
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatization-driven deconstructive functionalization of spiro dihydroquinazolinones via dual photoredox/nickel catalysis†
Hong-Jie
Miao
,
Jin-Hua
Zhang
,
Wenke
Li
,
Wenpeng
Yang
,
Hong
Xin
,
Pin
Gao
 ,
Xin-Hua
Duan
and
Li-Na
Guo
*
,
Xin-Hua
Duan
and
Li-Na
Guo
*
School of Chemistry, Xi'an Key Laboratory of Sustainable Energy Material Chemistry, Engineering Research Center of Energy Storage Materials and Devices, Ministry of Education, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: guoln81@xjtu.edu.cn
First published on 8th May 2024
Abstract
Aromatization-driven deconstruction and functionalization of spiro dihydroquinazolinones via dual photoredox/nickel catalysis is developed. The aromatization effect was introduced to synergistically drive unstrained cyclic C–C bond cleavage, with the aim of overcoming the ring-size limitation of nitrogen-centered radical induced deconstruction of carbocycles. Herein, we demonstrate the synergistic photoredox/nickel catalyzed deconstructive cross-coupling of spiro dihydroquinazolinones with organic halides. Remarkably, structurally diverse organic halides including aryl, alkenyl, alkynyl, and alkyl bromides were compatible for the coupling. In addition, this protocol is also characterized by its mild and redox-neutral conditions, excellent functional group compatibility, high atom economy, and easy scalability. A telescoped procedure involving condensation and ring-opening/coupling was found to be accessible. This work provides a complementary strategy to the existing radical-mediated C–C bond cleavage of unstrained carbocycles.
In modern organic synthesis, the deconstruction and functionalization of carbocycles has aroused great interest among chemists, because it provides a straightforward approach to difunctionalized chain compounds.1 In this field, radical-mediated cyclic C–C bond cleavage represents an attractive strategy due to its mild conditions and versatile chemical transformations.2 For example, the alkoxyl radical-mediated ring-opening of cycloalkanols and their derivatives has been extensively studied by Zhu, Knowles, Zuo and others, providing efficient routes to functionalized chain ketones.3 At the same time, Zard, Uemura, Zhou, Shi and others have developed a series of iminyl radical-mediated ring-opening reactions that provide a cyanide-free approach to functionalized alkyl nitriles.4 Our group has also devoted our efforts to various inexpensive metal (Cu, Fe, and Ni) catalyzed radical-mediated C–C bond cleavage reactions.5 There, the thermodynamic driving force of C–C bond cleavage comes mainly from the formation of a strong C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bond and energetically stabilized carbon-centered radicals, as well as the strain release of ring systems. Over the past few years, Waser, Zheng and others have reported the nitrogen-centered radical-induced ring-opening of aminocyclopropanes, which can serve as a 1,3-synthon to participate in [3 + 2] annulation and 1,3-difunctionalization reactions (Scheme 1a).6 However, this strategy is limited to the strongly strained three and four-membered ring systems. The deconstruction of unstrained aminocycloalkanes remains challenging, probably due to the high reverse cyclization rate and the insufficient ring strain.7 Therefore, the exploration of new strategies for unstrained C–C bond cleavage is still important and desirable.
X bond and energetically stabilized carbon-centered radicals, as well as the strain release of ring systems. Over the past few years, Waser, Zheng and others have reported the nitrogen-centered radical-induced ring-opening of aminocyclopropanes, which can serve as a 1,3-synthon to participate in [3 + 2] annulation and 1,3-difunctionalization reactions (Scheme 1a).6 However, this strategy is limited to the strongly strained three and four-membered ring systems. The deconstruction of unstrained aminocycloalkanes remains challenging, probably due to the high reverse cyclization rate and the insufficient ring strain.7 Therefore, the exploration of new strategies for unstrained C–C bond cleavage is still important and desirable.
 | ||
| Scheme 1 Radical mediated cyclic C–C bond cleavage. | ||
Aromatization is recognized as one of the most important thermodynamic driving forces for many chemical transformations.8 Especially in radical chemistry, the aromatization-driven cleavage of chemical bonds is an important transformation. In this field, structurally diverse pre-aromatic (PA) compounds such as 1,4-DHPs,9 benzothiazolines,10 dihydroquinazolinones,11 and others12 are widely used as hydrogen atom transfer (HAT) reagents, functional group transfer reagents and radical precursors. For example, Dong and co-workers disclosed several unique deacylative C–C bond functionalizations of chain ketones,13 in which the in situ formed pre-aromatic intermediates underwent Ir(III)-catalyzed aromatization-driven C–C bond cleavage to achieve the deacetylation process. Recently, Martin,14 Zhu15 and others16 reported a series of photoredox-catalyzed radical alkylation reactions by using 2,3-dihydroquinazolinones as efficient alkyl radical precursors. However, large amounts of quinazolinones were formed as by-products, which may be against the concept of green synthesis. Despite these reports, the aromatization-driven C–C bond cleavage of unstrained carbocycles remains unexplored. As mentioned above, the cyclic C–C bond cleavage induced by nitrogen-centered radicals is usually limited to strained rings. Therefore, we set out to embed this aminocycloalkane skeleton in a latent aromatic molecule, hoping that the strong driving force of aromatization would facilitate the cleavage of the less strained C–C bond (Scheme 1b). Here, we demonstrate a dual photoredox- and nickel-catalyzed deconstructive cross-coupling of spiro 2,3-dihydroquinazolin-4(1H)-ones with organic halides via nitrogen-centered radical-triggered β-scission (Scheme 1c). The noteworthy features of this study include: (1) the mild and redox-neutral reaction conditions, the practical scalability and the use of inexpensive, commercially available 4CzIPN as the organic photocatalyst, which make this protocol of great potential in organic synthesis; (2) this method exhibits broad substrate scope (aryl, alkenyl, alkynyl and alkyl bromides) and excellent functional group compatibility; (3) most remarkably, the useful quinazolinone fragments generated in situ were successfully retained in the final product, exhibiting excellent atom efficiency.17
To verify our hypothesis, spiro 2,3-dihydroquinazolin-4(1H)-one 1a was first prepared by condensation of 2-methyl cyclopentanone with 2-aminobenzamide. Then, the reaction of 1a with aryl bromide 2a was chosen as a model reaction to find the optimal conditions (Table 1). We were pleased to find that the reaction proceeded successfully in the presence of 4CzIPN (1 mol%), NiCl2·DME (10 mol%), dtbpy (L2, 12 mol%), and K2CO3 in DMF under 10 W blue LED irradiation, yielding the desired ring-opening/coupling product 3a in 73% yield. The screening of nickel catalysts and ligands showed that the combination of NiCl2·DME and L2 was the most efficient, giving the best yield of 3a (entries 2–7). Solvent screening showed that polar solvents were more suitable, giving 3a in moderate to good yields and DMSO was the optimal solvent (entries 8 and 9). Both Ir-based complex and organic photocatalysts were evaluated for this transformation, with 4CzIPN still being the best (entries 10 and 11). Inorganic bases such as Na2CO3 and organic bases such as 2,4,6-colidine both gave a comparable yield of 3a (entries 12 and 13). Satisfactorily, increasing the amount of 1a from 1.2 equiv. to 1.5 equiv. led to an improved 90% yield of 3a (entry 14). Control experiments showed that the photocatalyst, nickel catalyst, ligand, base and visible-light irradiation were all essential for this transformation (entry 15). Finally, it should be mentioned that a certain amount of ring-opening/HAT product 3a’ was also detected during the optimization.
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yield of 3a/b% | Yield of 3a′/b% |
| a Reaction conditions: 1a (0.24 mmol, 1.2 equiv.), 2a (0.2 mmol, 1.0 equiv.), 4CzIPN (1.0 mol%), NiCl2·DME (10 mol%), dtbpy (12 mol%) and K2CO3 (1.0 equiv.) in DMF (2.0 mL) with 10 W blue LEDs irradiation at room temperature for 12 h under N2. b Isolated yields. c DMSO was used as solvent. | |||
| 1 | None | 73 | 9 |
| 2 | Ni(COD)2 instead of NiCl2·DME | 18 | 12 |
| 3 | NiCl2 instead of NiCl2·DME | 45 | 18 |
| 4 | L1 instead of L2 | 55 | 13 |
| 5 | L3 instead of L2 | 72 | 10 |
| 6 | L4 instead of L2 | 61 | 17 |
| 7 | L5 instead of L2 | 37 | 11 |
| 8 | DMSO as solvent | 81 | <5 |
| 9 | DMAc, NMP, and MeCN as solvent | 71/68/46 | 10 |
| 10 | 4CzPN instead of 4CzIPN | 75c | <5 |
| 11 | Ir[dF(CF3)ppy]2(dtbpy)PF6 as PC | 66c | <5 |
| 12 | Na2CO3 as the base | 83c | <5 |
| 13 | 2,4,6-Colidine as the base | 80c | <5 |
| 14 | 1.5 equiv. of 1a was used | 90c | <5 |
| 15 | No pc, [Ni], L2, base or in darkness | Tracec | Trace |

|
|||

With the optimized conditions established, the generality and limitations of aryl bromides for this deconstructive arylation reaction were first evaluated. As shown in Table 2, a number of aryl bromides reacted efficiently with 1a under standard conditions to afford the target products 3a–l in good to excellent yields. The electronic and steric effects on the aromatic moiety showed a significant influence on the reaction efficiency. In general, aryl bromides with strong electron-withdrawing groups on the para position of the aromatic ring gave better yields (3a–hvs.3i and 3j). Satisfactorily, a wide range of functional groups including acetyl (3a and 3k), methylsulfonyl (3b), ester (3c), formyl (3d), cyano (3e), trifluoromethyl (3f), benzoyl (3g) and chloro (3h and 3l) groups were well tolerated in this reaction. 5-Bromoindanone and 5-bromophthalide also afforded the desired products 3m and 3n in good yields. Heteroaromatic bromides, such as 3-bromoquinoline, 6-bromoquinoline and 4-bromopyridine, were also amenable and afforded the target products 3o–q in moderate yields. In addition to aryl bromides, vinyl bromide (3s) and alkynyl bromide (3t) were also applicable to this protocol. Remarkably, aryl bromides masked by natural products or drugs also participated well in this coupling reaction, giving the expected products (3v–z) in satisfactory yields. These results highlighted the application potential of this protocol in the diversification of natural products and pharmaceuticals.
| a Reaction conditions: spiro dihydroquinazolinone 1a (0.3 mmol, 1.5 equiv.), aryl bromides 2 (0.2 mmol, 1.0 equiv.), 4CzIPN (1 mol%), NiCl2·DME (10 mol%), L2 (12 mol%), K2CO3 (1.0 equiv.), and DMSO (2.0 mL) with 10 W blue LEDs irradiation at room temperature for 12 h under N2. Isolated yields. |
|---|

|
Subsequently, the scope of spiro dihydroquinazolinones derived from various cycloalkanones and 2-aminobenzamides was investigated using aryl bromide 2a as a model coupling partner. As shown in Table 3, various dihydroquinazolinones derived from 2-substituted cyclopentanones were all efficiently engaged in this C–C bond cleavage/coupling to afford the products 5a–l in 55–91% yields. Substrates derived from norcamphors and loxoprofen also afforded the desired products 5e–g in good yields. Dihydroquinazolinones with a methyl protecting group on the amide moiety showed better reaction efficiency, giving the product 5h in 91% yield. Dihydroquinazolinones with different substituents on the aromatic rings also reacted efficiently with 2a to afford the products 5i–l in moderate yields. Meanwhile the substrate with a 5-Me group on the aromatic ring gave the desired product 5j in only 20% yield, due to low conversion. It was found that the existence of a 2-substituent on the aliphatic ring is very crucial for the success of this transformation, probably due to its stabilizing effect on the alkyl radical intermediates. Substrates derived from simple cyclopentanone and 2,2-dimethylcyclopentanone both failed to give the product. For the former, the rapid reverse 5-exo cyclization was probably the main reason. For the latter, the steric hindrance of the tertiary radical generated in situ made the coupling reaction more difficult. Notably, the substrate derived from cyclobutanone was efficient, giving the desired product 5m in 88% yield. For the six-membered ring systems, the 2-phenoxy-substituted substrate was successfully converted to the expected product 5n in 80% yield, while the 2-methyl analogue was inactive.
| a Reaction conditions: dihydroquinazolinone 1 (0.3 mmol, 1.5 equiv.), aryl halides 2a (0.2 mmol, 1.0 equiv.), 4CzIPN (1 mol%), NiCl2·DME (10 mol%), L2 (12 mol%), K2CO3 (1.0 equiv.), and DMSO (2.0 mL) with 10 W blue LEDs irradiation at room temperature for 12 h under N2. Isolated yields. |
|---|

|
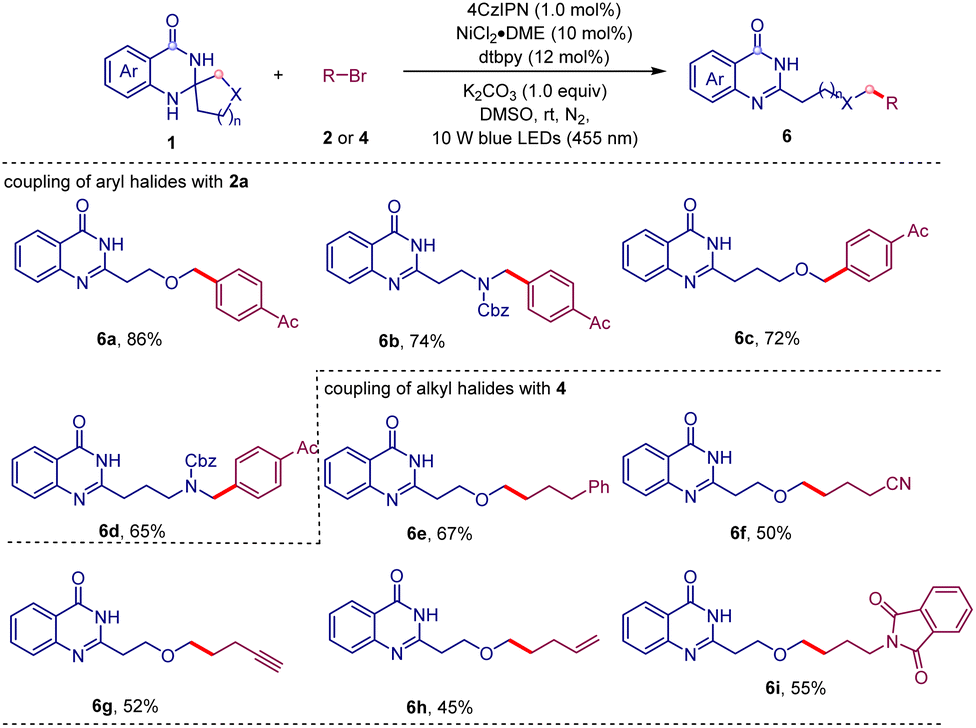
It is known that the nucleophilicity and lifetime of alkyl radicals can be influenced by the electronegativity and the π-bonding capability of the α-heteroatom.18 Therefore, we intend to evaluate the influence of the heteroatom on this C–C bond cleavage/coupling reaction (Table 4). We introduced oxygen and nitrogen atoms into aliphatic ring systems, respectively. Fortunately, the substrate derived from dihydrofuran-3(2H)-one successfully underwent this ring-opening/coupling reaction to give the expected product 6a in 86% yield with excellent regioselectivity. The substrate generated from N-Cbz-pyrrolidin-3-one also worked well with 2a to afford the product 6b in 74% yield. In addition to the five-membered system, the six-membered analogues were also compatible for this transformation (6c and 6d). Remarkably, the coupling partners can be further extended to alkyl halides (6e–i). A variety of functional groups such as cyano (6f), alkynyl (6g), alkenyl (6h), and phthalimide (6i) were all tolerated in this transformation.
| a Reaction conditions: dihydroquinazolinone 1 (0.3 mmol, 1.5 equiv.), aryl halides 2 or 4 (0.2 mmol, 1.0 equiv.), 4CzIPN (1 mol%), NiCl2·DME (10 mol%), L2 (12 mol%), K2CO3 (1.0 equiv.), and DMSO (2.0 mL) with 10 W blue LEDs irradiation at room temperature for 12 h under N2. Isolated yields. |
|---|
|
To demonstrate the applicability of this protocol, the gram-scale synthesis of 3a and its derivatizations were carried out (Scheme 2a). When the model reaction was scaled up to the 5.0 mmol scale, the reaction still proceeded smoothly to give 3a in a slightly reduced but acceptable yield (1.2 g, 72% yield). The derivatization of 3a was then investigated. 3a can undergo nucleophilic substitution with 2,4-dinitrofluorobenzene to give the N-aryl product 7a in 64% yield. It also reacted with Morita–Baylis–Hillman ester to give the N-allylic product 8a in 88% yield. In addition, quinazolin-4(3H)-one 3b underwent a HMDS-mediated amination reaction with benzylamine to afford 4-aminoquinazoline 9a in 84% yield. Finally, the treatment of 3a with NaBH4 afforded the unexpected dearomatized product 10a in 77% yield as the sole product. To further increase the synthetic utility of this protocol, we telescoped the formation of spiro dihydroquinazolinones and the ring-opening/coupling reaction without purification (Scheme 2b). Satisfactorily,the 2-methylcyclopentanone efficiently underwent this two-step process and afforded 3a in 71% isolated yield.
 | ||
| Scheme 2 Application investigation. | ||
To gain mechanistic insight into this reaction, some control experiments were performed (Scheme 3). It was found that the reaction of 1a and 2a was totally inhibited by the addition of 1.0 equiv. of TEMPO. In this case, the TEMPO adduct 11a was isolated in 92% yield. The addition of 1.0 equiv. of BHT also completely suppressed the reaction (Scheme 3a). These results indicated that the reaction proceeded via a radical pathway. When (1-cyclopropylvinyl)benzene was added to the reaction system, in addition to 31% of product 3a, the three-component product 12a was isolated in 20% yield, providing further evidence for a radical pathway (Scheme 3b). Stern–Volmer fluorescence quenching experiments showed that the excited 4-CzIPN* was effectively quenched by the dihydroquinazolinone 1a rather than the bromide 2a, suggesting that an excited state charge transfer occurred between 4-CzIPN* and 1a (Scheme 3c). Light on and off experiments showed that continuous irradiation is essential for this transformation (Scheme 3d and see also the ESI†). To elucidate the Ni catalytic cycle, an isolated Ni(II) complex C1 was prepared in advance and then treated with 1a under the standard conditions. As expected, the product 3f could be obtained in 42% yield. Furthermore, the use of Ni(II) complex C1 as a catalyst for the reaction of 1a and 2a also led to the formation of 3a in 85% yield, along with 12% yield of 3f (Scheme 3e). These results suggest that the Ar–Ni(II) complex is a key intermediate in the Ni-catalytic cycle.
 | ||
| Scheme 3 Mechanism studies. | ||
Based on the above experimental results and previous literature,19 a possible mechanism for this ring-opening/coupling is proposed (Scheme 4). First, 4CzIPN is photoexcited to its excited state 4CzIPN*, which is a sufficient SET oxidant. Then, single electron oxidation of dihydroquinazolinone 1a by the photoexcited 4CzIPN* yields the aminyl radical cation I and the radical anion [4CzIPN]˙−. In the presence of a base, the radical cation intermediate I undergoes aromatization driven β-scission to give the alkyl radical II. Concurrently, the oxidative addition of aryl bromide 2a to active Ni(0)Ln produces the Ni(II) species III. Radical recombination of the Ni(II) species III with the alkyl radical II yields the Ni(III) species IV, which undergoes reductive elimination to give the target product 3a and the LnNi(I)Br species V. Finally, the two catalytic cycles are completed simultaneously by a single electron transfer between the [4CzIPN]˙− radical anion and LnNi(I)Br V, regenerating both the active Ni(0)Ln species and 4CzIPN.
 | ||
| Scheme 4 Proposed Mechanism. | ||
Conclusions
In summary, we have developed a novel aromatization-driven deconstructive cross-coupling of spiro dihydroquinazolinones via dual photoredox/nickel catalysis. This is the first example of aromatization-driven nitrogen-centered radical-induced cyclic C–C bond cleavage. In contrast to previous reports, this strategy allows the aromatic fragments formed in situ via C–C bond cleavage to be successfully retained in the product. Under the synergistic photoredox/nickel catalysis, the deconstructive coupling of spiro dihydroquinazolinones with various organic halides, including aryl, alkenyl, alkynyl and alkyl bromides, proceeded efficiently to afford a series of useful functionalized quinazolinones in good yields with excellent functional group tolerance. An in-depth mechanism study revealed that this reaction proceeded via a radical-metal crossover pathway. This work provides a complementary strategy for the radical-mediated C–C bond cleavage of unstrained carbocycles.Data availability
All experimental and characterization data including NMR spectra are available in the ESI.†Author contributions
H.-J. Miao performed all the experiments and prepared the manuscript and ESI.† J.-H. Zhang, W. Li W. Yang and H. Xin performed the preparation of raw materials. X.-H. Duan and L.-N. Guo directed this project and revised the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 22171220, 21971201) and the Fundamental Research Funds of the Central Universities (No. xtr072022003) is greatly appreciated. We also thank Mr Zhang, Miss Feng and Miss Bai at the Instrument Analysis Center of Xi'an Jiaotong University for their assistance with NMR and HRMS analysis.Notes and references
- For selected reviews, see: (a) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC; (b) Y. Xue and G. Dong, Acc. Chem. Res., 2022, 55, 2341–2354 CrossRef CAS PubMed; (c) F. Song, B. Wang and Z.-J. Shi, Acc. Chem. Res., 2023, 56, 2867–2886 CrossRef CAS PubMed.
- For selected reviews, see: (a) X. Wu and C. Zhu, Chem. Commun., 2019, 55, 9747–9756 RSC; (b) P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC; (c) E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC; (d) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066–1083 CrossRef CAS PubMed; (e) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (f) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (g) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- For recent examples: (a) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490–3493 CrossRef CAS PubMed; (b) H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS PubMed; (c) L. Huang, T. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532–3539 CrossRef CAS PubMed; (d) S. Sakurai, A. Matsumoto, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2020, 142, 19017–19022 CrossRef CAS PubMed; (e) Y. Chen, J. Du and Z. Zuo, Chem, 2020, 6, 266–279 CrossRef CAS; (f) L. Wen, J. Ding, L. Duan, S. Wang, Q. An, H. Wang and Z. Zuo, Science, 2023, 382, 458–464 CrossRef CAS PubMed.
- For selected examples: (a) J. Boivin, E. Fouquet and S. Z. Zard, J. Am. Chem. Soc., 1991, 113, 1057–1059 CrossRef; (b) T. Nishimura, T. Yoshinaka, Y. Nishiguchi, Y. Maeda and S. Uemura, Org. Lett., 2005, 7, 2425–2427 CrossRef CAS PubMed; (c) L. Li, H. Chen, M. Mei and L. Zhou, Chem. Commun., 2017, 53, 11544–11547 RSC; (d) B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 12727–12731 CrossRef CAS PubMed; (e) X.-Y. Yu, J.-R. Chen, P.-Z. Wang, M.-N. Yang, D. Liang and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738–743 CrossRef CAS PubMed; (f) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS PubMed; (g) J. Chen, Y.-J. Liang, P.-Z. Wang, G.-Q. Li, B. Zhang, H. Qian, X.-D. Huan, W. Guan, W.-J. Xiao and J.-R. Chen, J. Am. Chem. Soc., 2021, 143, 13382–13392 CrossRef CAS PubMed.
- (a) S. Wang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Lett., 2015, 17, 4798–4801 CrossRef CAS PubMed; (b) Y.-R. Gu, X.-H. Duan, L. Yang and L.-N. Guo, Org. Lett., 2017, 19, 5908–5911 CrossRef CAS PubMed; (c) J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161–166 RSC; (d) J.-C. Yang, L. Chen, F. Yang, P. Li and L.-N. Guo, Org. Chem. Front., 2019, 6, 2792–2795 RSC; (e) L. Chen, L.-N. Guo, S. Liu, L. Liu and X.-H. Duan, Chem. Sci., 2021, 12, 1791–1795 RSC; (f) L. Liu, X.-H. Duan and L.-N. Guo, Synthesis, 2021, 57, 4375–4388 Search PubMed; (g) S. Liu, P. Ma, L. Zhang, S. Shen, H.-J. Miao, L. Liu, K. N. Houk, X.-H. Duan and L.-N. Guo, Chem. Sci., 2023, 14, 5220–5225 RSC; (h) Q.-C. Shan, Y. Zhao, S.-T. Wang, H.-F. Liu, X.-H. Duan and L.-N. Guo, ACS Catal., 2024, 14, 2144–2150 CrossRef CAS.
- (a) S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222–226 CrossRef CAS PubMed; (b) S. A. Morris, J. Wang and N. Zheng, Acc. Chem. Res., 2016, 49, 1957–1968 CrossRef CAS PubMed; (c) D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC; (d) Y. Cai, J. Wang, Y. Zhang, Z. Li, D. Hu, N. Zheng and H. Chen, J. Am. Chem. Soc., 2017, 139, 12259–12266 CrossRef CAS PubMed; (e) M.-M. Wang and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 13880–13884 CrossRef CAS PubMed; (f) M.-M. Wang, T. V. T. Nguyen and J. Waser, Chem. Soc. Rev., 2022, 51, 7344–7357 RSC.
- (a) J.-W. Zhang, Y.-R. Wang, J.-H. Pan, Y.-H. He, W. Yu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 3900–3904 CrossRef CAS PubMed; (b) C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- A. Bhunia and A. Studer, Chem, 2021, 7, 2060–2100 CAS.
- (a) Á. Gutierrez-Bonet, J. C. Tellis, J. K. Matsui, B. A. Vara and G. A. Molander, ACS Catal., 2016, 6, 8004–8008 CrossRef PubMed; (b) K. Nakajima, S. Nojima, K. Sakata and Y. Nishibayash, ChemCatChem, 2016, 8, 1028–1032 CrossRef CAS; (c) W. Chen, Z. Liu, J. Tian, J. Li, J. Ma, X. Cheng and G. Li, J. Am. Chem. Soc., 2016, 138, 12312–12315 CrossRef CAS PubMed; (d) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (e) H.-M. Huang, P. Bellotti, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 2464–2471 CrossRef CAS PubMed; (f) Y. Wei, B. Ben-zvi and T. Diao, Angew. Chem., Int. Ed., 2021, 60, 9433–9438 CrossRef CAS PubMed.
- (a) T. Uchikura, K. Moriyama, M. Toda, T. Mouri, I. Ibáñez and T. Akiyama, Chem. Commun., 2019, 55, 11171–11174 RSC; (b) T. Uchikura, M. Toda, T. Mouri, T. Fujii, K. Moriyama, I. Ibáñez and T. Akiyama, J. Org. Chem., 2020, 85, 12715–12723 CrossRef CAS PubMed; (c) S.-C. Lee, L.-Y. Li, Z.-N. Tsai, Y.-H. Lee, Y.-T. Tsao, P.-G. Huang, C.-K. Cheng, H.-B. Lin, T.-W. Chen, C.-H. Yang, C.-C. Chiu and H.-H. Liao, Org. Lett., 2022, 24, 85–89 CrossRef CAS PubMed; (d) T. Uchikura, N. Kamiyama, T. Mouri and T. Akiyama, ACS Catal., 2022, 12, 5209–5216 CrossRef CAS.
- (a) P. P. Mondal, A. Pal, A. K. Prakash and B. Sahoo, Chem. Commun., 2022, 58, 13202–13205 RSC; (b) X.-Y. Lv, R. Abrams and R. Martin, Angew. Chem., Int. Ed., 2023, 62, e202217386 CrossRef CAS PubMed.
- (a) M. Tian, X. Shi, X. Zhang and X. Fan, J. Org. Chem., 2017, 82, 7363–7372 CrossRef CAS PubMed; (b) S.-C. Chen, Q. Zhu, Y. Cao, C. Li, Y. Guo, L. Kong, J. Che, Z. Guo, H. Chen, N. Zhang, X. Fang, J.-T. Lu and T. Luo, J. Am. Chem. Soc., 2021, 143, 14046–14052 CrossRef CAS PubMed; (c) H. A. Sakai and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6185–6192 CrossRef CAS PubMed; (d) S.-C. Chen, Q. Zhu, H. Chen, Z. Chen and T. Luo, Chem.–Eur. J., 2023, 29, e202203425 CrossRef CAS PubMed.
- (a) Y. Xu, X. Qi, P. Zheng, C. C. Berti, P. Liu and G. Dong, Nature, 2019, 567, 373–378 CrossRef CAS PubMed; (b) X. Zhou, Y. Xu and G. Dong, Nat. Catal., 2021, 4, 703–710 CrossRef CAS PubMed; (c) X. Zhou, Y. Xu and G. Dong, J. Am. Chem. Soc., 2021, 143, 20042–20048 CrossRef CAS PubMed; (d) X. Zhou, T. Yu and G. Dong, J. Am. Chem. Soc., 2022, 144, 9570–9575 CrossRef CAS PubMed.
- (a) X.-Y. Lv, R. Abrams and R. Martin, Nat. Commun., 2022, 13, 2394–2412 CrossRef CAS PubMed; (b) F. Cong, R. S. Mega, J. Chen, C. S. Day and R. Martin, Angew. Chem., Int. Ed., 2023, 62, e202214633 CrossRef CAS PubMed.
- L. Li, L. Fang, W. Wu and J. Zhu, Org. Lett., 2020, 22, 5401–5406 CrossRef CAS PubMed.
- (a) P. P. Mondal, S. Das, S. Venugopalan, M. Krishnan and B. Sahoo, Org. Lett., 2023, 25, 1441–1446 CrossRef CAS PubMed; (b) H. Wu, S. Chen, D. Xiao, F. Li, K. Zhou, X. Yin, C. Liu, X. He and Y. Shang, Org. Lett., 2023, 25, 1166–1171 CrossRef CAS PubMed.
- Quinazolinone fragments were widely existed in bioactive natural products and drugs, for selected examples, see: (a) R. Bouley, D. Ding, Z. Peng, M. Bastian, E. Lastochkin, W. Song, M. A. Suckow, V. A. Schroeder, W. R. Wolter, S. Mobashery and M. Chang, J. Med. Chem., 2016, 59, 5011–5021 CrossRef CAS PubMed; (b) L. Hudson, J. Mui, S. Vázquez, D. M. Carvalho, E. Williams, C. Jones, A. N. Bullock and S. Hoelder, J. Med. Chem., 2018, 61, 7261–7272 CrossRef CAS PubMed.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem, 2021, 5, 486–499 CrossRef CAS PubMed.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–437 CrossRef CAS PubMed; (b) A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01111b |
| This journal is © The Royal Society of Chemistry 2024 |