
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstitutional adaptation to pKa modulation by remote ester hydrolysis†
Ferran
Esteve
 *,
Tanguy
Rieu
and
Jean-Marie
Lehn
*
*,
Tanguy
Rieu
and
Jean-Marie
Lehn
*
Laboratoire de Chimie Supramoléculaire, Institut de Science et d’Ingénierie Supramoléculaires (ISIS), Université de Strasbourg, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: estevefranch@unistra.fr; lehn@unistra.fr
First published on 11th April 2024
Abstract
The mechanisms through which environmental conditions affect the expression of interconnected species is a key step to comprehending the principles underlying complex chemical processes. In Nature, chemical modifications triggered by the environment have a major impact on the structure and function of biomolecules and regulate different reaction pathways. Yet, minimalistic artificial systems implementing related adaptation behaviours remain barely explored. The hydrolysis of amino acid methyl esters to their corresponding amino acids leads to a drastic change in pKa (ca. 7 and 9, respectively) that protonates the free amino group at physiological conditions. Dynamic covalent libraries (DCvLs) based on amino acid methyl esters and aldehydes respond to such hydrolysis and lead to constitutional adaptation. Each of the libraries studied experiences a DCvL conversion allowing for constituent selection due to the silencing of the zwitterionic amino acids towards imine formation. The selective action of different enzymes on the DCvLs results in states with simplified constitutional distributions and transient chirality. When additional components (i.e., scavengers) that are not affected by hydrolysis are introduced into the dynamic libraries, the amino acid methyl ester hydrolysis induces the up-regulation of the constituents made of these scavenging components. In these systems, the constituent distribution is resolved from a scrambled mixture of imines to a state characterized by the predominance of a single aldimine. Remarkably, although the final libraries display higher “simplexity”, the different transient states present an increased complexity that allows for the emergence of organized structures [micelle formation] and distributions [up-regulation of two antagonistic constituents]. This reactive site inhibition by a remote chemical modification resembles the scenario found in some enzymes for the regulation of their activity through proximal post-translational modifications.
Introduction
Cross-talks between amino acid residues located at different positions in proteins control essential processes like molecular recognition and catalysis.1 In addition, selective post-translational modifications (PTMs) on amino acid sidechains offer a powerful biological toolkit to expand the polypeptide functional repertoire.2–4 These chemical modifications, occurring in well-defined microenvironments, commonly lead to large perturbations in the pKa of surrounding ionizable groups that govern the function of biomolecules.5 For instance, the phosphorylation of a serine residue induces changes in the pKa of a nearby histidine sidechain and modulate the reactivity of cofilin proteins (Fig. 1A).6 Therefore, chemical systems that can adapt to analogous remote transformations deserve to be thoroughly explored as they might enlighten the mode-of-action underlying such biological processes.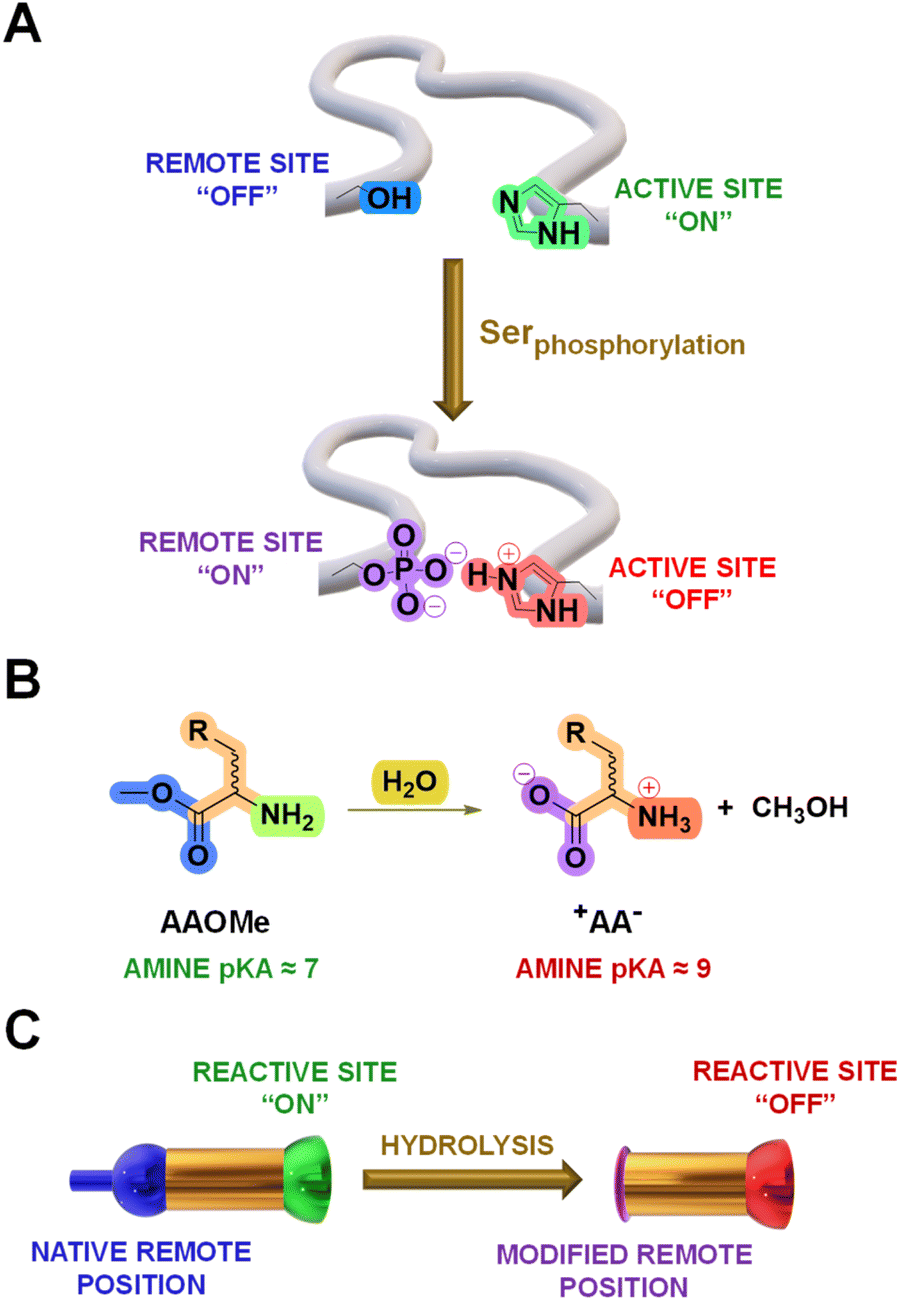 | ||
| Fig. 1 (A) Representation of a decrease in activity at the catalytic site of a protein by a PTM (serine phosphorylation) accompanied by a change in the pKa of a nearby residue (histidine protonation). (B) Hydrolysis of AAOMe (terminal –NH2 site reactive in imination) to yield +AA− (terminal –N+H3 site silent in imination) and a molecule of CH3OH. (C) Representation of the proposed remote modification leading to the inhibition of the components of DCvLs (colour code has been maintained for comparison). | ||
An extensive amount of studies discussing adaptive systems rely on Constitutional Dynamic Chemistry (CDC) at either the molecular (Dynamic Covalent Chemistry, DCvC) or supramolecular level (Dynamic Non-Covalent Chemistry, DNCvC).7 In DCvC, the reversible covalent bonds involved allow for error-checking and rearrangement processes under thermodynamic control.8–11 Hence, the distribution of diversified Dynamic Covalent Libraries (DCvLs) can be directed to a certain state upon exposure to external stimuli or chemical effectors.12,13 For example, the sorting of “messy” DCvLs has been attained using both physical (precipitation,14 distillation,15 adsorption16) and chemical (oxidation,17 templates,18 catalysis,19 coupled enzymatic reactions20) methods. In a similar manner, Constitutional Dynamic Networks (CDNs) are composed of products (i.e., constituents) constitutionally linked by exchangeable building blocks (i.e., components) that adapt their distribution through agonistic and antagonistic relationships.21,22 Substantial research efforts have been devoted to build-up relatively simple CDNs mimicking complex biological behaviours.23–25 Related dynamic networks mediate the communication between biomolecules (e.g., DNA, RNA, proteins), leading to programmed reaction patterns that regulate key intracellular processes like signal transduction and enzymatic activity.26 However, the design of DCvLs, and their underlying CDNs, able to undergo structural and/or constitutional adaptation in response to pKa variations triggered by remote chemical modifications remains unexplored.
The hydrolysis of amino acid methyl esters (AAOMe) to their corresponding zwitterionic amino acids (+AA−) leads to a drastic pKa change in the remote –NH2 group, resulting in its protonation at neutral pH (predicted Nα pKa values of ca. 9 and 7, respectively; Fig. 1B and Table S1†).27 Thus, we envisaged that DCvLs based on the condensation of AAOMe and water-soluble aldehydes should experience constitutional adaptation in response to the hydrolysis of the remote ester group since the resulting zwitterionic +AA− entity should be “poisoned” towards imine formation through reactive site protonation (Fig. 1C). After first devising the most reactive components towards imination, we optimized the conditions for the hydrolysis of the AAOMe ester group. Then, the time evolution of the DCvLs was investigated, observing that they resulted in states of higher “simplexity” (simplification of the output of the system)28 through transient states of higher complexity [enzymatic selection/transient chirality/sorting] and diversity [increased number of components/constituents]. Furthermore, such constituent selection induced emerging behaviours in the final simplified states of the libraries: micelle formation and antagonistic sorting (see below).
Results and discussion
Component screening
The first step was to select the reaction components by screening a series of water-soluble reactive aldehydes and different amines. Imines are generally disfavoured in water due to their propensity to hydrolyse back into the reactants.29 Nevertheless, a rational design of the system (increased aldehyde electrophilicity and amine nucleophilicity) can assist to overcome this issue and foster relatively high yields.30The imine yields for the reaction between different aldehydes, namely A1–6, and ArgOMe were evaluated at room temperature and pD = 7.0 (PBS 50 mM). 1H NMR signal integration after 1 h of equilibration showed that the highest imine formation was observed when using A1, likely as a result of the strong electron-withdrawing capacity of the two sulfonate groups (Fig. 2A).
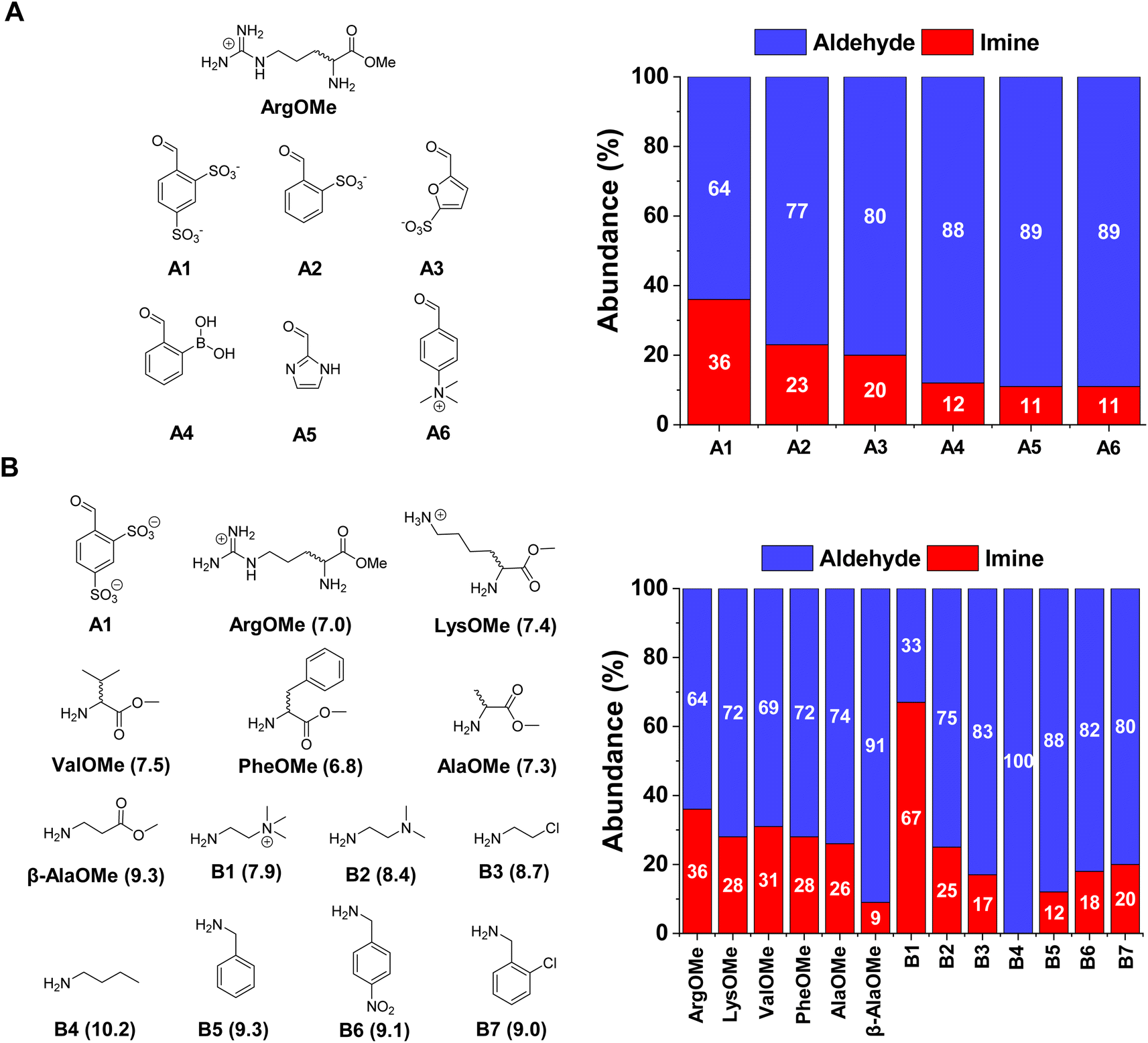 | ||
| Fig. 2 (A) Aldehyde screening using ArgOMe as the model amine. (B) Amine screening using best-performing A1 as aldehyde. Left: Chemical structures of the different reagents employed. Right: Component abundances obtained after equilibration (1 h at room temperature). The hydrate detected was included in the aldehyde composition. Amine pKa values (calculated using MarvinSketch) have been included in brackets. All abundances were calculated using 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 50 mM, 293 K). Initial concentration of reagents 5 mM each in all cases. Error in abundance: 5%. | ||
In terms of the amine screening, the studied α-AAOMe gave imine yields of ca. 30% (Fig. 2B). In contrast, the yield attained when using β-AlaOMe as a nucleophile was 9%, in good agreement with the higher pKa calculated for this derivative (Table S1†). Several commercially available amines were also assayed to select a nucleophile with similar reactivity to that of the α-AAOMe. The most reactive amine was found to be B1 which gave a remarkable imine yield of 67%, assigned to its low degree of protonation arising from the presence of a positive charge in close proximity.31 A similar scenario was found in the case of B2, but with a lower imine formation (25% abundance). The amines presenting pKa values between 8.7–9.3 (viz., B3, B5, B6, and B7) resulted in imine yields of ca. 15%. As predicted, amine B4 (pKa = 10.2) did not react with aldehyde A1 as its amino group is completely protonated at pD = 7.0. To increase the imine yields, the reaction between A1 and ArgOMe was performed at higher concentrations maintaining a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between components. The reaction at 80 mM gave 65% of imine A1ArgOMe (Fig. S1†). These results indicated that the low specificity of imine formation in water can be overcome at high concentrations and with adequate substrate selection, paving the way for the study of dynamic covalent reactions and libraries (see sections below).
:1 ratio between components. The reaction at 80 mM gave 65% of imine A1ArgOMe (Fig. S1†). These results indicated that the low specificity of imine formation in water can be overcome at high concentrations and with adequate substrate selection, paving the way for the study of dynamic covalent reactions and libraries (see sections below).
Hydrolysis of AAOMe
Next, we decided to investigate the ester hydrolysis of AAOMe. It is well established that AAOMe are hydrolysed to the corresponding amino acids (+AA−) under basic conditions.32 However, the pH must be optimized to attain the best conditions in terms of hydrolysis rates as well as imination efficiency and selection. Although higher pH values would favour imine formation and ester hydrolysis, undesired aldimines with +AA− would also be formed at high pH (Table S2†). Thereby, three experiments were run at pD values ranging from 6.2–7.8 to reveal the effect on the aforementioned variables.33 The reaction course could be easily monitored by 1H NMR spectroscopy, observing the disappearance of the signals assigned to ArgOMe and the appearance of those of +Arg− and methanol (Fig. S2†). As expected, the fastest hydrolysis rates were obtained under basic conditions, with the reaction at pD = 7.8 being slightly favoured over the one at pD = 7.0 (Fig. S3 and Table S3†). Notwithstanding, the –Nα/βH2 degree of protonation calculated revealed that, at pD = 7.8, the +Arg− inhibition towards imination would not be complete, as a significant amount of the reactive site would be in its nucleophilic non-protonated form (10%; see Table S2†). Separated reaction of ArgOMe and +Arg− with A1 at these different pD values corroborated this assumption (Table S4†). Although the highest imine yields were obtained at pD = 7.8 (44%), the undesired reaction between +Arg− and A1 was also detected (3% yield of A1+Arg−). On the other hand, slightly lower A1ArgOMe yields (36%) were noted at pD = 7.0, but with complete silencing of A1+Arg− (entry 2, Table S4†). Therefore, the optimal pD was found to be 7.0.We then evaluated the effect of the substrate structure on the hydrolysis rate, both in terms of the sidechain nature and the distance between the carbonyl unit and the amino group (i.e., α- or β-AAOMe). Results suggest that β-AAOMe hydrolyses at slower rates than the α-derivatives. For instance, the hydrolysis of β-AlaOMe was much slower than that of AlaOMe, as shown by the yields of 16 and 44% observed after 48 h, respectively (Fig. S4 and Table S5†). In terms of the sidechain effect, the α-AAOMe presenting the highest hydropathy,34 namely ValOMe, was barely hydrolysed after 48 h (≈20% CH3OH yield; Fig. S4†). This could be related to a less efficient attack of water molecules on the ester group because of the hydrophobic environment. LysOMe presented the fastest hydrolysis rate, and the other α-AAOMe (viz.ArgOME and PheOMe) gave similar profiles (Table S5†). Thus, LysOMe was selected for the subsequent experiments (see sections below).
Previous reports proved that the hydrolysis of esters in water could be catalysed by imidazole-derivatives and by enzymes with esterase activity.35,36 Acetylcholine esterase (AChE) is a conserved enzyme that presents superior activity in the hydrolysis of acetylcholine to choline (Fig. S5A†). The active site is composed of two well-defined functionalities: the esteratic site (containing a serine residue which cleaves the ester bonds) and the peripheral site (anionic groups that fix the substrate in the optimal position through supramolecular interactions).37 We envisaged that AChE could selectively catalyse the hydrolysis of the AAOMe that contained a positively charged unit in the sidechain, namely ArgOMe and LysOMe, as they present a relatively similar chemical structure to that of acetylcholine (Fig. S5B†). The model hydrolysis of ArgOMe was then studied in the presence of imidazole and AChE. Whereas the imidazole-based approach was not successful even at high base concentrations (200 mol%), up to a 3.2-fold increase in hydrolysis rate could be observed with increasing catalytic amounts of AChE (Fig. S6 and Table S6†). As expected, the catalytic activity of AChE was notably dependent on the structure of the AAOMe, since it only accelerated the hydrolysis of ArgOMe and LysOMe (Fig. S7 and Table S7†). In view of these results, two additional AAOMe hydrolases were tested: trypsin and chymotrypsin. Trypsin was found to selectively catalyse the depletion of LysOMe, in a similar manner to that of AChE, but with faster rates (Fig. S8 and Table S8†). In contrast, chymotrypsin showed excellent selectivity for the catalytic hydrolysis of PheOMe (Fig. S8 and Table S8†).
Considering the chiral nature of the enzymatic active sites, we also surmised that AChE could preferentially catalyse the hydrolysis of one of the enantiomers of LysOMe. Indeed, the rate of hydrolysis of (D)LysOMe was not significantly affected by the enzyme (Fig. S9 and Table S9†). On the other hand, the use of 0.01 mol% of AChE induced complete hydrolysis of (L)LysOMe in less than 16 h.
Therefore, AChE, trypsin and chymotrypsin are suitable catalysts for the selective kinetic simplification of complex mixtures of AAOMe (vide infraDCvL1), as well as for challenging enantioselective kinetic resolutions of AAOMe racemic mixtures (vide infraDCvL3).
Constitutional adaptation to AAOMe hydrolysis in a dynamic covalent reaction
Several reports show how peptide-based supramolecular systems are able to assemble/disassemble in response to ester/anhydride hydrolysis, as a result of solubility differences between the generated carboxylate groups and the parent units.38–41 However, the change in the reactivity of vicinal functionalities in response to such ester hydrolysis (e.g., potential decrease in the nucleophilicity of surrounding groups by supramolecular cross-talks, see biological example in Fig. 1A) has been quite overlooked, as well as its implementation in dynamic covalent libraries.Once the reaction conditions were optimized (see sections above), the effect of AAOMe hydrolysis on the imine yields was monitored for the reaction between AAOMe and A1. Considering that the –Nα/βH2 group of the hydrolysis product +AA− is protonated, this component should not react with the released aldehyde (see entry 2 in Table S4†), so that the final composition of the system is expected to consist predominantly of free A1 and +AA−.
The reaction between LysOMe and A1 (5 mM each) at room temperature in the absence of enzyme was monitored for 120 h using 1H NMR spectroscopy (Fig. 3). The aldimine A1LysOMe was formed quite fast, reaching its maximum concentration of ca. 1.5 mM (30% yield) after 1 h of reaction. However, its concentration progressively decreased over time, as the hydrolysis of LysOMe occurred (see CH3OH yield in Fig. 3B). As predicted, the final component distribution was +Lys− (ca. 5 mM) and A1 (ca. 5 mM), with aldimine A1+Lys− not being detected. Subsequently, the adaptive response – and emergence of new properties – in more complex DCvLs was investigated (see following sections).
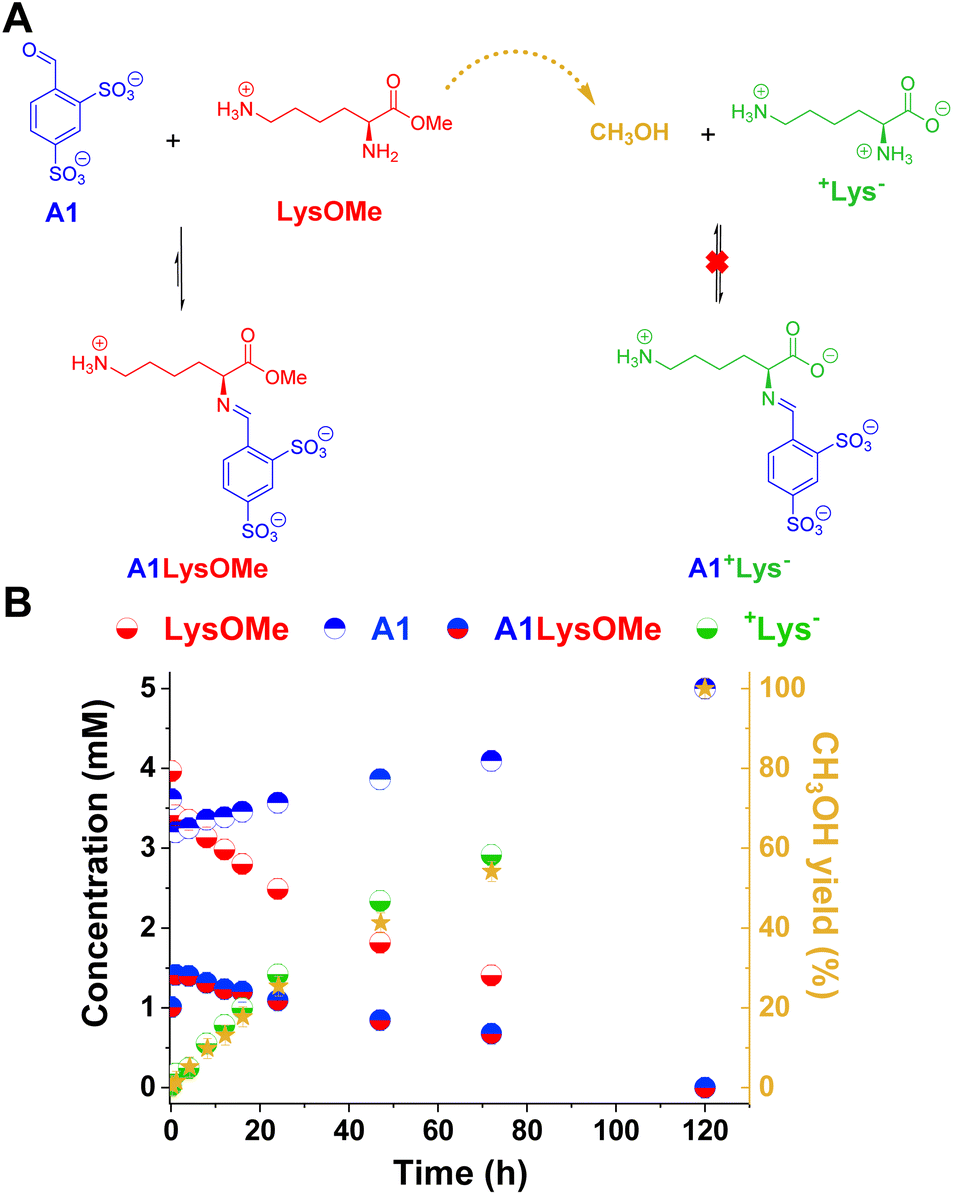 | ||
| Fig. 3 (A) Chemical representation of the LysOMe reaction with A1 influenced by the hydrolysis of LysOMe to produce +Lys−. (B) Concentration profile for the different species detected in solution over 120 h of reaction. The right Y-axis corresponds to the yield of methanol (dark yellow stars) calculated. Concentration and yields determined by 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 50 mM, 293 K). The hydrate detected was included in the aldehyde concentration (mM). Initial concentration of reagents: 5 mM. Error bars correspond to 5% of the measurement. | ||
Dynamic covalent libraries (DCvLs)
:LysOMe:PheOMe gave after 1 h a mixture of 2.9 mM A1, 4.1 mM LysOMe, and 3.8 mM PheOMe, together with 1.2 mM A1LysOMe and 0.7 mM A1PheOMe (DCvL1, Fig. 4A, B and S10†).
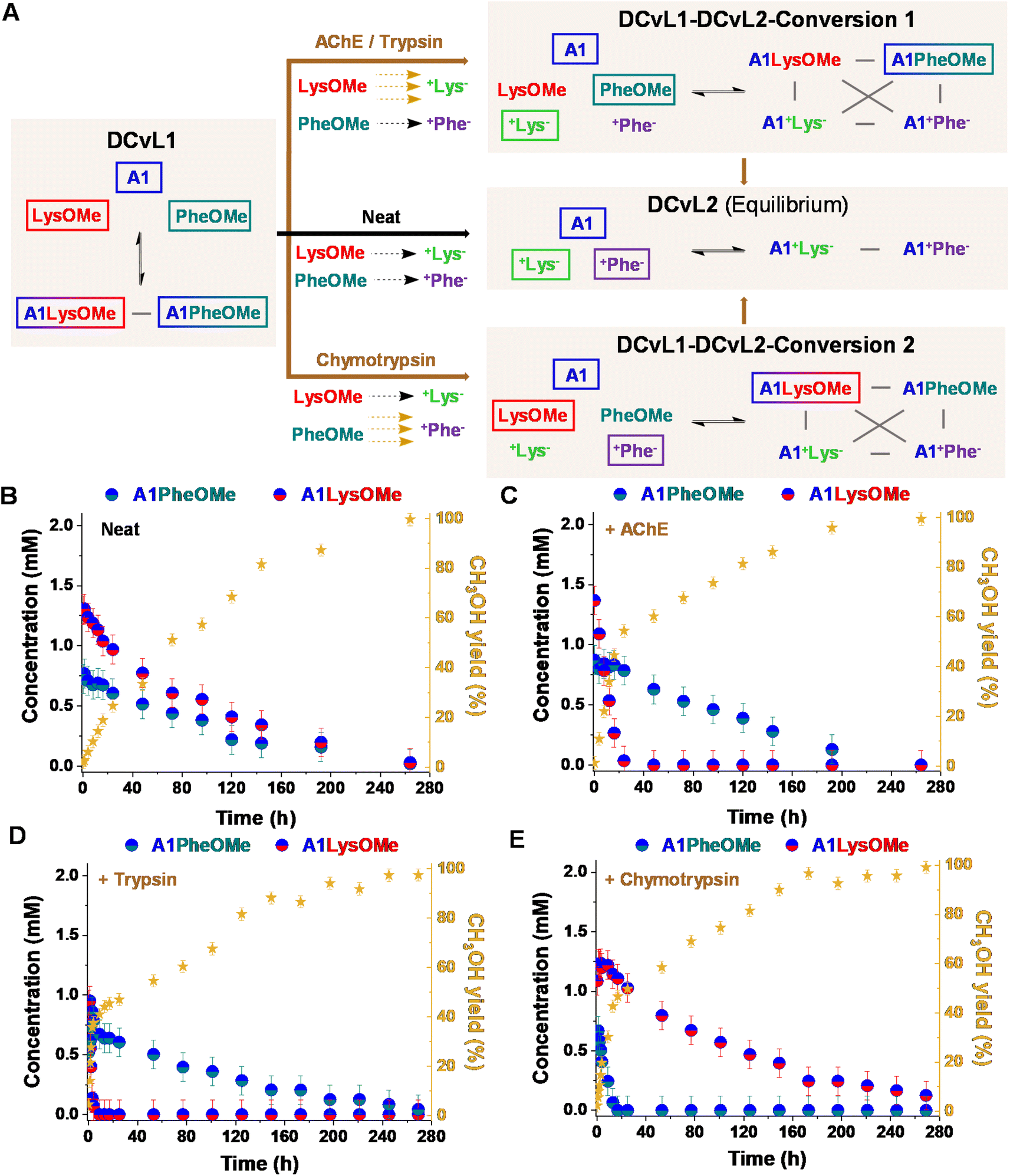 | ||
| Fig. 4 (A) Reaction between A1, LysOMe, and PheOMe in the absence (black path) and presence of AChE, trypsin and chymotrypsin (brown paths, below). The DCvL1-DCvL2-Conversion 1/2 (5 components, 4 constituents) from DCvL1 (3 components, 2 constituents) to DCvL2 (3 components, 2 constituents) have also been introduced. Predominant compounds (>0.7 mM) are marked with a coloured rectangle. Grey lines correspond to antagonistic relationships; the brown and black arrows represent the time evolution of DCvL1 to reach DCvL2, in the presence and absence of enzymes, respectively. Concentration profile for the different constituents detected in solution over 264 h of reaction in the absence (B) and presence of (C) AChE, (D) trypsin, and (E) chymotrypsin. Concentration and yields determined by 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 50 mM, 293 K). Initial concentration of reagents: 5 mM. | ||
The hydrolysis of both AAOMe to their corresponding +AA− derivatives converted DCvL1 (3 components: A1:LysOMe:PheOMe, 2 constituents: A1LysOMe:A1PheOMe) into the DCvL2 (3 components: A1:+Lys−:+Phe−, 2 constituents: A1+Lys−:A1+Phe−) through a transient state of higher constitutional complexity (5 components: A1:LysOMe:PheOMe:+Lys−:+Phe−, 4 constituents: A1LysOMe:A1PheOMe:+Lys−:+Phe−). After 264 h of reaction, the system reached the equilibrium characterized by the absence of imines and the presence of A1, +Lys−, and +Phe− (ca. 5 mM each, Fig. S10 and S11†). In the absence of an enzyme, a scrambled mixture of aldimines was always observed due to the subtly similar hydrolysis rates for LysOMe and PheOMe, with the concentration of constituent A1LysOMe being subtly higher than that of A1PheOMe (Fig. 4B and S4; see also Table S5†). However, when 0.01% of AChE was added to the mixture, a different scenario was found since this enzyme was able to selectively catalyse the hydrolysis of LysOMe (Fig. S7 and Table S7†). A1LysOMe completely vanished within the first 24 h, because of the very efficient hydrolysis of LysOMe to +Lys− catalysed by the enzyme. The concentration of these species after 20 h was of ca. 0.1 mM and 4.9 mM, respectively (Fig. S12 and S13†). Once the hydrolysis of LysOMe was completed, a drastic negative change in the slope CH3OH yield was observed, accompanied by a minor up-regulation of A1PheOMe (Fig. 4C).42 The fast and selective consumption of LysOMe resulted in a product distribution unattainable in the absence of enzyme, with A1PheOMe being the predominant aldimine constituent during the DCvL1-DCvL2-Conversion 1. DCvL1 was also subjected to trypsin and chymotrypsin to study the distinct adaptive behaviour (i.e., change in sorting selectivity) of this dynamic system. In the presence of trypsin (Fig. 4D), the new DCvL1-DCvL2-Conversion 1 species distribution after 5 h was of LysOMe (<0.1 mM), PheOMe (3.8 mM), A1LysOMe (<0.1 mM), A1PheOMe (0.7 mM), +Lys− (5 mM), and +Phe− (0.4 mM), stressing that the only aldimine constituent during this higher-in-complexity state was A1PheOMe (Fig. S14†). Alternatively, chymotrypsin accelerated the hydrolysis of PheOMe (Fig. 4E), as evidenced by the predominance of the imine A1LysOMe (ca. 1 mM after 24 h) during the transient DCvL1-DCvL2-Conversion 2 (Fig. 4A and E; see also Fig. S15†). With time, the imino-species that were not affected by the enzymes were also depleted, reaching in all cases the final DCvL2 equilibrium state after ca. 264 h.
One notes that the lifetime of the sorted distribution might be modulated with the enzyme concentration. The DCvL1 was thus studied in the presence of varying amounts of chymotrypsin. Whereas the use of 0.01% of the enzyme resulted in a transient sorting (A1LysOMe predominant aldimine) lasting for 250 h (Fig. S16B†), the introduction of 0.006 and 0.003% chymotrypsin resulted in sorted states for 240 h and 204 h, respectively (Fig. S16B and C†). Alternatively, when 0.03% of the enzyme was added, the A1PheOMe imine vanished in less than 4 h and the sorting was observed for 260 h (Fig. S16D†). These results inferred that the selectivity and lifetime of the sorted transient states can be easily controlled by the nature and the amount of enzyme introduced.
We also evaluated the possibility to build-up a similar library but based on a more challenging process: the selective chiral resolution of a racemic mixture induced by a remote modification. AChE was found to catalyse the hydrolysis of (L)LysOMe but it barely affected the hydrolysis rate of (D)LysOMe (Fig. S8 and Table S8†). The DCvL3 using A1:(L)LysOMe:(D)LysOMe as components was then assayed (5 mM each, Fig. 5A). In this case, 1H NMR analysis of the reaction course was less useful as it did not allow for differentiation between each pair of enantiomeric components, not even in the presence of water-soluble chiral shifting reagent [(R)-1,2-diaminopropane-N,N,N′,N′-tetraacetato]samarate(III).43 Yet, one can estimate the abundance of each species considering the rates of hydrolysis for each separated reaction and the yield of CH3OH monitored for the kinetic resolution of the racemic mixture (see ESI† for more details). The time evolution for the different species revealed the conversion of the DCvL3 to DCvL4 (Fig. 5A). After 16 h of reaction, a severe deacceleration in CH3OH production rate was observed, indicating that all (L)LysOMe had been consumed (Fig. S17†). At this point, the composition of the system was: (L)LysOMe (<0.1 mM), (D)LysOMe (2.5 mM), A1(L)LysOMe (<0.1 mM), A1(D)LysOMe (1.3 mM), +(L)Lys− (5 mM), and +(D)Lys− (2.1 mM) (Fig. 5B). Therefore, this higher-in-complexity DCvL3-DCvL4-Conversion (5 components, 4 constituents) presented remarkable transient chirality,44 with the highest s factor of ca. 30 being attained after 24 h (Fig. 5C).45,46 After 120 h, the complete consumption of both enantiomers of LysOMe was achieved, with the DCvL reaching its equilibrium state (DCvL4): a racemic mixture of +(L)Lys− and +(D)Lys− together with A1 (5 mM each; Fig. S18†). These results emphasize how the selective action of different enzymes on DCvLs can drastically change their constitution, allowing for transient states of higher constitutional complexity with sorted distributions.
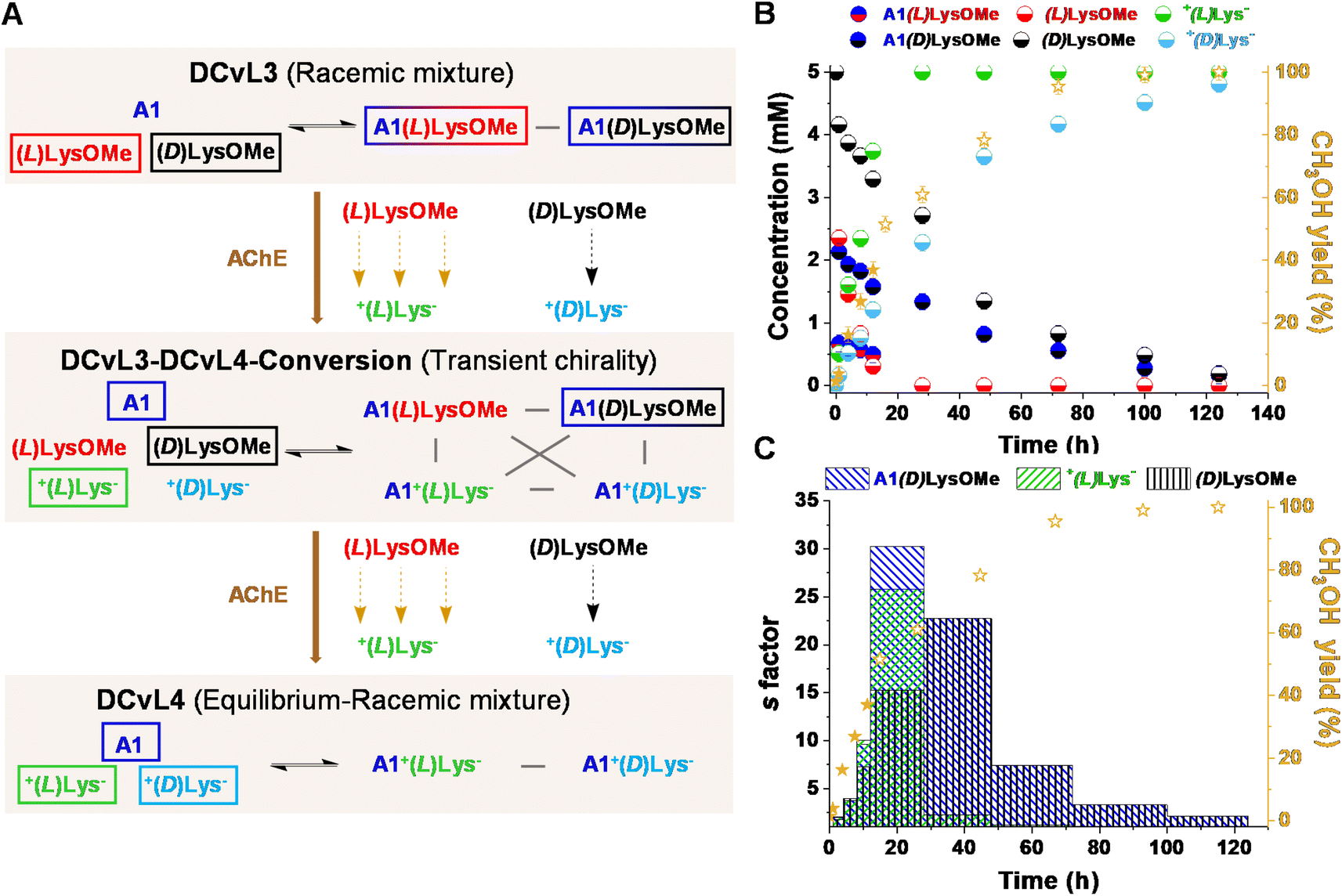 | ||
| Fig. 5 (A) Reaction between (D)LysOMe, (L)LysOMe and A1 in the presence of AChE (0.01 mol%). DCvL3-DCvL4-Conversion (5 components, 4 constituents) from DCvL3 (3 components, 2 constituents) to DCvL4 (3 components, 2 constituents) has also been introduced. Predominant compounds (>1.5 mM) are marked with a coloured rectangle. Grey lines correspond to antagonistic relationships; the brown arrows represent the time evolution of DCvL3 to reach DCvL4. (B) Concentration profile for the different constituents detected in solution over 140 h. Concentration and yields determined by 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 50 mM, 293 K). Initial concentration of reagents: 5 mM. (C) Time evolution of the s factors calculated for A1(D)LysOMe, +(L)Lys−, and (D)LysOMe over 126 h. | ||
[1 × 2] DCvL. In the systems described above, the ester hydrolysis gave rise to the unreactive +AA− component and the up-regulation of free aldehyde A1. It can be noted that if the competition experiments are performed in the presence of an amino component (B2) which can scavenge A1, the concentration of the resulting imine (A1B2) should be amplified with time as the silencing of the competing component AAOMe occurs by its conversion into +AA−. Initially, DCvL5 (reaction between A1, B2, and LysOMe; Fig. 6A and S19A†) was studied at 5 mM concentration for each reagent. Nevertheless, only a minor increase in the concentration of A1B2 was observed with time as the component LysOMe was hydrolysed to the inhibited +Lys− (Fig. S19B and S20†). Based on previous results (Fig. S1†), we evaluated the AAOMe hydrolysis and imination reactions of A1 and AAOMe/+AA− at 80 mM. At such high concentrations, the kinetic profiles for the hydrolysis of AAOMe were comparable to the ones observed at 5 mM (Fig. S21 and Table S10 vs. Fig. S4 and Table S5†), but the reaction between +AA− and A1 could not be completely inhibited at 80 mM as some A1+AA− aldimine was observed (ca. 5–10%, see Table S11†).
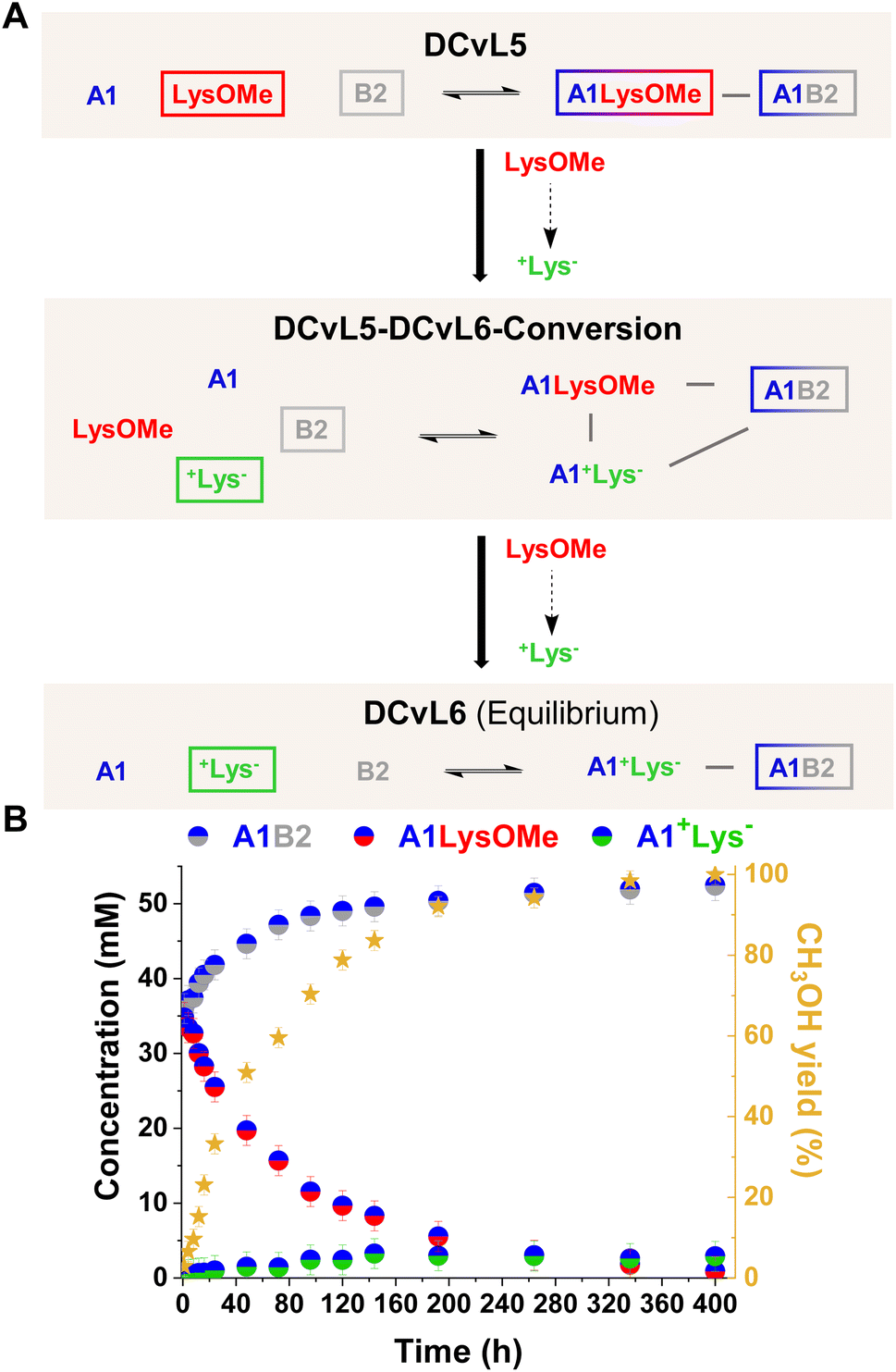 | ||
| Fig. 6 (A) Reaction between A1, LysOMe, and B2. DCvL5-DCvL6-Conversion (5 components, 4 constituents) from DCvL5 (3 components, 2 constituents) to DCvL6 (3 components, 2 constituents) has also been represented. Predominant compounds (>35 mM) are marked with a coloured rectangle. Grey lines correspond to antagonistic relationships; the black arrows represent the time evolution of DCvL5 to reach DCvL6. (B) Concentration profile for the different constituents detected in solution over 400 h of reaction. Concentration and yields determined by 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 500 mM, 293 K). Initial concentration of reagents: 80 mM. | ||
DCvL5 was then assessed at 80 mM to see the effect on both selection and activity. As expected, a more pronounced up-regulation of A1B2 was noted, with a concentration increase of 17 mM after 400 h (Fig. 6B; see also Fig. S19B† for comparison). During the hydrolysis of LysOMe, the system experienced a conversion from DCvL5 to DCvL6, with the corresponding increase in complexity during the DCvL5-DCvL6-Conversion: from 3 to 4 components and from 2 to 3 constituents (Fig. 6A).28,47,48 The final composition of DCvL6 was: 52 mM A1B2, 26 mM A1, 3 mM A1+Lys−, 77 mM +Lys−, and 28 mM B2 (Fig. S22 and S23†). Remarkably, as the LysOMe hydrolysis occurred, the system evolved from a scrambled mixture of two constituents in DCvL5 (ca. 35 mM of A1B2 and A1LysOMe) to a sorted outcome in DCvL6 containing A1B2 as the main aldimine constituent.49–51
This chemically-driven adaptation in DCvL5 was further studied in the absence of buffer (i.e. neat D2O). The presence of the quite basic component B2 led to an initial pD value of 8.3, resulting in slightly higher imine abundances than in the buffered system at pD 7.0 (ca. 40 mM and 35 mM, respectively; see Fig. 6B and S24†). The LysOMe to +Lys− conversion triggered a similar constitutional rearrangement to that observed in the presence of buffer, but at subtly slower rates in this case. The equilibrium state of DCvL6 was characterized by the absence of A1LysOMe and the predominance of A1B2 (ca. 60 mM). During the DCvL5-DCvL6-Conversion, the pD of the system dropped 1.2 units as a result of the ester hydrolysis, reaching a final pD value of 7.1 (Fig. S25†). Thus, such noteworthy decrease in pD values regulated the expression of the A1+Lys− constituent (in terms of imine formation) and allowed for a sorted state with higher fidelity in DCvL6.
[1 × 3] DCvL. We decided to study the behaviour of related [1 × 3] systems on addition of a further adaptive step by combining the up-regulation of the unaffected constituent to the generation of a macroscopic property. To this end, a new family of amphiphilic pseudopeptidic compounds with different chain lengths (AlaNHCX, Scheme 1) was synthesized following reported procedures.52 These amines were selected since their aldimine derivatives formed upon reaction with a charged aldehyde (e.g., A1) should present a prominent tendency to self-assemble in water and form micelles.531H NMR at 80 mM indicated yields of ca. 50% for the imines A1AlaNHCX (see Table S12†). These values, despite being slightly lower than those of AAOMe under the same conditions, showed that the pseudopeptides were suitable for competition experiments. Moreover, the concentration of the aldimines A1AlaNHCX was not altered over time, indicating that the amide bond was stable to hydrolysis for at least 1 month.
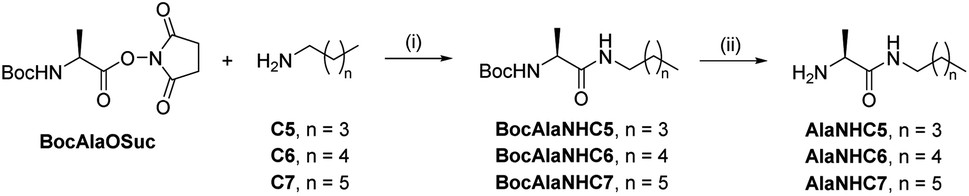 | ||
| Scheme 1 Two-step synthesis of amphiphilic pseudopeptidic compounds AlaNHCX. (i) DCM, r.t., 16 h. (ii) TFA, DCM, r.t., 3 h. | ||
The self-assembling properties of the different compounds were then evaluated by fluorescence emission spectroscopy using nile red (NR) as a hydrophobic probe.54 SDS was used as a control for the fluorescence emission values of a system containing micelles. The parent amines AlaNHCX barely formed micelles even at high concentrations (80 mM, Fig. S26†). In contrast, aldimine derivatives were found active in micellization. For instance, A1AlaNHC7 seemed to self-assemble into micellar structures, as evidenced by the intense fluorescence response observed (Fig. S27†). The CMC for A1AlaNHC7 was in the 7–12 mM range. DLS analyses of the A1AlaNHC7 sample at 12 mM revealed the presence of micelles with an average diameter of 7.9 nm (Fig. S28†).
The hydrolysis reactions occurring in the DCvL7 (A1, LysOMe, ArgOMe and AlaNHC7; 1 eq. each) should result in the decrease of A1LysOMe and A1ArgOMe together with an up-regulation of A1AlaNHC7 over time (Fig. 7A). When the concentration of A1AlaNHC7 increases from below to above CMC, the spontaneous formation of micelles (A1AlaNHC7-mic) should be noted in the solution. Reaction between A1:LysOMe:ArgOMe:AlaNHC7 (35 mM each) resulted in the DCvL7 with a constituent concentration of 10, 9, and 6 mM respectively for A1LysOMe, A1ArgOMe, and A1AlaNHC7. Since the concentration of A1AlaNHC7 was below the CMC, the relative fluorescence response observed was of 0.3, in good agreement with the absence of micelles.
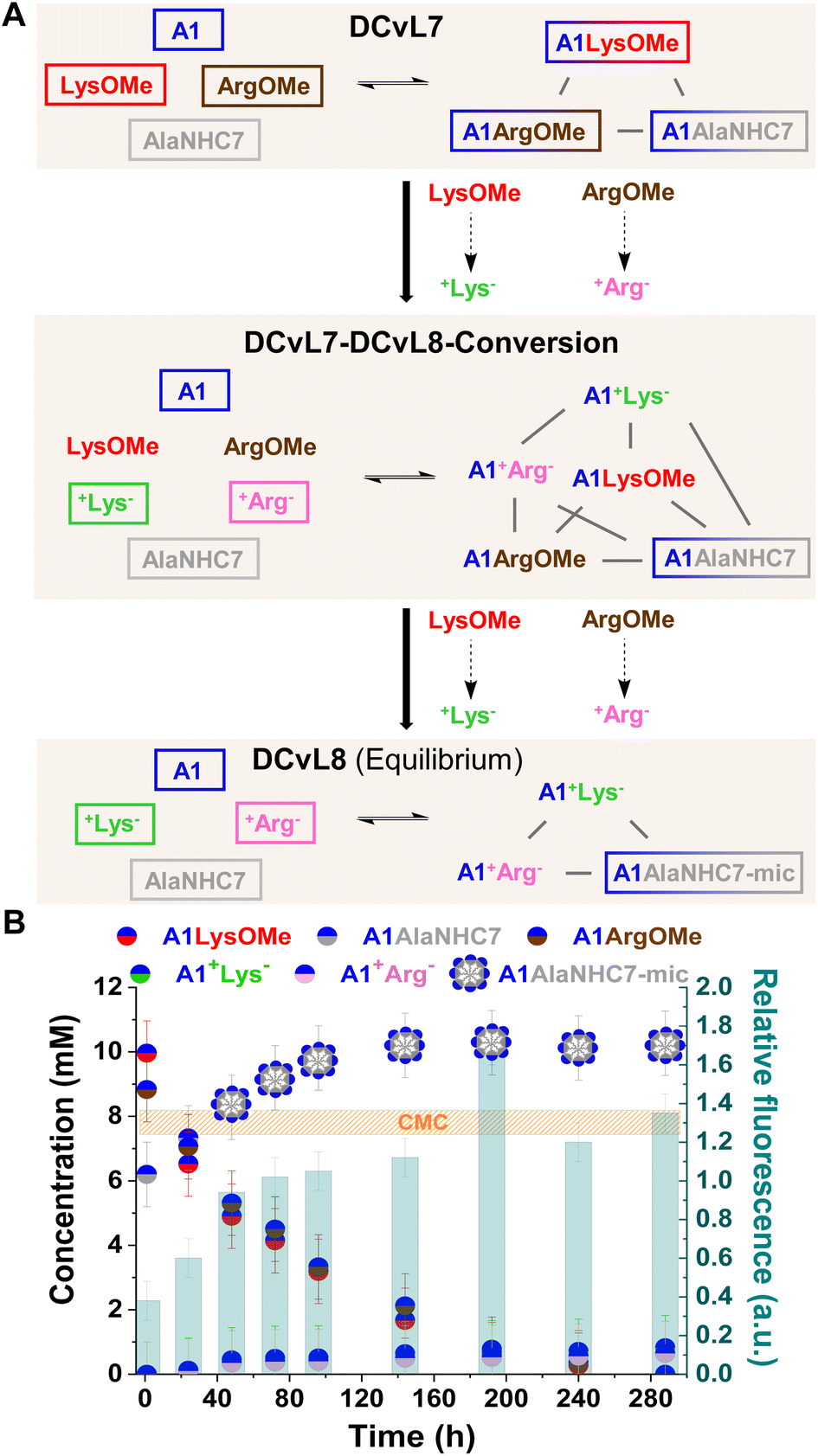 | ||
| Fig. 7 (A) Reaction between A1, LysOMe, ArgOMe, and AlaNHC7. DCvL7-DCvL8-Conversion (6 components, 5 constituents) from DCvL7 (4 components, 3 constituents) to DCvL8 (4 components, 3 constituents) has also been represented. Predominant compounds (>5 mM) are marked with a coloured rectangle. Grey lines correspond to antagonistic relationships; the black arrows represent the time evolution of DCvL7 to reach DCvL8. (B) Concentration profile for the different constituents detected in solution over 290 h of reaction. Concentrations determined by 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 500 mM, 293 K). Initial concentration of reagents: 35 mM. CMC determined by fluorescence spectroscopy. | ||
The hydrolysis of ArgOMe and LysOMe to their corresponding +AA− released A1 into the medium, as the hydrolysed amino acids were silent towards imination due to protonation. AlaNHC7 scavenged the generated aldehyde and led to an amplification of A1AlaNHC7. After 90 h of reaction, the concentration of A1AlaNHC7 was of ca. 10 mM, with a relative fluorescence emission >1 (Fig. 7B). Therefore, A1AlaNHC7 assemblies (namely A1AlaNHC7-mic) were present in the solution. Such high relative fluorescence values were maintained over time, indicating that the micelles were stable under the conditions assayed. DLS analyses for the final sample revealed the presence of micelles with a hydrodynamic diameter of 7.2 nm. The observed DCvL7-DCvL8-Conversion presented a higher complexity (6 components, 5 constituents) than the initial and final states (DCvL7 and DCvL8, respectively, both with 4 components and 3 constituents). The equilibrium distribution of the DCvL8 was of 10 mM A1AlaNHC7-mic, 1 mM A1+Lys−, 1 mM A1+Arg−, 25 mM A1, 34 mM +Lys− and 34 mM +Arg− (Fig. S29 and S30†). Therefore, DCvL7 experiences a triple-state adaptation: DCvL conversion + selective constituent amplification + formation of self-assembled dynamic micelles; resulting in the emergence of a property neither contained in the components nor in the initial library.
[2 × 2] DCvL. Finally, the adaptive behaviour of a [2 × 2] DCvL was also studied. As reported, [2 × 2] CDNs respond to constitutional relationships between species upon addition of certain effectors.55–57 The up-regulation of the most thermodynamically-favoured constituent of a network (e.g., AB) is accompanied by the amplification of its agonistic – and least fitted – A′B′ constituent, at expenses of the antagonistic AB′ and A′B constituents.7,26 We surmised that building-up a [2 × 2] library in which one of the competing amines is silenced by hydrolysis should result in a simultaneous increase in concentration of the two antagonistic constituents that are not hydrolysed.
The initial distribution of DCvL9, built-up from LysOMe, B2, A1 and A3 (Fig. 8A), revealed that the aldimines derived from A1 were present in higher concentrations (ca. 35 mM) than those of A3 (ca. 20 mM) as a result of its higher reactivity (see Fig. 2A and B). As the LysOMe ester hydrolysis proceeded, DCvL9 converted into DCvL10 through the higher-in-complexity DCvL9-DCvL10-Conversion, as seen from the 1H NMR spectra by the appearance of +Lys− signals. This hydrolysed component barely reacted with the aldehydes, resulting in the up-regulation of A1B2 and A3B2. Despite their antagonistic relationship -both constituents contain B2 as building block-, an increase in concentration of ca. 15 and 10 mM was noted for A1B2 and A3B2, respectively (Fig. 8B). The species distribution after 264 h of reaction was: 50 mM A1B2, 28 mM A3B2, 3 mM A1+Lys−, < 1 mM A3+Lys−, 29 mM A1, 48 mM A3, 77 mM +Lys− and 2 mM B2 (Fig. S31 and S32†). Thus, the DCvL conversion, triggered by ester hydrolysis, led to the constitutionally-disfavoured up-regulation of two antagonistic constituents in DCvL10 (see Fig. 8A). This behaviour resulted from the high degree of protonation of the competing amine (i.e., +Lys−) that stimulated the scavenging of A1 and A3 by B2.
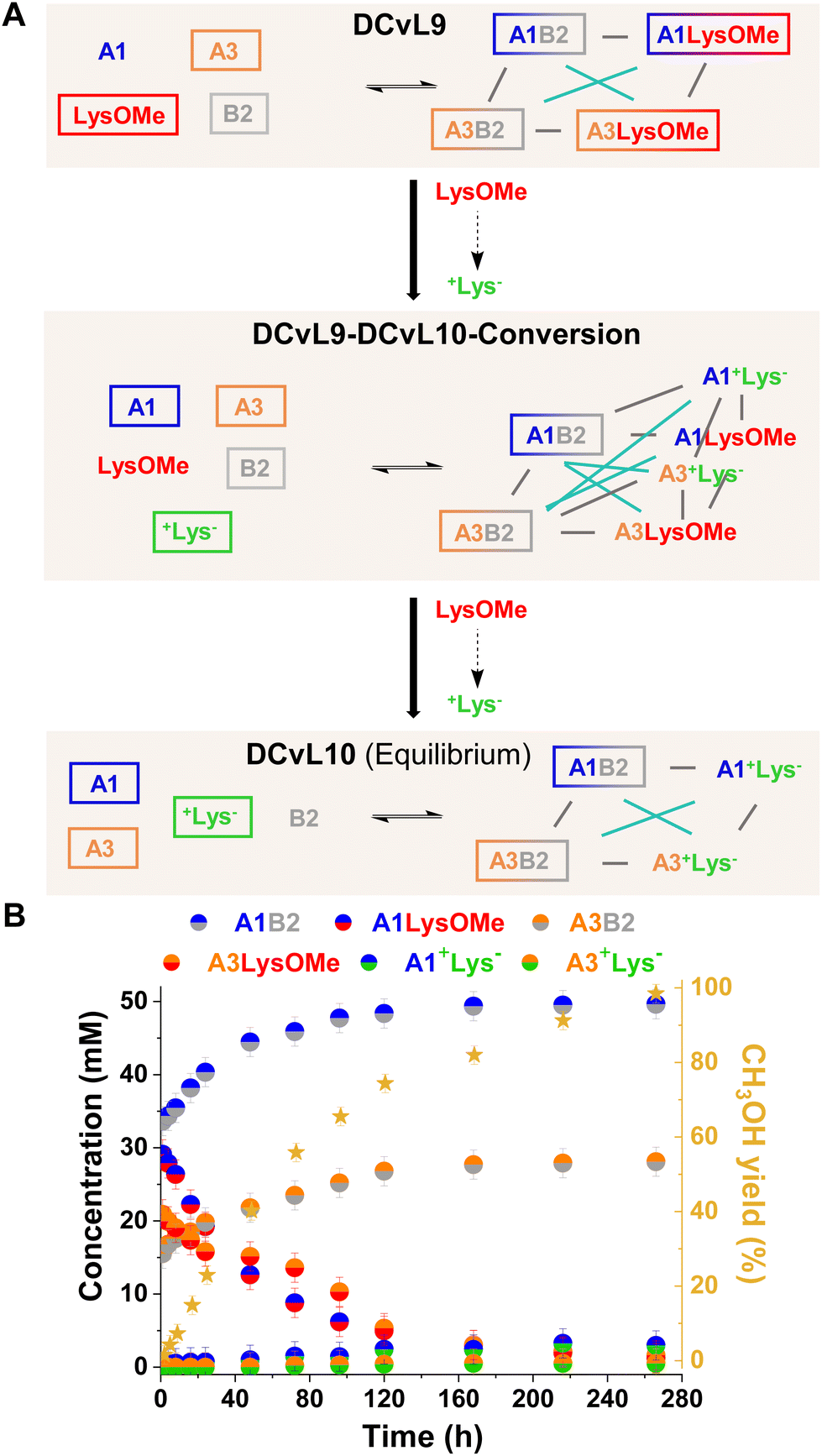 | ||
| Fig. 8 (A) Reaction between A1, A3, LysOMe, and B2. DCvL9-DCvL10-Conversion (5 components, 6 constituents) from DCvL9 (4 components, 4 constituents) to DCvL10 (4 components, 4 constituents) has also been represented. Predominant compounds (>15 mM) are marked with a coloured rectangle. Grey and cyan lines correspond to antagonistic and agonistic relationships, respectively; the black arrows represent the time evolution of DCvL9 to reach DCvL10. (B) Concentration profile for the different constituents detected in solution over 264 h of reaction. Concentration and yields determined by 1H NMR spectroscopy (500 MHz, D2O, pD = 7.0, PBS buffer 500 mM, 293 K). Initial concentration of reagents: 80 mM. | ||
Conclusions
The hydrolysis of different AAOMe constituents to their corresponding +AA− in a series of DCvLs affects imine formation with several aldehydes. This remote chemical modification increases the protonation degree of the Nα/β amino group due to the lower pKa of the hydrolysed species, thus hampering imine formation.Monitoring the reaction between LysOMe and A1 showed the expected depletion of A1LysOMe as +Lys− was produced, with the aldimine A1+Lys− not being observed due to the protonation of +Lys− (inhibited component). The kinetic simplification of two dynamic libraries (DCvL1 and DCvL3) in the presence of different enzymes (i.e., AChE, trypsin and chymotrypsin) was attained, with the catalysed remote chemical modification leading to selective silencing of components, sorted states and transient chirality. In [1 × 2] and [1 × 3] DCvLs, containing a component that was not hydrolysed (i.e., the scavenger B2 or AlaNHC7), the hydrolysis of the AAOMe resulted in the up-regulation of the constituents made of these components. Such DCvLs experienced a chemically-driven constitutional sorting, evolving from an almost statistical distribution (2 and 3 aldimines for DCvL5 and DCvL7, respectively) to a state characterized by the predominance of a single imine. In addition, the DCvL7 displayed a triple-state adaptation based on DCvL conversion, selective constituent up-regulation and supramolecular self-assembly with emergence of a macroscopic property that was not present in the initial library: the generation of micelles. A [2 × 2] library was also constructed in which one of the amino components was selectively poisoned by hydrolysis. The conversion from DCvL9 to DCvL10 resulted in a concentration increase for two constituents with an antagonistic relationship. Thus, all the DCvLs studied present transient states of higher constitutional complexity that adapt and evolve with time to states of higher “simplexity”. In addition, the interplay between the action of different enzymes and the libraries dictates the selectivity and the lifetime of the transient sorted states (up-regulated constituent and/or chirality).
In a broader sense, such adaptive responses in simple dynamic networks offer an evolutionarily relevant scenario were a remote side-reaction triggered by the environment (i.e., solvent) amplifies the population of the “surviving” constituents at the expense of the “poisoned” ones which undergo the detrimental chemical modification. These results will pave the way for the design of related dynamic systems of higher functionality with emerging functions. Furthermore, the observed silencing process based on reactive site protonation showcases the mode-of-action found in some proteins for the regulation of their activity through specific PTMs that lead to pKa modulations. Therefore, this work will also contribute to our knowledge on sophisticated biological mechanisms governed by supramolecular cross-talks between remote sites.
Data availability
Data available upon request from the authors.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Conceptualization: F. E., J. M. L. Data curation: F. E., T. R. Formal analysis: F. E. Funding acquisition: J. M. L. Investigation: T. R. (supporting), F. E. (lead). Methodology: F. E. Project administration: J. M. L. Resources: J. M. L. Supervision: F. E., J. M. L. Validation: F. E. Visualization: F. E. Writing original draft: F. E. Writing – review and editing: T. R. (supporting), F. E., J. M. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC (Advanced Research Grant SUPRADAPT 290585), the University of Strasbourg Institute for Advanced Study (USIAS), and Fondation Jean-Marie Lehn. F. E. acknowledges Fundación Ramón Areces for a postdoctoral fellowship. T. R. thanks the CSC Graduate School funded by the French National Research Agency (CSC-IGS ANR-17-EURE-0016) for a PhD fellowship. The authors thank Dr J. L. Schmitt, Dr A. Osypenko, Dr Z. Yang, Dr S. Jung, B. Kozibroda, M. J. Aguilera, and C. Antheaume for helpful discussions.Notes and references
- W. P. Jencks, Catalysis in Chemistry and Enzymology, McGraw-Hill, New York, 1969 Search PubMed.
- V. Csizmok and J. D. Forman-Kay, Complex regulatory mechanisms mediated by the interplay of multiple post-translational modifications, Curr. Opin. Struct. Biol., 2018, 48, 58–67 CrossRef CAS PubMed.
- Z. A. Wang and P. A. Cole, The Chemical Biology of Reversible Lysine Post-translational Modifications, Cell Chem. Biol., 2020, 27, 953–969 CrossRef CAS PubMed.
- A. V. Probst, E. Dunleavy and G. Almouzni, Epigenetic inheritance during the cell cycle, Nat. Rev. Mol. Cell Biol., 2009, 10, 192–206 CrossRef CAS PubMed.
- D. G. Isom, C. A. Castañeda, B. R. Cannon and B. García-Moreno, Large shifts in pKa values of lysine residues buried inside a protein, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- A. Schonichen, B. A. Webb, M. P. Jacobson and D. L. Barber, Considering Protonation as a Post-translational Modification Regulating Protein Structure and Function, Annu. Rev. Biophys., 2013, 42, 289–314 CrossRef CAS PubMed.
- J.-M. Lehn, Constitutional Dynamic Chemistry, Top. Curr. Chem., 2012, 322, 1–32 CAS.
- J.-M. Lehn, Perspectives in Chemistry-Steps towards Complex Matter, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Disulfide exchange: exposing supramolecular reactivity through dynamic covalent chemistry, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- K. Acharyya and P. S. Mukherjee, Organic Imine Cages: Molecular Marriage and Applications, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed.
- B. T. Herrera, S. R. Moor, M. McVeigh, E. K. Roesner, F. Marini and E. V. Anslyn, Rapid Optical Determination of Enantiomeric Excess, Diastereomeric Excess, and Total Concentration Using Dynamic-Covalent Assemblies: A Demonstration Using 2-Aminocyclohexanol and Chemometrics, J. Am. Chem. Soc., 2019, 141, 11151–11160 CrossRef CAS PubMed.
- A. Chevalier, A. Osypenko, J.-M. Lehn and D. Meyer, Phase transfer of metal cations by induced dynamic carrier agents: biphasic extraction based on dynamic covalent chemistry, Chem. Sci., 2020, 11, 11468–11477 RSC.
- B. Liu, M. A. Beatty, C. G. Pappas, K. Liu, J. Ottelé and S. Otto, Self-Sorting in Dynamic Combinatorial Libraries Leads to the Co-Existence of Foldamers and Self-Replicators, Angew. Chem., Int. Ed., 2021, 60, 13569–13573 CrossRef CAS PubMed.
- R. C. Lirag, K. Osowska and O. S. Miljanic, Precipitation-driven self-sorting of imines, Org. Biomol. Chem., 2012, 10, 4847–4850 RSC.
- K. Osowska and O. S. Miljanic, Self-sorting of dynamic imine libraries during distillation, Angew. Chem., Int. Ed., 2011, 50, 8345–8349 CrossRef CAS PubMed.
- C.-W. Hsu and O. S. Miljanic, Adsorption-driven self-sorting of dynamic imine libraries, Angew. Chem., Int. Ed., 2015, 54, 2219–2222 CrossRef CAS PubMed.
- K. Osowska and O. S. Miljanic, Oxidative kinetic self-sorting of a dynamic imine library, J. Am. Chem. Soc., 2011, 133, 724–727 CrossRef CAS PubMed.
- S. Gambaro, C. Talotta, P. Della Sala, A. Soriente, M. De Rosa, C. Gaeta and P. Neri, Kinetic and Thermodynamic Modulation of Dynamic Imine Libraries Driven by the Hexameric Resorcinarene Capsule, J. Am. Chem. Soc., 2020, 142, 14914–14923 CrossRef CAS PubMed.
- F. Schaufelberger and O. Ramström, Kinetic Self-Sorting of Dynamic Covalent Catalysts with Systemic Feedback Regulation, J. Am. Chem. Soc., 2016, 138, 7836–7839 CrossRef CAS PubMed.
- P. Vongvilai, M. Angelin, R. Larsson and O. Ramström, Dynamic Combinatorial Resolution: Direct Asymmetric Lipase-Mediated Screening of a Dynamic Nitroaldol Library, Angew. Chem., Int. Ed., 2007, 46, 948–950 CrossRef CAS.
- G. Men and J.-M. Lehn, Multiple adaptation of constitutional dynamic networks and information storage in constitutional distributions of acylhydrazones, Chem. Sci., 2019, 10, 90–98 RSC.
- S. Wang, L. Yue and I. Willner, Enzyme-Guided Selection and Cascaded Emergence of Nanostructured Constitutional Dynamic Networks, Nano Lett., 2020, 20, 5451–5457 CrossRef CAS PubMed.
- C. Wang, L. Yue and I. Willner, Controlling biocatalytic cascades with enzyme–DNA dynamic networks, Nat. Catal., 2020, 3, 941–950 CrossRef CAS.
- C. Wang, M. P. O'Hagan, E. Neumannn, R. Nechushtai and I. Willner, Integration of photocatalytic and dark-operating catalytic biomimetic transformations through DNA-based constitutional dynamic networks, Nat. Commun., 2021, 12, 4224 CrossRef CAS.
- Y. Ouyang, P. Zhang and I. Willner, Dynamic Catalysis Guided by Nucleic Acid Networks and DNA Nanostructures, Bioconjugate Chem., 2023, 34, 51–69 CrossRef CAS PubMed.
- L. Yue, S. Wang, Z. Zhou and I. Willner, Nucleic Acid Based Constitutional Dynamic Networks: From Basic Principles to Applications, J. Am. Chem. Soc., 2020, 142, 21577–21594 CrossRef CAS PubMed.
- Calculated using MarvinSketch; see Table S1† for predicted pKa values and –Nα/βH2 protonation degree (%) of AAOMe and +AA−.
- A. Osypenko, R. Cabot, J. J. Armao IV, P. Kovaricek, A. Santoro and J.-M. Lehn, Behavior of Constitutional Dynamic Networks: Competition, Selection, Self-sorting in Cryptate Systems, ChemistryEurope, 2023, 1, e202300017 CrossRef.
- M. S. Li, Y. W. Dong, M. Quan and W. Jiang, Stabilization of Imines and Hemiaminals in Water by an Endo-Functionalized Container Molecule, Angew. Chem., Int. Ed., 2022, 61, e202208508 CrossRef CAS PubMed.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, Structure-Stability Correlations for Imine Formation in Aqueous Solution, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- A. Bencini, A. Bianchi, E. García-España, M. Micheloni and J. A. Ramirez, Proton coordination by polyamine compounds in aqueous solution, Coord. Chem. Rev., 1999, 188, 97–156 CrossRef CAS.
- P. A. Khalaf-Alla, M. M. Shoukry, A. A. Jbarah and R. van Eldik, Base hydrolysis of α-amino acid esters catalysed by [Pd(N-ethylethylenediamine)(H2O)2]2+. Kinetic study and DFT calculations, Inorg. Chim. Acta, 2017, 458, 181–189 CrossRef CAS.
- Considering that pH = pD + 0.4, the pH values monitored were: 6.6, 7.4, and 8.2. These are the values at which the –Nα/βH2 degree of protonation was calculated (Table S2†).
- J. Kyte and R. F. Doolittle, A simple method for displaying the hydropathic character of a protein, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
- M. Tanihara and Y. Imanishi, Highly Efficient and Enantiomer-Selective Hydrolysis of α-Amino Acid Active Ester Hydrochlorides by a Cyclic Hexapeptide Containing Histidines, Polym. J., 1983, 15, 499–507 CrossRef CAS.
- T. Takahashi, Y. Yamazaki and K. Kato, Substrate Specificity of an a-Amino Acid Ester Hydrolase Produced by Acetobacter turbidans A.T.C.C. 9325, Biochem. J., 1974, 137, 497–503 CrossRef CAS PubMed.
- T. Bunyapaiboonsri, O. Ramström, S. Lohmann, J.-M. Lehn and M. Goeldner, Dynamic Deconvolution of a Pre-Equilibrated Dynamic Combinatorial Library of Acetylcholinesterase Inhibitors, ChemBioChem, 2001, 2, 438–444 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. Van Esch, Transient assembly of active materials fueled by a chemical reaction, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- M. Tena-Solsona, B. Rieb, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Non-equilibrium dissipative supramolecular materials with a tunable lifetime, Nat. Commun., 2017, 8, 15895 CrossRef CAS PubMed.
- B. Rieb, C. Wanzke, M. Tena-Solsona, R. K. Grötsch, C. Maity and J. Boekhoven, Dissipative assemblies that inhibit their deactivation, Soft Matter, 2018, 14, 4852–4859 RSC.
- A. M. Bergmann, J. Bauermann, G. Bartolucci, C. Donau, M. Stasi, L. Holtmannspötter, F. Jülicher, C. A. Weber and J. Boekhoven, Liquid spherical shells are a non-equilibrium steady state of active droplets, Nat. Commun., 2023, 14, 6552 CrossRef CAS PubMed.
- In a system where the imine reactions do not have such low yields, the up-regulation will be more pronounced.
- A. Inamoto, K. Ogasawara, K. Omata, K. Kabuto and Y. Sasaki, Samarium(III)-propylenediaminetetraacetate complex: A water-soluble chiral shift reagent for use in high-field NMR, Org. Lett., 2000, 23, 3543–3545 CrossRef PubMed.
- Y. Sakata, S. Chiba and S. Akine, Transient chirality inversion during racemization of a helical cobalt(III) complex, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2113237119 CrossRef CAS PubMed.
- A. Kamal, M. A. Azhar, T. Krishnaji, M. S. Malik and S. Azeeza, Approaches based on enzyme mediated kinetic to dynamic kinetic resolutions: A versatile route for chiral intermediates, Coord. Chem. Rev., 2008, 252, 569–592 CrossRef CAS.
- M. D. Greenhalgh, J. E. Taylor and A. W. Smith, Best practice considerations for using the selectivity factor, s, as a metric for the efficiency of kinetic resolutions, Tetrahedron, 2018, 74, 5554–5560 CrossRef CAS.
- P. Kovaricek, A. C. Meister, K. Flidrova, R. Cabot, K. Kovarickova and J.-M. Lehn, Competition-driven selection in covalent dynamic networks and implementation in organic reactional selectivity, Chem. Sci., 2016, 7, 3215–3226 RSC.
- Z. Yang, F. Esteve, C. Antheaume and J.-M. Lehn, Dynamic covalent self-assembly and self-sorting processes in the formation of imine-based macrocycles and macrobicyclic cages, Chem. Sci., 2023, 14, 6631–6642 RSC.
- P. Howlader and P. S. Mukherjee, Face and edge directed self-assembly of Pd12 tetrahedral nano-cages and their self-sorting, Chem. Sci., 2016, 7, 5893–5899 RSC.
- V. Abet, F. T. Szczypinski, M. A. Little, V. Santolini, C. D. Jones, R. Evans, C. Wilson, X. Wu, M. F. Thorne, M. J. Bennison, P. Cui, A. I. Cooper, K. E and A. G. Slater, Inducing Social Self-Sorting in Organic Cages To Tune The Shape of The Internal Cavity, Angew. Chem., Int. Ed., 2020, 59, 16755–16763 CrossRef CAS PubMed.
- A. Kumar and P. S. Mukherjee, Multicomponent Self-Assembly of PdII/PtII Interlocked Molecular Cages: Cage-to-Cage Conversion and Self-Sorting in Aqueous Medium, Chem.–Eur. J., 2020, 26, 4842–4849 CrossRef CAS PubMed.
- M. Allan, S. Manku, E. Therrien, N. Nguyen, S. Styhler, M.-F. Robert, A.-C. Goulet, A. J. Petschner, G. Rahil, A. R. MacLeod, R. Déziel, J. M. Besterman, H. Nguyen and A. Wahhab, N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1), Bioorg. Med. Chem. Lett., 2009, 19, 1218–1223 CrossRef CAS PubMed.
- C. B. Minkenberg, L. Florusee, R. Eelkema, G. J. M. Koper and J. H. van Esch, Triggered Self-Assembly of Simple Dynamic Covalent Surfactants, J. Am. Chem. Soc., 2009, 131, 11274–11275 CrossRef CAS PubMed.
- M. C. A. Stuart, J. C. van de Pas and J. B. F. N. Engberts, The use of Nile Red to monitor the aggregation behavior in ternary surfactant–water–organic solvent systems, J. Phys. Org. Chem., 2005, 18, 929–934 CrossRef CAS.
- M. He and J.-M. Lehn, Time-dependent switching of constitutional dynamic libraries and networks from kinetic to thermodynamic distributions, J. Am. Chem. Soc., 2019, 141, 18560–18569 CrossRef CAS PubMed.
- J.-F. Ayme, S. Dhers and J.-M. Lehn, Triple Self-Sorting in Constitutional Dynamic Networks: Parallel Generation of Imine-Based CuI, FeII, and ZnII Complexes, Angew. Chem., Int. Ed., 2020, 59, 12484–12492 CrossRef CAS PubMed.
- J.-F. Ayme and J.-M. Lehn, Self-sorting of two imine-based metal complexes: balancing kinetics and thermodynamics in constitutional dynamic networks, Chem. Sci., 2020, 11, 1114–1121 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, main text support figures and tables, copies of 1H, 13C NMR, and HRMS spectra. See DOI: https://doi.org/10.1039/d4sc01288g |
| This journal is © The Royal Society of Chemistry 2024 |