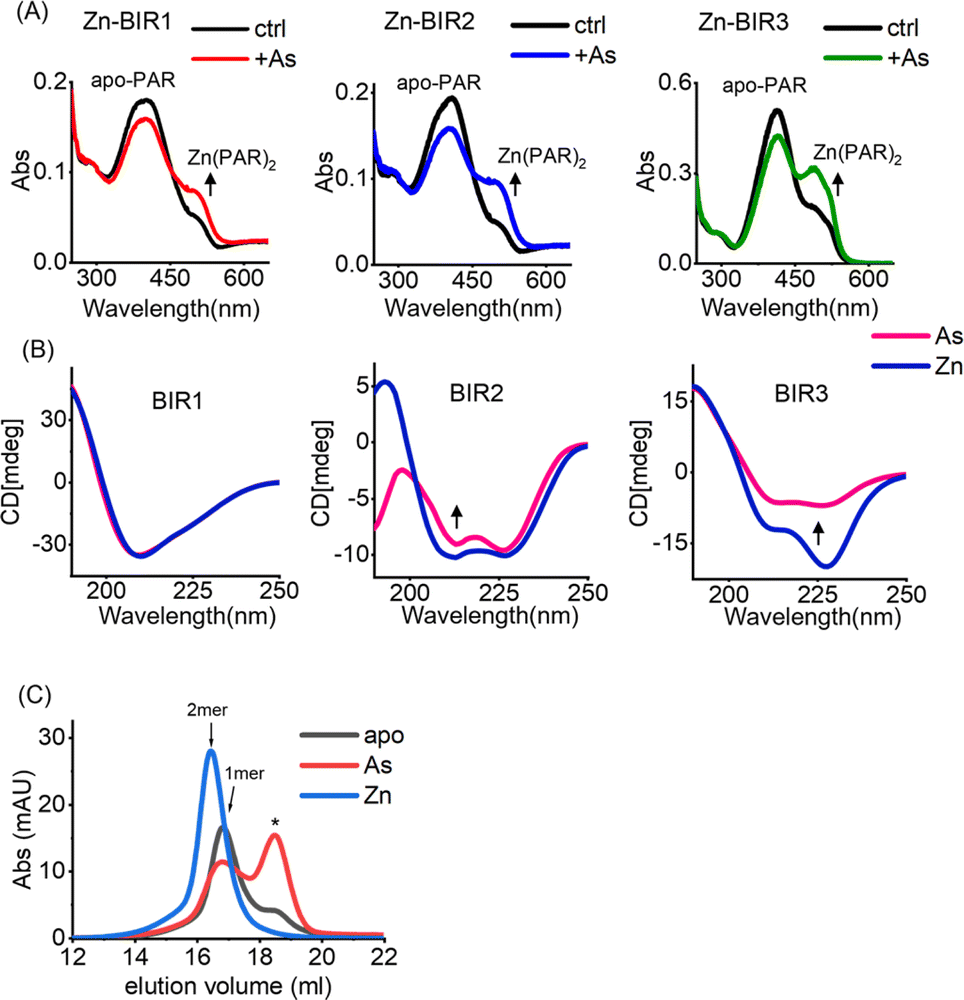 | ||
| Fig. 3 Arsenic induces Zn2+ release from BIR domains and a conformational change in XIAP. (A) UV-vis absorption spectra of 100 μM PAR and Zn-bound BIR1, BIR2 and BIR3 protein mixture with or without addition of arsenic trioxide in the presence of 5 mM GSH; (B) CD spectra of the Zn- and As-bound BIR1/BIR2/BIR3 domain of XIAP. (C) Analytical gel filtration analysis of the apo-, Zn- and As-bound BIR3 domain of XIAP. The indicated apo-BIR3 at a concentration of 20 μM was incubated with zinc or arsenic and applied to a Superdex 200 gel filtration column. *Impurity or fluctuation of the absorbance due to buffer. | ||
In view of the critical role of zinc ions in maintaining the overall structure of ZF,40 we next investigated the effect of arsenic-binding on the secondary structure of BIR domains by CD spectroscopy. Surprisingly, we found that arsenic binding had a limited effect on the structure of BIR1 (Fig. 3B and S7A†). However, we observed a significant change in the CD spectra of BIR2 and BIR3 upon arsenic binding as compared to those of Zn-bound forms (Fig. 3B). Furthermore, deconvolution analysis of CD spectra41 revealed that arsenic-binding led to a significant reduction of helix content from 26.9% to 18.3% in BIR2 and 54.4% to 19.5% in BIR3, respectively (Fig. S8†), indicating the structure changes induced by arsenic. Additionally, we observed that ATO mediated disruption of Zn-dependent dimerization of BIR3 by size-exclusion chromatography (Fig. 3C). These data collectively demonstrate that arsenic binding disrupts the ZF structure of XIAP, particularly in BIR2 and BIR3 domains.
Arsenic inhibits the anti-apoptosis functions of BIR domains
The integrity of the ZF structure is critical to its normal function.42,43 It is well known that XIAP targets caspase-3 and caspase-9 directly for inhibition.44,45 In view of that arsenic binding induces a conformational change in XIAP (Fig. 3), we therefore reasoned that arsenic might disrupt XIAP-caspase-3 or -caspase-9 interaction. To validate this, we accessed the interaction between XIAP and caspase-3/caspase-9 by Flag pull-down experiments, where both active caspase-3 and caspase-9 co-precipitated with recombinant XIAP-Flag protein (Fig. 4A and S9†). Strikingly, when XIAP-Flag was pre-loaded with arsenic, less XIAP-caspase-3/-9 binding occurred (Fig. 4A and S9†), confirming that arsenic disrupts XIAP binding to caspase-3/-9.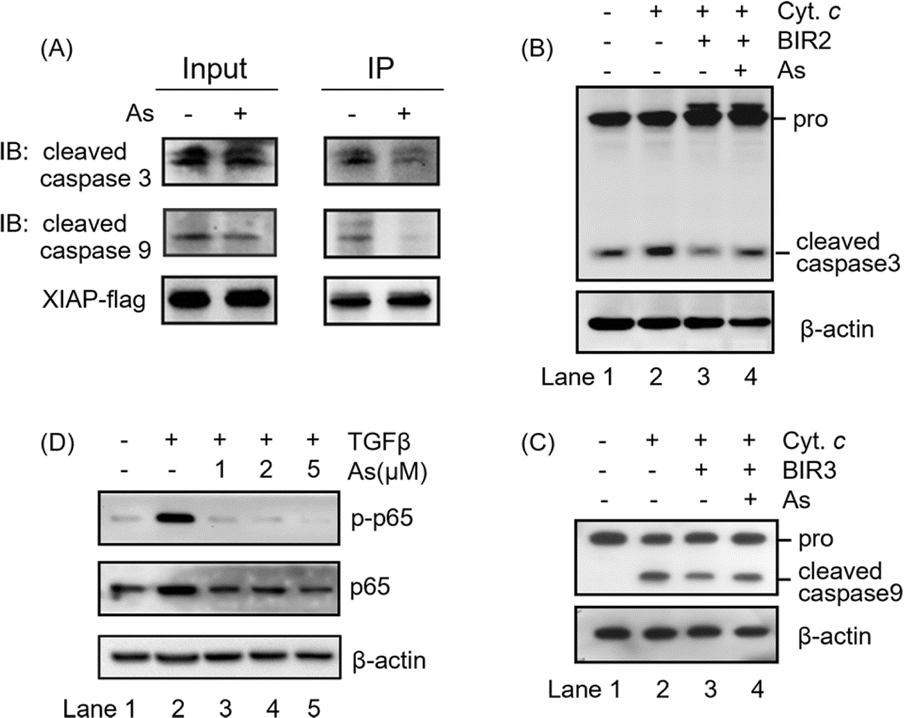 | ||
| Fig. 4 Arsenic inhibits the anti-apoptosis activity of all three BIR domains. (A) Flag pull down assay to assess XIAP-caspase-3/-9 interaction. (B) Western blot analysis of pro-caspase-3 and cleaved caspase-3 in cell lysates. β-Actin served as the loading control. Zn-BIR2 or As-BIR2 was added to the reactions. Cytochrome c and dATP were used to induce the processing of pro-caspase-3 in vitro. Reactions were stopped by using additional 5× SDS loading buffer. (C) Western blot analysis of pro-caspase-9 and cleaved caspase-9 in cell lysates. β-Actin served as the loading control. Zn-BIR3 or As-BIR3 was added to the reactions. Cytochrome c and dATP were used to induce the processing of pro-caspase-9 in vitro. Reactions were stopped by using additional 5× SDS loading buffer. (D) Representative immunoblots of AGS cells showing the effect of arsenic on the TGFβ-induced NF-κB activation. XIAP-mediated NF-κB signaling in AGS cells was initiated by addition of TGFβ into the cultured medium. Phosphorated and total p65 were revealed by immunoblotting with anti-p-p65 and anti-p65 antibodies, respectively. | ||
The BIR2 domain of XIAP directly blocks the activation of caspase-3,46 while the BIR3 domain inhibits caspase-9 activation;47,48 we next investigated whether arsenic relieves XIAP-mediated caspase inhibition by an in vitro caspase activation assay. Cytochrome c was used to initiate the cleavage of caspase-3 and caspase-9 in cell lysates, as indicated by the increased level of cleaved caspase 3 or 9 (Fig. 4B and C, lane 2; Fig. S9†). Supplementation of recombination Zn-bound BIR2 or BIR3 protein attenuated the processing of caspase-3 and caspase-9 respectively (Fig. 4B and C, lane 3; Fig. S9†). However, the presence of arsenic resulted in an increased cleaved caspase level compared to that of the control group, indicative of the restored activation of caspase-3 and -9 by arsenic (Fig. 4B and C, lane 4; Fig. S9†). These results collectively suggest that arsenic binding is able to suppress the function of BIR2 and BIR3 of XIAP, similar to the Smac peptide-like inhibitor.1
The BIR1 domain of XIAP is involved in survival signaling in various cancer cells by mediating NF-κB activation during TGFβ receptor signaling conduction.7 A previous study showed that the BIR1-mediated NF-κB activation depends on its interaction with TAB1.7 Although, unlike our expectation, arsenic did not disrupt BIR1-TAB1 interaction in vitro as shown by size-exclusion chromatography using recombination BIR1 and TAB1 proteins (Fig. S10†), we did observe that arsenic targeted BIR1 in cells (Fig. S5A†) and induced Zn release from BIR1 (Fig. 3A). We therefore further investigated whether arsenic blocks BIR1-dependent NF-κB activation in cells. We first stimulated the BIR1-dependent NF-κB activation in AGS cells with TGFβ treatment. We observed the increased p-p65 level upon addition of TGFβ into the culture medium by western blot analysis, indicating the activation of the NF-κB pathway (Fig. 4D, lane 2; Fig. S9†), whereas the addition of arsenic almost completely abolished the phosphorylation of p65 in a dose-dependent manner, suggesting that arsenic prevents the BIR1-mediated NF-κB signaling in cells (Fig. 4D, lane 3–5; Fig. S9†). Collectively, we demonstrate that arsenic blocks the anti-apoptosis activity of all three BIR domains.
Arsenic overcomes drug resistance in cancer cells and patient-derived colon cancer organoids
Given the critical role in regulation of cell apoptosis, XIAP has been an attractive target in anticancer therapy to overcome the resistance to chemotherapy-induced cell death.3 We show that arsenic acts as an effective XIAP antagonist by functionally blocking the BIR domains (Fig. 4), and degrading the whole XIAP protein (Fig. 1). We next evaluated the potential of ATO to overcome drug resistance in cancer cells using cisplatin (CDDP) as a showcase. CDDP is an important chemotherapy drug for the treatment of many advanced cancers via mediating DNA damage.49 However, the occurrence and rapid progression of cisplatin resistance has been a challenge for therapeutic success.49 We examined the dose–response of arsenic, cisplatin and their combination in AGS, HeLa and HCT116 cell lines. These cell lines responded to arsenic treatment (48 h) differently with IC50 values of ca. 6.3, 2.2 and 9.1 μM (Table 1, Fig. S4†), and showed a relatively high intrinsic resistance to cisplatin (48 h) with IC50 values of 55.7, 22.3 and 142.6 μM, respectively (Table 1). We examined the effect of arsenic on the anticancer activity of cisplatin in these cancer cells. As shown in Fig. 5, arsenic at 2 μM, a concentration that could be safely achieved in patients,35 significantly increased the sensitivity of AGS to cisplatin with an IC50 value of 2.5 μM (Table 1, Fig. 5A-left panel). Simultaneously, a reduction of the XIAP level by more than 90%, and increased cleaved caspase-3/caspase-9 could be observed in AGS cells (Fig. 5B-left panel; Fig. S11†). Similar phenomena could be observed in HeLa cells (Fig. 5A, 5B-middle panel). Arsenic significantly reduced the IC50 values of cisplatin from 22.3 to 16.8 μM (Table 1) in HeLa cells. In the case of HCT116 cells, consistent with the findings depicted in Fig. 1, relatively high levels of XIAP (30–50%) were still detectable even after arsenic treatment (Fig. 5B-right panel; Fig. S11†). This observation aligns with the inherent higher resistance of the HCT116 cell line to arsenic when compared to the other two cancer cell lines used in this study (Table 1, Fig. S4†). However, it is worth noting that despite this resistance, arsenic still significantly reduced the IC50 value of cisplatin from 142.6 μM to 88.8 μM for the HCT116 cell line (Table 1, Fig. 5A-right panel).| AGS | HeLa | HCT116 | |
|---|---|---|---|
| As | 6.3 ± 2.7 | 2.2 ± 0.4 | 9.1 ± 1.2 |
| CDDP | 55.7 ± 13.5 | 22.3 ± 3.8 | 142.6 ± 43.3 |
| CDDP (+As) | 2.5 ± 0.4 | 16.8 ± 5.8 | 88.8 ± 22.3 |
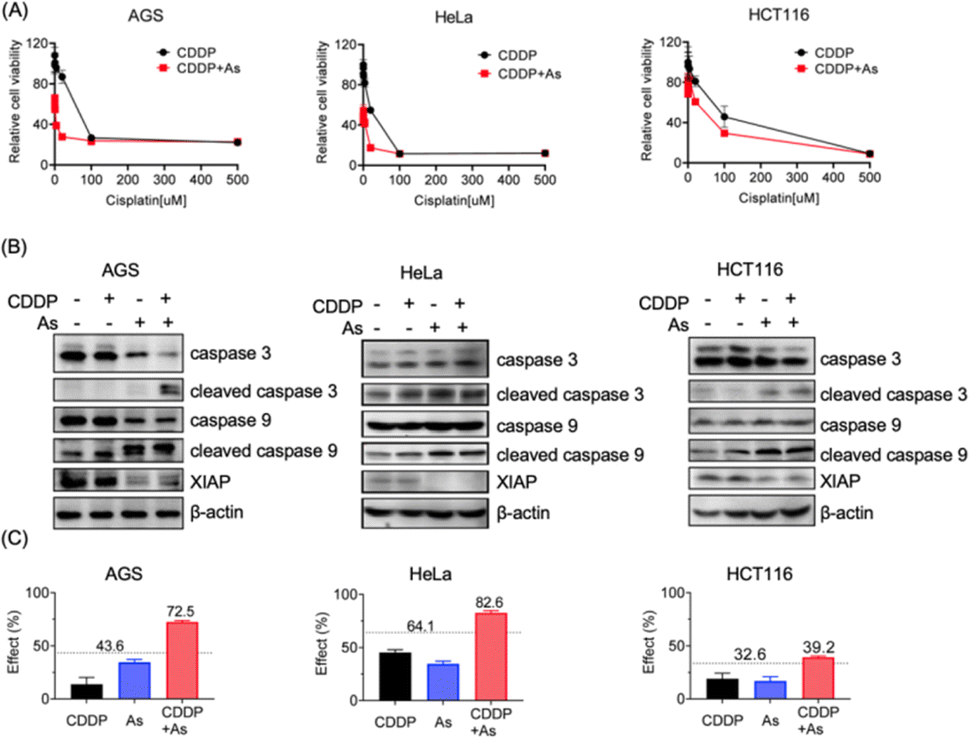 | ||
| Fig. 5 Arsenic sensitizes cancer cells with intrinsic drug resistance to cisplatin via XIAP targeting. (A) Cell viability of cancer cells upon the treatment of cisplatin with or without arsenic. Cancer cells AGS, HeLa and HCT116 were cultured for 48 h with various concentrations of cisplatin (500, 100, 20, 4, 0.8, 0.16, 0.032, and 0 μM) with or without arsenic. The percentage of viable cells relative to the control was examined by MTT assay (mean + SE, n = 3). (B) Representative immunoblots showing the protein level of endogenous caspase-3, caspase-9 and XIAP in AGS, HeLa and HCT116 upon the treatment with cisplatin and arsenic. (C) The Bliss independence model indicates a synergistic effect of the arsenic (2 μM) and cisplatin (20 μM) combination. The Bliss independence threshold was labelled and is shown as a dotted line. | ||
Furthermore, we examined the potential synergy between arsenic and cisplatin by using the Bliss independence model. The arsenic (2 μM) and cisplatin (20 μM) combination showed an inhibitory effect of 72.5% on AGS cells, 82.6% on HeLa cells and 39.2% on HCT116 cells, which all exceed the synergy threshold of 43.6%, 64.1% and 32.6% respectively, demonstrating a strong synergistic action between these two drugs (Fig. 5C). Collectively, these results indicate that arsenic is able to sensitize cancer cells with intrinsic resistance to cisplatin-induced apoptosis via XIAP depletion.
To further validate the potential of arsenic in overcoming acquired apoptosis resistance of cancer cells, we generated and characterized the cisplatin resistant Hep 3B hepatocellular carcinoma cell line (named H3B-R). Interestingly, we observed that the XIAP protein level is low in original Hep 3B (H3B) cells, whereas a significantly higher XIAP level could be detected in H3B-R cells in western blot analysis (Fig. 6A and S12†). Consistently, H3B-R cells showed almost 2-fold higher XIAP in mRNA level (Fig. 6B), in line with the previous report that XIAP overexpression contributes to drug resistance in cancer cells.17 H3B showed a relatively low resistance to cisplatin with an IC50 value of ca. 7.6 μM, compared to H3B-R with an IC50 value of ca. 298.5 μM (Table 2, Fig. 6E and S14†). Both H3B and H3B-R showed a high resistance to the arsenic treatment with IC50 values of ca. 28.9 μM and 14.9 μM respectively. No apparent cell death could be observed at the concentrations of 2 μM (Table 2, Fig. S13†), whereas arsenic treatment led to both dose- and time-dependent reduction in the level of XIAP in H3B-R (Fig. 6C, D and S12†). Remarkably, similar to cells with intrinsic resistance to cisplatin, arsenic also significantly reduced the IC50 values of cisplatin from almost 300 to 30.8 μM (Table 2) in H3B-R. The arsenic (2 μM) and cisplatin (64 μM) combination showed an inhibitory effect of 53.7%, which exceeds the synergy threshold of 6.83% (Fig. 6F). Collectively, these data demonstrate the high potential of arsenic in overcoming acquired apoptosis resistance of cancer cells.
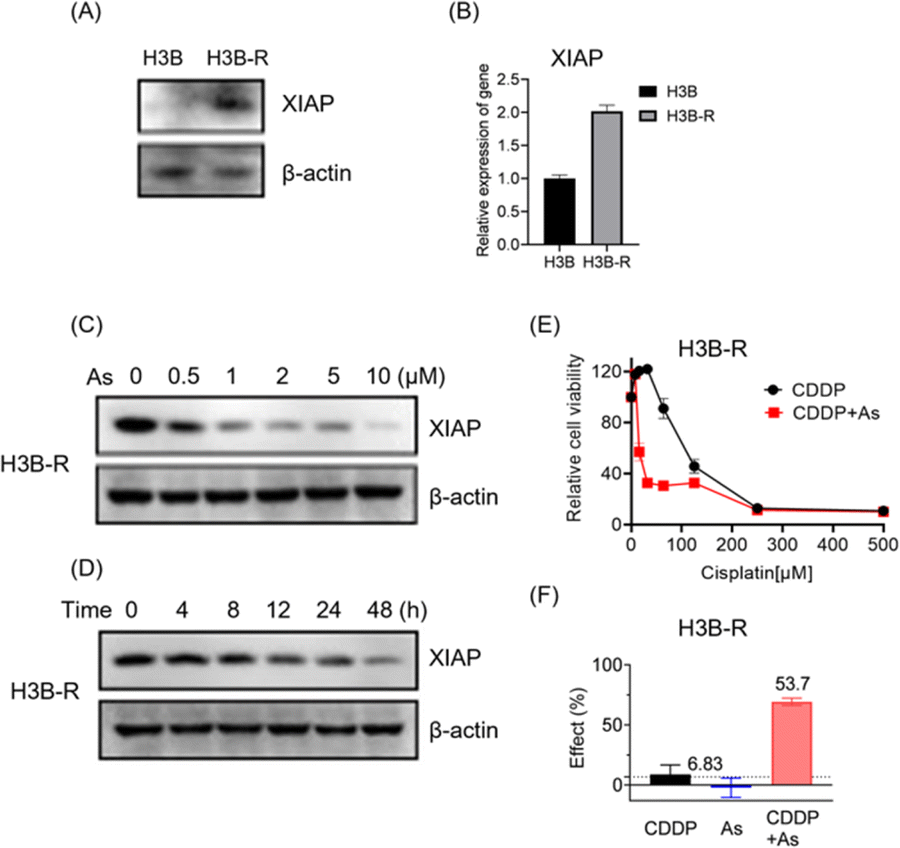 | ||
| Fig. 6 Arsenic sensitizes cancer cells with acquired drug resistance to cisplatin targeting via XIAP targeting. (A) Representative immunoblots showing the protein level of endogenous XIAP in H3B and H3B-R cells. (B) Quantitative real-time PCR analysis of XIAP expression in H3B and H3B-R cells. (C) Arsenic induces depletion of XIAP protein in a dose-dependent manner in H3B-R cells. (D) Arsenic induces depletion of XIAP protein in a time-dependent manner in H3B-R cells. XIAP levels were revealed by immunoblotting with anti-XIAP antibody. (E) Cell viability of H3B-R cells upon the treatment with cisplatin with or without arsenic (mean + SE, n = 3). (F) The Bliss independence model indicates a synergistic effect of the arsenic (2 μM) and cisplatin (64 μM) combination. The Bliss independence threshold was labelled and is shown as a dotted line. | ||
| H3B | H3B-R | |
|---|---|---|
| As | 28.9 ± 9.2 | 14.9 ± 5.1 |
| CDDP | 7.6 ± 2.4 | 298.5 ± 93.4 |
| CDDP (+As) | 7.2 ± 1.1 | 30.8 ± 9.2 |
Moreover, we validated the potential of arsenic in overcoming drug resistance of cancer cells in a scenario with more clinical relevance; we established patient-derived colon cancer primary cultures based on the clinical biopsies obtained from gastrointestinal endoscopy (Fig. 7A). We also observed the reduction of XIAP in colorectal cancer organoids upon arsenic treatment (Fig. S15†). We then treated colorectal cancer organoids with cisplatin (0, 6.25, and 12.5 μM) with or without 1 μM of arsenic and found that, similar to the results obtained from cancer cell lines, arsenic significantly increased the cytotoxicity of cisplatin to primary colon cancer organoids (Fig. 7B). In the case of donor 1, the colorectal cancer cells showed a high resistance to the cisplatin treatment, with no apparent cell death observed even at concentrations of 6.25 and 12.5 μM, compared with untreated controls. Nonetheless, the combination of arsenic and cisplatin resulted in a significant cell death of almost 50% (Fig. 7B) and showed a remarkable synergy with effect values of ca. 45.5% (vs. 19.4% threshold) (Fig. 7C and S16†). Similar phenomena were also observed in other donors. Additionally, in the organoid formation assay, the combination of arsenic and cisplatin led to significantly poorer organoid formation, compared to arsenic or cisplatin alone (Fig. 7D). Collectively, our results demonstrate that the combination of an arsenic drug (ATO) in cancer therapy represents an attractive option for overcoming apoptosis-resistance in cancer cells.
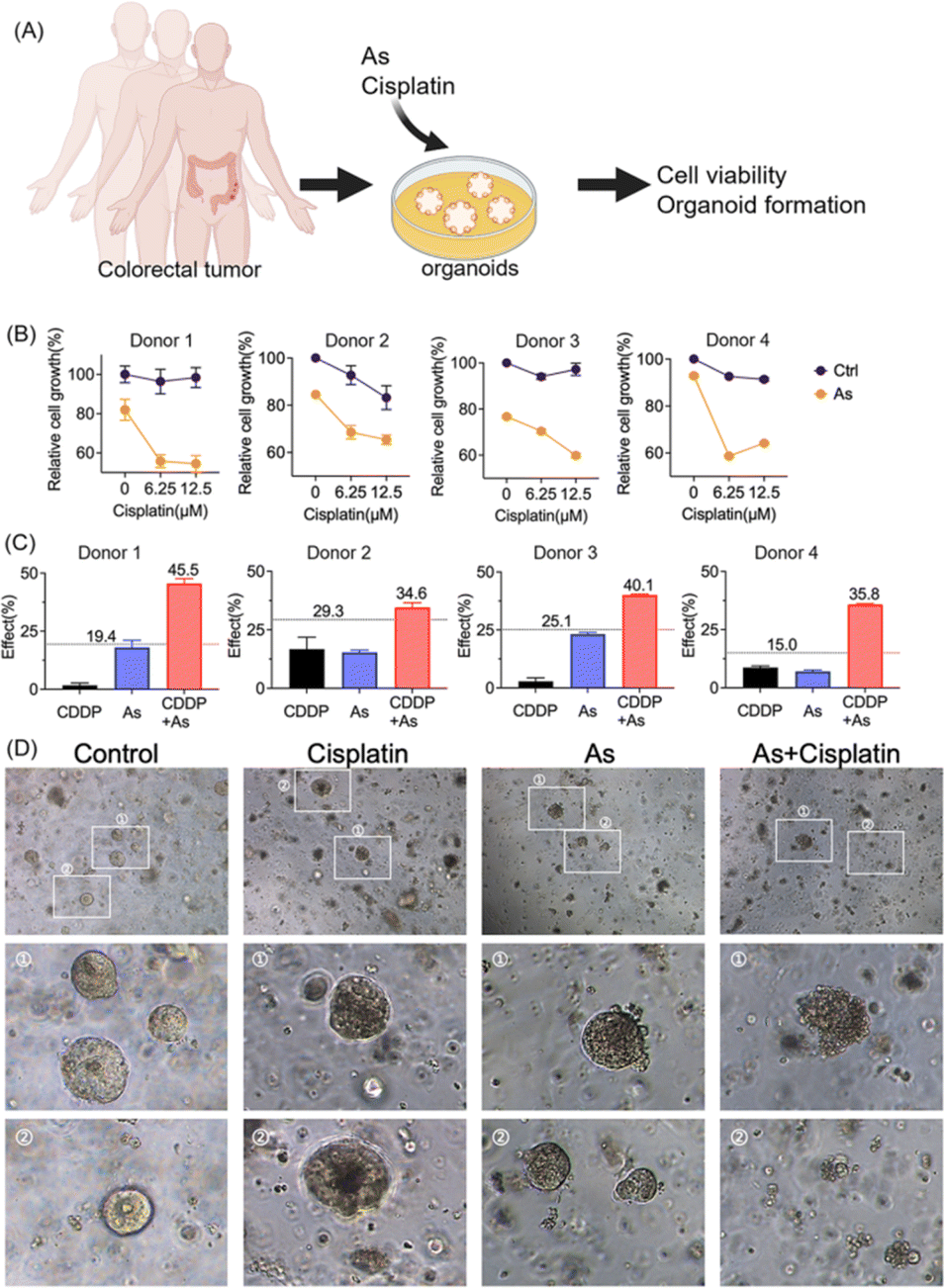 | ||
| Fig. 7 Arsenic overcomes apoptosis resistance in patient-derived colon cancer organoids. (A) Schematic overview for the assessment of drug sensitivity in a patient-derived colon cancer organoid model. (B) Cell viability of patient-derived colon cancer organoids from four donors. Patient-derived cancer organoids were cultured with various concentrations of cisplatin as indicated, with or without 1 μM arsenic. The cell viability relative to that of the control was examined by using a 3D cell viability assay kit (mean + SE, n = 3). (C) The Bliss independence model indicates a synergistic effect for the arsenic (1 μM) and cisplatin (12.5 μM) combination. The Bliss independence threshold was labeled and is shown as a dotted line. (D) Representative images for the formation of colon cancer organoids with or without the treatment with arsenic, cisplatin and their combination. | ||
Discussion and conclusion
Acquired apoptosis resistance due to tumor cell adaptation to persistent therapeutic stress is an unmet challenge in clinical settings. Overexpression of XIAP has been suggested to be accountable for the resistance to apoptosis in a variety of cancer cells.10 We report here that arsenic targets the BIR domains of XIAP, and enhances the sensitivity of cancer cells, including patient-derived colon cancer organoids, to the chemotherapy drug (Fig. 8). This study provides solid evidence for the arsenic-based drug ATO as an attractive agent for combination therapy against apoptosis-resistance in cancer.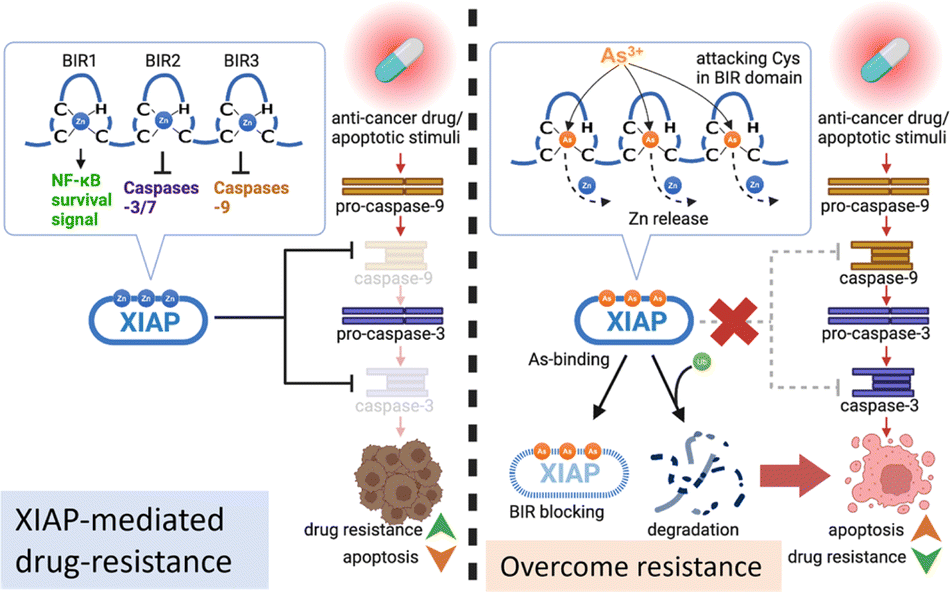 | ||
| Fig. 8 Proposed mechanism by which the arsenic-based drug overcomes apoptosis resistance in cancer cells via targeting XIAP. Arsenic (As3+) replaces Zn2+ from the BIR domains and disrupts XIAP anti-apoptosis functions via a dual-action mechanism, (a) blocking the anti-apoptosis functions of solo BIR domains and (b) inducing the ubiquitination-mediated degradation of XIAP whole protein. | ||
Great efforts have been made to develop XIAP antagonists, either to reduce the whole protein level or to block BIR action, for therapeutic purposes. Many of these inhibitors show great efficiency in preclinical development.3 Whereas, unfortunately, up to now, no XIAP antagonist has been approved for clinical use. Interestingly, in the present study, we found that arsenic targets XIAP via binding to the Cys residues in the ZF structure of BIR domains. Although XIAP contains a C-terminal RING domain with a ZF structure, based on a previous study,50 XIAP lacking RING was even more effective at limiting apoptosis than wild-type XIAP. Thus, we only focused on arsenic-BIR interaction in this study, considering that the anti-apoptosis function of XIAP heavily depends on its BIR domains. We further showed that the arsenic-BIR binding inhibits XIAP through two distinct actions by the direct and rapid blocking of BIR domains functionally, similar to the Smac peptide (Fig. 4), and by depleting the XIAP whole protein via promoting the ubiquitination-mediated degradation (Fig. 1). This dual-action mechanism of arsenic against XIAP may be an advantage of arsenic to induce cytotoxicity in cancer cells.
Arsenic trioxide (ATO), which transforms into As(OH)3 in aqueous solution,39 is highly toxic. However, despite its high cytotoxicity, it has shown great potential in treating cancer. Previous reports have demonstrated that arsenic is more toxic to cancer cells than normal cells.51,52 In line with these reports, we also observed greater cytotoxicity of arsenic in cancer cells compared to normal cells (Fig. S17†). The underlying mechanism for the promising anti-cancer activity of arsenic is still not fully understood. Nevertheless, the trivalent arsenic ion (As3+) from ATO was reported to alter different pathways or bind to different targets depending on the type of malignancy studied.23,29 In breast cancer, arsenic has been reported to target the active site of Pin1.31 Moreover, the glycolysis pathway was found to be a key target of arsenic in HK2-overexpressing cancer cells.28 Interestingly, arsenic has been found to restore the response of drug-resistance leukemia cells to the anti-cancer drug dexamethasone, at least in part (as the authors claimed), through reducing Akt signaling to downregulate the Akt-dependent XIAP level.53 However, in our study on solid tumor cells, we didn't observe a correlation between Akt signaling suppression and XIAP depletion (Fig. S18†). In contrast, we found that arsenic at subtoxic levels resulted in the activation of the Akt pathway (Fig. S18†). Alternatively, we found that arsenic targeted XIAP in cells directly (Fig. 2) and showed a dual inhibitory effect on XIAP in solid tumor cells. Consistent with our finding, a previous study reported that a small molecule that chelates zinc ions from BIR domains induced a rapid depletion of XIAP.54 Nevertheless, the potential of arsenic to affect different pathways via various targets may reflect context-dependent diversity in key pathways/proteins relied on by cancer cells, and suggests the potential advantage of arsenic-based drugs with such a broad target range.
Due to the potential of XIAP antagonists to overcome drug-resistance, these small molecules have been heavily explored by combination with standard chemotherapy.10 Smac peptides combined with cisplatin or the tumor necrosis factor-alpha-related apoptosis inducing ligand (TRAIL) have been found to suppress lung cancer cells and glioma xenografts, respectively.55,56 In our study, we found that arsenic, as an efficient XIAP inhibitor, greatly enhanced the sensitivity of a variety of cancer cells to anticancer drugs using cisplatin as a showcase (Fig. 5 and 6). Most importantly, the combination of arsenic–cisplatin showed promising anti-cancer effect in a drug test model based on the patient-derived colon cancer organoid (Fig. 7). Patient-derived organoids have emerged as a powerful tool for drug discovery, due to their great advantage of providing a more accurate test platform which correlates organoid responses and patient responses to cancer treatments.33 In this study, the promising action of arsenic to overcome apoptosis-resistance in cancers by targeting XIAP in the organoid model demonstrates the great potential of the ATO-based therapeutic strategy in clinical practice.
Materials and methods
Chemical reagents
Cisplatin was purchased from Beyotime. Kanamycin sulfate and ampicillin were obtained from MDbio, Luria–Bertani (LB) broth powder and LB agar were purchased from Invitrogen, and all other chemicals were sourced from Thermo Scientific, unless otherwise stated. Arsenic stocks were prepared in accordance with safety standards in an aqueous buffer containing 20 mM Tris at pH 7.4.Cancer cell line culture and maintenance
AGS (Purchased from Pricella Biotech.) was cultured in the RPMI 1640 medium (ThermoFisher) and HEK293t/HCT116/HeLa cells were cultured in the DMEM high-sugar medium (Corning) plus 10% of fetal bovine serum (WISENT) and penicillin-streptomycin (ThermoFisher). HeLa and HCT116 cells were obtained from the National Collection of Authenticated Cell Cultures, and HEK293t and Hep 3B cells were gifts from Prof. Fei XIAO (Sun Yat-sen University). All cells were maintained in a humidified cell culture incubator with a 5% CO2 gas level at 37 °C.Organoid model for drug sensitivity assay
Colon cancer organoids were established according to a previous report (37). Donors 1–4 represent four different patients. Cultured organoids were spliced into single cells and suspended in Matrigel (Corning). For cisplatin sensitivity assay, organoids were seeded with the appropriate culture medium for 2 days before the drug treatment, to allow the organoids to establish themselves, ensuring a uniform growth environment. Cisplatin was added into the culture medium to reach final concentrations of 6.25 or 12.5 μM, which are achievable in patients following cisplatin dosing,57 with or without arsenic (1 μM). According to a previous report, the serum concentration of arsenic could reach 1–2 μM when administering ATO at a dosage of 0.15 mg kg−1 per day through a 3 hour intravenous infusion during their study.35 For donors 3 and 4, 2 μM arsenic alone caused a significant reduction in cell viability of almost 50%. Therefore, with the aim to observe the potential synergy between arsenic and CDDP, we chose 1 μM arsenic for all four donors. The formation of organoids was recorded with microscopy (Leica). The organoid viability was determined by using a CellTiter-Lumi™ luminescent 3D II cell viability assay kit (Beyotime). The Bliss independence approach was used to examine the potential synergy between arsenic and cisplatin. The drug effects from single and combination treatments were identified in the form of probability (0 ≤ E ≤ 1), and the expected combined effect (Threshold value) was defined as ECDDP+As = ECDDP + EAs(1 − ECDDP), where EAs and ECDDP represent the observed effects of arsenic and cisplatin, respectively, and ECDDP+As the effect of cisplatin combined with arsenic.58 Colon cancer tissues were obtained from patients through colonoscopy at the Department of Gastroenterology (The Fifth Affiliated Hospital, Sun Yat-sen University). Prior donor consent was obtained from all patients. Approval from the Research Ethics Committee (The Fifth Affiliated Hospital, Sun Yat-sen University) was obtained for this study.Western blot analysis
Total protein samples from cancer cell lysates were separated by SDS-PAGE, transferred to a PVDF membrane (Bio-Rad), and probed with antibodies against XIAP, p-65, and p65 (cell signaling technology), or antibodies against Flag, HA, caspase-3, caspase-9, and β-actin (Abmart) respectively. The membranes were incubated in ECL solution (GeneSion) and the results were recorded with a ChemiDoc MP imaging system (Bio-Rad). The intensity of target bands was analyzed with ImageJ.Recombination protein preparation
To prepare BIR1, BIR2 and BIR3 recombination proteins, E. coli BL21(DE3) harboring pET28a-GST-BIR1, pET28a-GST-BIR2 or pET28a-GST-BIR3 (home-made, pET28a derivatives) was inoculated into an LB medium supplied with 50 μg ml−1 kanamycin and grown at 37 °C. Protein expression was induced by the addition of 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 20 °C overnight (∼16 h) after OD600 of the bacterial culture reached ca. 0.8. Targeted proteins with Hisx6-GST tag were purified by using a Ni(II)-loaded HiTrap column (GE Healthcare) according to the manufacturer's protocol. The Hisx6-GST-tag was cleaved by adding 20 NIH units of thrombin (ThermoFisher) at 4 °C for 4 h with mild shaking and removed by passing through the Ni(II)-NTA column again. The collected targeted proteins were further purified using a HiLoad 16/60 Superdex 200 pg gel filtration column. To prepare apo-BIR1, BIR2 and BIR3, protein samples were incubated with 20 mM EDTA in the presence of 1 mM TCEP at 4 °C overnight (∼16 h) to remove the metal ions from protein. A 5 ml Hitrap desalting column (GE Healthcare) was employed to further purify the apo-form protein. The metal content of protein samples was determined via ICP-MS. All protein samples were kept in reduced buffer containing TCEP to protect cysteine residues of the apo-BIR proteins.MALDI-TOF-mass spectrometry
The binding of arsenic to BIR1, BIR2, and BIR3 was examined by matrix assisted laser ionization time of flight mass spectrometry (MALDI-TOF-MS) (ABI 4800). Apo-form BIR1 (20–99), BIR2 (120–240), and BIR3 (241–365) proteins (30 μM) were incubated with different molar equivalents of arsenic trioxide at 4 °C for 2 h. For MALDI-TOF-MS analysis, protein samples were co-crystallized with sinapinic acid (SA) as the matrix. 1 μl of the protein sample was mounted on a modified stainless-steel sample plate. The plate was then introduced into the mass spectrometer. The mass resolution of ion peaks was determined in a TOF mass analyzer.Cellular thermal shift assay (CETSA)
Home-made vectors pcDNA3.1-XIAP-Flag, pCMV-N-Flag-pur-BIR1-2-3, pCMV-N-Flag-pur-BIR1, pCMV-N-Flag-pur-BIR2 and pCMV-N-Flag-pur-BIR3 are derivatives of pCDNA3.1-Flag-C (Miaoling) or pCMV-N-Flag-pur (beyotime). HEK293t cells expressing full length XIAP-Flag, truncated XIAP-Flag or solo BIR domain-Flag were treated with or without arsenic in the culture medium for 4 h and harvested. Equal volumes of cell suspensions from control and arsenic-treated groups were aliquoted into PCR strip-tubes. The heating procedure was carried out in a PCR cycler (BioRad) with a gradient-temperature program (35–80 °C) for 3 min followed by immediate cooling on ice. After three freeze–thaw cycles using liquid nitrogen, the samples were centrifuged at 20000×g for 30 min at 4 °C to pellet the denatured protein precipitate. The soluble XIAP proteins in the supernatant were analyzed by following western blot analysis using anti-Flag antibody (Abmart) or Coomassie blue staining.
UV-vis analysis
UV-vis spectra were collected at room temperature on a Nanodrop spectrometer (Thermo Scientific). To study arsenic-BIR domain interaction, aliquots of arsenic trioxide were added to 100 μM apo-BIR3 in HEPES buffer (50 mM HEPES, 0.1 M NaCl, pH 7.4). Spectra were recorded between 200 and 400 nm after equilibrium at 4 °C. We failed to obtain the UV absorption spectra for arsenic-BIR1 and -BIR2 binding, due to the serious protein precipitation during arsenic titration. For Zn release assay, Zn-BIR1/2/3 samples were premixed with PAR solution in the presence of 5 mM GSH, arsenic trioxide was titrated into Zn-BIR1/2/3-PAR solution with gentle mixing, and the UV-vis spectra (250–650 nm) were recorded after a 20 min incubation.In vitro caspase assay
HeLa cell extracts were prepared in HEPES buffer. Recombination Zn-bound GST-BIR2 or GST-BIR3 protein (0.2 μM) with or without arsenic treatment was added into the cell lysate to inhibit potential caspase activation. Cytochrome c (20 μg ml−1) and dATP (1 mM) were used to initiate the activation of caspase-3 and caspase-9 in cell lysates, in which the level of cleaved caspase 3 or 9 was used as an indicator for caspase activation. The reaction was terminated by the addition of 5× SDS loading buffer. The samples were subjected to western blot analysis using active/pro-caspase-3 or caspase-9 antibody (Abmart).Flag-tag pull-down experiments
Cytochrome c (20 μg ml−1) and dATP (1 mM) were used to induce the activation of caspase-3 and caspase-9 in HeLa cell lysates. Recombinant Flag-fused XIAP protein was extracted from HEK293t cells and treated with or without arsenic (in the presence of 5 mM GSH). XIAP-Flag (Zn-bound or As-bound form) and cell lysate containing excess active caspase-3 and caspase-9 were incubated with Flag-beads followed by three washes. The bound proteins were eluted and were subjected to western blot analysis using active-caspase-3 or caspase-9 antibody (Cell Signaling Technology).Circular dichroism spectroscopy
Circular dichroism (CD) spectra of BIR1, BIR2 and BIR3 were collected on a Jasco-810 spectrophotometer. Approximately 10 μM apo or metal-bound protein samples were prepared in 20 mM HEPES buffer. CD spectra were recorded from 200 to 650 nm at a scan rate of 50 nm min−1 in a 0.1 cm quartz cuvette at room temperature. Four scans were averaged for each spectrum, and the reference spectrum of the buffer was subtracted. The secondary structure contents of BIR proteins were calculated according to a previous report.41Size-exclusion chromatography
Size-exclusion chromatography (SEC) was performed with a Tricorn Superdex 75 10/300 GL analytical column (GE Healthcare) at 4 °C. The column was pre-equilibrated with Tris–HCl buffer (20 mM Tris–HCl, 150 mM NaCl, pH 7.4). The protein samples were prepared in the same buffer supplemented with 0.1 mM TCEP. Protein samples were preincubated with zinc or arsenic for 2 h at 4 °C before loading to the column at a flow rate of 0.5 ml min−1.Data availability
All data have been included in the manuscript and ESI.†Author contributions
H. Z. S. and X. M. Y. designed the research; L. W. S., J. L., X. X., and X. M. Y. performed the research; X. L., X. H. L., and J. Z. assisted in the experiments; L. W. S., J. L., and X. F. L. analysed the data, and H. Z. S., H. Y. L., and X. M. Y. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Guangdong-Hong Kong-Macao University Joint Laboratory of Interventional Medicine Foundation of Guangdong Province (2023LSYS001), the Research Grants Council (RGC) of Hong Kong (SRFS2122-7S04, T11-709/21N, C7034-20E, and 17306323) and Zhuhai municipal Bureau of Science-Industry-Trade and Information Technology (20181117A010062). We thank Prof. Dan Li and Dr Zhuangzhuang Zhang (Sun Yat-sen University) for helpful comments. We thank Prof. Ji'an PAN (Sun Yat-sen University) for the LPC-Ub-HA vector as a gift. We thank all staff at the Molecular Imaging Center (Sun Yat-sen University) for helpful support. We are also grateful to Ms. Chung Yan Sherry Chan for critical reading of the manuscript.References
- S. Fulda and D. Vucic, Targeting IAP proteins for therapeutic intervention in cancer, Nat. Rev. Drug Discovery, 2012, 11, 109–124 CrossRef CAS PubMed.
- B. P. Eckelman, G. S. Salvesen and F. L. Scott, Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family, EMBO Rep., 2006, 7, 988–994 CrossRef CAS PubMed.
- A. D. Schimmer, S. Dalili, R. A. Batey and S. J. Riedl, Targeting XIAP for the treatment of malignancy, Cell Death Differ., 2006, 13, 179–188 CrossRef CAS PubMed.
- S. Kesavardhana, R. K. S. Malireddi and T. D. Kanneganti, Caspases in Cell Death, Inflammation, and Pyroptosis, Annu. Rev. Immunol., 2020, 38, 567–595 CrossRef CAS PubMed.
- E. Burstein, L. Ganesh, R. D. Dick, B. van De Sluis, J. C. Wilkinson, L. W. Klomp, C. Wijmenga, G. J. Brewer, G. J. Nabel and C. S. Duckett, A novel role for XIAP in copper homeostasis through regulation of MURR1, EMBO J., 2004, 23, 244–254 CrossRef CAS PubMed.
- A. R. Mufti, E. Burstein, R. A. Csomos, P. C. Graf, J. C. Wilkinson, R. D. Dick, M. Challa, J. K. Son, S. B. Bratton, G. L. Su, G. J. Brewer, U. Jakob and C. S. Duckett, XIAP Is a copper binding protein deregulated in Wilson's disease and other copper toxicosis disorders, Mol. Cell, 2006, 21, 775–785 CrossRef CAS PubMed.
- M. Lu, S. C. Lin, Y. Huang, Y. J. Kang, R. Rich, Y. C. Lo, D. Myszka, J. Han and H. Wu, XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization, Mol. Cell, 2007, 26, 689–702 CrossRef CAS PubMed.
- P. J. Jost and D. Vucic, Regulation of Cell Death and Immunity by XIAP, Cold Spring Harbor Perspect. Biol., 2020, 12, a036426 CrossRef CAS PubMed.
- Y. Akizuki, M. Morita, Y. Mori, A. Kaiho-Soma, S. Dixit, A. Endo, M. Shimogawa, G. Hayashi, M. Naito, A. Okamoto, K. Tanaka, Y. Saeki and F. Ohtake, cIAP1-based degraders induce degradation via branched ubiquitin architectures, Nat. Chem. Biol., 2022, 19, 311–322 CrossRef PubMed.
- E. C. LaCasse, D. J. Mahoney, H. H. Cheung, S. Plenchette, S. Baird and R. G. Korneluk, IAP-targeted therapies for cancer, Oncogene, 2008, 27, 6252–6275 CrossRef CAS PubMed.
- V. Vetma, C. Guttà, N. Peters, C. Praetorius, M. Hutt, O. Seifert, F. Meier, R. Kontermann, D. Kulms and M. Rehm, Convergence of pathway analysis and pattern recognition predicts sensitization to latest generation TRAIL therapeutics by IAP antagonism, Cell Death Differ., 2020, 27, 2417–2432 CrossRef CAS PubMed.
- M. Hashimoto, Y. Saito, R. Nakagawa, I. Ogahara, S. Takagi, S. Takata, H. Amitani, M. Endo, H. Yuki, J. A. Ramilowski, J. Severin, R. I. Manabe, T. Watanabe, K. Ozaki, A. Kaneko, H. Kajita, S. Fujiki, K. Sato, T. Honma, N. Uchida, T. Fukami, Y. Okazaki, O. Ohara, L. D. Shultz, M. Yamada, S. Taniguchi, P. Vyas, M. de Hoon, Y. Momozawa and F. Ishikawa, Combined inhibition of XIAP and BCL2 drives maximal therapeutic efficacy in genetically diverse aggressive acute myeloid leukemia, Nat. Cancer, 2021, 2, 340–356 CrossRef CAS.
- B. A. Carneiro and W. S. El-Deiry, Targeting apoptosis in cancer therapy, Nat. Rev. Clin. Oncol., 2020, 17, 395–417 CrossRef PubMed.
- R. Xiao, Y. An, W. Ye, A. Derakhshan, H. Cheng, X. Yang, C. Allen, Z. Chen, N. C. Schmitt and C. Van Waes, Dual Antagonist of cIAP/XIAP ASTX660 Sensitizes HPV(−) and HPV(+) Head and Neck Cancers to TNFα, TRAIL, and Radiation Therapy, Clin. Cancer Res., 2019, 25, 6463–6474 CrossRef CAS PubMed.
- J. Dittmann, T. Haydn, P. Metzger, G. A. Ward, M. Boerries, M. Vogler and S. Fulda, Next-generation hypomethylating agent SGI-110 primes acute myeloid leukemia cells to IAP antagonist by activating extrinsic and intrinsic apoptosis pathways, Cell Death Differ., 2020, 27, 1878–1895 CrossRef CAS PubMed.
- M. Daoud, P. N. Broxtermann, F. Schorn, J. P. Werthenbach, J. M. Seeger, L. M. Schiffmann, K. Brinkmann, D. Vucic, T. Tüting, C. Mauch, D. Kulms, P. Zigrino and H. Kashkar, XIAP promotes melanoma growth by inducing tumour neutrophil infiltration, EMBO Rep., 2022, 23, e53608 CrossRef CAS PubMed.
- O. Berezovskaya, A. D. Schimmer, A. B. Glinskii, C. Pinilla, R. M. Hoffman, J. C. Reed and G. V. Glinsky, Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells, Cancer Res., 2005, 65, 2378–2386 CrossRef CAS.
- M. Chawla-Sarkar, S. I. Bae, F. J. Reu, B. S. Jacobs, D. J. Lindner and E. C. Borden, Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis, Cell Death Differ., 2004, 11, 915–923 CrossRef CAS.
- Q. S. Tong, L. D. Zheng, L. Wang, F. Q. Zeng, F. M. Chen, J. H. Dong and G. C. Lu, Downregulation of XIAP expression induces apoptosis and enhances chemotherapeutic sensitivity in human gastric cancer cells, Cancer Gene Ther., 2005, 12, 509–514 CrossRef CAS.
- X. W. Zhang, X. J. Yan, Z. R. Zhou, F. F. Yang, Z. Y. Wu, H. B. Sun, W. X. Liang, A. X. Song, V. Lallemand-Breitenbach, M. Jeanne, Q. Y. Zhang, H. Y. Yang, Q. H. Huang, G. B. Zhou, J. H. Tong, Y. Zhang, J. H. Wu, H. Y. Hu, H. de Thé, S. J. Chen and Z. Chen, Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML, Science, 2010, 328, 240–243 CrossRef CAS.
- H. Maeda, S. Hori, H. Ohizumi, T. Segawa, Y. Kakehi, O. Ogawa and A. Kakizuka, Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine, Cell Death Differ., 2004, 11, 737–746 CrossRef CAS.
- A. J. Murgo, Clinical trials of arsenic trioxide in hematologic and solid tumors: overview of the National Cancer Institute Cooperative Research and Development Studies, Oncologist, 2001, 6(Suppl 2), 22–28 CrossRef CAS.
- Q. Q. Wang, Y. Jiang and H. Naranmandura, Therapeutic strategy of arsenic trioxide in the fight against cancers and other diseases, Metallomics, 2020, 12, 326–336 CrossRef CAS.
- B. Chen, F. Cao, X. Lu, S. Shen, J. Zhou and X. C. Le, Arsenic speciation in hair and nails of acute promyelocytic leukemia (APL) patients undergoing arsenic trioxide treatment, Talanta, 2018, 184, 446–451 CrossRef CAS PubMed.
- Y. Maimaitiyiming, H. H. Zhu, C. Yang and H. Naranmandura, Biotransformation of arsenic trioxide by AS3MT favors eradication of acute promyelocytic leukemia: revealing the hidden facts, Drug Metab. Rev., 2020, 52, 425–437 CrossRef CAS PubMed.
- P. Bercier, Q. Q. Wang, N. Zang, J. Zhang, C. Yang, Y. Maimaitiyiming, M. Abou-Ghali, C. Berthier, C. Wu, M. Niwa-Kawakita, T. Dirami, M. C. Geoffroy, O. Ferhi, S. Quentin, S. Benhenda, Y. Ogra, Z. Gueroui, C. Zhou, H. Naranmandura, H. de The and V. Lallemand-Breitenbach, Structural basis of PML/RARA oncoprotein targeting by arsenic unravels a cysteine rheostat controlling PML body assembly and function, Cancer Discovery, 2023, 13(12), 2548–2565 CrossRef PubMed.
- J. H. Mao, X. Y. Sun, J. X. Liu, Q. Y. Zhang, P. Liu, Q. H. Huang, K. K. Li, Q. Chen, Z. Chen and S. J. Chen, As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21683–21688 CrossRef CAS PubMed.
- H. N. Zhang, L. Yang, J. Y. Ling, D. M. Czajkowsky, J. F. Wang, X. W. Zhang, Y. M. Zhou, F. Ge, M. K. Yang, Q. Xiong, S. J. Guo, H. Y. Le, S. F. Wu, W. Yan, B. Liu, H. Zhu, Z. Chen and S. C. Tao, Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15084–15089 CrossRef CAS PubMed.
- X. Hu, H. Li, T. K. Ip, Y. F. Cheung, M. Koohi-Moghadam, H. Wang, X. Yang, D. N. Tritton, Y. Wang, Y. Wang, R. Wang, K. M. Ng, H. Naranmandura, E. W. Tse and H. Sun, Arsenic trioxide targets Hsp60, triggering degradation of p53 and survivin, Chem. Sci., 2021, 12, 10893–10900 RSC.
- M. P. Martelli, I. Gionfriddo, F. Mezzasoma, F. Milano, S. Pierangeli, F. Mulas, R. Pacini, A. Tabarrini, V. Pettirossi, R. Rossi, C. Vetro, L. Brunetti, P. Sportoletti, E. Tiacci, F. Di Raimondo and B. Falini, Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells, Blood, 2015, 125, 3455–3465 CrossRef CAS PubMed.
- S. Kozono, Y. M. Lin, H. S. Seo, B. Pinch, X. Lian, C. Qiu, M. K. Herbert, C. H. Chen, L. Tan, Z. J. Gao, W. Massefski, Z. M. Doctor, B. P. Jackson, Y. Chen, S. Dhe-Paganon, K. P. Lu and X. Z. Zhou, Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells, Nat. Commun., 2018, 9, 3069 CrossRef PubMed.
- X. Yan, J. Li, Q. Liu, H. Peng, A. Popowich, Z. Wang, X. F. Li and X. C. Le, p-Azidophenylarsenoxide: An Arsenical “Bait” for the In Situ Capture and Identification of Cellular Arsenic-Binding Proteins, Angew Chem. Int. Ed., 2016, 55, 14051–14056 CrossRef CAS PubMed.
- E. Driehuis, K. Kretzschmar and H. Clevers, Establishment of patient-derived cancer organoids for drug-screening applications, Nat. Protoc., 2020, 15, 3380–3409 CrossRef CAS.
- G. Tang, M. Cho and X. Wang, OncoDB: an interactive online database for analysis of gene expression and viral infection in cancer, Nucleic Acids Res., 2022, 50, D1334–d1339 CrossRef CAS PubMed.
- P. Rousselot, J. Larghero, B. Arnulf, J. Poupon, B. Royer, A. Tibi, I. Madelaine-Chambrin, P. Cimerman, S. Chevret, O. Hermine, H. Dombret, J. Claude Brouet and J. Paul Fermand, A clinical and pharmacological study of arsenic trioxide in advanced multiple myeloma patients, Leukemia, 2004, 18, 1518–1521 CrossRef CAS PubMed.
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundbäck, P. Nordlund and D. Martinez Molina, The cellular thermal shift assay for evaluating drug target interactions in cells, Nat. Protoc., 2014, 9, 2100–2122 CrossRef CAS PubMed.
- X. Zhou, X. Sun, K. L. Cooper, F. Wang, K. J. Liu and L. G. Hudson, Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs, J. Biol. Chem., 2011, 286, 22855–22863 CrossRef CAS PubMed.
- Y. Hong, Y. T. Lai, G. C. Chan and H. Sun, Glutathione and multidrug resistance protein transporter mediate a self-propelled disposal of bismuth in human cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3211–3216 CrossRef CAS PubMed.
- Q. Zhang, M. R. Vakili, X. F. Li, A. Lavasanifar and X. C. Le, Polymeric micelles for GSH-triggered delivery of arsenic species to cancer cells, Biomaterials, 2014, 35, 7088–7100 CrossRef CAS PubMed.
- A. Weiss, C. C. Murdoch, K. A. Edmonds, M. R. Jordan, A. J. Monteith, Y. R. Perera, A. M. Rodríguez Nassif, A. M. Petoletti, W. N. Beavers, M. J. Munneke, S. L. Drury, E. S. Krystofiak, K. Thalluri, H. Wu, A. R. S. Kruse, R. D. DiMarchi, R. M. Caprioli, J. M. Spraggins, W. J. Chazin, D. P. Giedroc and E. P. Skaar, Zn-regulated GTPase metalloprotein activator 1 modulates vertebrate zinc homeostasis, Cell, 2022, 185, 2148–2163.e2127 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y. H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra, Nucleic Acids Res., 2018, 46, W315–w322 CrossRef CAS PubMed.
- G. D. Shimberg, K. Ok, H. M. Neu, K. E. Splan and S. L. J. Michel, Cu(I) Disrupts the Structure and Function of the Nonclassical Zinc Finger Protein Tristetraprolin (TTP), Inorg. Chem., 2017, 56, 6838–6848 CrossRef CAS.
- K. Ok, M. R. Filipovic and S. L. J. Michel, Targeting Zinc Finger Proteins with Exogenous Metals and Molecules: Lessons learned from Tristetraprolin, a CCCH type Zinc Finger, Eur. J. Inorg. Chem., 2021, 2021, 3795–3805 CrossRef CAS PubMed.
- Y. Huang, Y. C. Park, R. L. Rich, D. Segal, D. G. Myszka and H. Wu, Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain, Cell, 2001, 104, 781–790 CAS.
- E. N. Shiozaki, J. Chai, D. J. Rigotti, S. J. Riedl, P. Li, S. M. Srinivasula, E. S. Alnemri, R. Fairman and Y. Shi, Mechanism of XIAP-mediated inhibition of caspase-9, Mol. Cell, 2003, 11, 519–527 CrossRef CAS PubMed.
- Q. L. Deveraux, E. Leo, H. R. Stennicke, K. Welsh, G. S. Salvesen and J. C. Reed, Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases, EMBO J., 1999, 18, 5242–5251 CrossRef CAS PubMed.
- R. Takahashi, Q. Deveraux, I. Tamm, K. Welsh, N. Assa-Munt, G. S. Salvesen and J. C. Reed, A single BIR domain of XIAP sufficient for inhibiting caspases, J. Biol. Chem., 1998, 273, 7787–7790 CrossRef CAS PubMed.
- Q. L. Deveraux, N. Roy, H. R. Stennicke, T. Van Arsdale, Q. Zhou, S. M. Srinivasula, E. S. Alnemri, G. S. Salvesen and J. C. Reed, IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases, EMBO J., 1998, 17, 2215–2223 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Molecular mechanisms of cisplatin resistance, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- Y. Yang, S. Fang, J. P. Jensen, A. M. Weissman and J. D. Ashwell, Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli, Science, 2000, 288, 874–877 CrossRef CAS PubMed.
- S. K. Chow, J. Y. Chan and K. P. Fung, Inhibition of cell proliferation and the action mechanisms of arsenic trioxide (As2O3) on human breast cancer cells, J. Cell. Biochem., 2004, 93, 173–187 CrossRef CAS.
- N. Sadaf, N. Kumar, M. Ali, V. Ali, S. Bimal and R. Haque, Arsenic trioxide induces apoptosis and inhibits the growth of human liver cancer cells, Life Sci., 2018, 205, 9–17 CrossRef CAS.
- B. C. Bornhauser, L. Bonapace, D. Lindholm, R. Martinez, G. Cario, M. Schrappe, F. K. Niggli, B. W. Schäfer and J. P. Bourquin, Low-dose arsenic trioxide sensitizes glucocorticoid-resistant acute lymphoblastic leukemia cells to dexamethasone via an Akt-dependent pathway, Blood, 2007, 110, 2084–2091 CrossRef CAS PubMed.
- P. Makhov, K. Golovine, R. G. Uzzo, J. Rothman, P. L. Crispen, T. Shaw, B. J. Scoll and V. M. Kolenko, Zinc chelation induces rapid depletion of the X-linked inhibitor of apoptosis and sensitizes prostate cancer cells to TRAIL-mediated apoptosis, Cell Death Differ., 2008, 15, 1745–1751 CrossRef CAS PubMed.
- L. Yang, T. Mashima, S. Sato, M. Mochizuki, H. Sakamoto, T. Yamori, T. Oh-Hara and T. Tsuruo, Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide, Cancer Res., 2003, 63, 831–837 CAS.
- S. Fulda, W. Wick, M. Weller and K. M. Debatin, Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo, Nat. Med., 2002, 8, 808–815 CrossRef CAS PubMed.
- K. Hanada, K. Nishijima, H. Ogata, S. Atagi and M. Kawahara, Population pharmacokinetic analysis of cisplatin and its metabolites in cancer patients: possible misinterpretation of covariates for pharmacokinetic parameters calculated from the concentrations of unchanged cisplatin, ultrafiltered platinum and total platinum, Jpn. J. Clin. Oncol., 2001, 31, 179–184 CrossRef CAS PubMed.
- P. Jaaks, E. A. Coker, D. J. Vis, O. Edwards, E. F. Carpenter, S. M. Leto, L. Dwane, F. Sassi, H. Lightfoot, S. Barthorpe, D. van der Meer, W. Yang, A. Beck, T. Mironenko, C. Hall, J. Hall, I. Mali, L. Richardson, C. Tolley, J. Morris, F. Thomas, E. Lleshi, N. Aben, C. H. Benes, A. Bertotti, L. Trusolino, L. Wessels and M. J. Garnett, Effective drug combinations in breast, colon and pancreatic cancer cells, Nature, 2022, 603, 166–173 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01294a |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |