
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation and reactivity of a unique M⋯C–H interaction stabilized by carborane cages†
Xin-Ran
Liu‡
a,
Peng-Fei
Cui‡
a,
Yago
García-Rodeja
b,
Miquel
Solà
 b and
Guo-Xin
Jin
*a
b and
Guo-Xin
Jin
*a
aState Key Laboratory of Molecular Engineering of Polymers, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Department of Chemistry, Fudan University, 2005 Songhu Road, Shanghai, 200433, P. R. China. E-mail: gxjin@fudan.edu.cn
bInstitut de Química Computacional i Catàlisi, Departament de Química, Universitat de Girona, C/Maria Aurèlia Capmany, 69, 17003 Girona, Spain
First published on 16th May 2024
Abstract
Broadening carborane applications has consistently been the goal of chemists in this field. Herein, compared to alkyl or aryl groups, a carborane cage demonstrates an advantage in stabilizing a unique bonding interaction: M⋯C–H interaction. Experimental results and theoretical calculations have revealed the characteristic of this two-center, two-electron bonding interaction, in which the carbon atom in the arene ring provides two electrons to the metal center. The reduced aromaticity of the benzene moiety, long distance between the metal and carbon atom in arene, and the upfield shift of the signal of M⋯C–H in the nuclear magnetic resonance spectrum distinguished this interaction from metal⋯C π interaction and metal–C(H) σ bonds. Control experiments demonstrate the unique electronic effects of carborane in stabilizing the M⋯C–H bonding interaction in organometallic chemistry. Furthermore, the M⋯C–H interaction can convert into C–H bond metallization under acidic conditions or via treatment with t-butyl isocyanide. These findings deepen our understanding regarding the interactions between metal centers and carbon atoms and provide new opportunities for the use of carboranes.
Introduction
Icosahedral carboranes (C2B10H12) are carbon–boron molecular clusters,1 serving as useful building blocks in various applications.2–7 The carborane substituents have been widely used to tune ligand properties. Because of the unique electronic effects of carboranes,8–12 they are often recognized as three-dimensional inorganic benzene analogs.13,14 Based on this property, many studies have explored compounds such as carborynes (1,2-dehydro-ortho-carboranes),15 transition metal–carboryne complexes,16 and o-carborane-fused borirans.17 However, being a large steric, electron-deficient group, carborane also exhibits considerable differences compared to the benzene ring. These differences contribute to the stabilization of radicals18–20 and intermediates.21 However, whether carborane can stabilize specific bonding interactions is yet to be discovered. Currently, considerable effort in carborane chemistry is primarily devoted to the research of metal centers and B–H bonds.22–27 The advantages of carborane itself in stabilizing special interactions, especially the metal⋯C–H interaction, are unclear and under investigation.Because species containing a metal–arene bonding interaction are known to be the key intermediates in synthetic chemistry,28–30 considerable efforts have been devoted to understanding this bonding interaction.31,32 Among these species, the metal⋯C–H interaction of arenes is easily confused with other interactions, such as metal⋯C π interactions,33,34 metal–C(H) σ bonds (Wheland intermediate),29,35–37 metal⋯H–C agostic interactions,38–40 and the van der Waals interactions41 (Chart 1). Structural similarity and unclear concept of metal⋯C–H interaction are the root causes of this confusion. Moreover, the unique M⋯C–H interaction is unstable and can easily metallize the C–H bond, making its stabilization more difficult. Therefore, the M⋯C–H interaction is mainly stabilized by additional intramolecular restraints. Such restraints can be introduced by incorporating the arene moiety into an appropriately sized macrocycle.41,42 However, it is unclear whether the metal⋯C–H interaction can be stabilized by regulating the intramolecular electronic effect.42 A clear definition of this bonding interaction has also not yet been proposed.
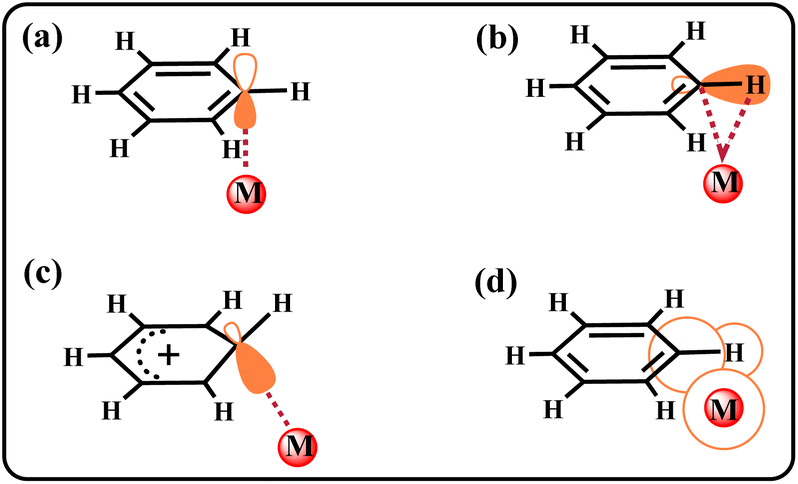 | ||
| Chart 1 Examples of bonding interactions between a metal centre and arenes. The (a) metal⋯C π interaction, (b) metal⋯H–C agostic interaction, (c) metal–C(H) σ bond, and (d) van der Waals interaction. | ||
To investigate the stabilizing effect and bonding characteristics of the metal⋯C–H interaction, tridentate coordinating pincer-type ligands containing carborane, phenyl, or n-butyl groups were designed and synthesized. The results reveal that the special electronic effects of the carborane cage can stabilize this bonding interaction. Theoretical calculations were also performed to gain insights into the bonding model of this special metal⋯C–H interaction. Further investigations indicate that the unique metal⋯C–H interaction can achieve C–H bond activation under acidic conditions or in the presence of the strong coordination ligand t-butyl isocyanide (CN–But). These findings clearly define the characteristics of the metal⋯C–H interaction and broaden the prospect of carboranes in stabilizing special bonding interactions.
Results and discussion
Generally, the C–H bond of benzene in pincer-type ligands is easily activated by transition metals owing to strong chelation.43 Therefore, obtaining the metal⋯C–H bonding interaction in such complexes is difficult. To address this challenge, different pincer-type ligands were designed and synthesized (for more details, see the ESI†). Further, the reactions of the synthesized pincer-type ligands with [Cp*MCl2]2 (M = Ir or Rh) were explored (Scheme 1). | ||
| Scheme 1 Synthesis of complexes 1, 2, and 3. | ||
First, when ligand 1 was treated with [Cp*IrCl2]2 (0.5 equiv.), followed by the addition of AgOTf and potassium bis(trimethylsilyl)amide (KMDH), the 1H nuclear magnetic resonance (NMR) spectrum of complex 1 showed no singlet peak corresponding to the C5–H proton and the Cp* signal was observed at 1.64 ppm (Fig. S1†). This indicated that the C5–H bond on the benzene ring was directly activated. The 19F NMR spectrum of complex 1 showed a signal at −78.35 ppm (Fig. S6†), indicating the formation of an ionic complex; this result was also supported by electrospray ionization mass spectrometry (ESI-MS) analysis [(M − OTF)+ = 675.1479; calc. 675.1456; Fig. S7†]. These results confirm the metallization of the C5–H bond. After failing to achieve the metal⋯C–H interaction using ligand 1, it was determined if the interaction could be achieved by altering the electronic effects of the R group presented in Scheme 1. Then, ligand 2 was designed, where phenyl was replaced with n-butyl. However, a similar reaction as in case of ligand 1 occurred, forming complex 2. The structure of complex 2 was further confirmed via NMR and ESI-MS analysis (Fig. S8, S10 and S14†). The signals of Cp* and the N–H proton were observed at 1.52 and 10.94 ppm in the 1H NMR spectrum of complex 2 (Fig. S8†), respectively, indicating the presence of a C5–H bond-metallized complex such as complex 1. The ESI-MS analysis results [(M − OTF)+ = 635.2096; calc. 635.2083; Fig. S14†] also supported the formation of complex 2. These results indicate that the C–H bond of the benzene ring is easily activated in such a coordinated model.
To achieve the metal⋯C–H interaction, the charge density at the iridium center needs to be modified. Because phenyl and n-butyl can be considered as electron-donating groups, the effect of an electron-withdrawing group on the stability of the metal⋯C–H interaction needs to be explored. The carborane cage is a very competitive candidate as an electron-withdrawing group because of the electron deficiency of the B atom. Currently, carborane is used to stabilize radical complexes because of its large steric hindrance and unique electronic effects, but its use for stabilizing special bonding interactions is challenging. Ligand 3 was designed and reacted with [Cp*IrCl2]2 (0.5 equiv.) under conditions similar to those of ligands 1 and 2 (Scheme 1 and Fig. 1). The 1H NMR spectrum of complex 3a shows a singlet corresponding to the C5–H proton, and this peak shifted upfield (from δ = 7.51 to 5.66 ppm) from the normal region for aryl protons in complex 3a (Fig. 1c and d). This indicates that the metal⋯C–H interaction is observed in complex 3a. The 13C NMR spectra of complex 3a also showed that the signal of C5 appeared at δ = 41.71 ppm, which is in contrast to the other carbon atoms in the benzene ring, exhibiting peaks at 118.83–159.92 ppm (Fig. S29†). No signal was observed in the 19F NMR spectrum of complex 3a, indicating the formation of neutral complex 3a, consistent with the ESI-MS results [(M + H)+ = 807.4387; calc. 807.4412; Fig. 1b]. These results support the presence of a strong interaction between the metal center and the carbon atom in arene. Notably, van der Waals interactions and metal⋯C π interactions do not exhibit this phenomenon.44 Therefore, the NMR signal is one of the characteristic signals indicating the presence of the metal⋯C–H interaction. Furthermore, the NMR signals of the protons of C(7,9)–H were also shifted upfield from δ = 7.52 (in ligand 1) to 6.59 ppm (Fig. 1c and d). Additionally, cyclohexadienyl cations usually exhibit an absorption band at ∼420 nm in ultraviolet-visible (UV-vis) spectra;45 however, no corresponding resonance was observed for complex 3a (Fig. S34†). This shows the difference between the M⋯C–H interaction and the M–C(H) σ bond. As mentioned above, the metal⋯C–H interaction exhibits unique characteristics and may be an important intermediate in synthetic chemistry. Moreover, it is easily confused with other similar bonding interactions. It was assumed whether it was possible to come to an inductive definition of this bond, which requires as complete a characterization as possible.
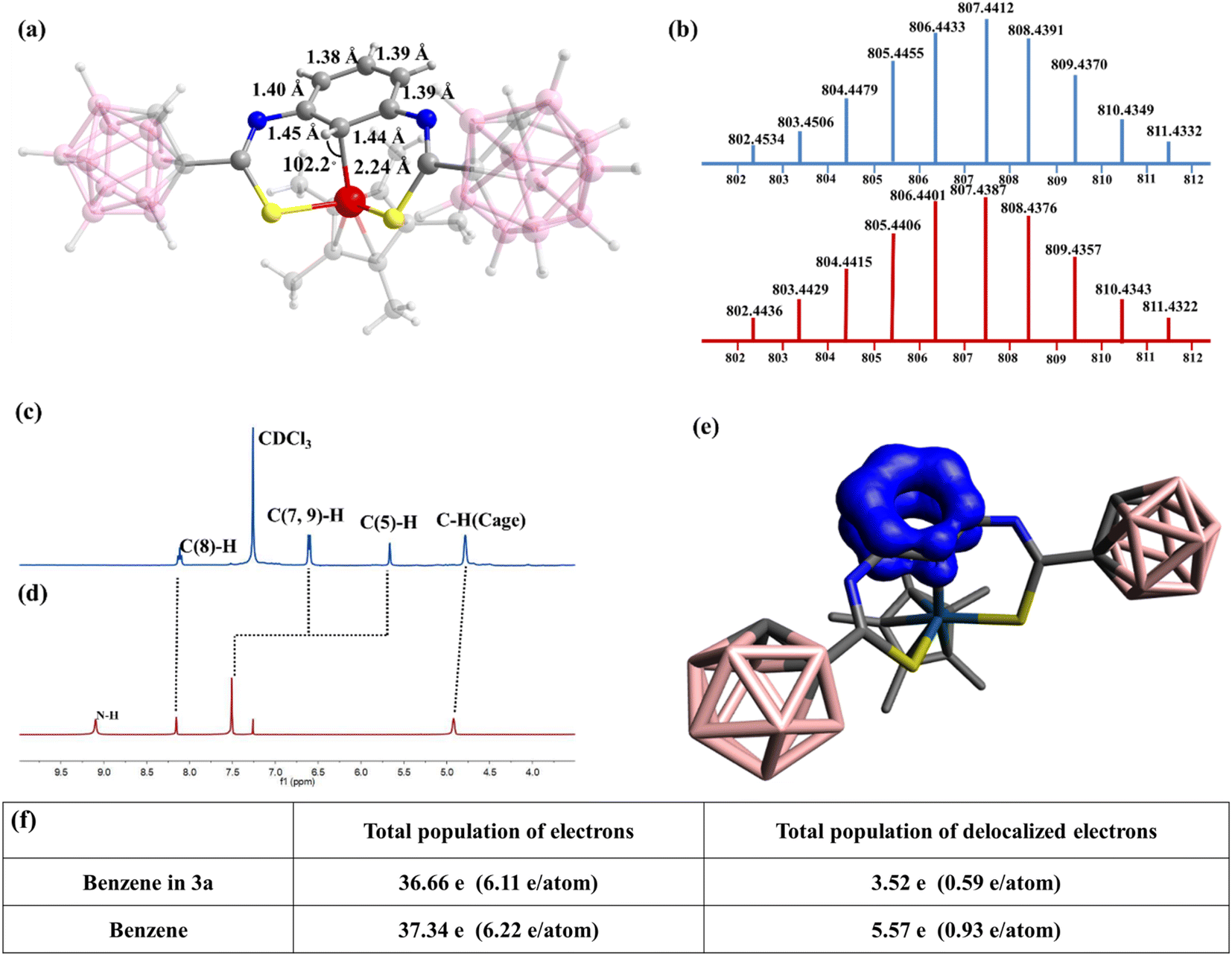 | ||
| Fig. 1 (a) Molecular structure of complex 3a: colour codes: Ir, red; S, yellow; N, blue; C, gray; B, pink; and H, light gray. The numbers indicate the bond lengths in the benzene ring and the angle of Ir–C5–H. (b) ESI-MS of complex 3a (bottom: experimental; top: theoretical). Comparison of partial 1H NMR (CDCl3, 298 K) spectra of complex 3a (c) and ligand 3 (d). The numbers indicate the carbon number on the benzene ring. (e) EDDB analysis of the complex 3a and benzene ring. (f) Comparison of the total population of electrons (from EDDB charges in electrons) and delocalized electrons in benzene and in the benzene ring of complex 3a. | ||
The X-ray single-crystal diffraction data of complex 3a were collected. The structure of complex 3a shows that the metal center remains trivalent and is coordinated with two S atoms, a Cp* ring, and a C5 atom (Fig. 1a). It indicates that the valence state of the benzene ring does not change in M⋯C–H bonding type. However, as for M–C(H) σ bonding type, the carbon atom can be viewed as sp3 carbon and the complex is a σ complex or Wheland intermediate. Thus, the benzene ring should be positive.29,45 The lengths of the C5–C6 and C5–C4 bonds in 3a are 1.45 and 1.44 Å, respectively, which are slightly longer than those of the other four bonds (∼1.39 Å) (Fig. 1a). However, the lengths of the C5–C6 and C5–C4 bonds do not correspond to the length of a C–C single bond (1.54 Å). Additionally, the six carbon atoms in the benzene ring remain planar (but not in the M–C (H) σ bonding type29,35) and the angle of H–C5–Ir is 102.2° (Fig. 1a). These results suggest a considerable but not complete disruption of the aromaticity in the benzene ring of 3a. Given the partial preservation of aromaticity, the Ir–C5 bond is expected to be somewhat longer than a comparable Ir–C σ bond. In line with this hypothesis, the observed Ir–C5 length of 2.24 Å (Fig. 1a) is consistent with the lengthening of the Ir–C σ bond (2.05 Å).
Natural bond order (NBO) analysis (Table S1†) verified the role of carboranes in stabilizing the M⋯C–H interaction. Because of the electron-withdrawing nature of the carborane cage, sulphur atoms exhibited lower charges (−0.089e and −0.121e) in ligand 3 than in ligands 1 and 2, where S atoms had a charge of approximately −0.2e. Therefore, in ligands 1 and 2, the lone pairs of electrons on the S atoms were more available for dative bonding compared with ligand 3. These differences favoured the formation of a neutral structure for ligand 3 and cationic structures for ligands 1 and 2. This is corroborated by the calculated Gibbs energies of the following reaction (see Fig. S66†):
| Complex n + NEt3H+ → complex np + NEt3 | (1) |
For ligand 3, the C–H activation from complex 3a is endergonic by 12.50 kcal mol−1, whereas for ligands 1 and 2, it is exergonic by 7.01 and 6.04 kcal mol−1, respectively.
Density functional theory calculations provide insights into this bonding interaction. The three-center electron sharing index (3c-ESIs) value is close to zero, which indicates the absence of a three-centre bond in complex 3a (see Table S4†).46 Therefore, almost no interaction occurs between Ir and the H atom on C5. This illustrates that the M⋯C–H interaction is a two-centre bond, which differs from the three-center bond, metal⋯H–C agostic interaction. Table S4† gathers the results for complex 3a and other types of interactions. M⋯C–H bond complexes are characterized by a relatively large M–C and a C–H bond order, a M–H bond order close to zero, and a relatively small 3c-ESI. The electron density of delocalized bonds (EDDB) was computed to determine the influence of M⋯C–H interaction on the aromaticity of the benzene ring.47 The delocalized electrons decreased from 5.6e for the benzene ring to 3.5e for complex 3a (Fig. 1e and f and Table S2†). Moreover, the computed out-of-plane component of the nucleus-independent chemical shift (NICS(1)zz) corroborate the results obtained by EDDB, with NICS(1)zz = −19.9 ppm for complex 3a and −27.7 for benzene (Table S3†). Therefore, the aromaticity of the benzene moiety is reduced but not completely disrupted. This is consistent with the aforementioned experimental results. To further understand the role of the C5 atom in this bonding interaction, NBO analysis was used,48 which indicates that the Ir–C5 bond is formed through the interaction between the p-orbital of the C5 atom and the d-orbital (5dz2) of the Ir. With this interaction, the C5 atom can be viewed as providing a pair of electrons for iridium to satisfy the 18-electron rule. So far, the basic characteristics of the M⋯C–H interaction have been clearly characterized. Its characteristics can be summarized as follows: (1) the M⋯C–H interaction is a two-centre, two-electron bond, and the C atom provides a pair of electrons to the metal centre. (2) The aromaticity of the benzene moiety is reduced but not completely disrupted, causing a slight increase in the C–C bond length in the arene ring and metal–C interaction, but the six carbon atoms in the benzene ring remain planar. (3) Owing to the strong interaction between the metal centre and carbon atom in arene, the signal of M⋯C–H in the NMR spectrum shifts considerably upfield. These characteristics provide a way for identifying the M⋯C–H interaction. Similarly, this interaction can also be formed by reacting [Cp*RhCl2]2 (0.5 equiv.) and ligand 3 (Scheme 1). The characteristics of complex 3b have been described in detail in the ESI (Fig. S35–S42 and S69†).
Then, the ligand 4 was designed and synthesized to observe the effect of substituents on the benzene ring. The reaction of Ligand 4 with [Cp*IrCl2]2 (1.5 equiv.) to form complex 4 in CH2Cl2 was explored (Fig. 2a). The ESI-MS analysis shows the synthesis of trinuclear complex 4 ((M + H)+ = 1659.7423; calc. 1659.7402) (Fig. 2b). The 1H NMR spectrum of complex 4 also exhibits a singlet at δ = 5.59 ppm (Fig. S43†), indicating M⋯C–H interaction formation. The X-ray single-crystal diffraction data of complex 4 confirm the unusual bonding pattern between the Ir center and C atom on one side and metallization of the B(3, 4) sites of the carborane cage on the other side (Fig. 2c). These results demonstrate that an M⋯C–H interaction can be stabilized by the carborane cage and is not affected by substituents on the para-position of the benzene ring.
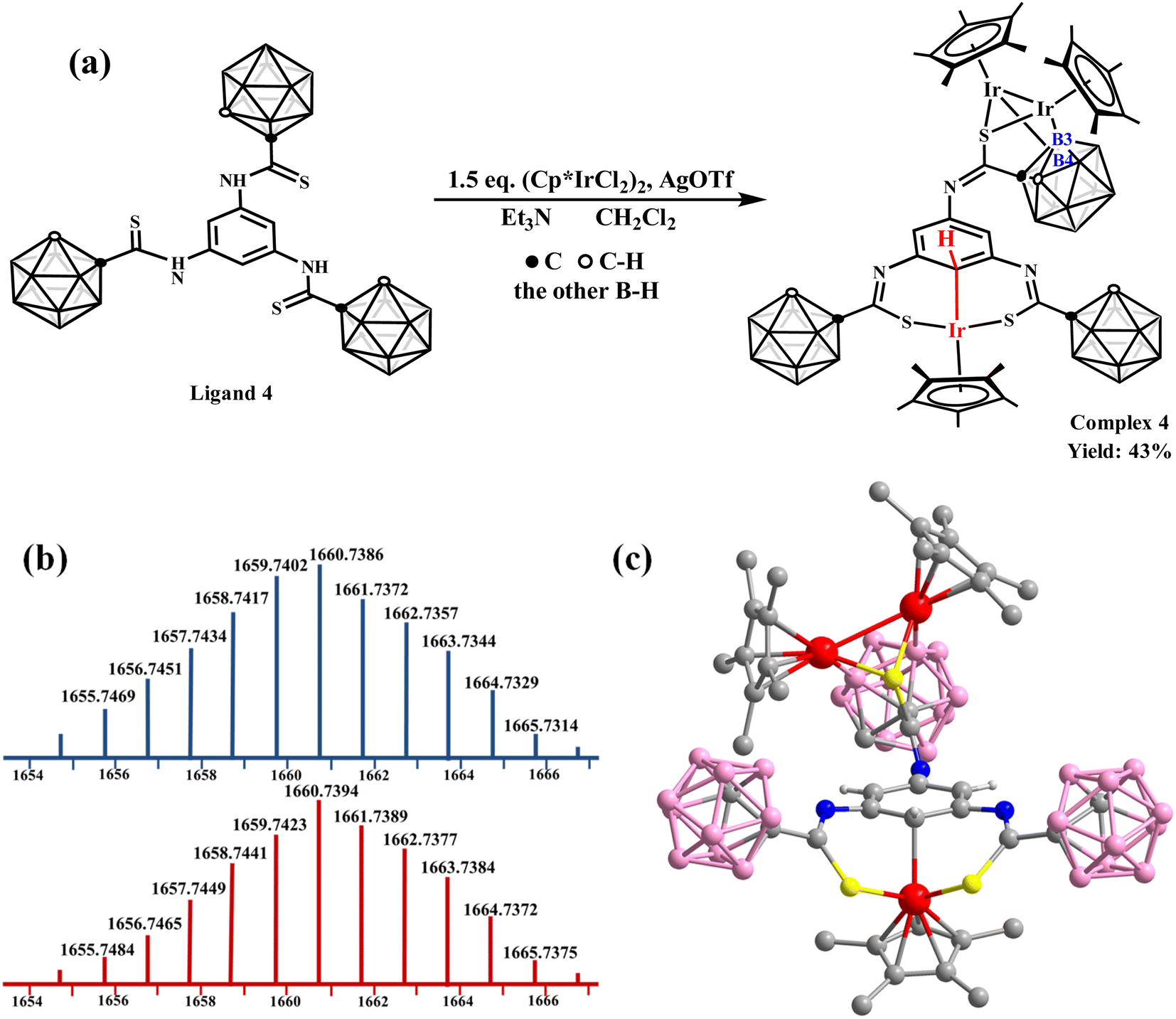 | ||
| Fig. 2 (a) Synthesis of complex 4. (b) ESI-MS of complex 4 (bottom: experimental; top: theoretical). (c) Molecular structure of complex 4. Colour code: Ir, red; S, yellow; N, blue; C, gray; B, pink; H, light gray. | ||
After achieving the controlled formation of M⋯C–H interaction, its reactivities attract our attention. The first challenge is whether it can be converted into C–H metallization products. Compared with complexes 1, 2, and 3, we found that if we could increase the charges of the S atoms, C–H bond metallization might be achieved. Hence, trifluoromethanesulfonic acid (HOTf) was added to complex 3 (Fig. 3a) to facilitate the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds, increasing the charges of the S atoms. In the 1H NMR spectrum of complex 5a, the singlet signal of the C5–H bond in complex 3 disappeared, indicating C–H activation. The proton of the C–H peak from the remaining benzene ring appears at approximately δ = 6.78 ppm (Fig. S48†). In the 13C NMR spectrum (Fig. S50†), in contrast to complex 3a (δ = 41.71 ppm), the signal of the carbon atom attached to Ir appears at δ = 108.35 ppm. The bond lengths of the C5–C6 and C5–C4 bonds are 1.40 and 1.39 Å (Fig. 3b), slightly shorter than those in complex 3a (1.43 Å and 1.44 Å). Moreover, the benzene ring in complex 5a exhibits considerable rotation (Fig. 1a and 3b), reducing the length of the Ir–C5 bond (2.24 Å in complex 3a and 2.05 Å in complex 5a) and activating the C–H bond.
S bonds, increasing the charges of the S atoms. In the 1H NMR spectrum of complex 5a, the singlet signal of the C5–H bond in complex 3 disappeared, indicating C–H activation. The proton of the C–H peak from the remaining benzene ring appears at approximately δ = 6.78 ppm (Fig. S48†). In the 13C NMR spectrum (Fig. S50†), in contrast to complex 3a (δ = 41.71 ppm), the signal of the carbon atom attached to Ir appears at δ = 108.35 ppm. The bond lengths of the C5–C6 and C5–C4 bonds are 1.40 and 1.39 Å (Fig. 3b), slightly shorter than those in complex 3a (1.43 Å and 1.44 Å). Moreover, the benzene ring in complex 5a exhibits considerable rotation (Fig. 1a and 3b), reducing the length of the Ir–C5 bond (2.24 Å in complex 3a and 2.05 Å in complex 5a) and activating the C–H bond.
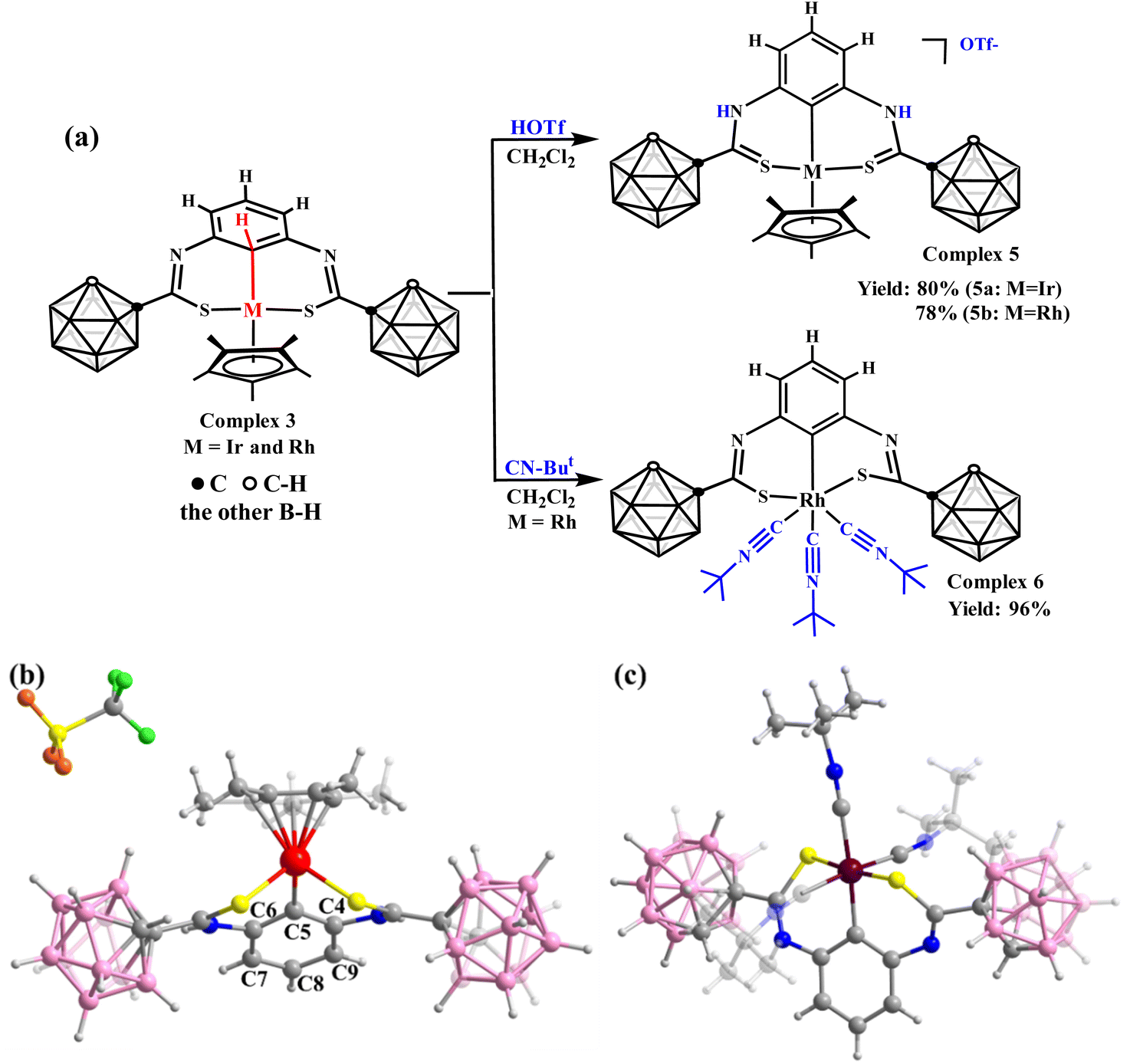 | ||
| Fig. 3 (a) Reactivities of complex 3. Molecular structure of complex 5a (b) and complex 6 (c). Colour code: Ir, red; Rh, dark red; S, yellow; N, blue; C, gray; B, pink; H, light gray; O, orange; F, green. The notes show the carbon number on the benzene ring. | ||
The abovementioned strategies to achieve C–H activation require changing the C–S bond to a CS bond. This raises the question of whether C–H bond activation can be achieved while retaining the C–S single bond. To achieve this, the oxidation state or coordination configuration of the metal centre must be changed. However, complex 3 dissociates when an oxidizer (I2) is added to it. This shows that C–H bond metallization cannot be achieved by changing the oxidation state of the metal centre. Subsequently, we applied a strong electron-donor ligand, CN–But. When CN–But is added to complex 3b, the Cp* ring leaves to form complex 6 (Fig. 3a). However, a similar reaction did not occur in complex 3a, possibly due to the stronger affinity of the Ir centre to the Cp* ring. The crystal structure of complex 6 (Fig. 3c) confirms that C–H-bond activation also causes a rotation of the benzene ring compared to complex 3. These results again illustrate the importance of charge density at the metal centre in stabilizing the M⋯C–H interaction.
Conclusions
In this study, compared to alkyl or aryl groups, the carborane cage presents a unique application prospect in stabilizing special chemical bonding interactions. Owing to the importance of the M⋯C–H interaction in synthetic chemistry, this interaction was characterized in detail through single-crystal analysis, NMR spectroscopy, UV-vis spectroscopy, ESI-MS analysis, and theoretical calculations to provide a definition of this interaction. This would also effectively reduce the confusion of the metal⋯C–H interaction with other interactions. The analysis of this bonding interaction type can help deepen our understanding of the interactions between a metal centre and carbon atoms and is beneficial to understand the C–H bond activation process.Data availability
Data supporting this study is available in the ESI† and further details are available from the authors on reasonable request.Author contributions
Xin-Ran Liu and Peng-Fei Cui contributed equally to this work. Xin-Ran Liu and Peng-Fei Cui designed and synthesized all the compounds. Guo-Xin Jin supervised the research. Yago García-Rodeja and Miquel Solà performed theoretical calculations. All authors discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (22031003, 22201043) and the Shanghai Science Technology Committee (19DZ2270100). P. F. C. thanks the China Postdoctoral Science Foundation (BX2021073, 2021M700801). M. S. and Y. G. R. are grateful to the Spanish Ministerio de Ciencia, Innovación y Universidades (MICIU/AEI/10.13039/501100011033) for projects PID2020-113711GB-I00 and the “European Union-Next Generation” Maria Zambrano grant to Y. G.-R., and the Generalitat de Catalunya for project 2021SGR623.Notes and references
- R. N. Grimes, Carboranes, Elsevier, Oxford, 3rd edn, 2016, pp. 283–502, ISBN: 978-0-12-801894-1 Search PubMed.
- (a) A. Saha, E. Oleshkevich, C. Viñas and F. Teixidor, Adv. Mater., 2017, 29, 1704238 CrossRef PubMed; (b) J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed.
- (a) D. Zhao and Z. Xie, Coord. Chem. Rev., 2016, 314, 14–33 CrossRef CAS; (b) P. F. Cui, Y. Gao, S. T. Guo, Y. J. Lin, Z. H. Li and G. X. Jin, Angew. Chem., Int. Ed., 2019, 58, 8129–8133 CrossRef CAS PubMed.
- (a) B. J. Eleazer, M. D. Smith, A. Popov and D. V. Peryshkov, J. Am. Chem. Soc., 2016, 138, 10531–10538 CrossRef CAS PubMed; (b) J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed; (c) C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed; (d) R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692–7711 CrossRef CAS PubMed.
- P. F. Cui, X. R. Liu, Y. J. Lin, Z. H. Li and G. X. Jin, J. Am. Chem. Soc., 2022, 144, 6558–6565 CrossRef CAS PubMed.
- P. F. Cui, X. R. Liu and G. X. Jin, J. Am. Chem. Soc., 2023, 145, 19440–19457 CrossRef CAS PubMed.
- I. B. Sivaev, Comprehensive Organometallic Chemistry IV, Elsevier, Amsterdam, 2022, vol. 9, pp. 196–262, ISBN: 9780128202067 Search PubMed.
- D. S. Tu, H. Yan, J. Poater and M. Solà, Angew. Chem., Int. Ed., 2020, 59, 9018–9025 CrossRef CAS PubMed.
- (a) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed; (b) R. R. Huang, C. Wang, D. Tan, K. Wang, B. Zou, Y. T. Shao, T. H. Liu, H. N. Peng, X. G. Liu and Y. Fang, Angew. Chem., Int. Ed., 2022, 61, e202211106 CrossRef CAS PubMed.
- X. R. Liu, P. F. Cui, S. T. Guo, Y. J. Lin and G. X. Jin, J. Am. Chem. Soc., 2023, 145, 8569–8575 CAS.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- Y. N. Ma, H. Ren, Y. Wu, N. Li, F. Chen and X. Chen, J. Am. Chem. Soc., 2023, 145, 7331–7342 CrossRef CAS PubMed.
- H. Y. Ren, P. Zhang, J. K. Xu, W. L. Ma, D. S. Tu, C. S. Lu and H. Yan, J. Am. Chem. Soc., 2023, 145, 7638–7647 CrossRef CAS PubMed.
- H. L. Gingrich, T. Ghosh, Q. R. Huang and Jr and M. Jones, J. Am. Chem. Soc., 1990, 112, 4082–4083 CrossRef CAS.
- Z. Qiu, S. Ren and Z. Xie, Acc. Chem. Res., 2011, 44, 299–309 CrossRef CAS PubMed.
- H. Zhang, J. Y. Wang, W. G. Yang, L. B. Xiang, W. C. Sun, W. B. Ming, Y. X. Li, Z. Y. Lin and Q. Ye, J. Am. Chem. Soc., 2020, 142, 17243–17249 CrossRef CAS PubMed.
- K. Jaiswal, N. Malik, B. Tumanskii, G. Ménard and R. Dobrovetsky, J. Am. Chem. Soc., 2021, 143, 9842–9848 CrossRef CAS PubMed.
- S. Li and Z. Xie, J. Am. Chem. Soc., 2022, 144, 7960–7965 CrossRef CAS PubMed.
- L. B. Xiang, J. Y. Wang, I. Krummenacher, K. Radacki, H. Braunschweig, Z. Y. Lin and Q. Ye, Chem.–Eur. J., 2023, 29, e202301270 CrossRef CAS PubMed.
- (a) S. Kim, J. W. Treacy, Y. A. Nelson, J. A. M. Gonzalez, M. Gembicky, K. N. Houk and A. M. Spokoyny, Nat. Commun., 2023, 14, 1671 CrossRef CAS PubMed; (b) S. L. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed.
- Y. Quan and Z. Xie, Chem. Soc. Rev., 2019, 48, 3660–3673 RSC.
- X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS.
- Y. Baek, S. Kim, J. Y. Son, K. Lee, D. Kim and P. H. Lee, ACS Catal., 2019, 9, 10418–10425 CrossRef CAS.
- F. R. Lin, J. L. Yu, Y. J. Shen, S. Q. Zhang, B. Spingler, J. Y. Liu, X. Hong and S. Duttwyler, J. Am. Chem. Soc., 2018, 140, 13798–13807 CrossRef CAS PubMed.
- L. Yang, B. B. Jei, A. Scheremetjew, R. Kuniyil and L. Ackermann, Angew. Chem., Int. Ed., 2021, 60, 1482–1487 CrossRef CAS PubMed.
- (a) W. Y. Man, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2016, 55, 4596–4599 CrossRef CAS PubMed; (b) Y. N. Ma, Y. Gao, Y. B. Ma, Y. Wang, H. Ren and X. Chen, J. Am. Chem. Soc., 2022, 144, 8371–8378 CrossRef CAS PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- A. J. Canty and G. V. Koten, Acc. Chem. Res., 1995, 28, 406–413 CrossRef CAS.
- W. D. Jones, Activation of Unreactive Bonds and Organic Synthesis, ed. S. Murai, H. Alper, R. A. Gossage, V. V. Grushin, M. Hidai, Y. Ito, W. D. Jones, F. Kakiuchi, G. Koten, Y. S. Lin, Y. Mizobe, S. Murai, M. Murakami, T. G. Richmond, A. Sen, M. Suginome and A. Yamamoto, Springer Berlin, Heidelberg, 1999, pp. 9–46, ISBN: 978-3-642-08436-2 Search PubMed.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed.
- J. D. Watson, L. D. Field and G. E. Ball, Nat. Chem., 2022, 14, 801–806 CrossRef CAS PubMed.
- C. C. Li, Z. Chen, Y. M. Huang, Y. R. Zhang, X.-Y. Li, Z. W. Ye, X. Xu, S. E. J. Bell and Y. K. Xu, Chem, 2022, 8, 2514–2528 CAS.
- S. Filipuzzi, P. S. Pregosin, M. J. Calhorda and P. J. Costa, Organometallics, 2008, 27, 2949–2958 CrossRef CAS.
- M. Albrecht, R. A. Gossage, A. L. Spek and G. V. Koten, J. Am. Chem. Soc., 1999, 121, 11898–11899 CrossRef CAS.
- D. M. Grove, G. V. Koten, J. N. Louwen, J. G. Noltes, A. L. Spek and H. J. C. Ubbels, J. Am. Chem. Soc., 1982, 104, 6609–6616 CrossRef CAS.
- T. Steinke, B. K. Shaw, H. Jong, B. O. Patrick, M. D. Fryzuk and J. C. Green, J. Am. Chem. Soc., 2009, 131, 10461–10466 CrossRef CAS PubMed.
- X. H. Lin, W. Wu and Y. R. Mo, Coord. Chem. Rev., 2020, 419, 213401 CrossRef CAS.
- P. L. Arnold, A. Prescimone, J. H. Farnaby, S. M. Mansell, S. Parsons and N. Kaltsoyannis, Angew. Chem., Int. Ed., 2015, 54, 6735–6739 CrossRef CAS PubMed.
- S. Murugesan, B. Stçger, E. Pittenauer, G. Allmaier, L. F. Veiros and K. Kirchner, Angew. Chem., Int. Ed., 2016, 55, 3045–3048 CrossRef CAS PubMed.
- M. Stepien, L. Latos-Grazynski, L. Szterenberg, J. Panek and Z. Latajka, J. Am. Chem. Soc., 2004, 126, 4566–4580 CrossRef CAS PubMed.
- (a) X. Ribas, C. Calle, A. Poater, A. Casitas, L. Gómez, R. Xifra, T. Parella, J. Benet-Buchholz, A. Schweiger, G. Mitrikas, M. Solà, A. Llobet and T. D. P. Stack, J. Am. Chem. Soc., 2010, 132, 12299–12306 CrossRef CAS PubMed; (b) C. Lepetit, J. Poater, E. Alikhani, B. Silvi, Y. Canac, J. Contreras-García, M. Solà and R. Chauvin, Inorg. Chem., 2015, 54, 2960–2969 CrossRef CAS PubMed; (c) M. Montag, L. Schwartsburd, R. Cohen, G. Leitus, Y. Ben-David, J. M. L. Martin and D. Milstein, Angew. Chem., Int. Ed., 2007, 46, 1901–1904 CrossRef CAS PubMed.
- Y. L. Wang, Z. D. Huang, G. X. Liu and Z. Huang, Acc. Chem. Res., 2022, 55, 2148–2161 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- (a) D. M. Brouwer, E. L. Mackor and C. MacLean, Carbonium Ions, ed. G. A. Olah and P. V. Schleyer, Wiley-Interscience, New York, 1970, pp. 837–897, ISBN978-1-4684-8541-7 Search PubMed; (b) R. Rathore, J. rgen Hecht and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 13278–13279 CrossRef CAS.
- Y. Garcıa-Rodeja, F. Feixas, E. Matito and M. Solà, Phys. Chem. Chem. Phys., 2022, 24, 29333–29337 RSC.
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawełek, T. M. Krygowski, H. Szatylowicz and M. Solà, Phys. Chem. Chem. Phys., 2017, 19, 28970–28981 RSC.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2303636 (complex 3b), 2303637 (complex 3a), 2303638 (complex 5a), 2303639 (complex 6), 2303640 (L3), 2303641 (complex 4), 2303643 (L4) and 2303644 (complex 5b). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01158a |
| ‡ X.-R. L. and P.-F. C. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |