
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective organocatalytic strategies to access noncanonical α-amino acids
Pietro
Pecchini
 a,
Mariafrancesca
Fochi
a,
Francesca
Bartoccini
b,
Giovanni
Piersanti
*b and
Luca
Bernardi
*a
a,
Mariafrancesca
Fochi
a,
Francesca
Bartoccini
b,
Giovanni
Piersanti
*b and
Luca
Bernardi
*a
aDepartment of Industrial Chemistry “Toso Montanari”, Center for Chemical Catalysis C3 & INSTM RU Bologna, V. Gobetti 85, 40129 Bologna, Italy. E-mail: luca.bernardi2@unibo.it
bDepartment of Biomolecular Sciences, University of Urbino Carlo Bo, Piazza Rinascimento 6, 61029 Urbino, PU, Italy. E-mail: giovanni.piersanti@uniurb.it
First published on 25th March 2024
Abstract
Organocatalytic asymmetric synthesis has evolved over the years and continues to attract the interest of many researchers worldwide. Enantiopure noncanonical amino acids (ncAAs) are valuable building blocks in organic synthesis, medicinal chemistry, and chemical biology. They are employed in the elaboration of peptides and proteins with enhanced activities and/or improved properties compared to their natural counterparts, as chiral catalysts, in chiral ligand design, and as chiral building blocks for asymmetric syntheses of complex molecules, including natural products. The linkage of ncAA synthesis and enantioselective organocatalysis, the subject of this perspective, tries to imitate the natural biosynthetic process. Herein, we present contemporary and earlier developments in the field of organocatalytic activation of simple feedstock materials, providing potential ncAAs with diverse side chains, unique three-dimensional structures, and a high degree of functionality. These asymmetric organocatalytic strategies, useful for forging a wide range of C–C, C–H, and C–N bonds and/or combinations thereof, vary from classical name reactions, such as Ugi, Strecker, and Mannich reactions, to the most advanced concepts such as deracemisation, transamination, and carbene N–H insertion. Concurrently, we present some interesting mechanistic studies/models, providing information on the chirality transfer process. Finally, this perspective highlights, through the diversity of the amino acids (AAs) not selected by nature for protein incorporation, the most generic modes of activation, induction, and reactivity commonly used, such as chiral enamine, hydrogen bonding, Brønsted acids/bases, and phase-transfer organocatalysis, reflecting their increasingly important role in organic and applied chemistry.
 Pietro Pecchini | Pietro Pecchini received his M.Sc. in Chemistry from the University of Bologna in 2022. During the master thesis internship with Prof. L. Bernardi, he worked on stabilized sulfoxonium ylides in aminocatalysis for enantioselective cyclopropanations. Currently, he is spending his second year of PhD in the Bernardi/Fochi group, focusing on sulfoxonium ylide reactivity in organocatalysis and heterogeneous asymmetric catalysis based on the exploitation of gels derived from renewable alginate biopolymers. |
 Mariafrancesca Fochi | Mariafrancesca Fochi received her master's degree in 1994 and PhD in Chemistry in 1998 from the University of Bologna with Prof. A. Ricci. She spent research periods as an academic visitor at the University of Nijmegen (The Netherlands) and at Oxford University (UK). In 2004, she was appointed assistant professor, and in 2019 associate professor, at the University of Bologna. Her scientific interests include acylsilane and thioacylsilane chemistry, chiral ferrocene derivatives, metal-mediated homogeneous asymmetric catalysis and finally asymmetric organocatalysis. She is a co-author of more than 80 papers published in international journals. |
 Francesca Bartoccini | Francesca Bartoccini received her master's degree in chemistry in 2001 from the University of Bologna under the supervision of Prof. Gianfranco Cainelli. She then moved to the University of Urbino Carlo Bo to join Prof. Giorgio Tarzia's research group. Since 2003, she has been working as a laboratory technician at the same University. In 2020, she was awarded with the National Scientific Habilitation as Associate Professor of Organic Chemistry. Her research interests include dehydroamino acids, organic compounds containing boron, non-canonical amino acids, natural products, and (bio)molecules with redox-active sulfhydryl function(s). In 2017, she co-founded the academic spin-off GLUOS srl. |
 Giovanni Piersanti | Giovanni Piersanti received his MS in Chemistry from the University of Bologna. He moved to the University of Urbino Carlo Bo, where he got his PhD. After a period at the University of California-Berkeley, Massachusetts Institute of Technology and Nerviano Medical Sciences, he returned to Urbino in 2006 to begin his independent career where he has remained since and is now Full Professor. His current research focus is on the synthesis of noncanonical amino acids/peptides and natural products in concert with the discovery and the development of new transformations including catalytic, asymmetric, practical and green processes. |
 Luca Bernardi | Luca Bernardi is currently associate professor at the University of Bologna, Italy, where he received an MSc in Industrial Chemistry in 2000, and PhD in 2004 (with Prof. A. Ricci). He was a post-doc in Aarhus (Denmark) and a visiting scientist in Groningen (The Netherlands). His research interests focus mainly on enantioselective organocatalysis, and on the valorisation of biopolymers from marine sources. He has been involved in several industrial activities and is serving as coordinator of the national PRIN2020 project “Natural products-assisted organic synthesis”. |
1. Introduction
α-Amino acids (AAs) are the fundamental biological building blocks of peptides and proteins; thus, they are of utmost importance in almost all biological processes and play essential roles in all living organisms.1 As a significant portion of the chiral pool, they are also extensively used in chemical processes, such as the preparation of chiral auxiliaries,2 catalysts,3 ligands for transition-metal catalysis,4 polymers/materials,5 and biologically active molecules including peptidomimetic drugs.6 Because of their distinct pharmacological profile, high specificity, low immunogenicity, and superior selectivity over larger biologics, synthetic peptides embedded with proteinogenic (canonical) AAs7 have seen a renaissance; nevertheless, some properties must be addressed because of their poor oral bioavailability, low plasma stability, and short lifetimes.8 While several chemical approaches have been developed to address these issues, one effective tactic that has been used is the selective insertion of nonproteinogenic AAs into the peptide backbone.9 In fact, nature provides 20 common AAs as building blocks, with side chains displaying polar, aromatic, or aliphatic groups. However, these 20 canonical AAs severely limit the types and functional applications of proteins, chiral/ligand catalyst design, and drugs owing to their low structural/stereochemical diversity and ensuing chemical properties; therefore, they are no longer sufficient to meet the most recent research needs of both biologists and chemists. To exploit the versatility offered by AAs in full, it is important to have efficient and straightforward methods to access structural motifs beyond those exhibited by natural AAs (Fig. 1).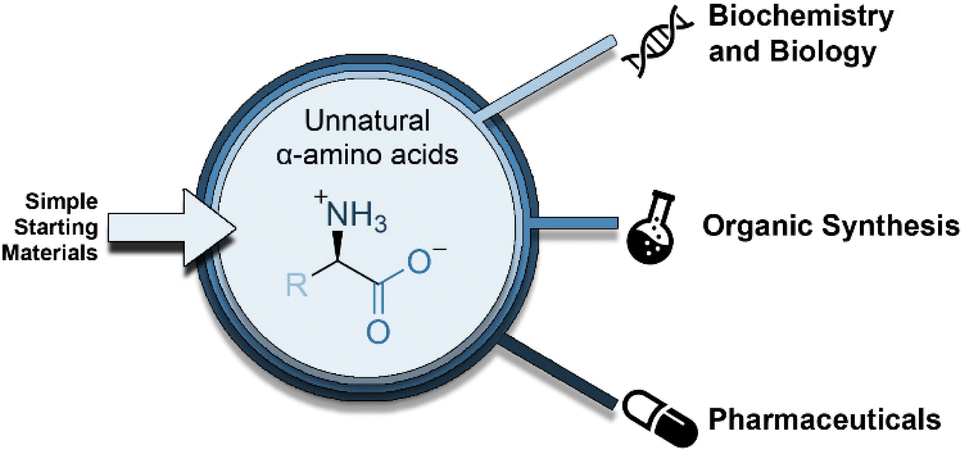 | ||
| Fig. 1 Unnatural, noncanonical amino acids (ncAAs): synthesis and utility. | ||
As such, novel enantiopure synthetic analogues of the canonical AAs, called noncanonical amino acids (ncAAs), increase the diversity available to acquire valuable information on the structure, dynamics, and function of peptides and proteins.10 ncAAs have found broad applications in biochemistry and biological sciences, thanks to their possibility of being genetically encoded in bacteria, yeast, and mammalian cells, i.e., via protein engineering and/or selective incorporation by solid-phase peptide synthesis into peptide-based drugs and short proteins.11 The latter technique can easily facilitate the introduction of ncAAs. While this method was previously limited to peptides of less than 50 AAs, recent developments have drastically increased that limit.12 These technologies allow chemists to probe as well as change the properties of proteins (e.g., increase their potency or induce unusual conformations), in vitro or in vivo, by directing novel, lab-synthesized chemical moieties specifically into any chosen site of any protein of interest, including antibody–drug conjugates. This enables the regulated synthesis of homogeneous, site-specific adducts. In the context of enzymatic processes, the introduction of ncAAs into new biocatalysts can change the activity of a native enzyme.13 There have also been significant and rapid developments in chemoselective reactions for labelling biological molecules in vitro and in cells, initiated by the development of so-called bioorthogonal chemistry.14 Additionally, ncAAs are often part of complex natural products such as vancomycin (a widely employed antibiotic) and are prominent features in small-molecule therapeutics used to treat many diseases (Fig. 2).15 Branched-chain AAs, such as L-tert-leucine, are the building blocks of several pharmaceutical development projects targeting various diseases such as cancer, rheumatic arthritis, and AIDS.16 In addition, AAs with silicon incorporation, e.g., (trimethylsilyl)alanine, have several attractive features that can be useful in drug discovery.17 Other outstanding and more recent examples of the powerful compliance-inducing impact of non-native characteristics include essential drugs like nirmatrelvir, the novel SARS-CoV-2 3C-like protease inhibitor discovered in 2020 by Pfizer scientists that comprises three ncAAs and is used to treat COVID-19,18 and telaprevir, a peptide inhibitor of the enzyme hepatitis C virus NS3 protease19 that embodies four ncAAs. More recently, ncAA-containing small cyclic peptides that are orally available have effectively targeted atypical biological targets like thrombin.20
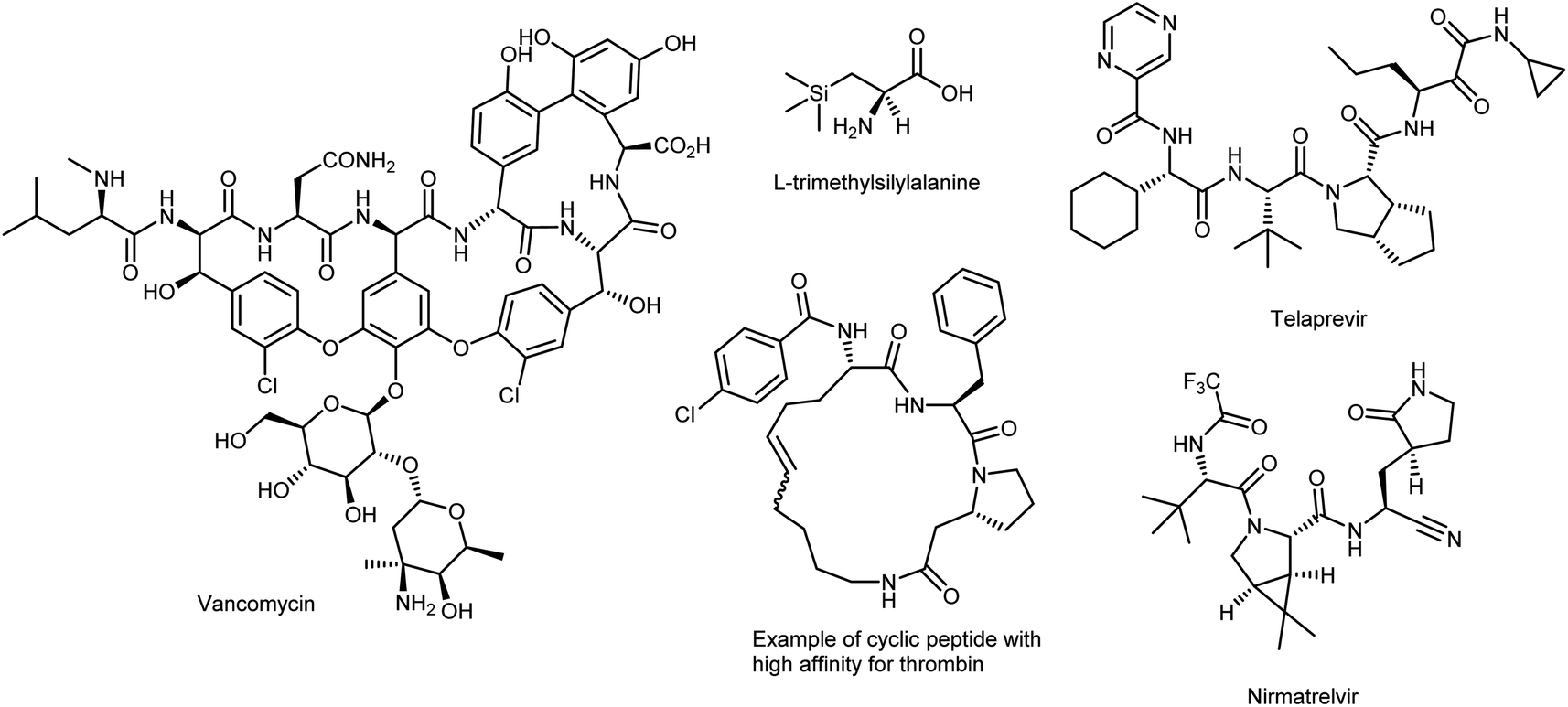 | ||
| Fig. 2 Biologically relevant ncAAs. | ||
The success of ncAAs in drug discovery is related, although not exclusively, to their resemblance to canonical AAs, typically comprising an amine, a carboxylate, a side chain, and an asymmetric carbon atom. ncAAs are not limited to the functional groups found in the 20 genetically encoded AAs but instead employ a myriad of chemical motifs. This diversity of possible functional groups, for example ketone, aldehyde, azide, alkyne, nitro, carbamate, boronate, silicon, tetrazine, cyclopropene, diazirine, etc., enables ncAAs to engage in new interactions, even covalent ones, within or between biological protein targets. Thus, ncAAs can be used to affect protein function, enhance protein stability, investigate protein–protein interactions, and improve pharmacological properties, thereby creating opportunities for drug development. ncAAs can also be readily transformed into other classes of chiral molecules, including β-amino alcohols, oxazolidines, and 1,2-diamines, which are widely used in asymmetric synthesis as ligands for asymmetric catalysts and building blocks leading to enantiopure natural products and drugs.21
The dramatically increasing demand for optically active ncAAs has continuously driven chemists to develop new and efficient methodologies, especially those based on asymmetric catalysis. Multiple catalytic technologies and sophisticated asymmetric catalyst designs have been reported, including the hydrogenation of olefins and imines, electrophilic amination of enolates, electrophilic alkylation of glycine derivatives, and, of course, hydrocyanation of an imine or imine equivalent (the venerable Strecker synthesis), and many more.22 These strategies afford enantioenriched ncAAs by assembling enantioselectively the AA portion of the molecule. However, an alternative yet powerful catalytic approach to ncAAs is based on the manipulation of the side chains of canonical AAs by exploiting their innate enantiopurity. In this context, metal-catalyzed and photochemical C–H activation reactions have recently become a fruitful platform for a range of functionalization reactions of the side-chains of canonical AAs. In some instances, these catalytic platforms have been implemented for the late-stage functionalization of AAs embedded in complex peptide/protein structures.23
In this perspective, we describe asymmetric organocatalysis according to the common themes of ncAA synthesis. Considering their key and rapidly growing roles in different scientific fields, these popular target compounds well illustrate the different facets of this asymmetric approach, including its considerable synthetic potential. Limiting the discussion to tertiary, noncyclic AA structures, we aim to highlight the early disclosures on the use of organocatalysts to promote the reactions of organic compounds to access enantioenriched ncAAs as well as to cover the stream of work that has appeared recently. For the most part, we have grouped reactions according to which atom or group is introduced, e.g., carboxyl group, amino group, side chain, or the hydrogen of the chiral center (Scheme 1). Moreover, we have sought to highlight the variety of reactive intermediates as well as particularly intriguing approaches (e.g., biomimetic) that may be accessed via this general reaction manifold. Although enzymes are valuable classes of enantioselective catalysts in terms of cost, enantioselectivity, and sustainability and numerous innovative advancements have been made in the last two decades, including recombinant DNA technologies, bioinformatics, and directed evolution, the coverage of biocatalytic ncAA production is beyond the scope of this work and we refer the reader to some other reviews.24 This could seem to be a bizarre choice, since (a) organocatalysis was originally bioinspired from enzymes; (b) recent advances in biocatalysis were also obtained with chemical modification; and (c) the constant input of new enzymes is largely due to the ncAA-expanded AA set. Similarly, chiral organometallic catalysts and general metal complexes containing chiral organic ligands will not be covered.
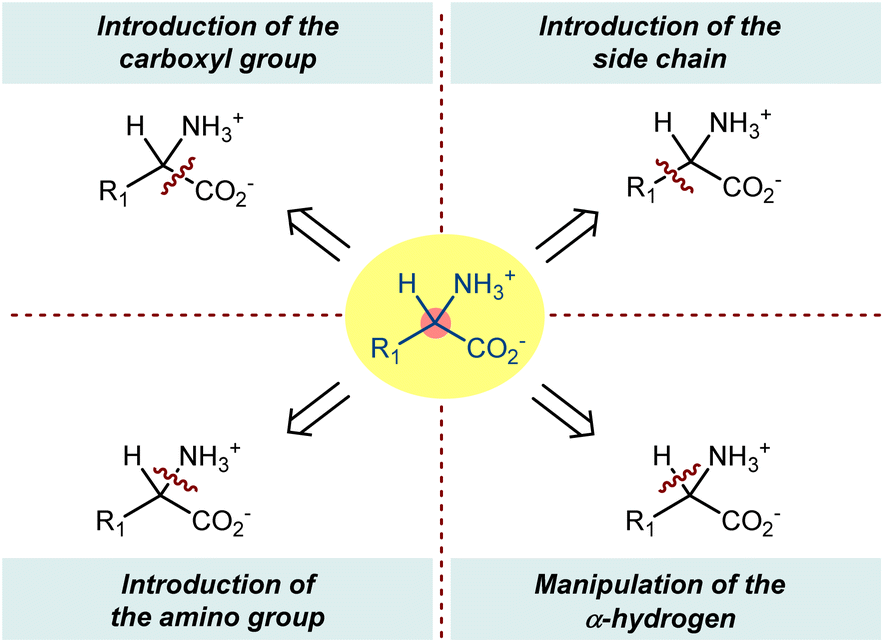 | ||
| Scheme 1 | ||
2. Introduction of the carboxyl group
This section describes the addition of synthetic equivalents of the carboxyl carbanion synthon to imines (Scheme 2). Cyanide, isocyanides, and nitroalkanes are discussed, giving organocatalytic enantioselective versions of the corresponding name reactions (e.g., Strecker, aza-Henry, and Ugi).25 The carboxylate functionality is unveiled through synthetic manipulations of the catalytic products. Since the reactions invariably employ N-substituted imines, AA derivatives carrying a substituent at their amino group are obtained.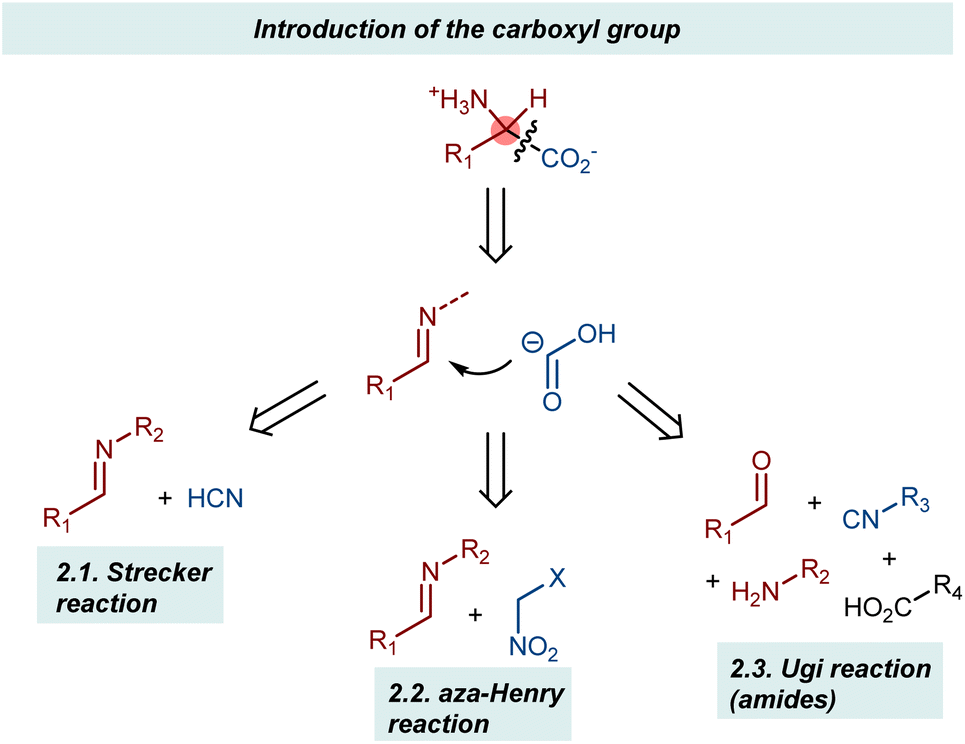 | ||
| Scheme 2 | ||
2.1. Strecker reaction
As a classical and convenient approach to AAs, investigations on asymmetric Strecker-type reactions started many years ago. From the outset, hydrogen-bond interactions were recognized as a useful tool to achieve stereocontrol. A seminal publication from 1996 by Lipton, taking inspiration from previous asymmetric cyanohydrin syntheses,26 introduced chiral diketopiperazine as a catalyst for this reaction.27 However, this article was recently retracted due to the fact that the enantioselectivity of the reaction could not be reproduced. In contrast, (thio)urea derivatives are without a doubt reliable and efficient catalysts for the reaction. The first example, which at the same time disclosed efficient asymmetric catalysis with chiral thioureas, was reported in 1998 and is the result of a serendipitous, but not entirely unexpected, discovery, considering the diketopiperazine above. While screening libraries of chiral ligands for the development of a Lewis acid-catalyzed Strecker reaction, Sigman and Jacobsen realized that the metal was in fact not necessary, with the ligand itself being sufficient for the promotion of the enantioselective reaction. The screening of a large library of Schiff base thioureas led to the identification of 1 as a competent structure (Scheme 3). The reaction could also be applied to challenging aliphatic imine substrates, affording good results.28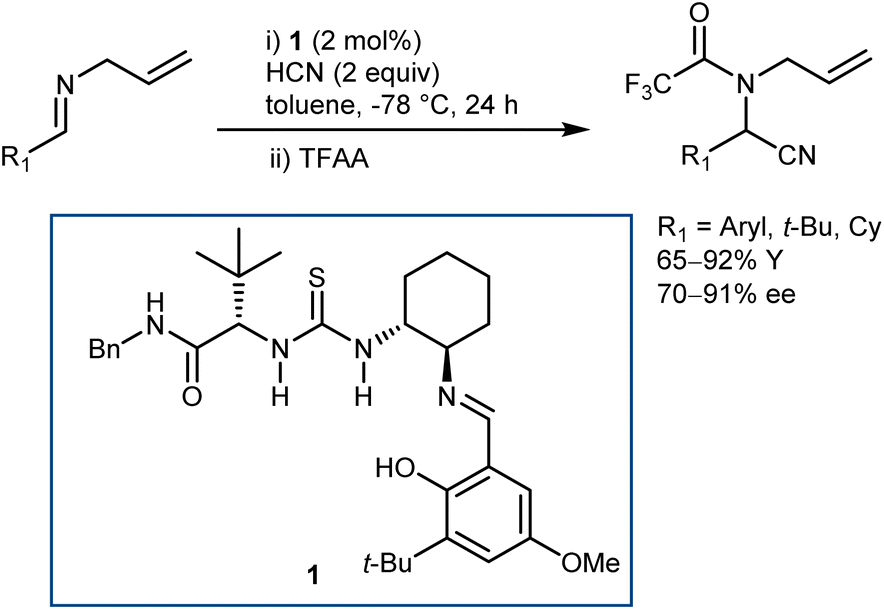 | ||
| Scheme 3 | ||
Other catalyst structures were explored with success in those years. Corey and Grogan reported that the basic guanidine 2 promotes the enantioselective reaction with N-benzhydryl imines and is also applicable to some aliphatic imines (Scheme 4).29 The latter substrates give an opposite face-selectivity compared to their aromatic counterparts, hinting to a different arrangement within the catalyst coordination sphere.
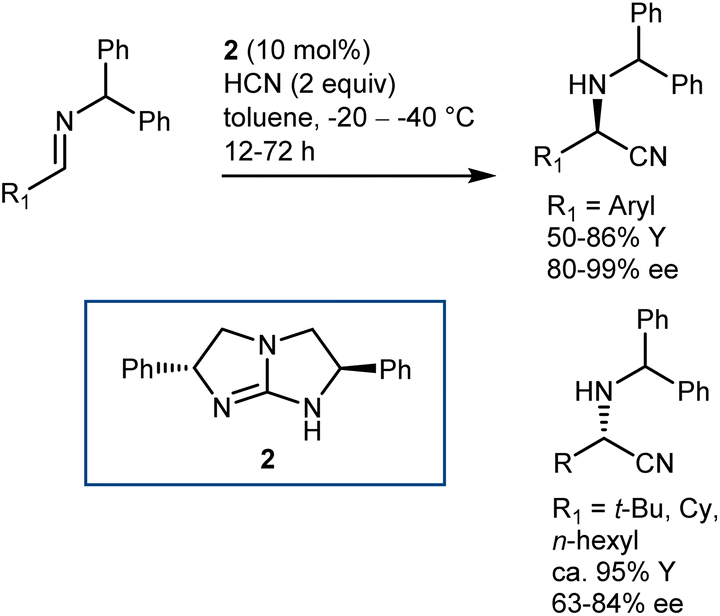 | ||
| Scheme 4 | ||
Irrespective of the remarkable results provided by guanidine 2 and other catalysts in these two-component Strecker reactions, it might be argued that the major advances that followed with N-alkyl imines arose from Jacobsen's systematic investigations on thiourea catalysts. Facilitated by their modular nature, screening of different catalysts recognized 3 as a simplified but more efficient structure (compared to 1, Schemes 3 and 5), proving very general for the addition of HCN (generated in situ from TMSCN and MeOH) to a wide range of N-benzhydryl imines. The lack of sensitive functional groups on this catalyst enabled the application of more practical cyanide sources (e.g., NaCN and KCN) under biphasic reaction conditions. The avoidance of cryogenic temperatures, low catalyst loading, and simplified downstream chemistry ultimately resulted in a scalable protocol to afford complex AAs in their synthetically useful N-Boc-protected form.30
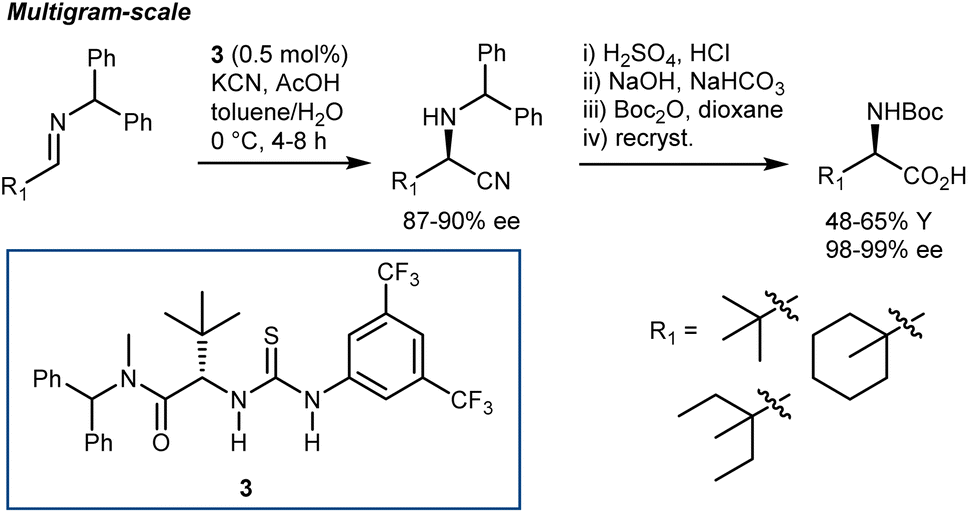 | ||
| Scheme 5 | ||
Detailed experimental and computational studies on this reaction and related processes suggested a fascinating parallelism between the mode of action of this catalyst and enzymatic ones. In essence, this catalyst coordinates with the H-bond network that is geometrically defined by its secondary structure, the (partially) charged transition state, and intermediates of the reaction, leading to the observed enantiomer of the product (Fig. 3).31 The departure from a “simple” imine activation by the Lewis-acidic thiourea followed by the sterically controlled addition of cyanide to one of its two prochiral faces and the analogy with the preorganized electrostatic field invoked for enzymatic catalysis32 are apparent.
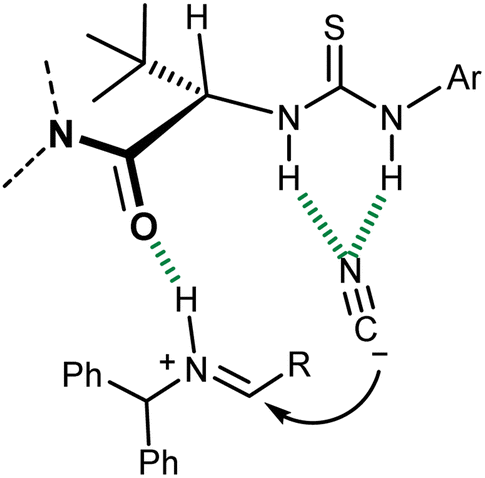 | ||
| Fig. 3 Transition state of the Strecker reaction catalyzed by thiourea 3. | ||
The application of this reaction to N-Boc-AAs via the three-step downstream sequence (Scheme 5) speaks for itself on the desirability of enantioselective Strecker reactions on N-Boc-protected and related imines. Our group reported a moderately enantioselective example under phase-transfer catalysis (PTC) conditions, using acetone cyanohydrin as the cyanide source and α-amido sulfones as convenient imine precursors.33 Only aliphatic substrates could be used. Afterwards, a more efficient protocol was disclosed by Song, using the chiral polyether 4 as the catalyst and KCN (Scheme 6).34 The reaction provides N-Boc-protected α-aminonitriles from aromatic, heteroaromatic, and secondary/tertiary aliphatic imines, delivering outstanding results.
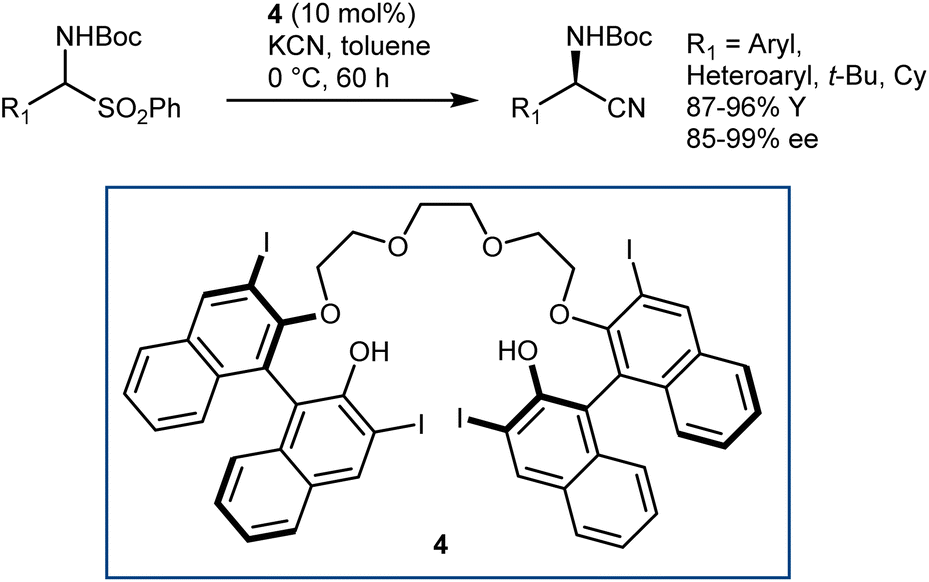 | ||
| Scheme 6 | ||
An obvious intrinsic issue of Strecker-type reactions is the toxicity of cyanide and its precursors. The cyanide sources highlighted in this subsection so far (HCN, silyl cyanides, NaCN, KCN, and acetone cyanohydrin) are all characterized by extreme toxicity, formalized by their H300, H310, and H320 statements (fatal if swallowed, in contact with skin, or inhaled). However, relatively less toxic cyanide sources,35 such as acetylcyanide (H301 and H331; toxic if swallowed or inhaled) and ethyl cyanoformate (H301, H311, and H331; toxic if swallowed, in contact with skin, or inhaled), have also been used in organocatalytic enantioselective Strecker-type reactions.
In more detail, the enantioselective acetylcyanation of N-benzyl imines with acetylcyanide was realized by List using catalyst 5, which is closely related to the archetypal thiourea 1 (Schemes 3 and 7).36 Furthermore, the implementation of this reaction in its three-component version was reported by the same laboratory; thanks to the presence of molecular sieves, the imine was generated in situ prior to the asymmetric reaction.37
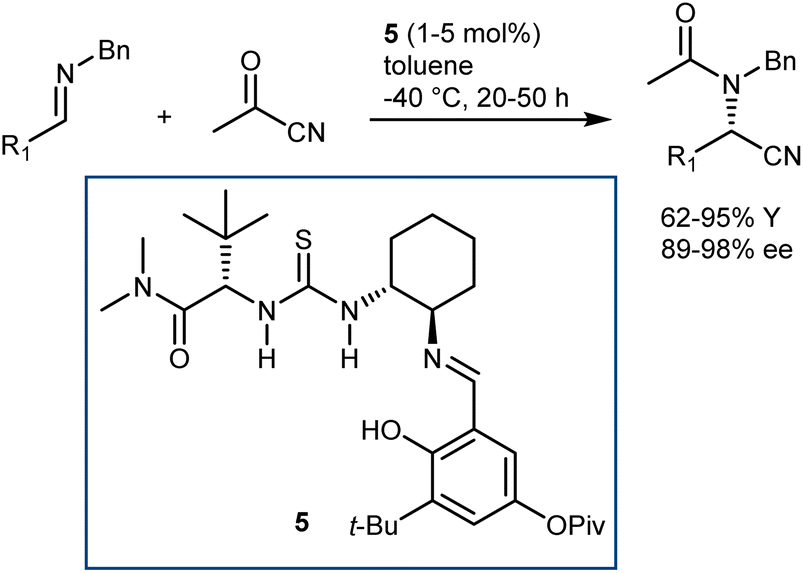 | ||
| Scheme 7 | ||
Conversely, Khan and coworkers reported that the relatively simple double H-bond donor 6 efficiently promoted the reaction with either N-benzhydryl or N-tosyl imines and ethylcyanoformate (Scheme 8).38
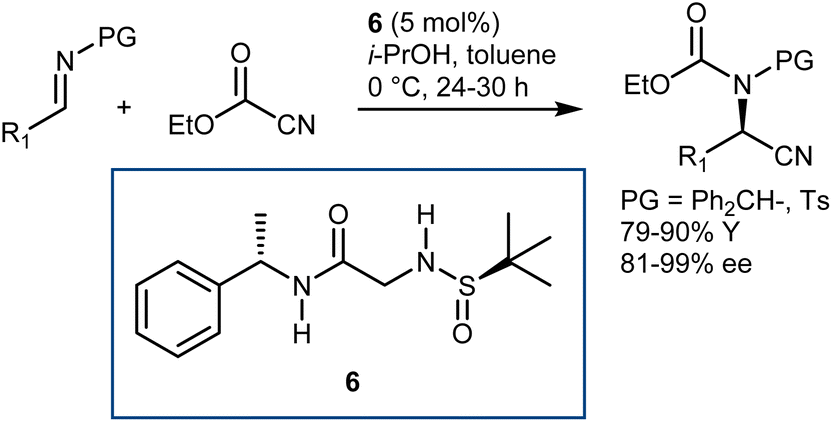 | ||
| Scheme 8 | ||
To conclude this subsection, despite the considerable number of examples, we note that the three-component version of the organocatalytic enantioselective Strecker reaction has received relatively little attention, with just one example dealing with secondary amines.39 For a comprehensive overview of the subject, please see the referenced review articles.40
2.2. Aza-Henry reaction
The enantioselective addition of nitromethane to imines (aza-Henry or nitro-Mannich reaction) followed by an oxidative Nef-type reaction represents another approach to optically active AA derivatives. Imines carrying electron-withdrawing groups at their nitrogen have been recognized as useful substrates for the reaction. These groups guarantee sufficient electrophilicity to the imine and stabilize the aza-Henry adduct, avoiding reversibility issues. The first examples of organocatalytic enantioselective aza-Henry reactions appeared in 2004–2006.41N-Phosphinoyl- and N-Boc-imines derived from (hetero)aromatic aldehydes reacted enantioselectively with nitromethane in the presence of catalysts working through H-bond interactions. Bifunctional catalysts, such as Takemoto's 7 and H-bond-donor Jacobsen-type catalysts, combined with an exogenous tertiary amine base (8) have been useful for these processes (Scheme 9). The nitronate stabilized by the H-bond network of the catalyst is likely the key intermediate.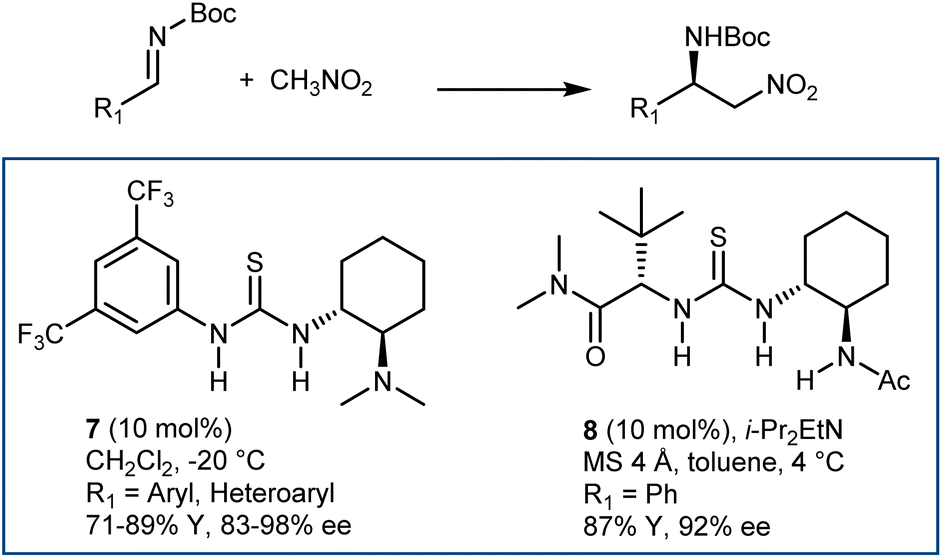 | ||
| Scheme 9 | ||
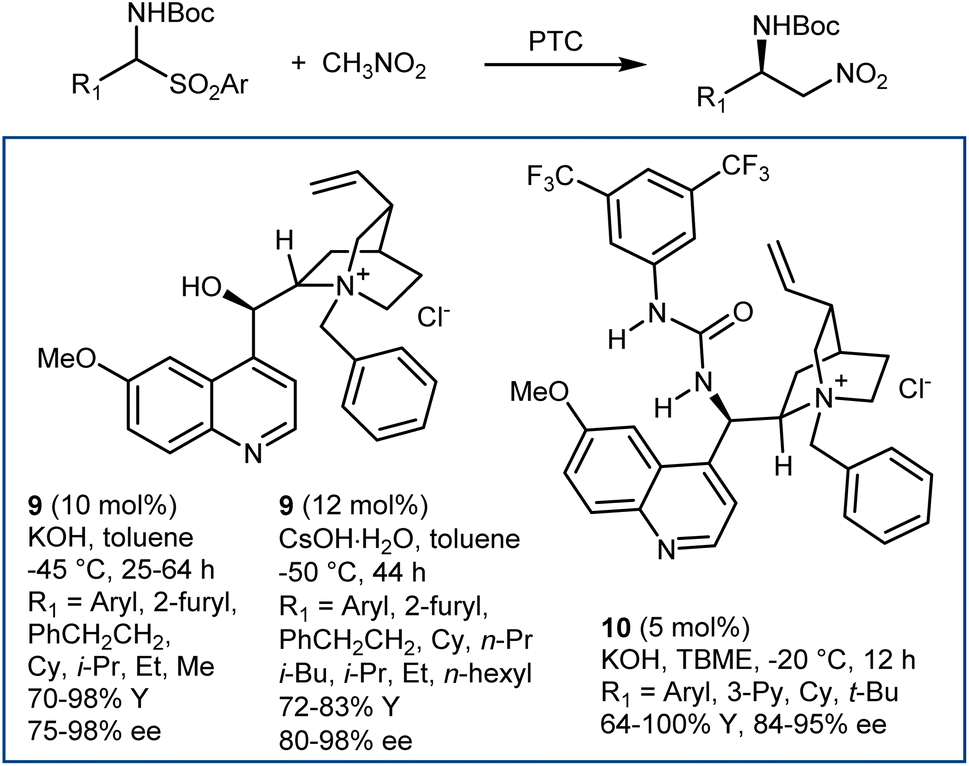
Application of PTC enabled the reaction generating the N-Boc-imine in situ from the corresponding α-amido sulfone. This innovation, reported in 2005 by our group and Palomo's research team,42 gave a practical dimension to the aza-Henry reaction overcoming the tedious preparation of the N-Boc-imines. At the same time, it enlarged its scope, allowing the employment of unstable imines derived from aliphatic, enolizable aldehydes. Phase-transfer catalysts from Cinchona alkaloids, in combination with strong inorganic bases, are useful for this purpose (Scheme 10).42,43 Here, the simple N-benzylquininium chloride 9 gives very good results with aliphatic substrates, while more elaborate structures like Dixon's 10 are more effective with (hetero)aromatic ones.
 | ||
| Scheme 10 | ||
These catalysts present H-bond donors such as the free OH group in 9 and the urea in 10, which are likely to interact with charged species during the reaction. A thorough computational study on the transition state of the reaction with catalyst 9, performed by Palomo, confirmed the binding of the hydroxyl group to the nitronate (Fig. 4).44 Furthermore, the disclosed model includes several key H-bond interactions between the imine and some relatively acidic C–H groups of the catalyst. This type of binding, where O–H and C–H H-bond donors exert a key role complementing electrostatics, is considered to be important in several PTC reactions.45
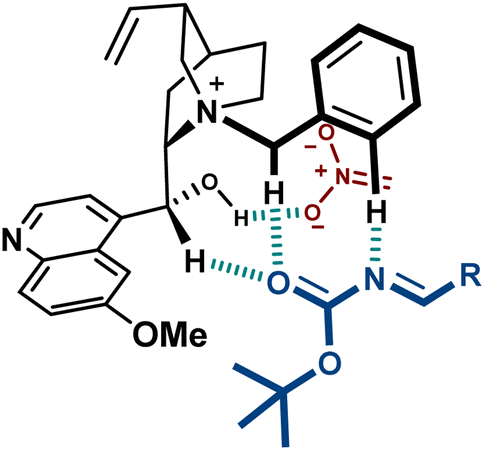 | ||
| Fig. 4 Interactions between catalyst 9 and substrates in the aza-Henry reaction. | ||
The conversion of the catalytic adduct to the AA derivative requires oxidative conditions and is typically performed using a combination of NaNO2 and acetic acid in DMSO, which gives a minimal loss of enantioenrichment (Scheme 11)41e,46 that is less than that caused by other Nef protocols.47 Alternatively, Hayashi reported that the use of molecular oxygen and iodine as oxidants in the presence of a trapping amine affords N-Boc-protected α-amino amides.48
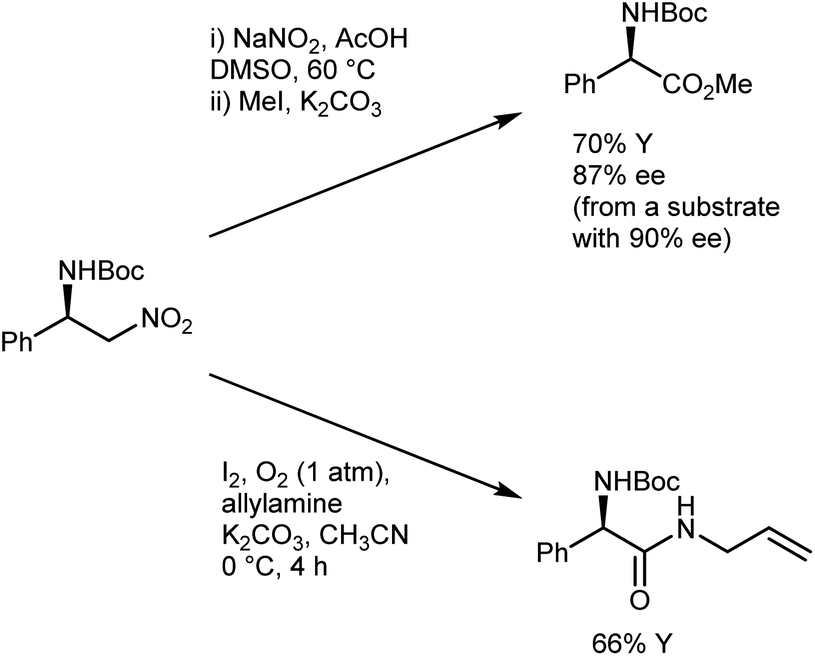 | ||
| Scheme 11 | ||
The latter transformation involves an α-iodonitroalkane intermediate and is similar to Johnston's Umpolung amide synthesis based on α-bromonitroalkanes.49 In fact, enantioselective aza-Henry reactions with bromonitromethane followed by amide bond formation have been developed by Johnston and coworkers. A first approach employed the PTC strategy with α-amidosulfones as imine precursors.50 However, bromonitromethane was poorly compatible with the highly basic PTC reaction conditions, and access to the two enantiomers of the product was in part impeded by the difference in selectivity between the pseudoenantiomeric catalysts derived from quinine and quinidine. Thus, an alternative protocol, still based on the in situ formation of the N-Boc-imine from the α-amido sulfone but using the homogeneous catalyst 11 for the efficient promotion of the reaction, was developed (Scheme 12).51
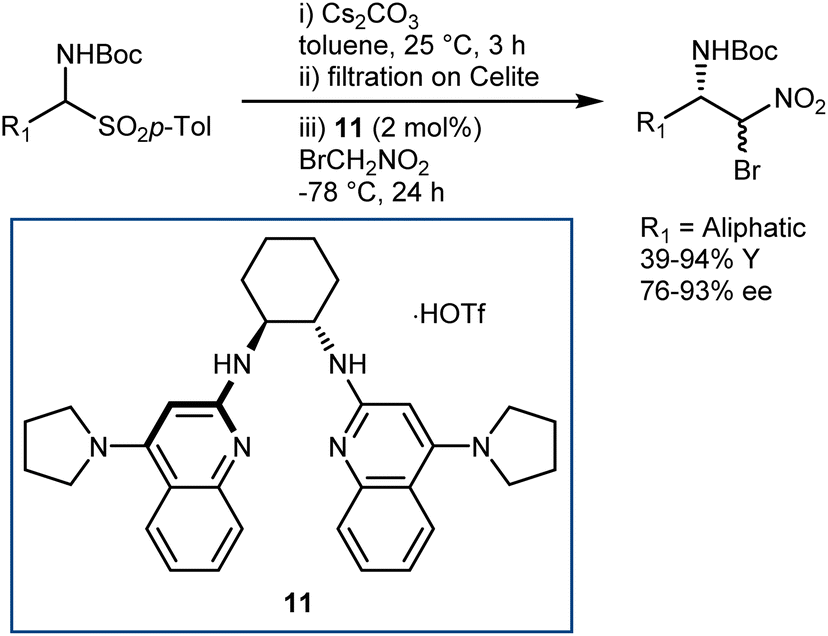 | ||
| Scheme 12 | ||
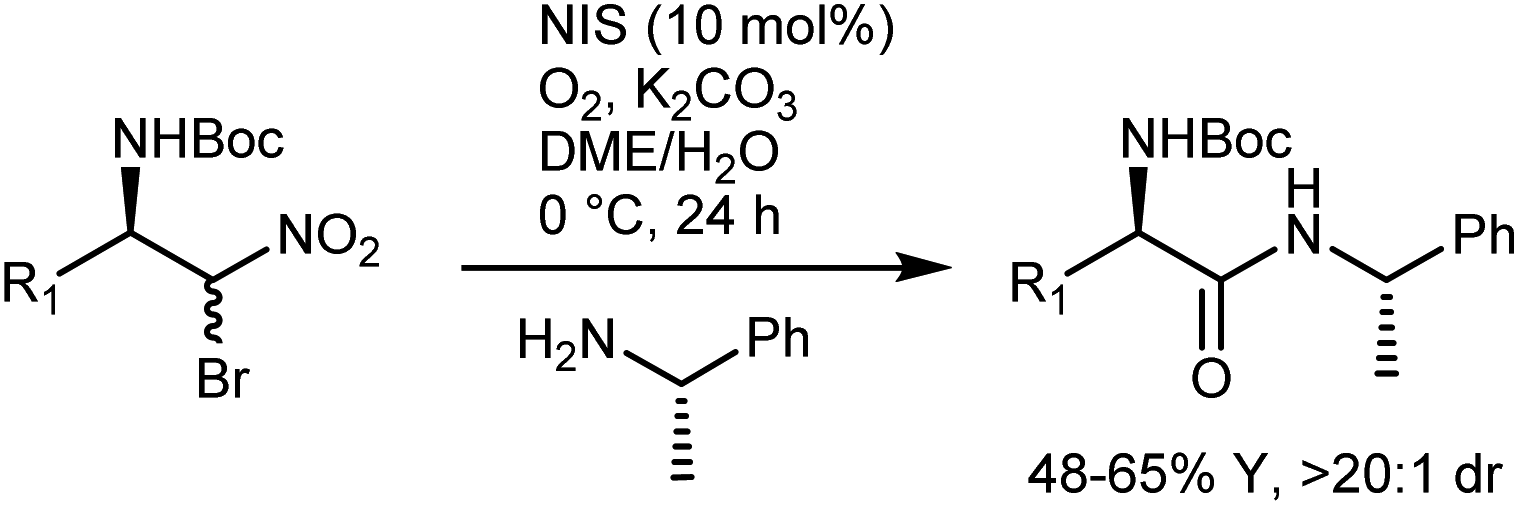
Treatment of the enantiomeric adducts, which were obtained using ent-11 as the catalyst and enantiopure (S)-α-methylbenzylamine under the Umpolung amide synthesis conditions, delivered the corresponding amides with high diastereomeric ratios, suggesting that racemization does not occur (Scheme 13, NIS = ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-iodosuccinimide).
N-iodosuccinimide).
 | ||
| Scheme 13 | ||
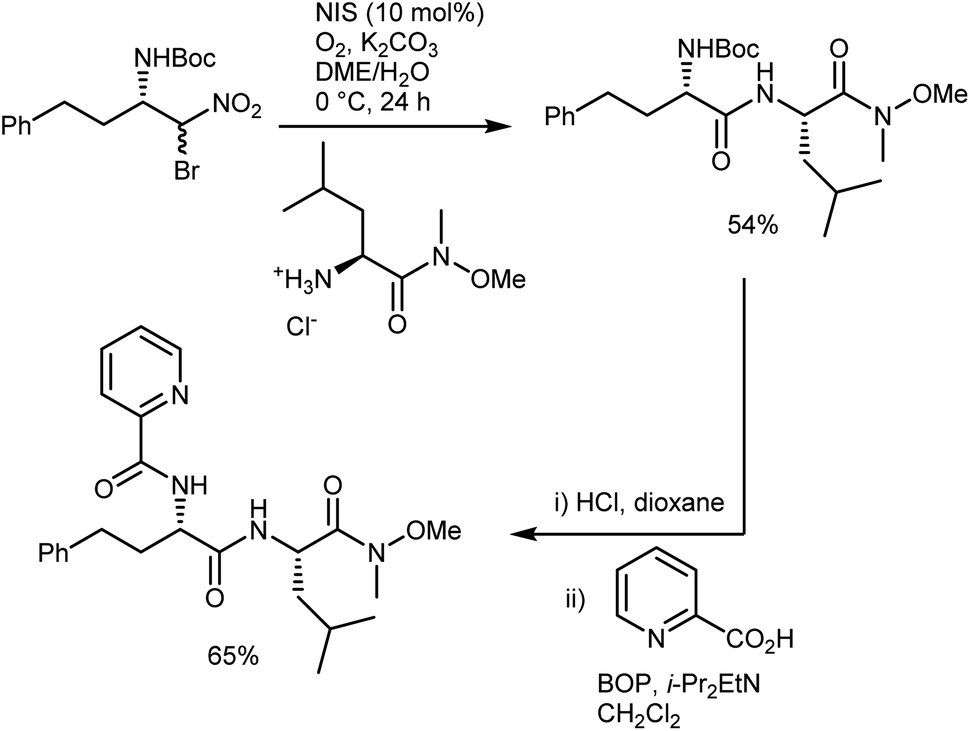
In short, the organocatalytic aza-Henry reaction, especially its versions based on α-amido sulfones, is currently a well-established and reliable tool to obtain a broad range of N-Boc-protected β-nitroamines in enantioenriched form.52,53 It can be argued that the main utility of the reaction concerns the preparation of 1,2-diamines via straightforward reduction of the nitro group. In fact, the Nef conversion to the AA does not appear fully convincing. However, its implementation with amide-forming processes (Schemes 11 and 13), which proceed without apparent racemization, has brought a new synthetic dimension to the aza-Henry reaction. For example, a target dipeptide that is effective in the reversal of P-glycoprotein-mediated resistance to carfilzomib can be readily prepared in three steps from an N-Boc-aza-Henry adduct (Scheme 14).51
 | ||
| Scheme 14 | ||
2.3. Ugi reaction
The Ugi reaction is one of the most important multicomponent reactions (MCRs). It delivers α-amido amides carrying four points of diversity arising from the four reaction components (aldehyde, amine, carboxylic acid, and isocyanide) in a fast manner. The realization of catalytic enantioselective versions of this reaction appears particularly challenging, considering the complexity of the reaction pathway and that the reaction usually proceeds in the absence of a catalyst/promoter. The first organocatalytic enantioselective Ugi reaction was reported in 2009 by Wang and Zhu. The reaction, promoted by chiral phosphoric acid (CPA) 12, employs an α-isocyanoacetamide (Scheme 15).54 Thus, the nitrilium ion formed upon addition of the isocyanide to the imine is intramolecularly trapped by the amide, resulting in an oxazole product. This strategy considerably simplifies the system by excluding the carboxylic acid component but reduces the appeal of the reaction, rendering it a “simple” three-component transformation. Products derived from aliphatic aldehydes, p-substituted anilines, and isocyanoacetamides were obtained with moderate-to-good enantioselectivities. However, aromatic aldehydes gave lower enantioselectivities.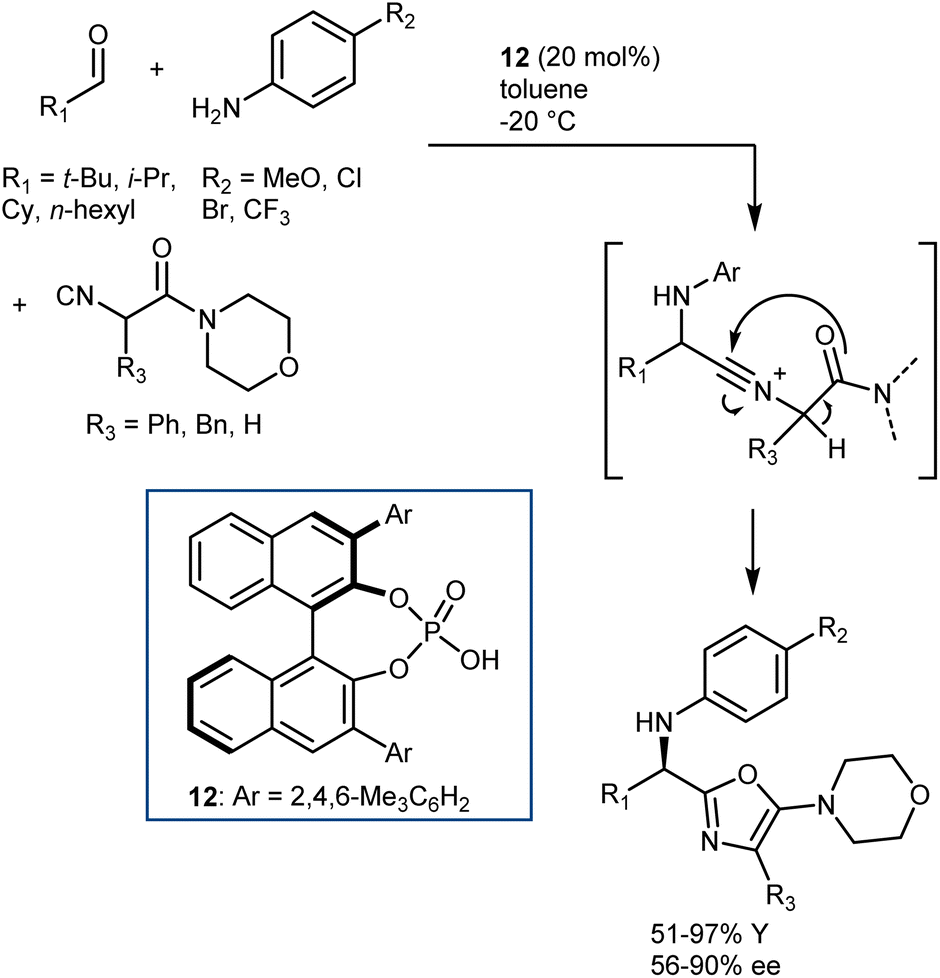 | ||
| Scheme 15 | ||
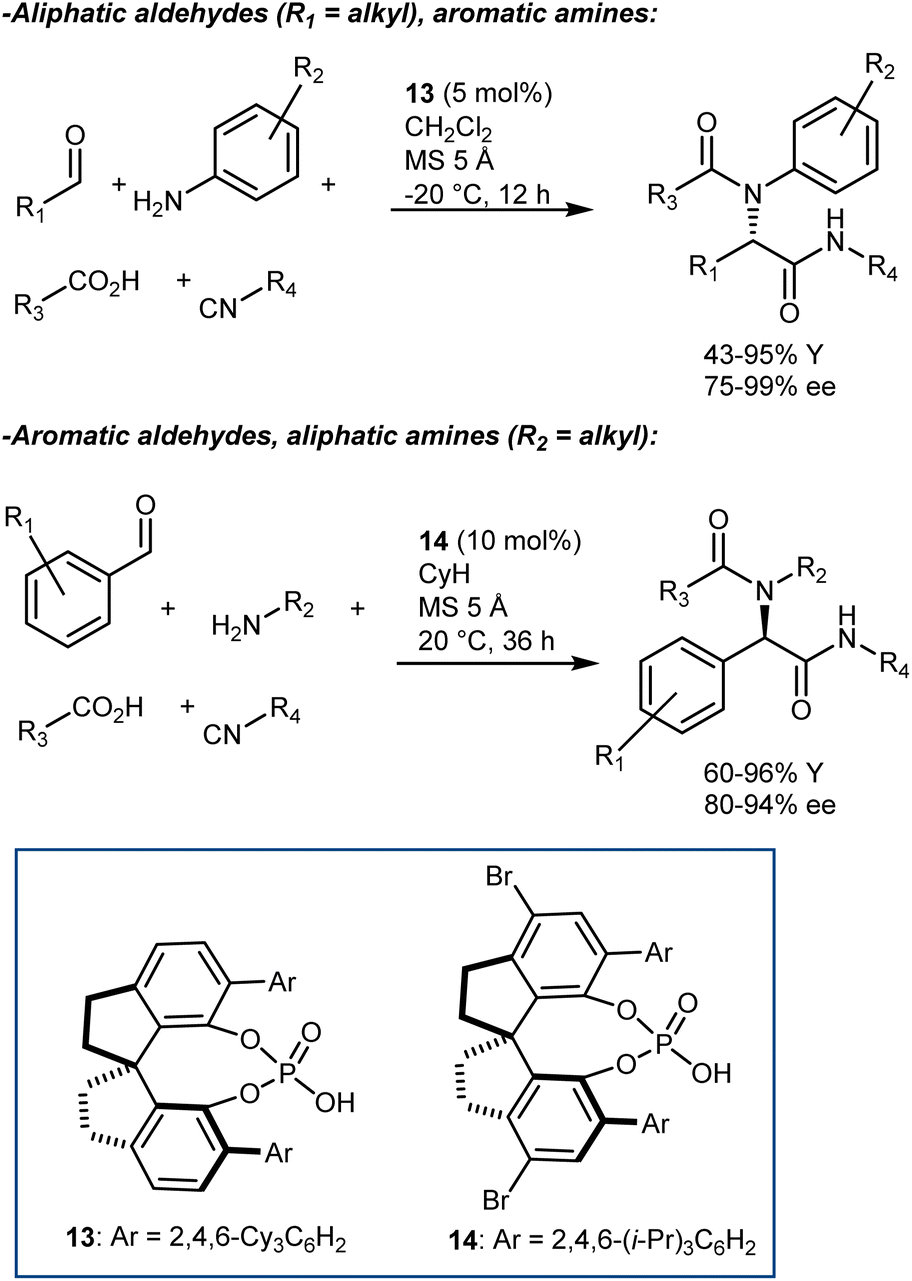
Although other variations of the Ugi reaction appeared in the literature in the following years,55 it was not until 2018 that Houk and Tan reported a genuine catalytic enantioselective four-component Ugi reaction (Scheme 16).56 The availability of a plethora of CPAs carrying different substituents was probably one of the keys to achieve high enantioselectivity in the reaction. Very good results were obtained by combining aromatic aldehydes with aliphatic amines, and vice versa. The two combinations required slightly different reaction conditions and catalysts (i.e., 13 for aliphatic aldehydes and 14 for aromatic ones). The reaction accommodates different carboxylic acids and isocyanides very well.
 | ||
| Scheme 16 | ||
Density functional theory (DFT) calculations on a simplified model confirmed the initial working hypothesis regarding the importance of the heterodimer formed between the catalyst and the carboxylic acid as well as the involvement of all reaction partners in the key rate- and stereo-determining transition state of the reaction (Scheme 17). This transition state was found to be lower in energy compared to an alternative transition state without the carboxylic acid or to the carboxylic acid-catalyzed reaction.
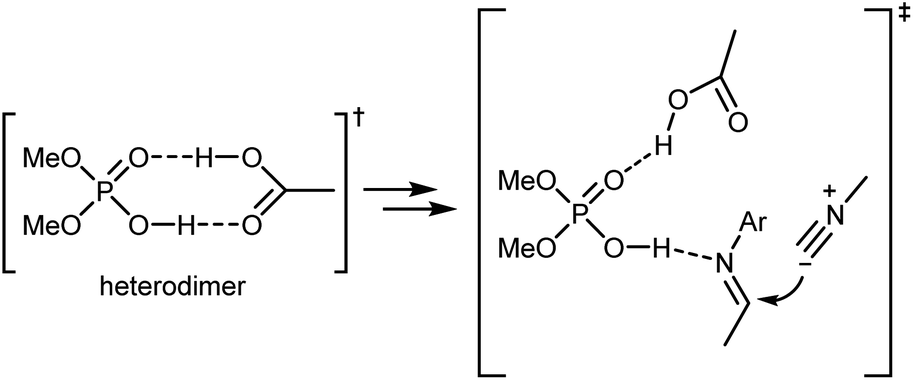 | ||
| Scheme 17 | ||
As shown in Scheme 16, the two aldehyde/amine combinations gave opposite face selectivity in the attack of the isocyanate to the imine, even though catalysts 13 and 14 are derived from the same chiral source ((R)-SPINOL) and are structurally quite similar. DFT calculations performed on the full system showed the importance of noncovalent interactions between the catalyst and the aromatic group of the substrate.
More recently, a catalytic, asymmetric, four-component Ugi reaction was developed by Cao and Liu using the chiral-at-metal anionic Co(III) complex 15 as the catalyst.57 While being metallic in nature, this catalyst exerts its function by acting as a chiral counteranion, coordinating reaction transition states and intermediates via weak interactions like H-bonds. The methodology was applied to a broad range of anilines, aromatic and aliphatic aldehydes, carboxylic acids, and isocyanides. Besides, the carboxylic acid component could be swapped for hydrazoic acid, generated in situ from sodium azide and acetic acid, thus giving the corresponding Ugi-azide products with very good results (Scheme 18).
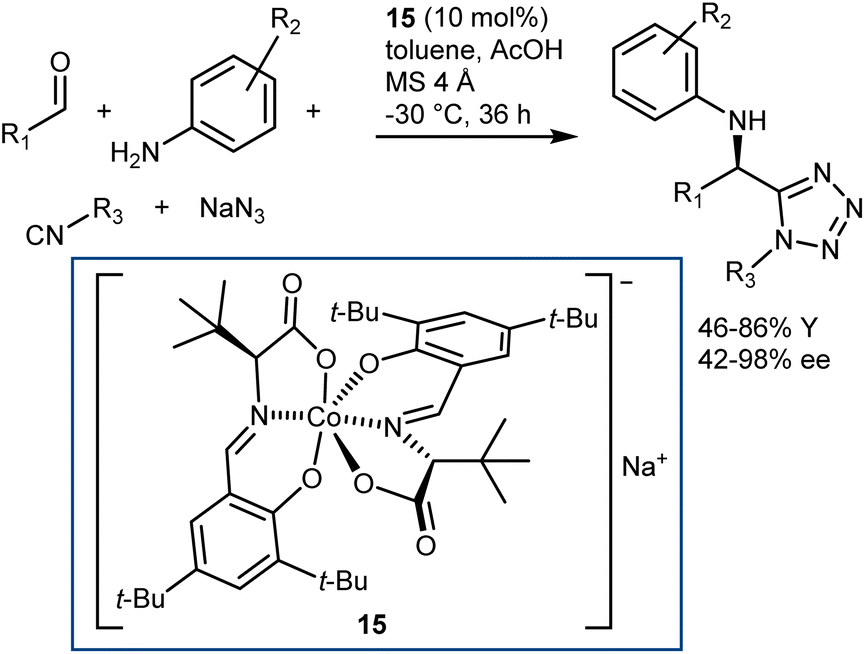 | ||
| Scheme 18 | ||
3. Introduction of the side chain
Two complementary disconnections lead to AA assembly by introducing the side chain based on the glycine α-anion and α-cation synthons, respectively (Scheme 19). The two glycine α-anion approaches discussed in this section exploit the acidification of the α-protons of glycine esters through their imines, either as preformed substrates (alkylation of glycine imines and related reactions) or as transient intermediates (aldehyde catalysis).58 Conversely, glyoxylate imines (α-imino esters) are typical synthetic equivalents of glycine α-cations. Their Mannich reactions will be presented in brief.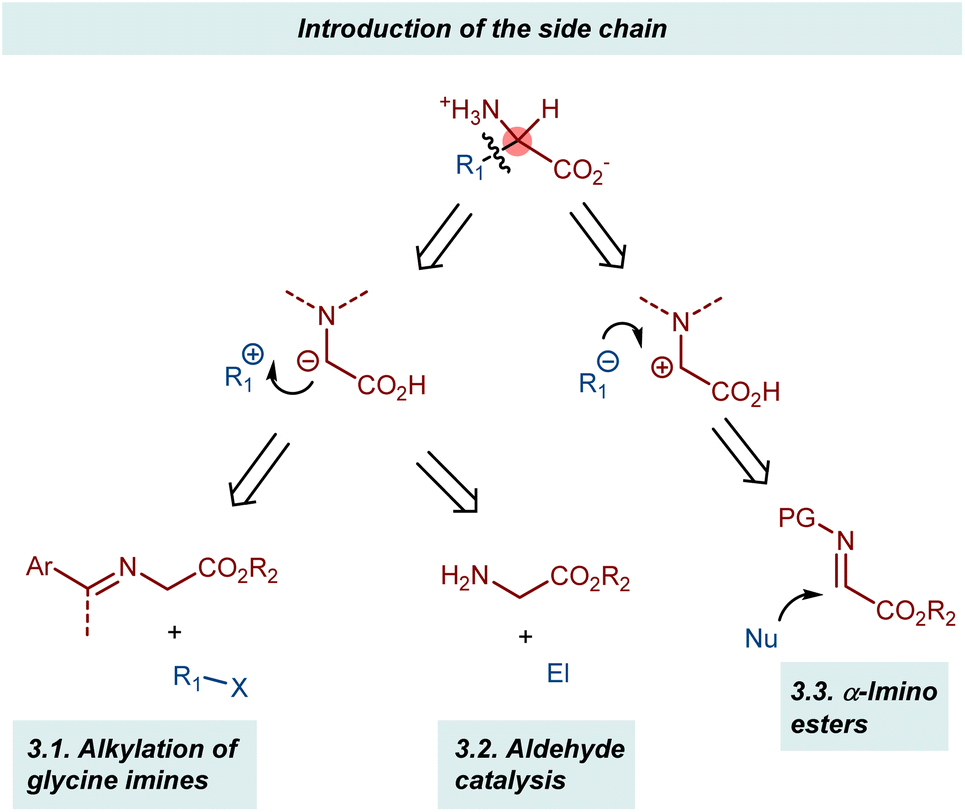 | ||
| Scheme 19 | ||
3.1. Alkylation of glycine imines
The disclosure of the enantioselective alkylation of the benzophenone imine derived from t-butyl glycine ester by O'Donnell in 1989 is a landmark of PTC and of asymmetric synthesis in general (Scheme 20).59 Initially, using a simple PTC catalyst (16), moderate enantioselectivities were obtained. It was found that deprotonation of the hydroxyl group of catalyst 16 under basic reaction conditions resulted in a lipophilic base, causing partial racemization of the product in the absence of the alkylating agent. In fact, the actual catalyst of the reaction is the O-alkylated counterpart of 16, formed in situ. This observation prompted the preparation and utilization of N,O-bisalkylated catalysts like 17.60 Conversely, increasing the size of the aryl group of the quinuclidine N-substituent resulted in the very efficient 9-anthracenylmethyl structures 18 and 19, disclosed in 1997 by Corey61 and Lygo,62 respectively. These early studies on this alkylation reaction highlight its broad scope. The reaction accommodates a range of alkylating agents (benzylic, allylic, and propargylic bromides, primary alkyl iodides, and other activated alkylating agents such as haloacetates). Besides, pseudoenantiomeric cinchonidinium and cinchoninium catalysts display similar enantioselectivities, favoring (S) and (R) products, respectively. This feature, which guarantees access to both AA enantiomers, is not common for PTC based on Cinchona alkaloids.63 Finally, hydrolyses of the benzophenone imine and of the t-butyl ester, unveiling the AA, are trivial.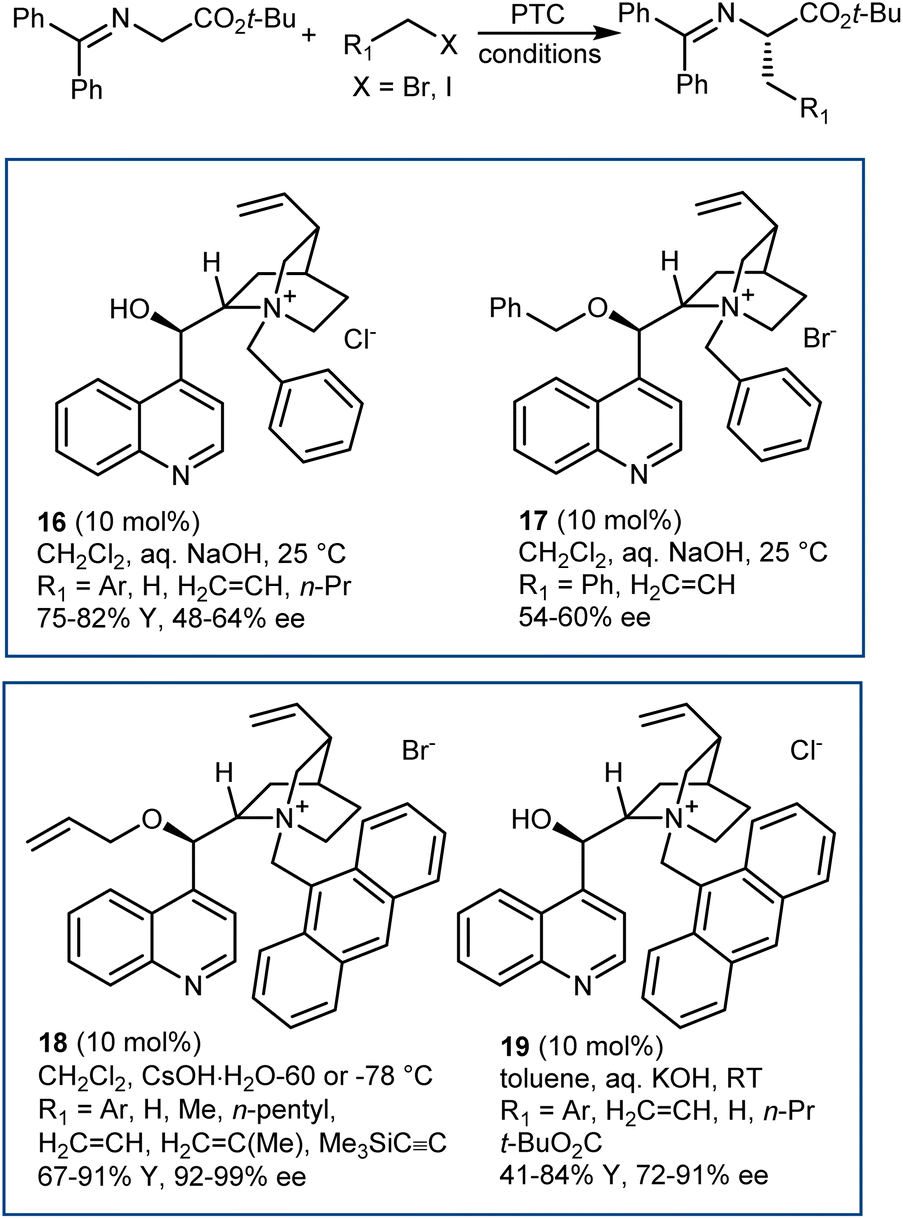 | ||
| Scheme 20 | ||
In fact, this reaction has become the benchmark for the development of new PTC catalysts. A plethora of structures, based on either Cinchona alkaloids or many other chiral scaffolds, capable of promoting this reaction with outstanding selectivity (ee > 90%) have appeared in the literature.64 Many of these catalysts proved to be useful for unrelated PTC reactions as well. Arguably, Maruoka catalysts, such as 20 and its simplified version 21, stand out for their efficiency and generality (Scheme 21).65 Catalyst 21 presents lower lipophilicity compared to 20. This makes it more efficient to extract the enolate from the interfacial region to the organic phase, considering that Mąkosza's interfacial mechanism66 is operative in this alkylation reaction.
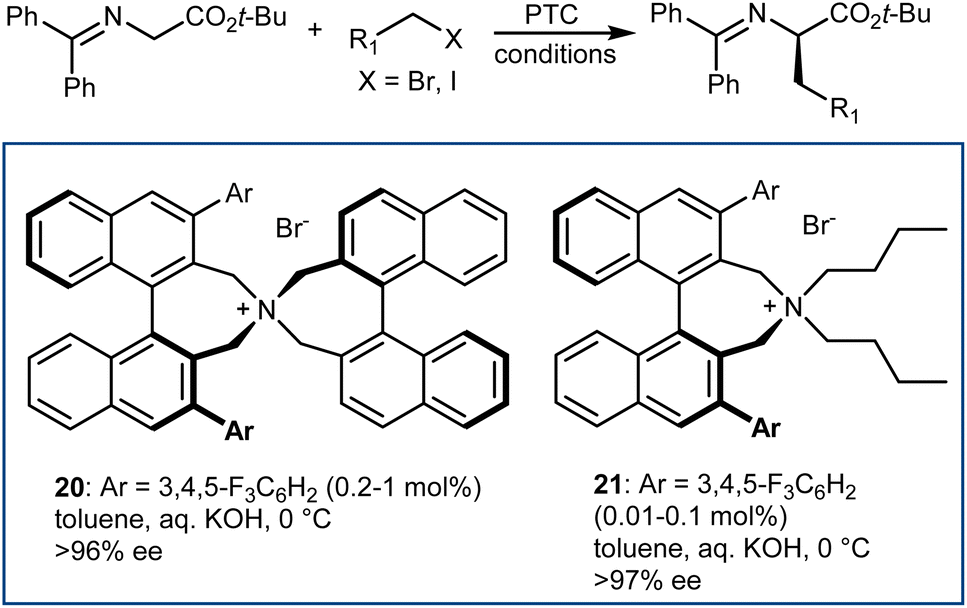 | ||
| Scheme 21 | ||
These binaphthyl-based catalysts are less prone to degradation via the Hoffman elimination pathway compared to quaternized Cinchona derivatives (Scheme 22),64a which also tend to lose their benzylic N-substituent under highly basic reaction conditions.67 Maruoka catalysts can thus be used at low loadings, which can even be improved by applying derivatives deuterated at their benzylic positions.68 In fact, these catalysts degrade via a Stevens rearrangement, which can be avoided, in part, by using stronger C–2H benzylic bonds.
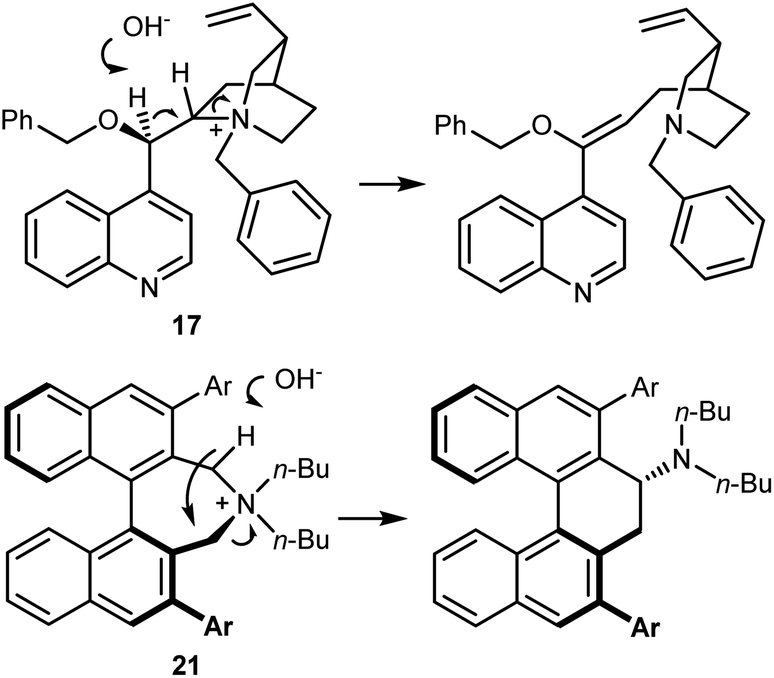 | ||
| Scheme 22 | ||
In industrial settings, the reliability of the PTC alkylation of glycine imines has rendered it a favorite method for the rapid development of routes to enantio-enriched AAs up to the kilogram scale.69 One of the most renowned examples is the synthesis of the AA component of denagliptin tosylate, a potent dipeptidyl peptidase IV inhibitor. The AA was prepared on a kilogram scale by alkylating the benzophenone imine with a benzhydryl alkylating agent in the presence of catalyst 18 (Scheme 23).67 The catalytic reaction, which proceeded with a modest enantiomeric excess (ee) of 60%, was followed by crystallization to improve the ee; a final hydrolysis delivered the target AA. A key parameter noted in the development process is the order of addition. Due to longer cooling times on a large scale, it is essential to add the aqueous base last to the cooled mixture containing all components in order to avoid catalyst degradation.
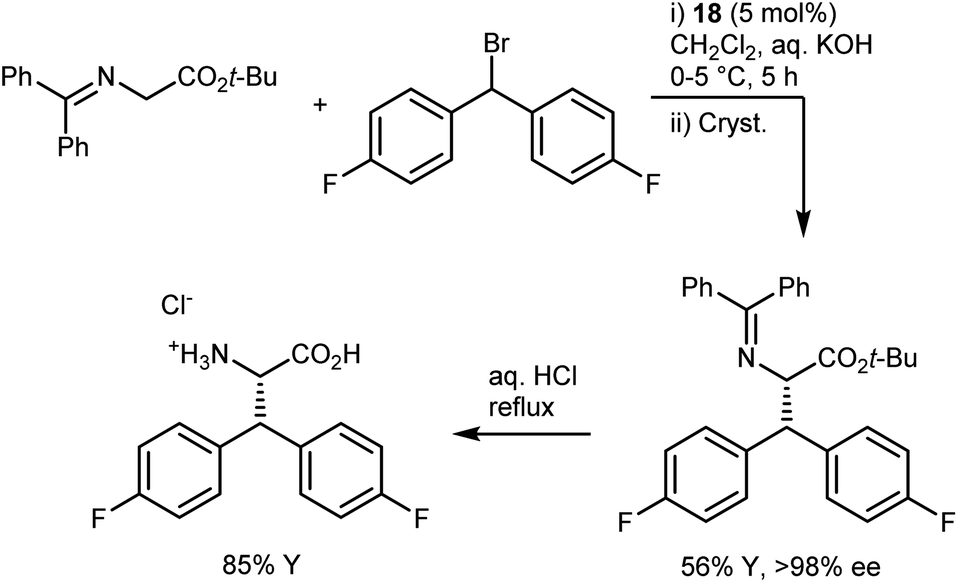 | ||
| Scheme 23 | ||
These reactions employ the benzophenone imine of t-butyl glycine ester. The t-butyl ester gives stability to the substrate under the reaction conditions and renders higher enantioselectivities, especially in reactions catalyzed by Cinchona PTCs.59 Conversely, the ketimine ensures a substantial difference in the pKa value, of about four units, between the glycine substrate and the alkylated product. This difference is due to the destabilization of the anion of the product because of the steric clash between one of the phenyl groups and the alkyl residue.70 Thus, deprotonation of the product with ensuing racemization or dialkylation is avoided. Indeed, to obtain quaternary AAs under enantioselective PTC conditions, aldimines—but not ketimines—derived from α-alkyl glycine esters are generally used.64,69 However, for the large-scale production of AAs, the use of a benzophenone ketimine and a t-butyl ester group is not optimal for cost reasons and poor atom economy. Aiming at the large-scale production of AAs, Maruoka and Ikunaka demonstrated that with the use of the binapthyl catalyst ent-21, a readily available benzaldimine derived from glycine ethyl ester could be employed (Scheme 24).71 Thus, surprisingly, a ketimine was not necessary to avoid racemization and/or bis-alkylation. In more detail, allylation of the aldimine substrate provided a synthetically versatile AA building block, and this process was implemented at the plant scale thanks to its cost-effectiveness. The same substrate could be used for a benzylation reaction with racemic 1-(1-bromoethyl)-4-fluorobenzene, delivering an AA carrying a second chiral center at the β-position with anti-selectivity (Scheme 24).
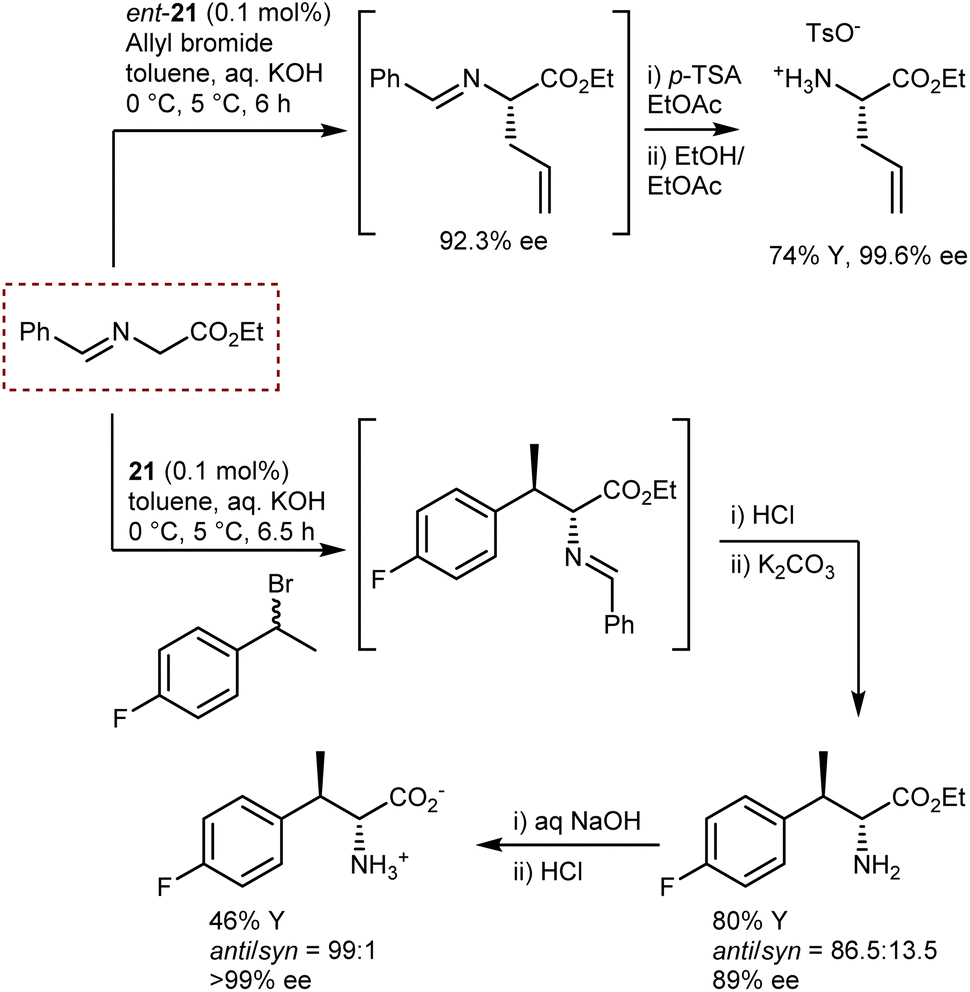 | ||
| Scheme 24 | ||
Despite the venerability of the alkylation reaction of the benzophenone imine derived from t-butyl glycine, new and highly competent catalysts still keep appearing in the literature. Fig. 5 depicts some of the most recent ones, which include Jurczak's Cinchona alkaloid derivative (22),72 Xu's and Bai's spirocyclic ammonium salt 23,73 and Lu's bifunctional phosphonium catalyst 24.74 Moreover, some catalysts based on a different transfer mechanism that works through complexation of the metal countercation of the enolate have appeared. These are represented by Della Sala's and Izzo's cyclopeptoid 25,75 Lacour's receptor 26,76 and Kondo's and Sasai's macrocycle 27, with an embedded azo switch.77 This latter species is able to switch between an active and an inactive state upon light irradiation at different wavelengths. Finally, we recall that asymmetric reactions of the benzophenone imine of glycine esters are certainly not limited to alkylations but also encompass Michael, Mannich, aldol, and other addition reactions.78 The use of PTC, with catalysts benchmarked in the alkylation, is one of the most popular but not the exclusive approach to realize these transformations.
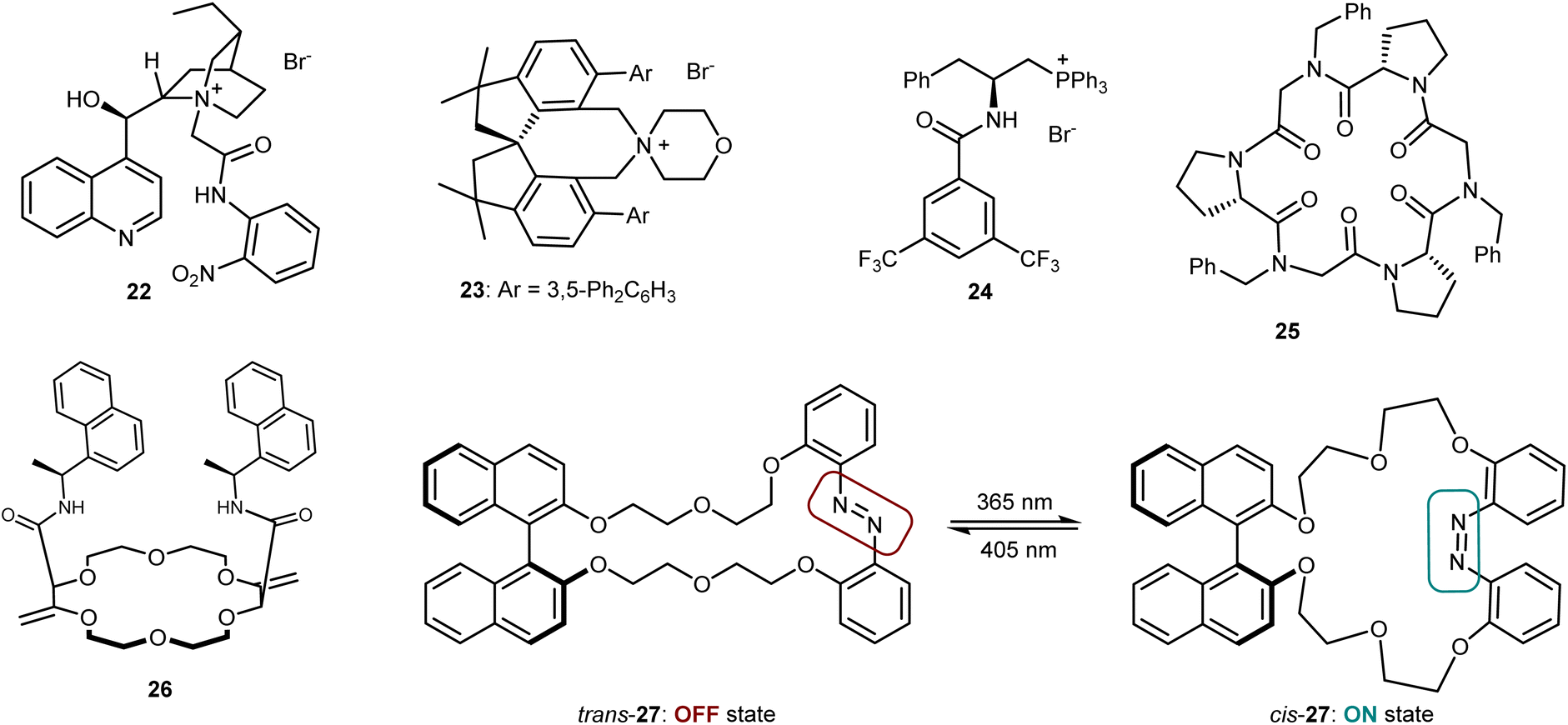 | ||
| Fig. 5 Recently disclosed phase-transfer catalysts 22–27. | ||
3.2. Aldehyde catalysis
The acidification of the α-protons of glycine esters can also be achieved through the formation of transient imine species, taking inspiration from vitamin B6-dependent enzymatic transformations (see also Section 4.2). Threonine aldolase enzymes catalyze the aldol reaction of glycine with aldehydes as well as the reverse retro-aldol reaction, according to the sequence of steps summarized in Scheme 25.79 In short, the pyridoxal phosphate cofactor is bound to the enzyme via a lysine residue. Glycine combines with the cofactor via transimination. These imines are additionally stabilized by coordination of their nitrogen atom with an o-hydroxy group. Removal of a glycine α-proton by a basic residue of the enzyme is facilitated by the electron-withdrawing effect of the pyridinium ion, resulting in the formation of a highly delocalized glycine enolate-like species; the quinonoid resonance form is shown in Scheme 25. This species undergoes aldol addition. Upon transimination, the aldol product is released and the lysine-bound cofactor is restored.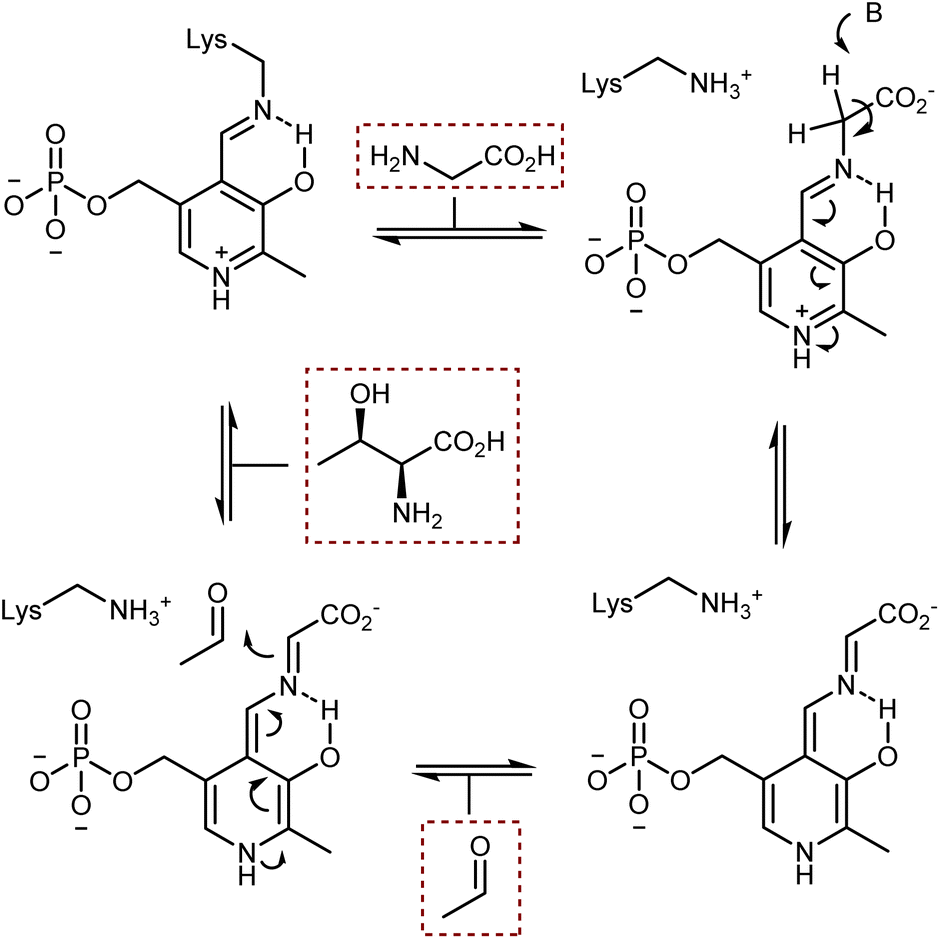 | ||
| Scheme 25 | ||
Attempts to mimic this enzymatic machinery using small synthetic molecules have mostly focused on designing chiral pyridoxal analogs rather than combining achiral pyridoxal cocatalysts with chiral catalysts, as evident in the enzymatic mechanism. Early contributions in the 1980s and 1990s by Kuzuhara,80 Breslow,81 and Murakami82 employed glycine in combination with chelating metals, as shown in Scheme 26, which depicts an example dealing with a planar chiral pyridoxal analog 28. Despite their conceptual appeal, these early approaches were far from being synthetically useful and the pyridoxal mimic is used in stoichiometric amounts.
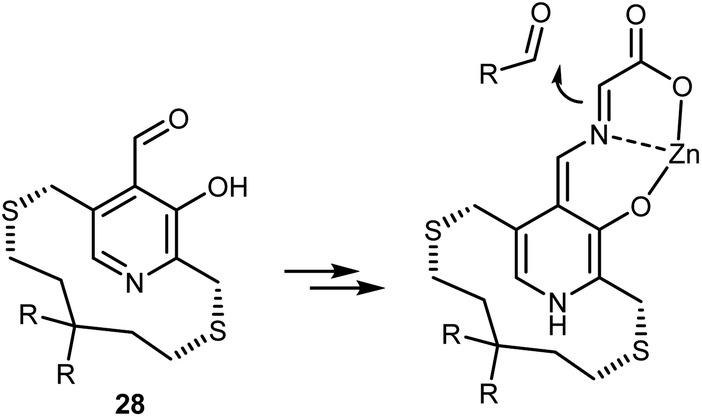 | ||
| Scheme 26 | ||
In recent years, this biomimetic approach to the functionalization of glycine has received renewed attention and has resulted in remarkable advancements. Mainly thanks to the contributions from Guo's83 and Zhao's84 laboratories, new pyridoxal mimics, effective at the functionalization of glycine esters and other amines with a variety of electrophiles, have been disclosed. The two research teams have developed structurally different aldehyde catalysts.
The catalysts proposed by Guo are based on BINOL derivatives carrying an aldehyde functionality. Their first disclosure dates back to 2014 (Scheme 27).85 Although aminomalonates, instead of glycine esters, were used, the activation of the α-protons of an amine via an aldehyde catalyst was clearly revealed. Thus, aminomalonates reacted with alkylideneindolenines, generated in situ by dehydration, in the presence of catalyst 29 and an acidic cocatalyst. Mass spectroscopic analysis detected some reaction intermediates, leading to the proposed transition state in which the catalyst also activates the alkylidene indolenine. This reaction has been used in some cases as a benchmark to demonstrate the utility of newly synthesized axially or planar chiral aldehydes.83
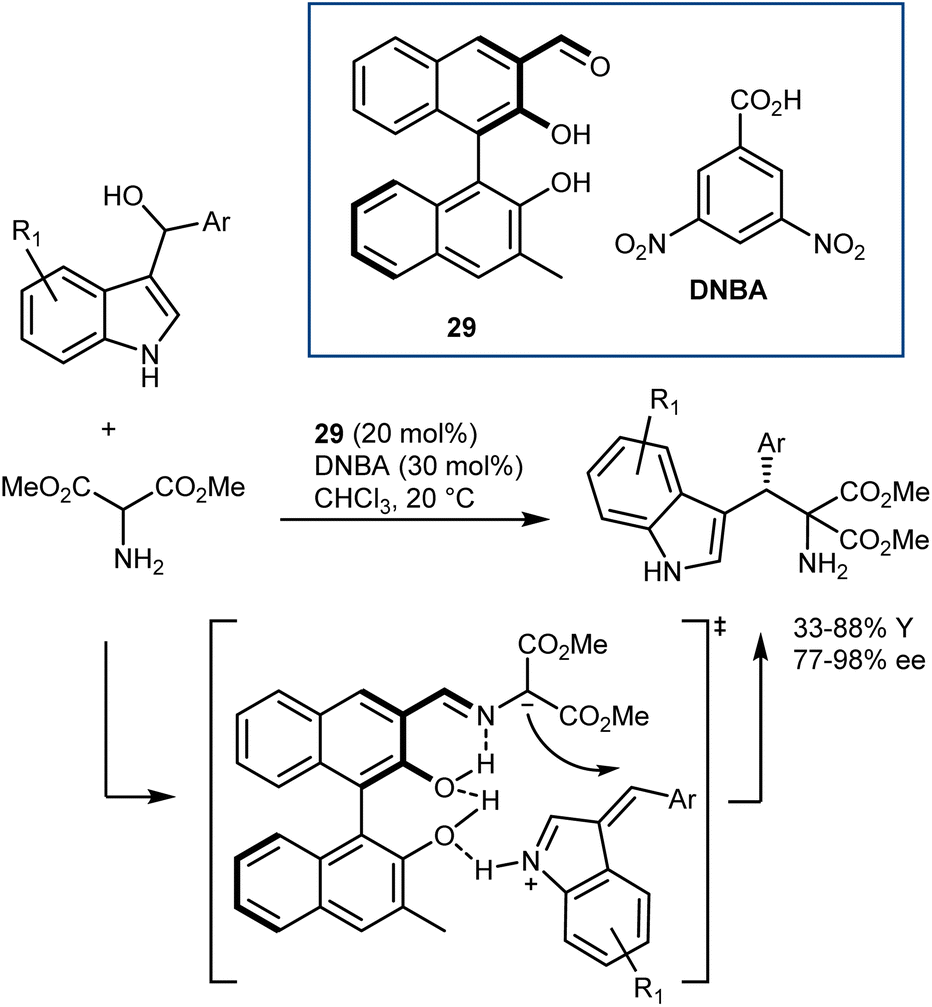 | ||
| Scheme 27 | ||
In fact, subsequent work demonstrated that this approach can be applied to glycine esters. For example, catalyst 30 promoted the conjugate addition of t-butyl glycine esters to alkylidene malonates (Scheme 28).86 The Michael addition reaction was followed by lactamization.
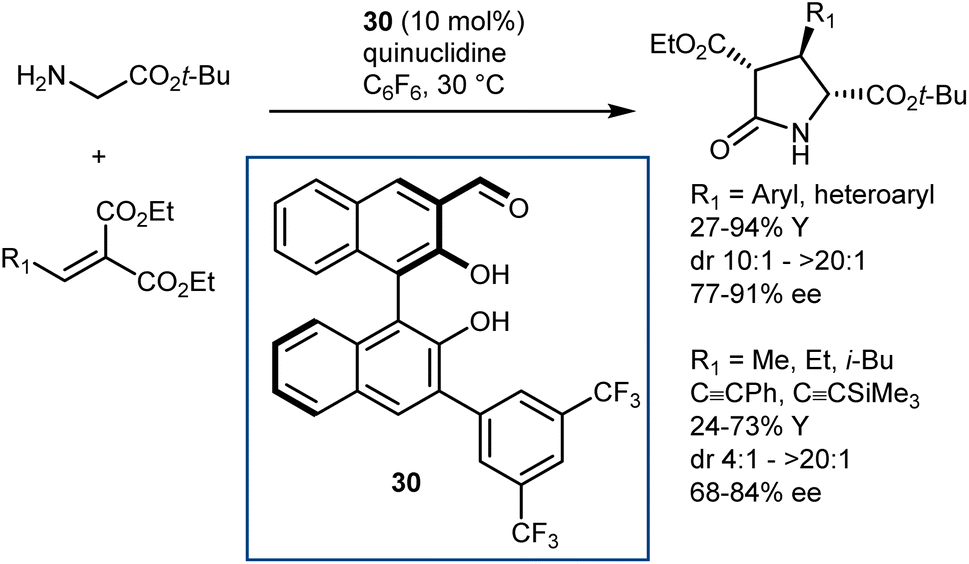 | ||
| Scheme 28 | ||
Concurrently, paralleling their studies on a related biomimetic transformation (transamination; see Section 4.2), Zhao and coworkers developed catalysts for glycine functionalization presenting a stunning resemblance with the pyridoxal cofactor. Their first disclosure, in 2018, described a Mannich reaction (Scheme 29).87 The catalyst 31 employed carries aldehyde and hydroxy functionalities on an N-methylpyridinium ion and bears a chiral amino alcohol side chain. A dramatic loss of activity was observed by using the corresponding pyridine derivative or by swapping any of the H-bond donors of the amino alcohol appendage with a methyl group. Thus, the pyridinium ion can stabilize the glycine enolate, analogously to the protonated pyridine of pyridoxal, while enantiocontrol is assisted by the coordination of the imine electrophile to the hydrogen bonds of the amino alcohol. The reaction is performed in a biphasic mixture containing an inorganic base, making the direct use of the glycine ester hydrochloride salt possible. Of note, outstanding diastereo- and enantioselectivities in the Mannich adducts could be achieved with low catalyst loading.
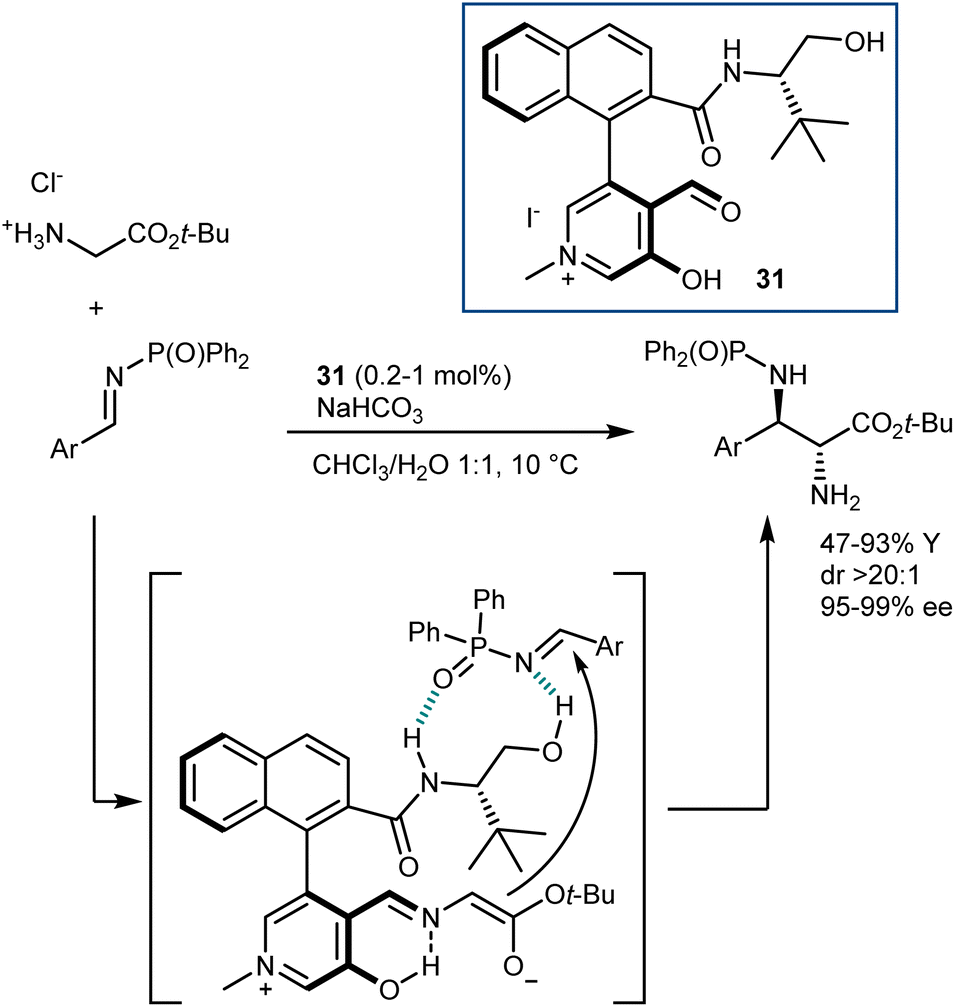 | ||
| Scheme 29 | ||
With both catalyst classes (Guo's BINOLs and Zhao's pyridines), this chemistry was rapidly extended to several other reactions, sometimes in combination with chelating metal salts.88 It can be safely concluded that “aldehyde catalysis” has been a fertile ground for unlocking new enantioselective α-functionalization reactions of glycine esters and other amines.83,84 Finally, a peculiarity of this strategy is that it leads to AA derivatives carrying the free amine (see Schemes 27 and 29) instead of the N-substituted products typically afforded by other approaches to AA synthesis.
3.3. α-Imino esters
Imines derived from glyoxylate esters are obvious synthetic equivalents of the glycine α-cation synthon. These substrates were introduced to asymmetric catalysis in the late 1990s, when their chelating properties were exploited in the framework of chiral Lewis-acid catalysis.89 Subsequently, they have found use in a variety of organocatalytic methodologies, encompassing different nucleophiles and activation modes. We refer to a review for a comprehensive overview of recent advancements on this subject.90 Here, we highlight a transformation that turned out to be especially fruitful, the Mannich reaction,91 and a few examples of other reactions.In the context of the sudden growth of proline catalysis, including the Mannich reaction,92 Barbas et al. recognized the potential of the reactions with α-imino esters as convenient routes to AAs. These early examples published in 2002 showed that both aldehydes and ketones can be engaged in the reaction with N-p-methoxyphenyl (PMP) glyoxylate imine, under the catalysis of the simple L-proline (32) (Scheme 30).93 The reactions afford the syn-adducts with very good stereoselectivities. In the case of unsymmetrical ketones, the more substituted regioisomer is formed as the major product, with the exception of fluoroacetone.
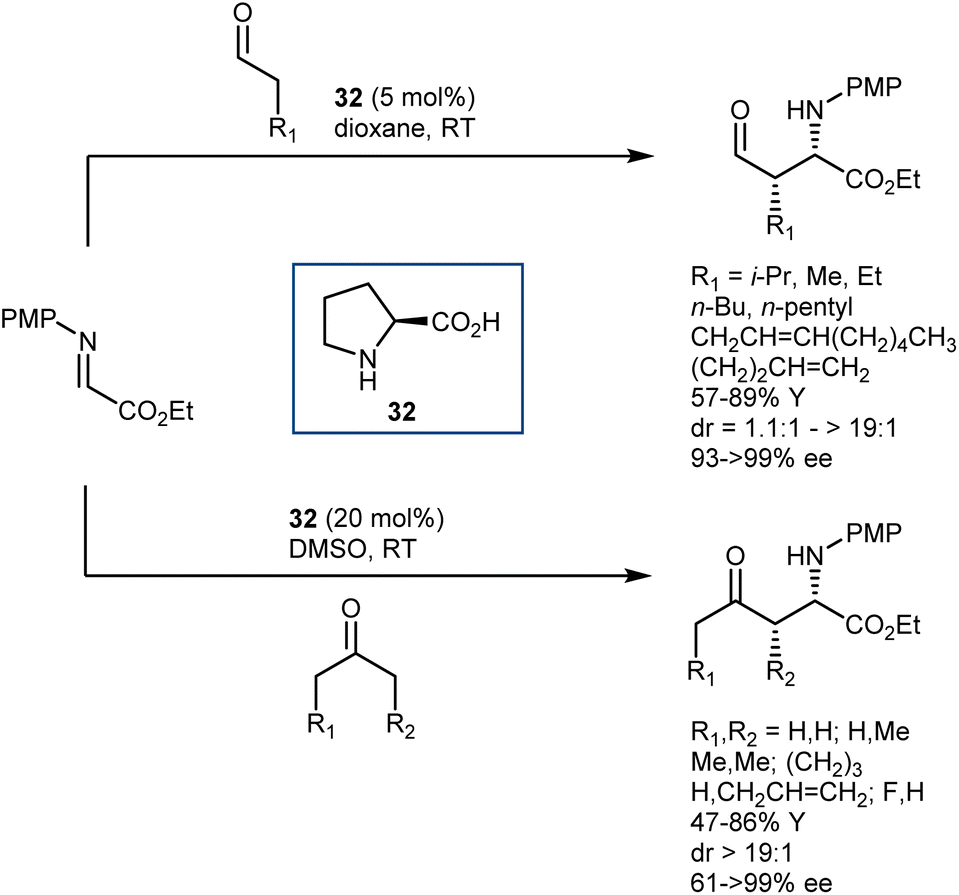 | ||
| Scheme 30 | ||
An interesting aspect of these and other aminocatalytic Mannich reactions is their predisposition to diastereodivergency by using different catalyst classes. Amongst the large number of examples,91aScheme 31 depicts the proposed transition-state arrangements and ensuing stereochemical outcomes for three catalysts representative of each class. Thus, in reactions with L-proline (32), coordination of the approaching imine by the carboxylic acid group affords the syn-isomer. Related transition states lead to the same isomer with other α-AAs as well as with pyrrolidines carrying an acidic group at the C2 position. On the other hand, the anti-counterparts can be accessed using alternative structures. By moving the coordinating group further (catalyst 33),94 a different transition-state arrangement that still involves coordination of the imine by the acidic group occurs. This leads to the formation of the anti-isomer. Finally, the anti-isomer is also the major product in reactions catalyzed by amines lacking an acidic coordinating group, such as the Jørgensen–Hayashi catalyst 34,95 where the reaction occurs via the open synclinal transition state shown.
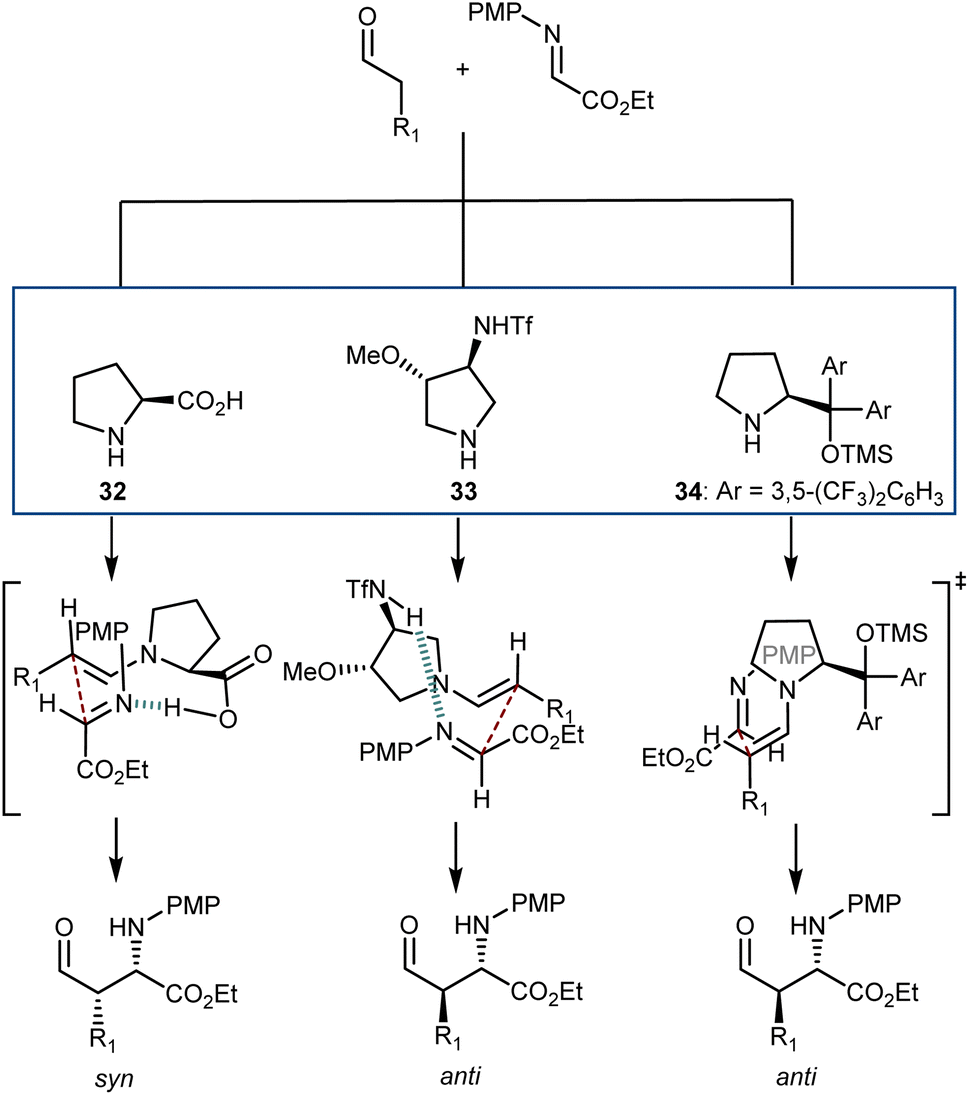 | ||
| Scheme 31 | ||
Another example of diastereodivergency is derived from the enzymatic reduction of the carbonyl group in the Mannich reaction with acetone, resulting in γ-hydroxynorvaline derivatives. Simon and Kroutil showed that by employing alcohol dehydrogenase enzymes with opposite stereoselectivities in this reduction, access to 1,3-cis- and 1,3-trans-isomers is possible (Scheme 32).96 Thus, using specific combinations of proline [L-proline (32) or D-proline (ent-32)] and enzyme (ADH-A or evo-1.1.200), all four possible stereoisomers are reachable. Thanks to the outstanding selectivities offered by proline and the two enzymes, the four γ-hydroxynorvalines, isolated as lactones upon cyclization, could be obtained with perfect stereoselectivity.
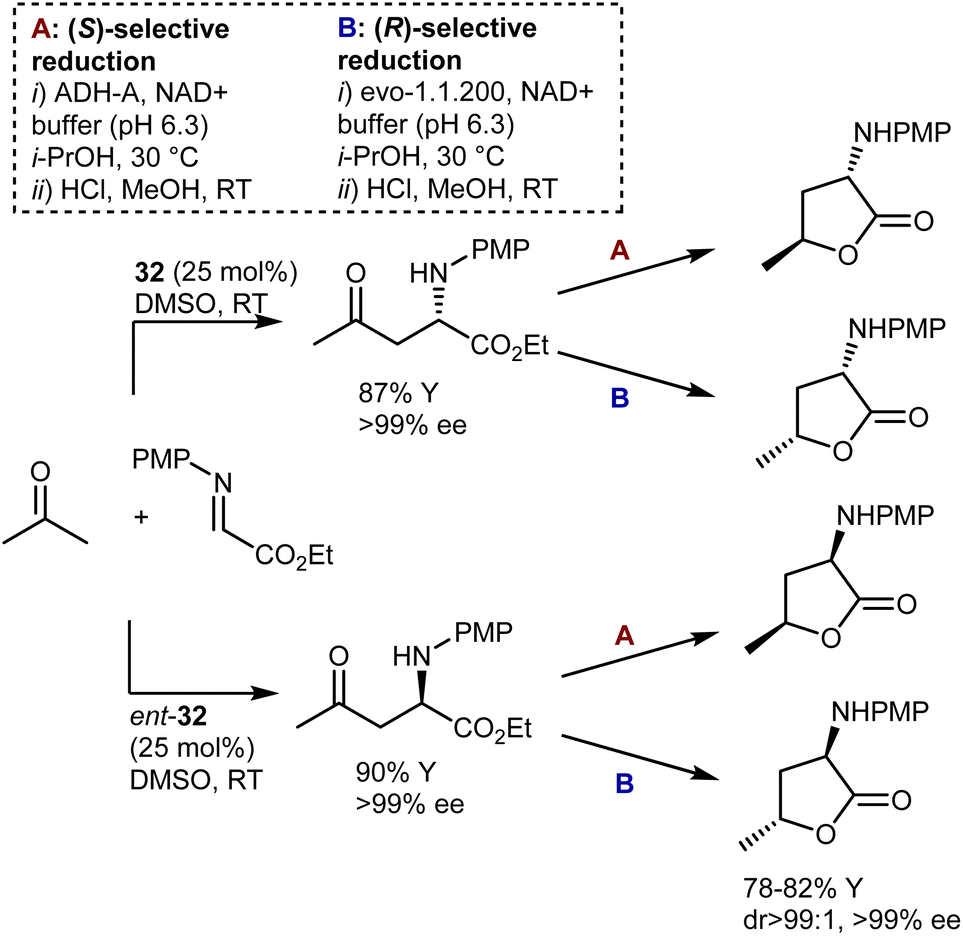 | ||
| Scheme 32 | ||
Despite the outstanding efficiency of these amino-catalyzed Mannich reactions, the deprotection of the products from the PMP group is not straightforward. For example, removal of this group from one of the lactones above required substantial optimization. Trichloroisocyanuric acid worked, but other standard methodologies (e.g., PhI(OAc)2, ceric ammonium nitrate, HIO6, and laccase) failed. Furthermore, another article defines the PMP deprotection of the Mannich adduct resulting from the L-proline (32)-catalyzed Mannich reaction with butanone as “problematic.”97 Ultimately, reverse addition of the adduct to ceric ammonium nitrate enabled deprotection and utilization of the Mannich reaction in the total synthesis of a lipid–peptide isolated from Streptomyces species, of interest for its antimalarial properties (Scheme 33). However, the two-step protecting group swap from PMP, required for the catalytic reaction, to Boc, required for the subsequent peptide couplings, was suboptimal in terms of efficiency.
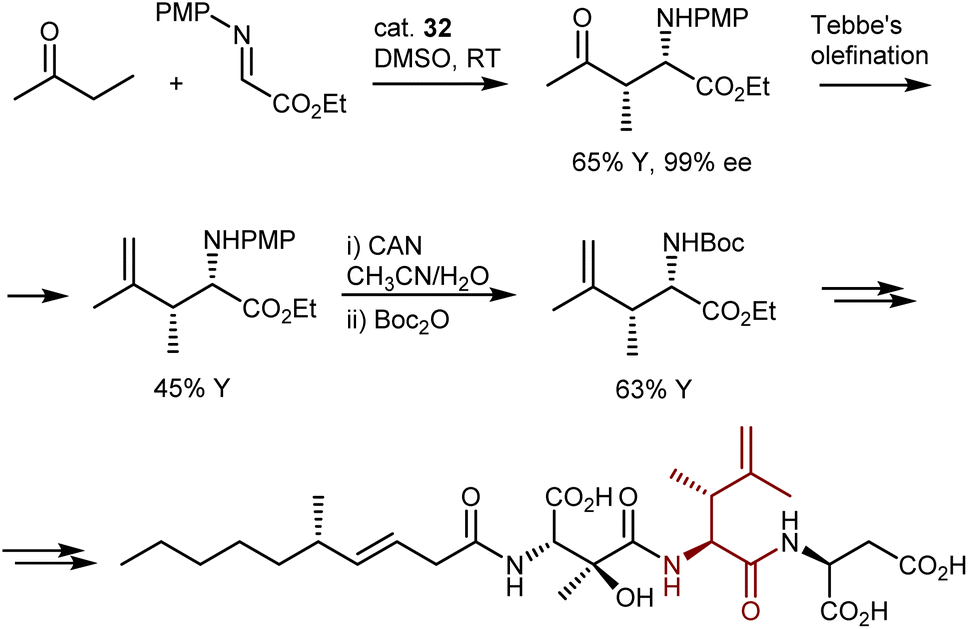 | ||
| Scheme 33 | ||
An alternative to the use of N-PMP imines in L-proline (32)-catalyzed Mannich reactions with ketones is the cyclic iminoglyoxylate shown in Scheme 34.98 This substrate, introduced by Glorius,98a gives the anti-isomers, thus providing an additional example of the diastereodivergency of the Mannich reaction. In contrast with N-PMP imines, the Mannich adducts can directly deliver the corresponding unprotected AAs upon hydrogenolysis. More recently, this substrate was applied by Zhang and Ma to a CPA-catalyzed reaction with enamides,98c giving access to additional AA structures.
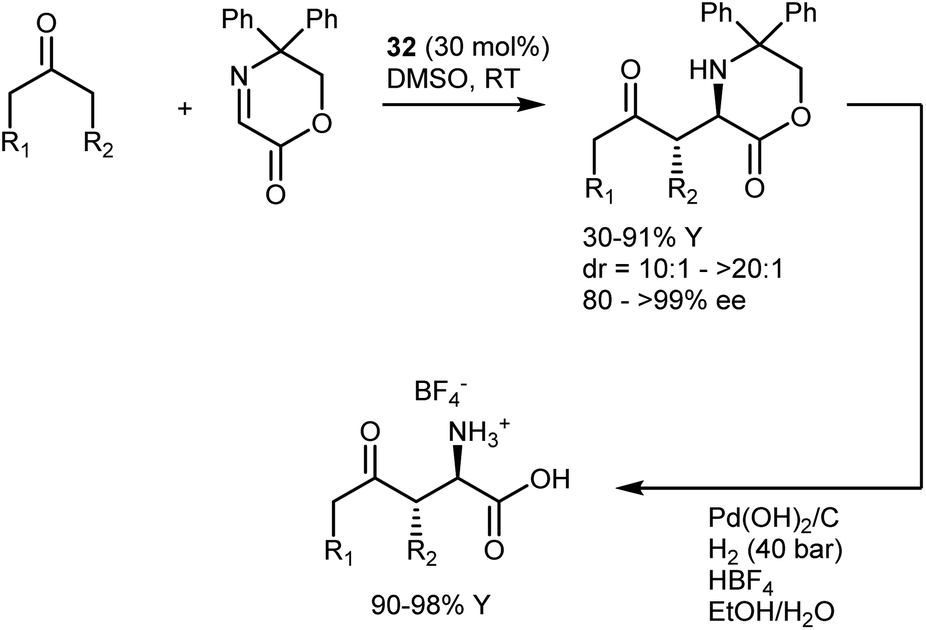 | ||
| Scheme 34 | ||
Nevertheless, with an eye on amide coupling, using an N-Boc- or N-carbamoyl-protected α-imino ester would considerably improve the synthetic appeal of these Mannich transformations. However, N-carbamoyl-protected α-imino esters are rather unstable and must be generated immediately before use.99 To circumvent this issue, researchers have developed methodologies allowing their generation in situ from different stable precursors for their engagement in several organocatalytic Mannich reactions.100
A first example was reported by Melchiorre in 2008, using N-Boc- and N-Cbz-α-amino sulfones as convenient precursors of the unstable imines (Scheme 35, top).101 Their Mannich reaction with aldehydes is catalyzed by the Jørgensen–Hayashi catalyst 34 and proceeds in the presence of KF, which serves as a superstoichiometric base to generate the imines. More recently, an imine precursor carrying a different leaving group (acetate) was used by Luo in a Mannich reaction with a range of 1,3-diketones, β-ketoesters, ketones, and an aldehyde (Scheme 35, bottom).102 In this case, the reaction pathway also involves an enamine intermediate, formed by the condensation of the carbonyl compounds with catalyst 35. However, no additive is required to form the imine or to neutralize the acetic acid coproduct, and the reaction is applicable to Fmoc-protected imine substrates as well.
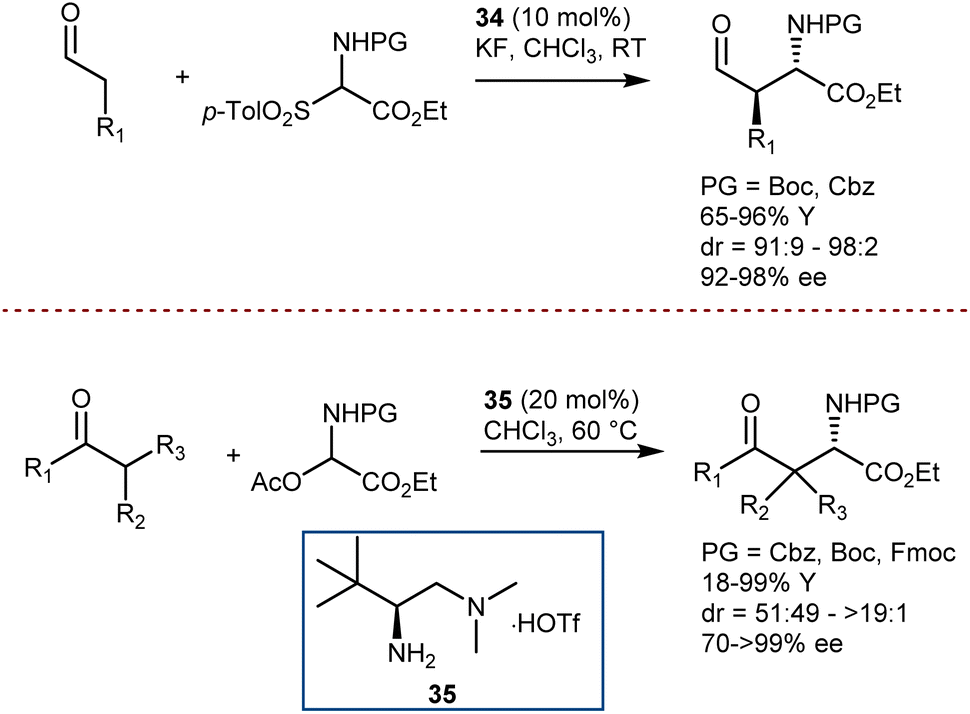 | ||
| Scheme 35 | ||
Catalytic strategies departing from enamine intermediates, and more specifically dealing with bifunctional catalysis, have been described, too.100 Roche and Jacobsen reported on the use of an α-chloroamine as the N-Cbz-imine precursor, in combination with Takemoto catalyst 7 (Scheme 36, top).103 Thanks to the combined action of its basic and acidic/halide binding functionalities, this catalyst can form an N-Cbz-iminium ion by halide abstraction (or an imine by elimination of HCl) and then promote the ensuing enantioselective addition of 1,3-diketones and β-ketoesters. In addition, Wang and He reported a somewhat related Mannich reaction using the bifunctional catalyst 36 (Scheme 36, bottom).104 A different and more stable imine precursor (N,O-bis(t-butoxycarbonyl)hydroxylamine) was used, which, under the reaction conditions, can form the unstable N-Boc-imine by the elimination reaction shown, releasing t-BuOH and CO2.
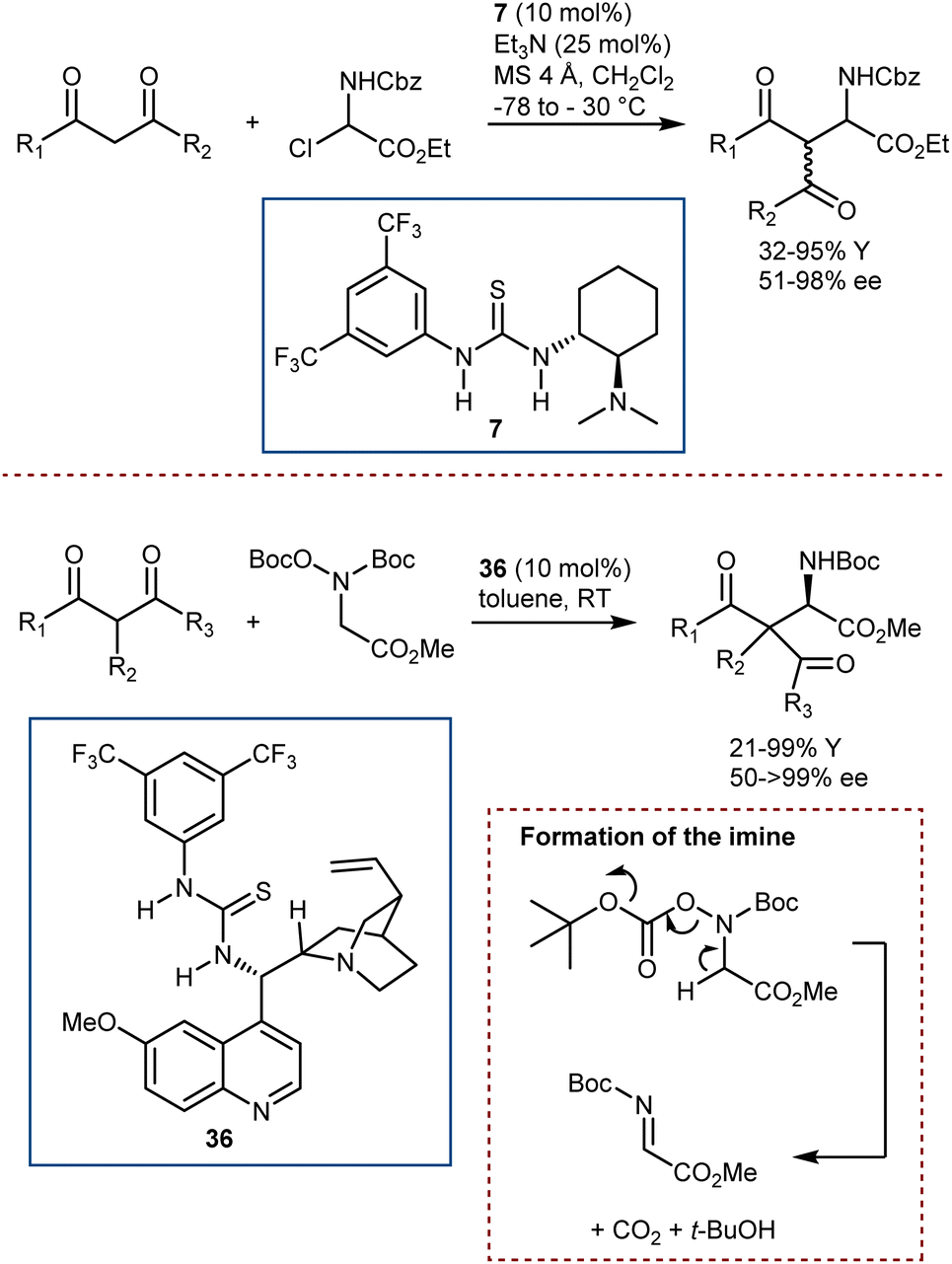 | ||
| Scheme 36 | ||
Departing from the Mannich reaction, we conclude this subsection by highlighting a recent example of a Friedel–Crafts addition to an α-imino ester. Catalyzed by chiral Brønsted or Lewis acids, this reaction is typically limited to electron-rich (hetero)aromatic compounds such as indoles, anisoles, anilines, etc.105 However, a remarkable version of this transformation was recently developed by List,106 using Luo's acetate imine precursor in combination with a strong and confined Brønsted acid catalyst, such as imidodiphosphorimidate 37 (Scheme 37). This combination allowed unactivated (hetero)aromatic compounds (e.g., alkylbenzenes) to be used for the first time in the asymmetric Friedel–Crafts reaction. The 37-catalyzed process employs the aromatic donor as the solvent and is proposed to proceed via a Wheland-type intermediate stabilized by the Fmoc carbamoyl. It shows excellent p-selectivity for monosubstituted benzenes and is also applicable to more activated substrates (e.g., anisoles and thiophene), using a catalyst related to 37 and pentane as the solvent.
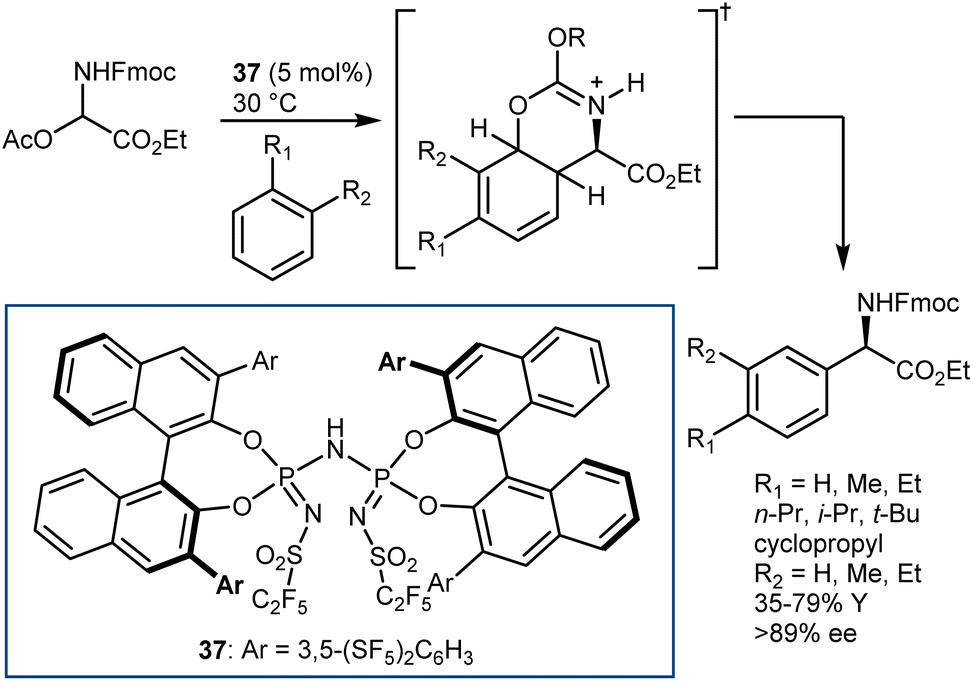 | ||
| Scheme 37 | ||
Finally, we recall that in the MCR platform based on the combination of diazocarbonyl compounds with the cooperative catalysis of rhodium and CPA species (see also Section 5.2),107N-aryl- and N-benzhydryl-α-imino esters have sometimes been engaged as trapping agents of the onium ylide intermediates.108Scheme 38 depicts one of these examples, a four-component reaction with α-diazoketones, water, ethyl glyoxylate, and anilines that affords γ-keto-β-hydroxy AA derivatives in the presence of Rh2(OAc)4 and CPA 38 as catalysts.108a
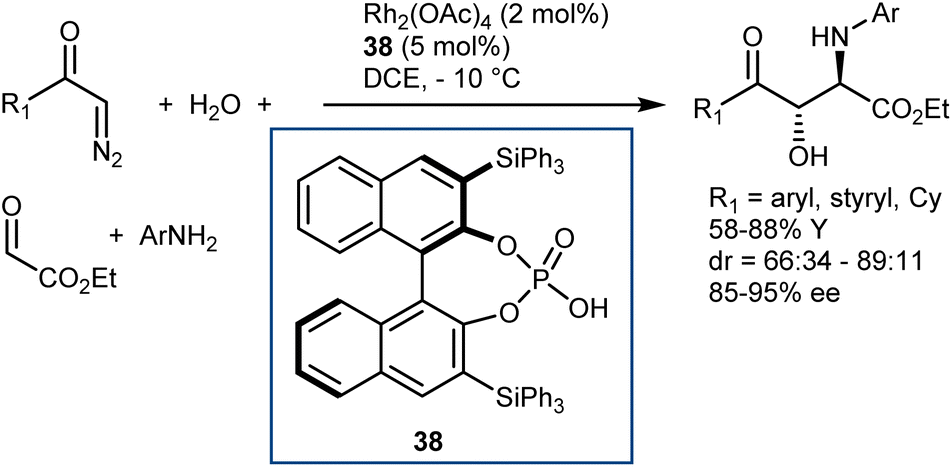 | ||
| Scheme 38 | ||
4. Manipulation of the α-hydrogen
Playing with the α-hydrogen of a substrate gives various possibilities to obtain AA derivatives in enantioenriched form (Scheme 39). A typical approach is the reduction of α-ketimino esters, where the hydrogen atom is introduced enantioselectively through the addition of a hydride equivalent. Somewhat related to this process is the transamination reaction. In this case, the hydrogen is transferred within the same molecule via a formal 1,3-H shift process. In these two reactions, the enzymatic machinery clearly serves as a source of inspiration. Another transformation in which the chiral center is established through the introduction of a hydrogen atom is the addition to dehydroalanine (DHA) derivatives. Here, an enantioselective protonation defines the configuration of the α-chiral center. The last two reaction categories discussed in this section involve various sorts of dynamic processes: the dynamic kinetic resolution (DKR) of azlactones and the recently described deracemizations. These latter reactions achieve the remarkable conversion of a racemic mixture of a compound into its enantioenriched form using light irradiation as the energy source.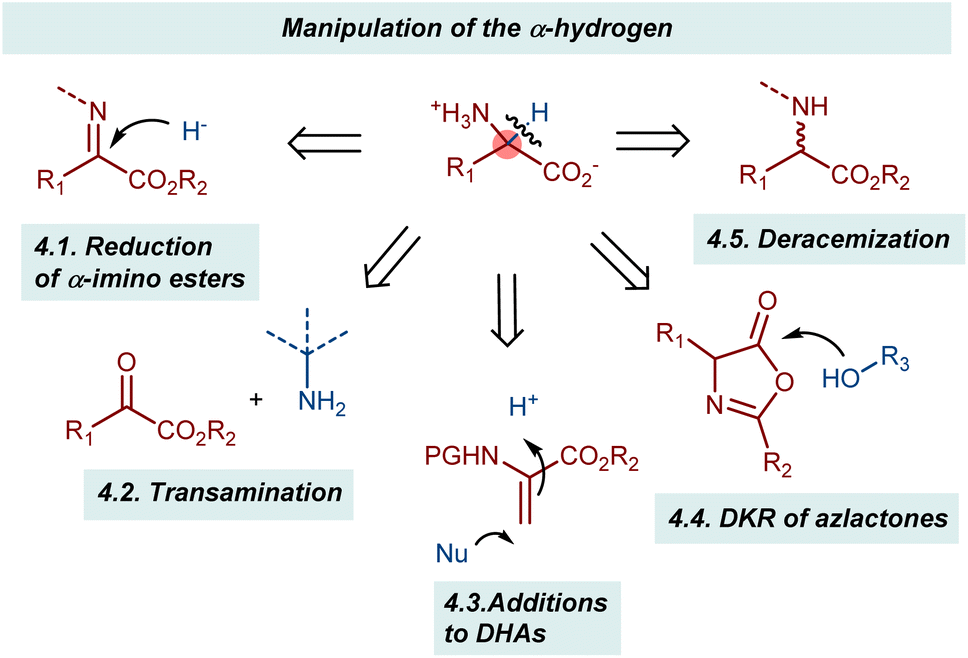 | ||
| Scheme 39 | ||
4.1. Reduction of α-imino esters
Perhaps the most straightforward means to produce AA derivatives is the direct reduction of α-imino esters, since they are readily produced from α-ketoesters in which the ketone and ester functionalities are on adjacent atoms and they are available with numerous substitution patterns. It can be considered a particular case of the more general enantioselective reduction of unsymmetrical ketimines, with the advantage of having highly electrophilic imines.Although a metal-free organocatalytic enantioselective hydrosilylation protocol of an α-imino ester (single example) to produce a phenylglycine derivative with moderate ee was reported in 2006,109 more convincing results portraying the success of this strategy were reported in the following year; these findings were based on the utilization of Hantzsch esters (HEs) for organocatalytic transfer hydrogenation reactions, an approach that was rapidly emerging at that time.110 HEs are analogs of the enzymatic cofactors NADH and NADPH. Amongst the multifarious enzymatic processes in which these cofactors play an essential role, the reductive amination of 2-ketoglutarate to afford L-glutamic acid111 presents a considerable resemblance with the chemistry developed using chiral Brønsted acids as small-molecule catalysts. The results are described in the following paragraphs.
In early 2007, Antilla reported that acyclic α-imino esters can be reduced to provide α-amino esters in high yield and with excellent enantioselectivity using the hindered (S)-VAPOL-derived CPA 39 and an ethyl HE as the hydrogen source (Scheme 40).112 A series of N-PMP- and N-phenyl-α-imino esters was evaluated, and it was shown that imino substrates derived from substituted aryl and alkyl α-keto esters could be reduced to the corresponding α-amino esters in excellent yields with ee values of 94–99%. In some cases, the imines were prepared in situ prior to the reduction, thus resulting in an overall reductive amination process.
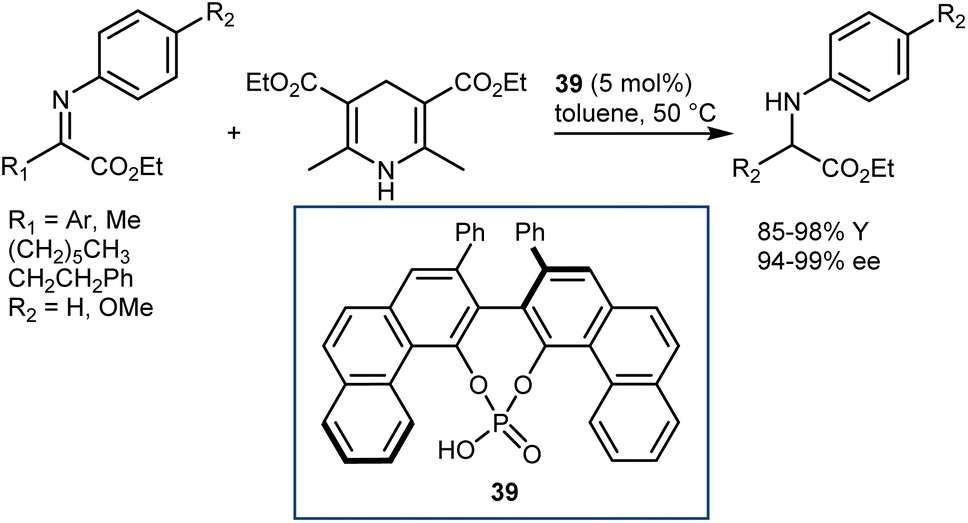 | ||
| Scheme 40 | ||
A few months later, You similarly reported excellent results with N-PMP-α-imino esters employing the same HE as the hydrogen donor but using a different catalyst: the CPA 40 derived from (S)-BINOL (Scheme 41).113 Besides the typical esters, the reaction accommodates an amide.
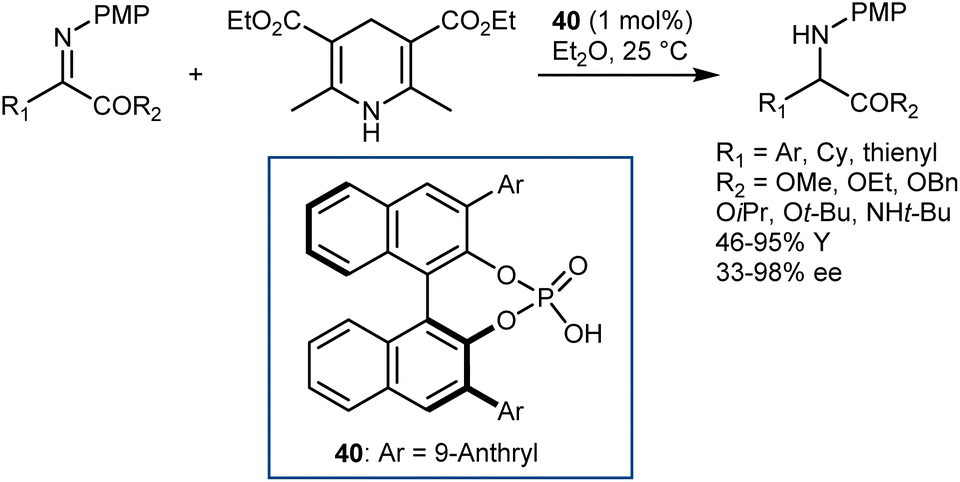 | ||
| Scheme 41 | ||
Using similar reaction conditions and catalyst system, Hu reported the application of this process to the more practical and reactive N-Cbz-aryl-α-imino esters, delivering arylglycine derivatives carrying a more convenient (compared to PMP) protecting group.114 In this work, the N-Cbz-imines were prepared from α-diazocarbonyl compounds, benzyl carbamate, and an oxidant (2,3-dichloro-5,6-dicyano-1,4-benzoquinone), via a rhodium-catalyzed reaction.
Conversely, an organocatalytic transfer hydrogenation of N-alkyl C-aryl α-imino esters that gives direct access to N-alkyl arylglycines was developed by Mazuela (Scheme 42).115 Excellent yields and enantiomeric ratios were achieved for a wide range of substrates, facilitating the preparation of more complex molecules as well as intermediates for active pharmaceuticals such as aprepitant. The reaction requires a catalyst different from a CPA, namely the disulfonimide 41, and Boc2O as a trapping agent for amine, in line with List's disclosures on other N-alkyl ketimines.116 The poor stability of the N-Boc-protected products led to the inclusion of a deprotection step at the end of the reaction to enable isolation.
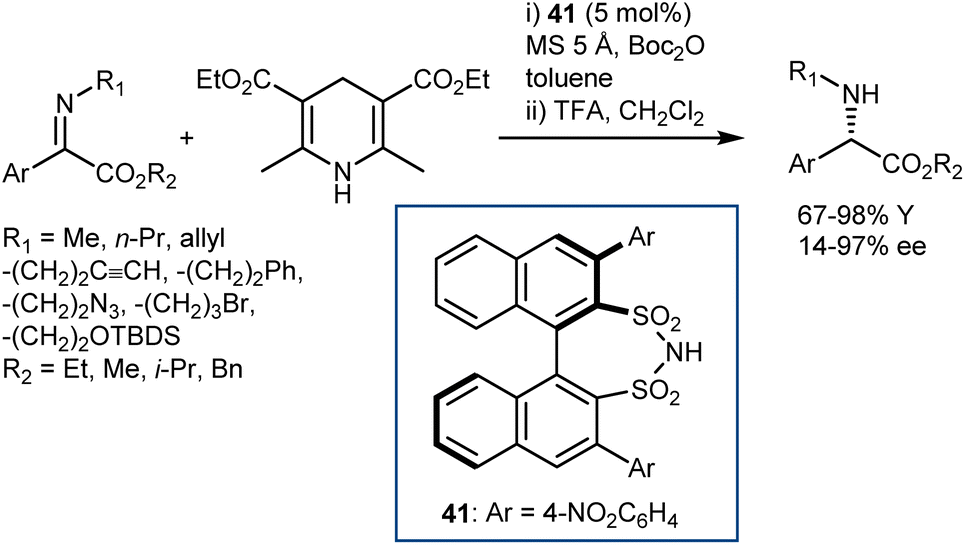 | ||
| Scheme 42 | ||
Instead of HEs, it is worth mentioning that 2H-benzothiazolines have been employed in these CPA-catalyzed reactions.117 Moreover, a 2-deuterated benzothiazoline counterpart can be used for asymmetric deuteration, a process that is still underexplored despite the great importance of deuterated compounds in various areas of chemistry.
Recent reviews comprehensively describe organocatalytic transfer hydrogenation reactions, including detailed mechanistic aspects and structural features of the organocatalysts.118
4.2. Transamination
In biological systems, AAs are also synthesized by the transamination of α-ketoacids from pyridoxamine 5′-phosphate, a vitamin B6 cofactor (see also Section 3.2), catalyzed by enzymes of the transaminase class, also known as aminotransferases. The transformation proceeds through a condensation followed by a 1,3-H shift assisted by a lysine residue of the enzyme, as depicted in Scheme 43.119 After transamination and hydrolysis of the resulting AA product, the cofactor is present in its pyridoxal 5′-phosphate form, bound to the enzyme via the lysine residue. The regeneration of the pyridoxamine requires an amine donor and occurs via mechanistically similar steps.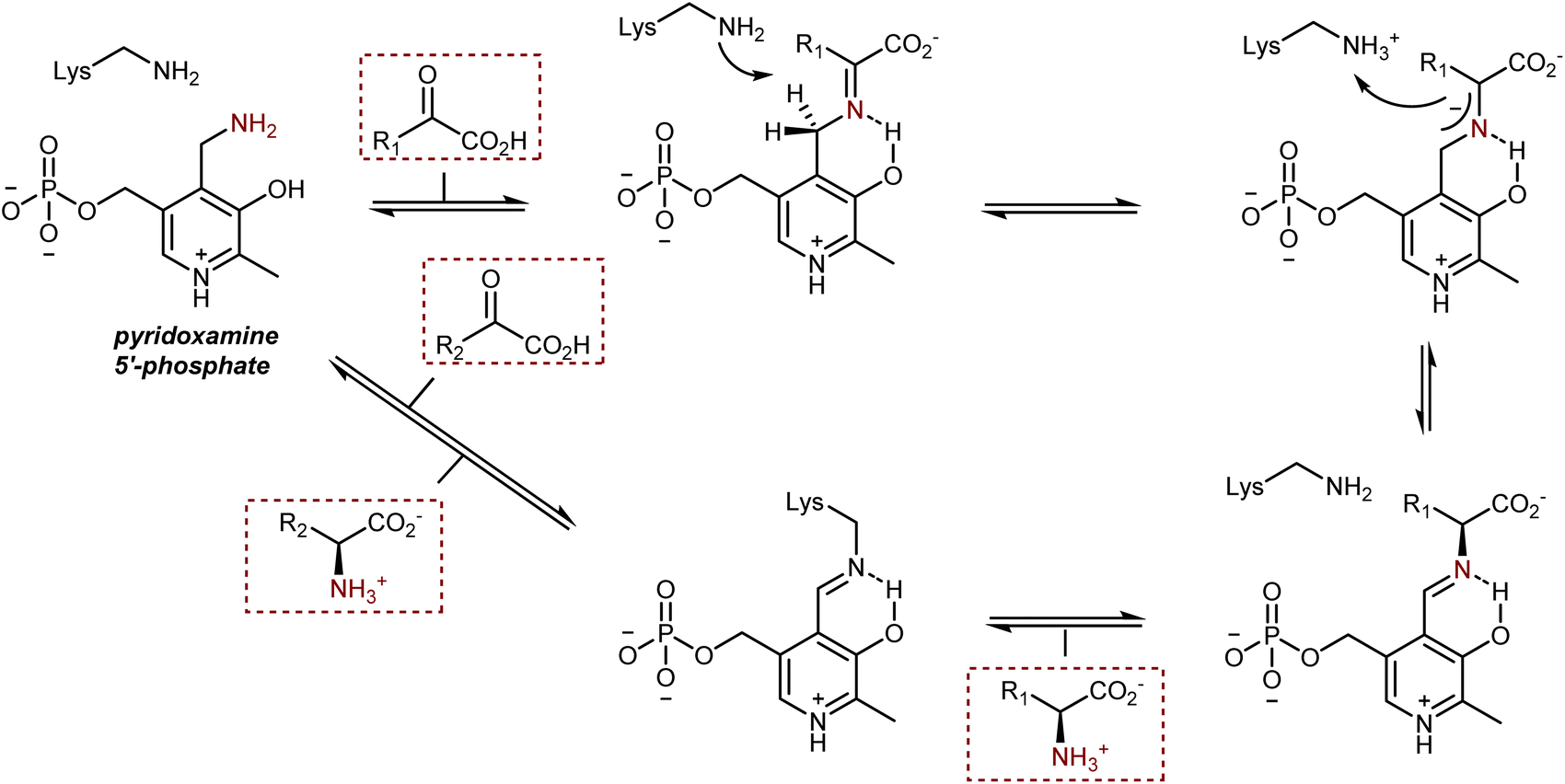 | ||
| Scheme 43 | ||
Two aspects of this enzymatic transformation stand out to the eye of a synthetic chemist. First, the imine carbon atom of the conjugated intermediate or, even better, the 2-azaallyl anion is deprotonated, which inverts the imine's natural reactivity because it now acts as a Brønsted base instead of being electrophilic. Second, a remarkably efficient and selective biosynthesis of AAs is produced by this base-catalyzed reversible isomerization, which proceeds with complete control of the newly formed stereogenic center.
As such, it is not surprising that the development of nonenzymatic asymmetric catalysts for this reaction proved to be a challenging task. Nevertheless, it was recently successful using two distinct approaches, both based on organocatalysis. The first approach combines an achiral stoichiometric amine, playing the role of the pyridoxamine cofactor, with a chiral catalyst exerting enantiocontrol during the 1,3-H shift. The second approach makes use of a chiral pyridoxamine mimic instead; thus, it requires a catalyst regeneration step after the enantioselective donation of the amine functionality.
A moderately enantioselective (up to 45% ee) example of the first approach, with a chiral guanidine catalyst and 9-aminothioxanthene 10,10-dioxide as the pyridoxamine equivalent, was reported in 2002 by Berg.120 This example demonstrates that the approach is possible and suggests that the reaction may proceed via a stepwise, bifunctional mechanism, thus providing valuable insight for the further development of more effective systems. The first highly enantioselective organocatalytic variation of the transamination reaction was more recently introduced by Shi in 2011; the best conditions in terms of both reactivity and enantioselectivity were determined to be cupreine derivative 42 as the catalyst and o-chlorobenzylamine as the nitrogen source for the in situ imine generation (Scheme 44).121 The fundamental feature discovered was the employment of a bifunctional chiral organocatalyst to make the key isomerization enantioselective. The corresponding 2-azaallyl anion—the real Umpolung step that turns the imine carbon atom nucleophilic—is meant to be formed more easily with the help of such a catalyst. Now that the protonated chiral catalyst and this achiral anion are ion-paired, face differentiation is possible in the subsequent protonation step, resulting in the chiral transamination product. The 6′-OH of the catalyst played a very important role in the transamination in terms of both reactivity and enantioselectivity, likely via H-bonding with the imine to facilitate the reaction and influence the enantioselectivity. An investigation on different H-bond donors at this position and on different alkyl chains at the 9-OH indicated that hindered sulfonamides and alkyl chains could lead to a more efficient catalyst 43.122 This elegant transamination tolerates a variety of α-keto esters, giving access to the corresponding amino esters with very high enantioselectivities; however, a large ester group is mandatory to reach high enantioselectivity. The transamination was also carried out on the gram scale, and both enantiomers were synthesized in high yields and with high ee values. More recently, Bierer and Wang developed the transamination with a catalyst related to 42 for the synthesis of β,β-difluoro amino amide derivatives.123
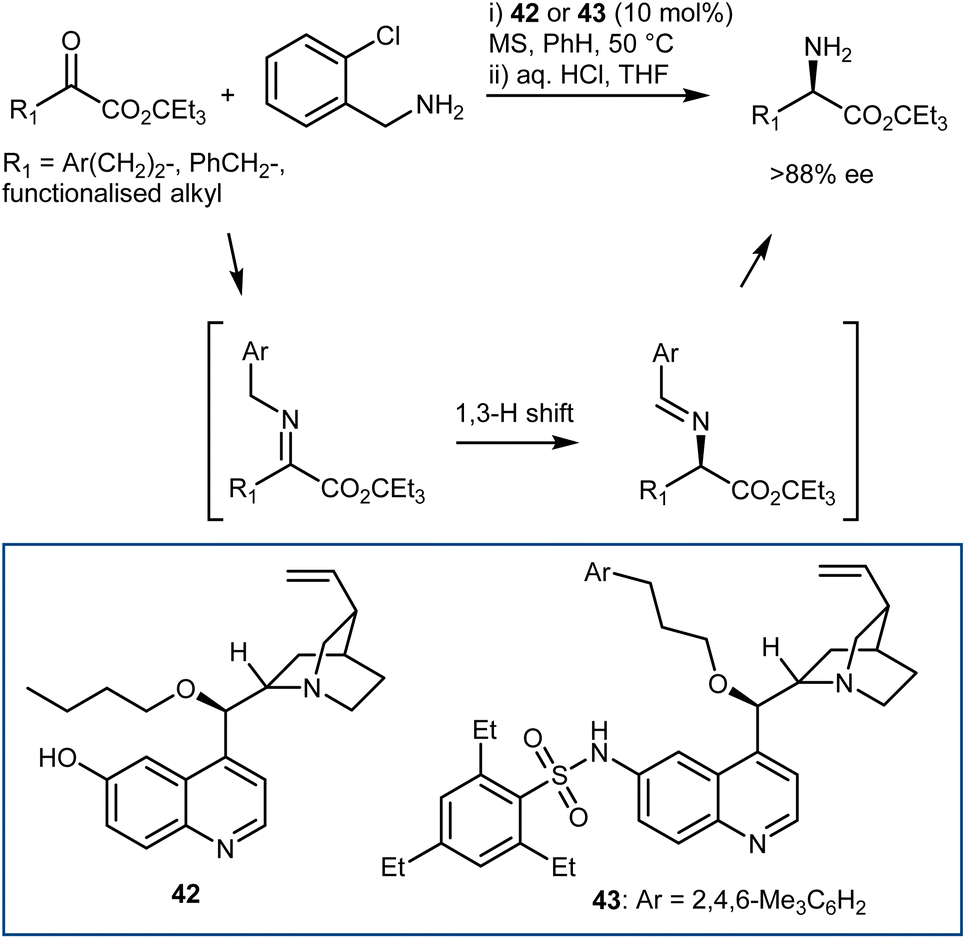 | ||
| Scheme 44 | ||
To check the feasibility of the asymmetric biomimetic transamination using a different catalyst type for the enantioselective 1,3-H shift process, Maruoka carried out the reaction of α-keto esters with p-nitrobenzylamine in the presence of 5 mol% of chiral quaternary ammonium carboxylates in toluene at room temperature.124 As expected, a quite extensive investigation of the counter-anion effect was necessary for base-free PTC conditions in order to increase the yield and ee value. Eventually, a bulky carboxylate counteranion afforded a good yield and enantioselectivity (up to 91% ee).
The second successful approach to the catalytic asymmetric transamination reaction is based on chiral pyridoxamine mimics. The series of equilibria of the enzymatic process (Scheme 43) suggests that full transamination with a pyridoxal mimic is challenging, since the catalyst must donate the amine to one substrate and receive it from another (similar) one after the asymmetric reaction.
In parallel with their studies on aldehyde catalysis (see Section 3.2), in 2016, Zhao disclosed the feasibility of this reaction with pyridoxamine mimics using a class of axially chiral biaryl pyridoxamines armed with a cooperative lateral amine chain as the catalyst 44 for the asymmetric transamination of a variety of α-ketoesters (Scheme 45).125 Thus, the reversibility challenges of the full transamination cycle were solved by resorting to a decarboxylative transamination with 2,2-diphenylglycine to regenerate the amine catalyst, that is, to a transamination releasing CO2 instead of relying on a 1,3-H shift. The catalyst comparison demonstrated that the enantioselectivity and evidently high activity were largely dependent on the NHMe lateral chain, building on early studies by Breslow, who showed the importance of such basic residues in stoichiometric transamination reactions with pyridoxamine mimics.126 The purpose of this basic group is to mimic the transaminases lysine residue (Scheme 43) to expedite the asymmetric transamination through cooperative catalysis. The chiral pyridoxamines exhibited high catalytic activity and allowed for the delivery of a variety of AAs with excellent activity and enantioselectivity (up to 99% yield and 94% ee) from a variety of α-keto acids (20 very diverse examples). Remarkably, the reaction directly affords AAs devoid of any protecting group.
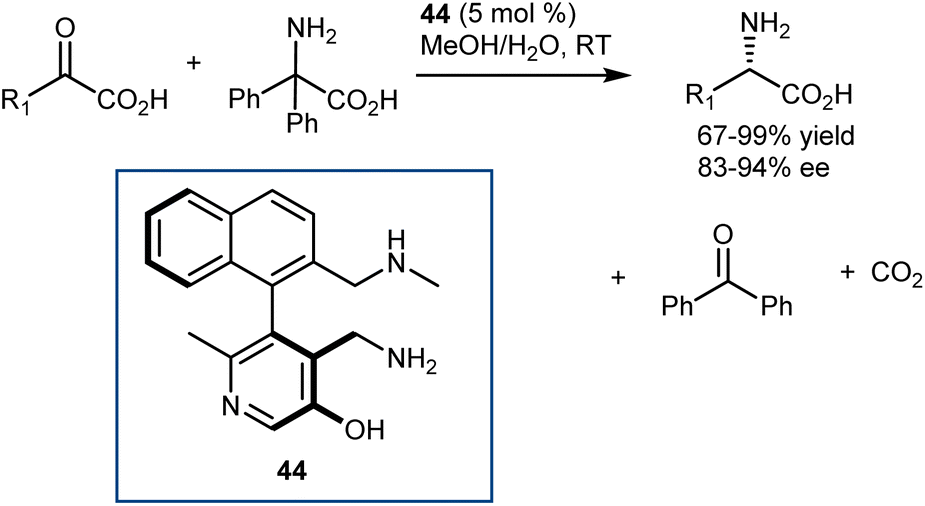 | ||
| Scheme 45 | ||
Using the same logic, lower reactive substrates such as α-keto amides were successfully transaminated in good yields with excellent enantio- and diastereoselectivities using N-quaternized chiral pyridoxamine 45 to make the pyridine ring more electron deficient for facilitating deprotonation of the ketimine formed between the α-keto amide and pyridoxamine. With this catalyst, a wide array of α-keto amides underwent asymmetric transamination to offer pharmaceutically and biologically crucial peptides in up to 90% yield with 98% ee or 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Intriguingly, a successive “condensation–transamination” strategy was employed to make the hexapeptide from a tetrapeptide (the methyl ester of the dipeptidyl peptidase 4 inhibitor diprotin A), exemplifying an efficient approach for peptide extension (Scheme 46).127 Recent reviews on asymmetric organocatalytic transamination reactions have been reported.84,128
:1 dr. Intriguingly, a successive “condensation–transamination” strategy was employed to make the hexapeptide from a tetrapeptide (the methyl ester of the dipeptidyl peptidase 4 inhibitor diprotin A), exemplifying an efficient approach for peptide extension (Scheme 46).127 Recent reviews on asymmetric organocatalytic transamination reactions have been reported.84,128
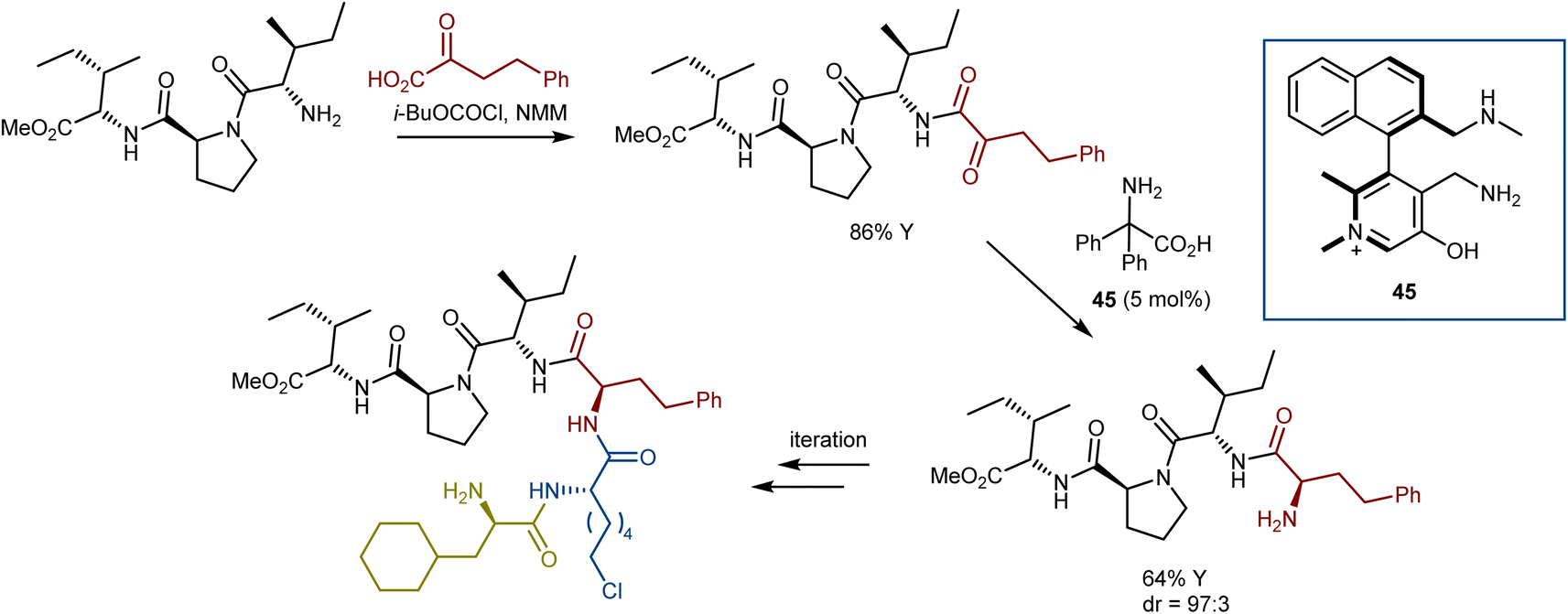 | ||
| Scheme 46 | ||
4.3. Additions to DHAs
A convergent and effective method for synthesizing enantioenriched ncAAs is the direct enantioselective catalytic transformation of prochiral DHAs. However, because of their high tendency to tautomerize to the ketimine form under acidic conditions, low inherent electrophilicity, and low propensity for catalyst activation, this class of Michael acceptors with α,β-unsaturated esters substituted with free or protected amino groups at the α-position has remained a persistent challenge in enantioselective/asymmetric catalysis to date.129To the best of our knowledge, the instances of organocatalytic enantioselective Michael additions to DHAs are limited to four examples. Tan reported the addition of thiols, thanks to a chiral bicyclic guanidine derivative 46 similar to structure 2 used in seminal work by Corey for the Strecker reaction (Scheme 47, top).130 In addition, Glorius showed that N-heterocyclic carbene 47 catalysis can be used for the Stetter addition of aldehydes (Scheme 47, bottom).131 Remarkably, in these two reactions, the catalysts give exquisite enantiocontrol in a challenging protonation reaction.
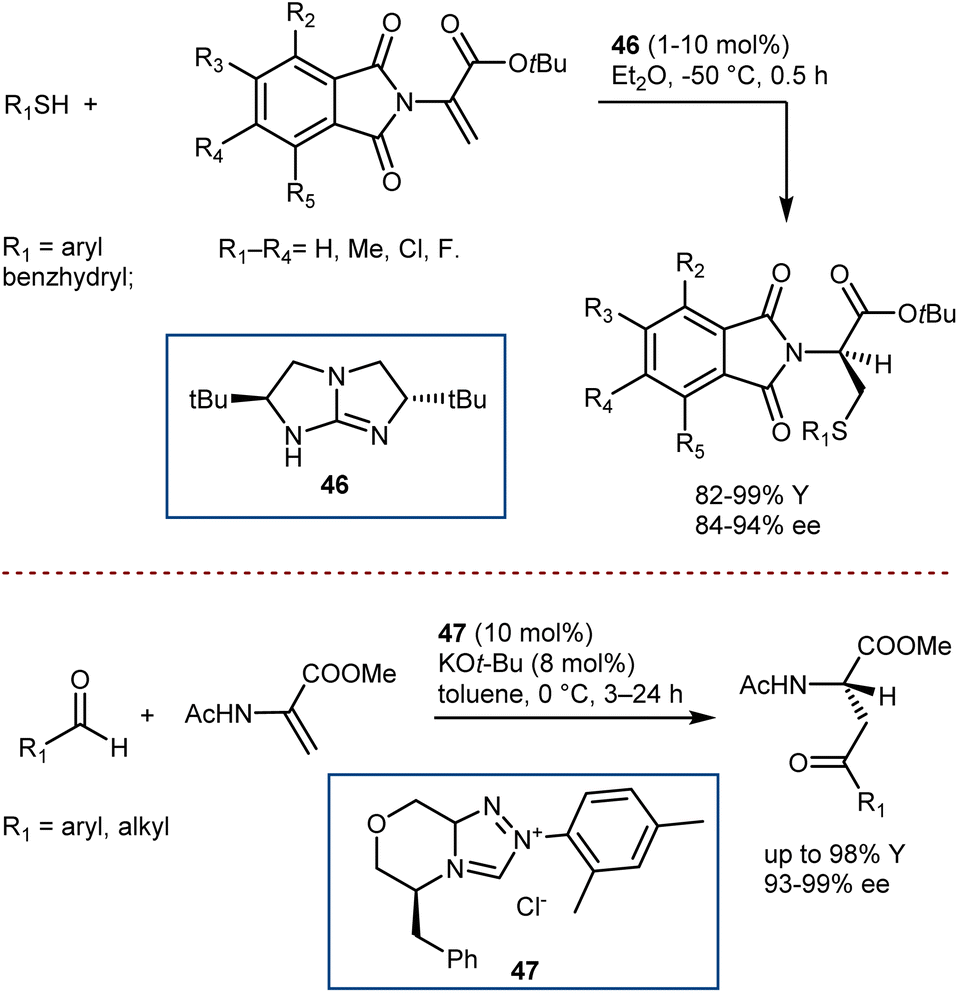 | ||
| Scheme 47 | ||
Conversely, Chen and Xiao reported the addition of 3-substituted oxindoles catalyzed by thiourea 36 (Scheme 48, top),132 and, more recently, our group developed the addition of cyclic ketones with the use of the chiral bifunctional primary amine/thiourea catalyst 48, which is capable of activating the ketone via a transient enamine intermediate and the DHA via H-bonding (Scheme 48, bottom).133
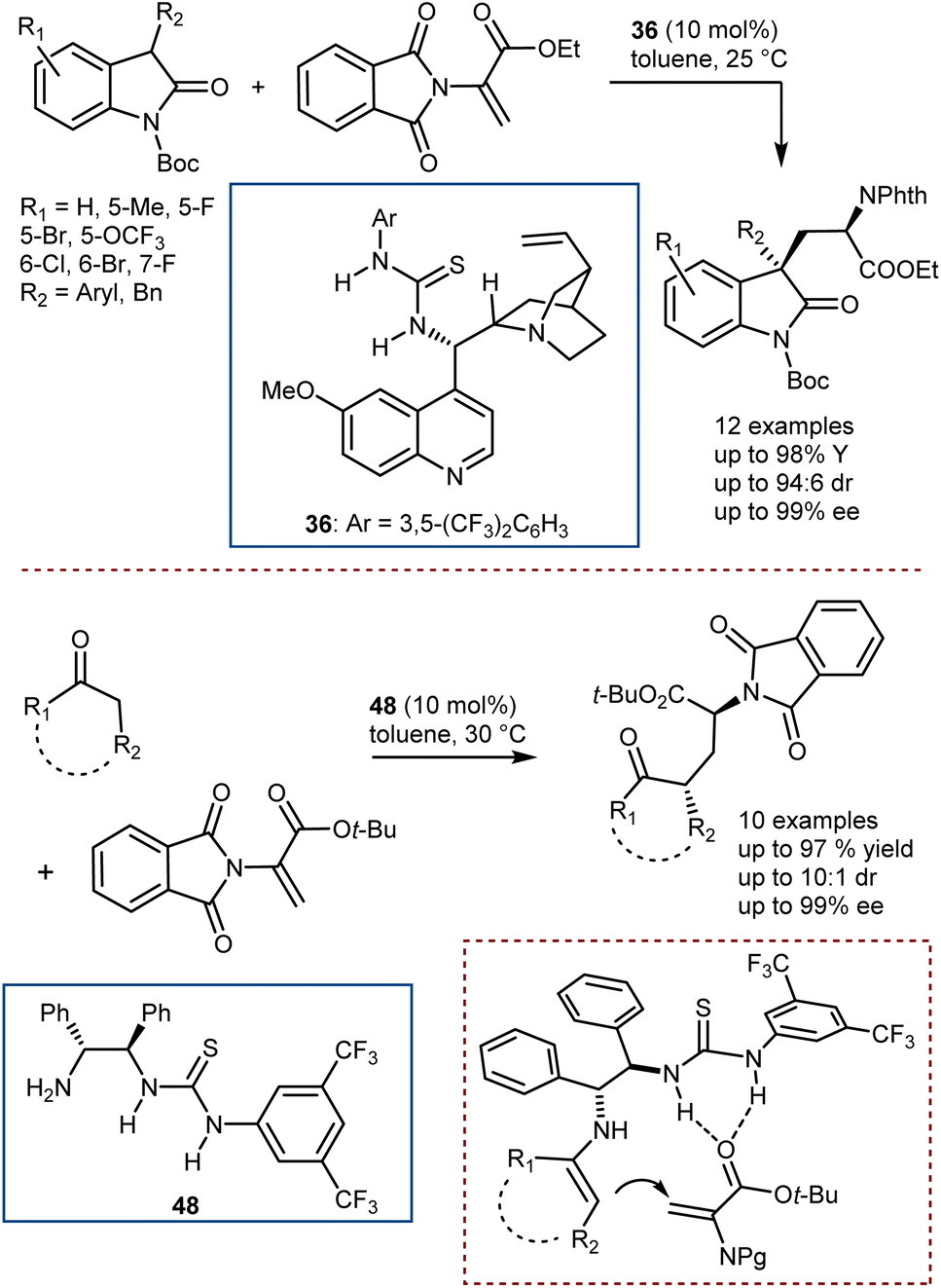 | ||
| Scheme 48 | ||
The unique electronic structure of DHA offers a plethora of chemical transformations for its modification. In fact, depending on the reaction conditions, polar nucleophilic additions can be performed with α- or β-selectivity. In slightly acidic media, the enamine character dominates and protonation of the β-position followed by nucleophilic addition to the ketoimine leads to quaternary AAs. However, due to the acidic conditions, the α-substitution is limited to nonbasic nucleophiles, e.g., electron-rich aryl compounds.134
4.4. DKR of azlactones
An elegant entry to enantioenriched AA derivatives is through the DKR of oxazol-5(4H)-ones (“azlactones”) via ring-opening (Scheme 49). It has attracted increasing attention from researchers in recent times because it enables the formal transformation of the racemic AA into the desired AA enantiomer. Moreover, the desired configuration and high enantioselectivity of the product may be achieved by the proper design of the catalysts.135 Azlactones are convenient substrates in DKR processes, because they are rapidly racemized by external bases – the pKa value of the hydrogen atom at the 4-position is about 9 – but autocatalytic racemisation can also occur.136 Curiously, these processes are usually an issue to avoid in peptide synthesis. Azlactones react readily with a variety of nucleophiles. As such, they are widely used for syntheses of optically active ncAAs of either configuration by the proper design of the organocatalysts.137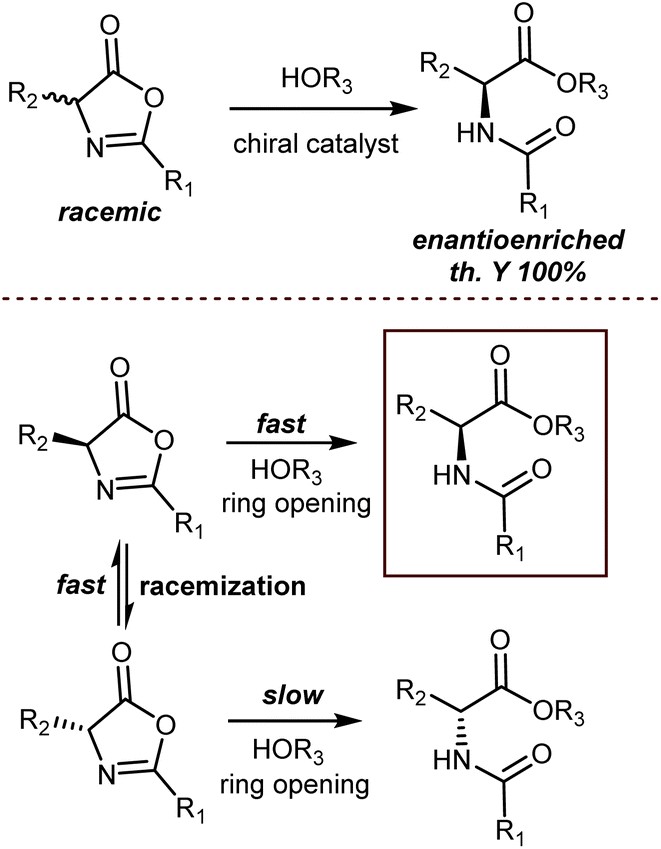 | ||
| Scheme 49 | ||
Seminal organocatalytic examples although with moderate to poor enantiomeric excess were reported by Leung back in 1999 using derivatives of dipeptides containing a His residue. These species catalyse the ring-opening of 2-phenyl-4-benzyl-5(4H)-oxazolone by methanol, ethanol and n-butanol, preferentially affording the N-benzoyl-L-phenylalaninates (20–39% ee).138 Interestingly, the mixture of cyclic dipeptides with L-diisopropyl tartrate, which possesses both a hydrogen-bond donor and a hydrogen-bond acceptor, is a more effective and enantioselective catalyst than the dipeptides alone. Around the same time, Fu investigated enantioselective methanolysis by racemisation/ring-opening of a range of azlactones catalyzed by a planar-chiral derivative of 4-(dimethylamino)pyridine, thereby affording protected ncAAs.139 This study furnished a benchmark for non-enzymatic enantioselective catalysis of this important process for a long time. In fact, for a broadly applicable organocatalytic method for the DKR of azlactones with high levels of enantioselectivity we had to wait until 2005 when Berkessel reported that organocatalysts 49 and 7,140 disclosed by Takemoto in those years, efficiently promote the reaction (Scheme 50). The method is based on bifunctional urea/thiourea–amine catalysts, bearing both a Lewis acidic (thio)urea moiety and a Brønsted basic tertiary amine group, which activate the substrate azlactones for rapid racemization followed by ring opening/nucleophilic attack through the formation of a hydrogen-bonded supramolecular aggregate.141 The nucleophilic attack occurs faster on the (R)-azlactone and selectively on the Re-face of the lactone. The latter selectivity is however inconsequential for the enantioenrichment of the product.
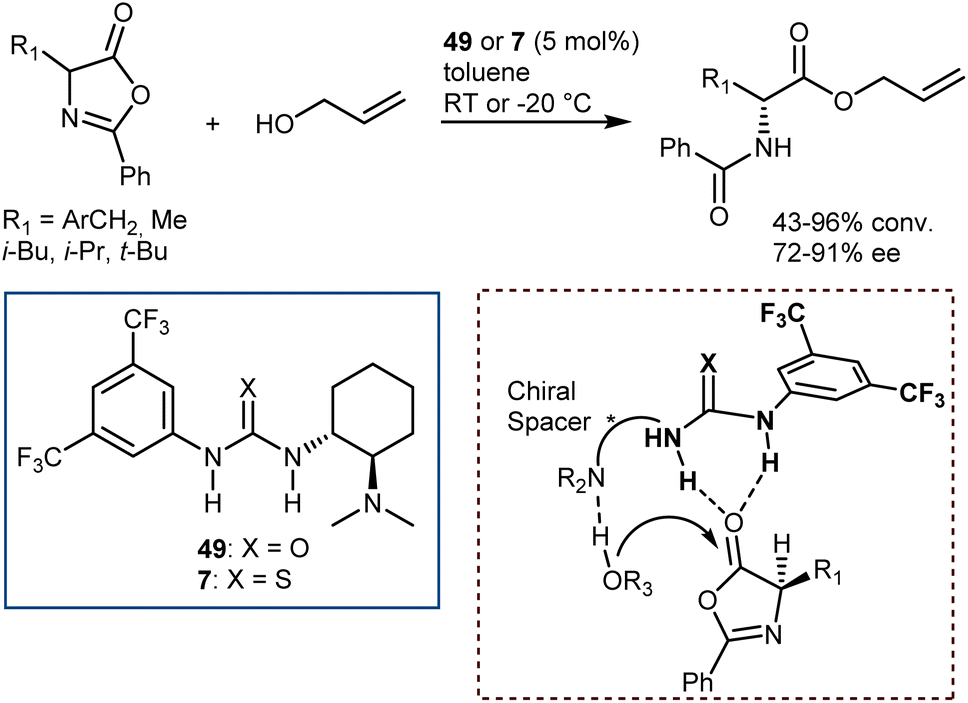 | ||
| Scheme 50 | ||
These catalysts can be easily prepared and have a modular structure. Thanks to this, the same research group reported structural optimization of bifunctional organocatalysts of the thiourea-tert-amine type, and carrying two “matched” elements of chirality. The second-generation catalyst 50 was disclosed (Fig. 6), that effected the alcoholytic DKR of a variety of azlactones with up to 95% ee.142 Subsequent work by Connon applied to the reaction the urea 51, closely related to Takemoto's catalyst 7 but built on the epi-9-amino-9-deoxy dihydroquinine chirality unit.143 Furthermore, the DKR was extended to the thiolysis with moderate results (up to 64% ee), that were later improved using as catalyst the arylated Cinchona52 (up to 73% ee), and the azo derivative 53 (up to 90% ee).144 In terms of enantioselectivity, generality, and reaction efficiency in the alcoholytic DKR of azlactones, significant improvements were reported by Song,145 who used the dimeric squaramide 54 as a catalyst in the reaction. Elegant studies based on detailed experimental, NMR spectroscopic studies and single crystal X-ray analysis demonstrated that these dimeric organocatalysts, as well as the related thioureas, do not form H-bonded self-aggregates in either solution or solid state, making possible the use of less diluted conditions compared to monomeric catalyst species.
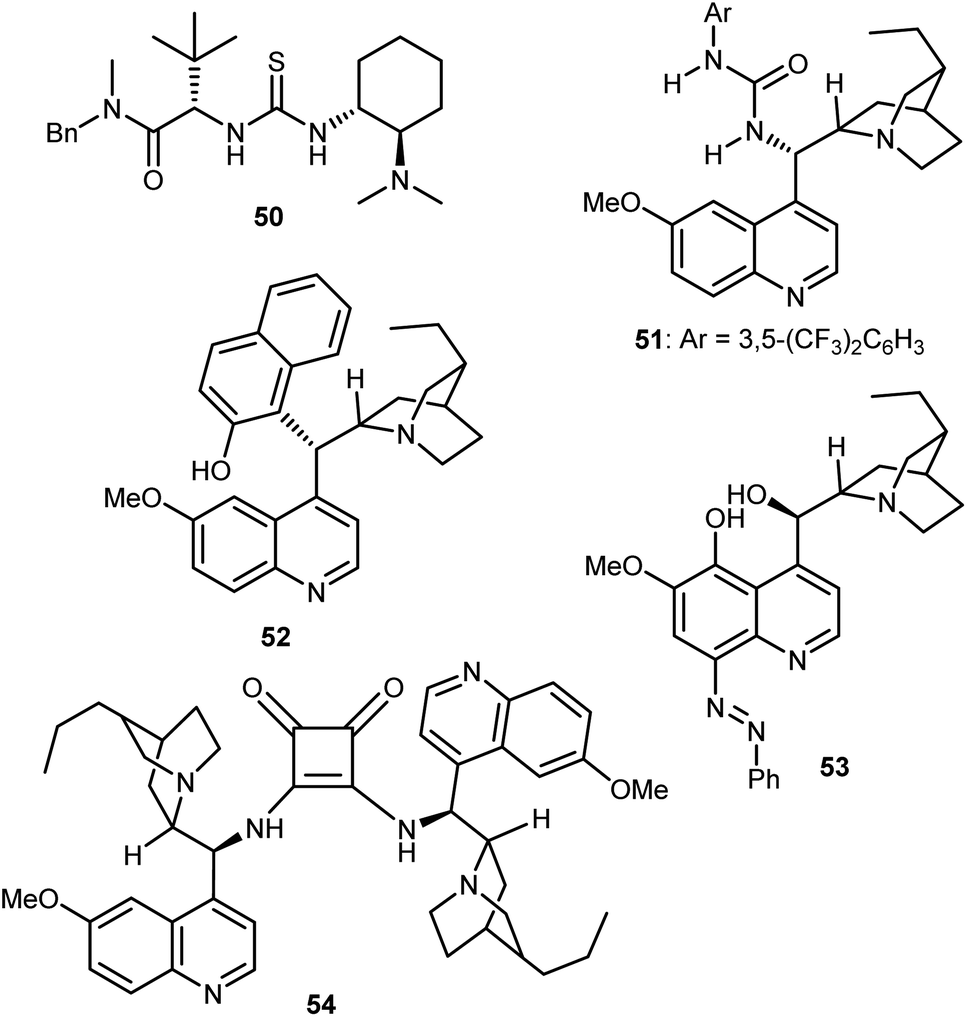 | ||
| Fig. 6 Bifunctional catalysts 50–54 used in the DKR of azlactones. | ||
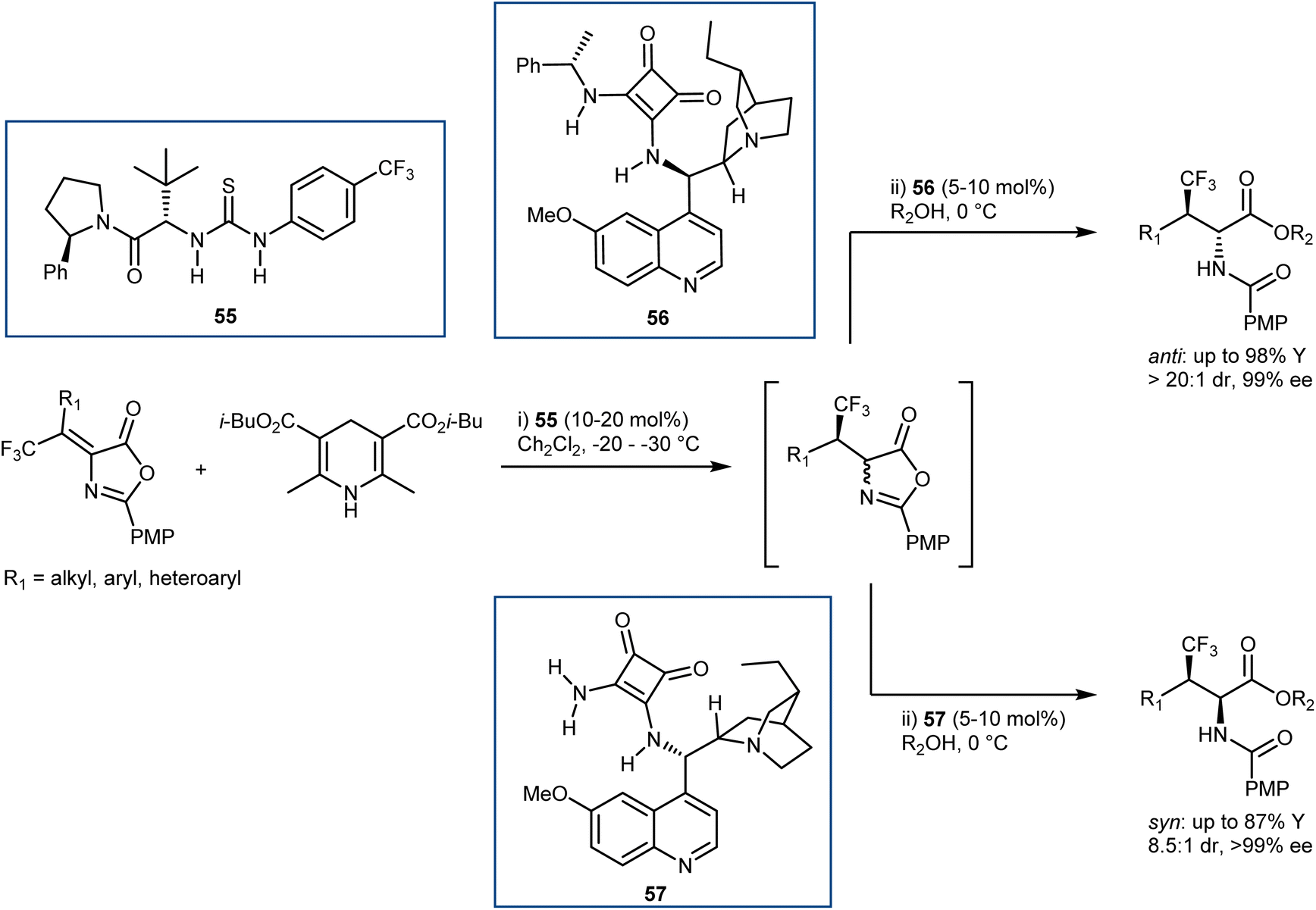
Relatively few organocatalytic methods can offer a unified route that can lead to all possible stereoisomers of ncAAs containing multiple contiguous stereocentres i.e. stereodivergent synthetic methodologies. Recently, our laboratories reported a conceptually new and stereodivergent approach to β-branched AAs by a sequential process involving the enantioselective transfer hydrogenation of Erlenmeyer–Plöchl substrates followed by the dynamic ring-opening of the resulting azlactones.146 The realization of this tactic with trifluoromethylated substrates has disclosed a one-pot entry to β-branched, β-trifluoromethyl AA derivatives (Scheme 51). The first step controls the configuration of the β-chirality centre and is performed using an HE and the Jacobsen-type thiourea 55. Leveraging the anti-bias exerted by the substrate in the ring-opening step, that governs the α-configuration, the anti-products are obtained with excellent stereoselectivities using the dihydroquinine-derived squaramide 56 (d.r. up to >20:1, ee ≥ 89%). The scope of this reaction includes examples where the traditional catalytic enantioselective hydrogenation on the corresponding α,β-dehydroamino acids (DHAAs) is known to be reluctant. In contrast, the obtainment of the syn-isomers proved to be more challenging, and required the development of a newly designed ammonia-derived squaramide catalyst (57), ultimately affording these products with variable diastereoselectivities (d.r. up to 8.5:1) and high enantioselectivities (ee ≥ 99%). It is worth noting that the syn-isomers cannot be accessed with the catalytic asymmetric hydrogenation, since the corresponding E-DHAA isomers, required due to the stereospecificity of the hydrogenation (E-DHAA → syn-AA), cannot be prepared.
 | ||
| Scheme 51 | ||
In general terms, the AA derivatives obtained from the alcoholysis of azlactones carry the amine protected as a benzamide, difficult to cleave without cleaving also the ester, thus limiting the versatility of these methodologies. To overcome this, Connon developed the DKR reaction of peculiar C2-substituted azlactones with benzyl alcohol as a nucleophile (Scheme 52).147 Upon treatment of the ring-opened amide with DABCO in a one-pot fashion, orthogonally protected N- and C-AAs are obtained. Furthermore, the electron-poor nature of the C2 substituent enhanced the efficiency of the DKR with dimeric catalyst 58.
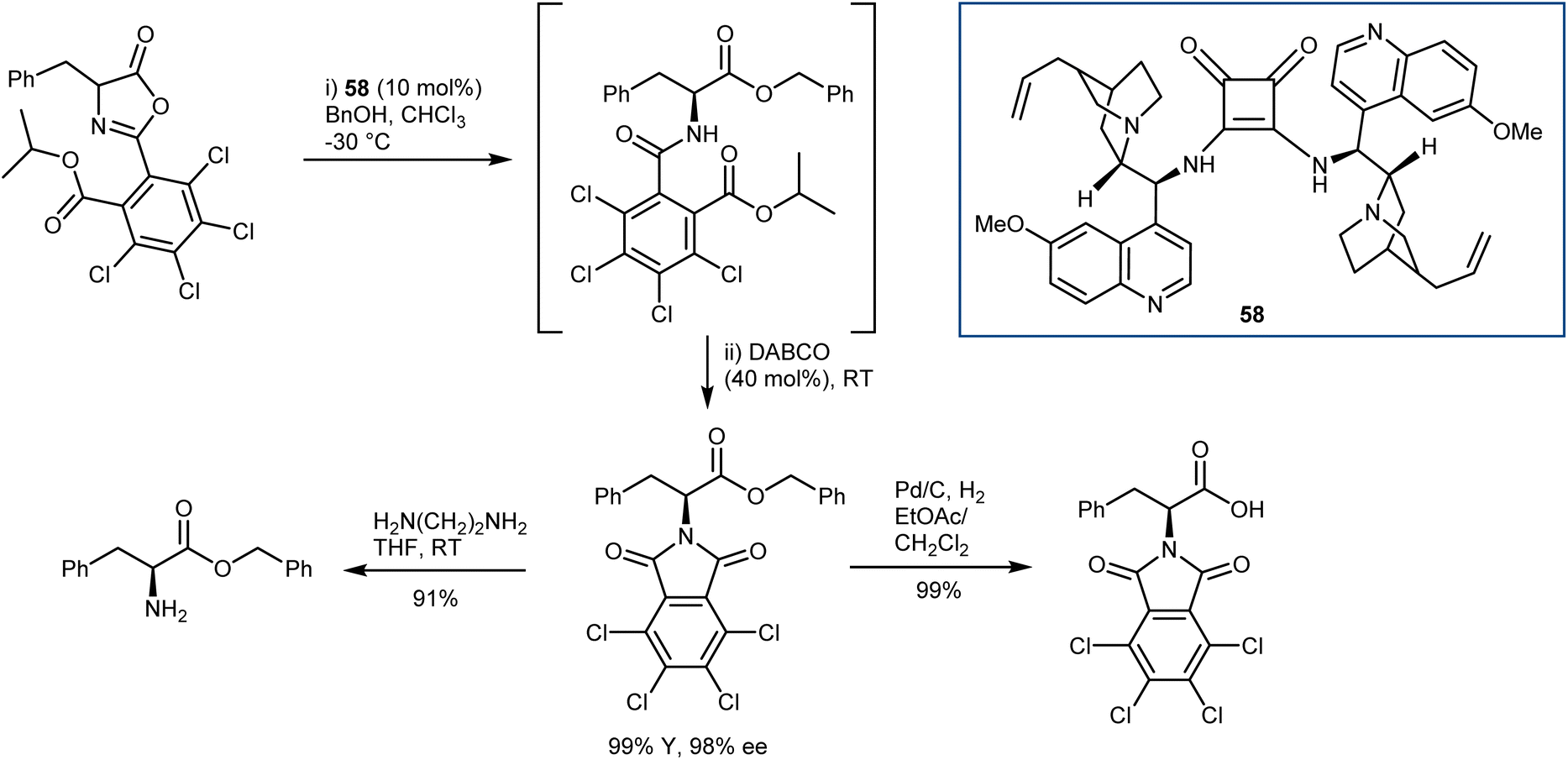 | ||
| Scheme 52 | ||
Several other methods to the DKR of azlactones via organocatalytic alcoholysis using chiral Brønsted acids,148 chiral nucleophilic catalysis,149 and small peptides150 have been reported, highlighting the potency of this method to access enantiomerically enriched AA derivatives. Among recent reports in this fruitful field, it is pertinent to note the work of Tokunaga who developed asymmetric alcoholysis of azlactones via PTC affording the corresponding α-chiral amido acid esters in up to 98% yield to 98% ee.151 A wide range of alcohols and azlactones are suitable for this method. For example, this catalysis was applied to the asymmetric alcoholysis with 1,1,1,3,3,3-hexafluoroisopropyl alcohol, providing the product with only moderate loss of yield (71% yield) and enantioselectivity (98:2 er) compared to more commonly employed alcohols.
4.5. Deracemization
A deracemization procedure can completely transform a racemic mixture, whose preparation is frequently less expensive and more efficient, into a single enantiomer. Catalytic deracemisation is a very attractive method of producing enantiomerically pure compounds as the target product and the substrate have the same molecular structure and it is not necessary to take the extra steps to remove the added/resolving agents from the products. Catalytic deracemization is atom- and step-economical, and continues to garner a lot of research interest via the methodological fusion of known asymmetric catalyses, such as that of enzymes, transition metals, and organocatalysts. It allows an efficient stereoediting of organic molecules despite an exogenous energy input procedure being needed. In fact, to overcome the thermodynamic and kinetic limitations of this perfect-almost-ideal process, selective energy input and careful reaction design are needed. First, deracemization is thermodynamically unfavoured and requires an energy supplement because it is an entropy-decreasing process without an enthalpy change (ΔG = +0.41 kcal mol−1 at 298 K). Second, any series of elementary steps where an enantiomer converts to its antipode share the same transition/activated state in both directions, which suggests that a chiral catalytic cycle that currently converts between two mirror-image isomers would likewise convert back in the same way. As a result, no net deracemization can be accomplished without an external perturbation that circumvents the principle of microscopic reversibility.152 Thus, a deracemization reaction must comprise two half-reactions that are opposite in reaction direction and have distinct mechanistic pathways. At least one of these should operate enantioselectively. This approach was devised as a comprehensive resolution for the deracemization of AAs. In AA chemistry, chemo-enzymatic deracemization is possible due to the ease of racemization of AAs and the numerous enantioselective catalytic systems operating on this class of compounds.One of the earliest instances concerning AA deracemization documented in the literature was demonstrated by Chibata in 1965 using a whole-cell system. A 24-hour incubation period of the racemic AA phenylalanine combined with a Pseudomonas fluorescens cell suspension could deracemize the racemate and yield L-phenylalanine.153 Subsequent investigations revealed that the reaction followed a non-selective oxidation pathway, resulting in phenyl pyruvate, and then an L-selective transamination reaction, resulting in a linear redox deracemization. In the other way around, Turner proposed a preparative chemoenzymatic method for deracemization of AAs by inclusion of a non-selective chemical reductant (amine-borane), and a selective D- or L-amino acid oxidase from Proteus myxofaciens.154 In this reaction, the non-selective reducing agent transforms back the intermediate imino acid produced by the selective enzyme to the racemic mixture of the original AA, thus allowing 100% conversion of the initial AA racemate. Starting from the racemate, a range of D-amino acids were obtained in yields of up to 90% and ee >99% using L-selective oxidase enzymes. This is the prototype of the cyclic deracemization that operates starting with a selective transformation of one enantiomer to a prochiral intermediate, followed by a non-selective reaction back to both mirror image isomers.
The catalytic deracemization cycle can now be disrupted by excited states thanks to the renaissance of photochemical synthesis and the radical polar cross-over strategy, where the cleavage of the desired bond (initial homolytic cleavage) and stereoselective polar bond re-formation proceed through distinct one- and two-electron processes, respectively.155
In fact, recently, Cao and Jiang combined well-developed CPAs such as 59 and 60, and organo-photocatalyst 61 to deracemize N-aryl α-amino esters (Scheme 53).156 The system for reaction was able to tolerate amino esters with different α-alkyl, α-aryl, and cyclic skeletons, each class of substrates requiring different reaction conditions and CPA catalyst (i.e.59 for arylglycine substrates and 60 for alkyl ones). Deuteration could also be accomplished in the presence of excess D2O without compromising the enantioselectivity. For amino esters to be amenable to undergoing the electron-transfer to the excited photocatalyst, an electron-rich N-protecting group is needed.
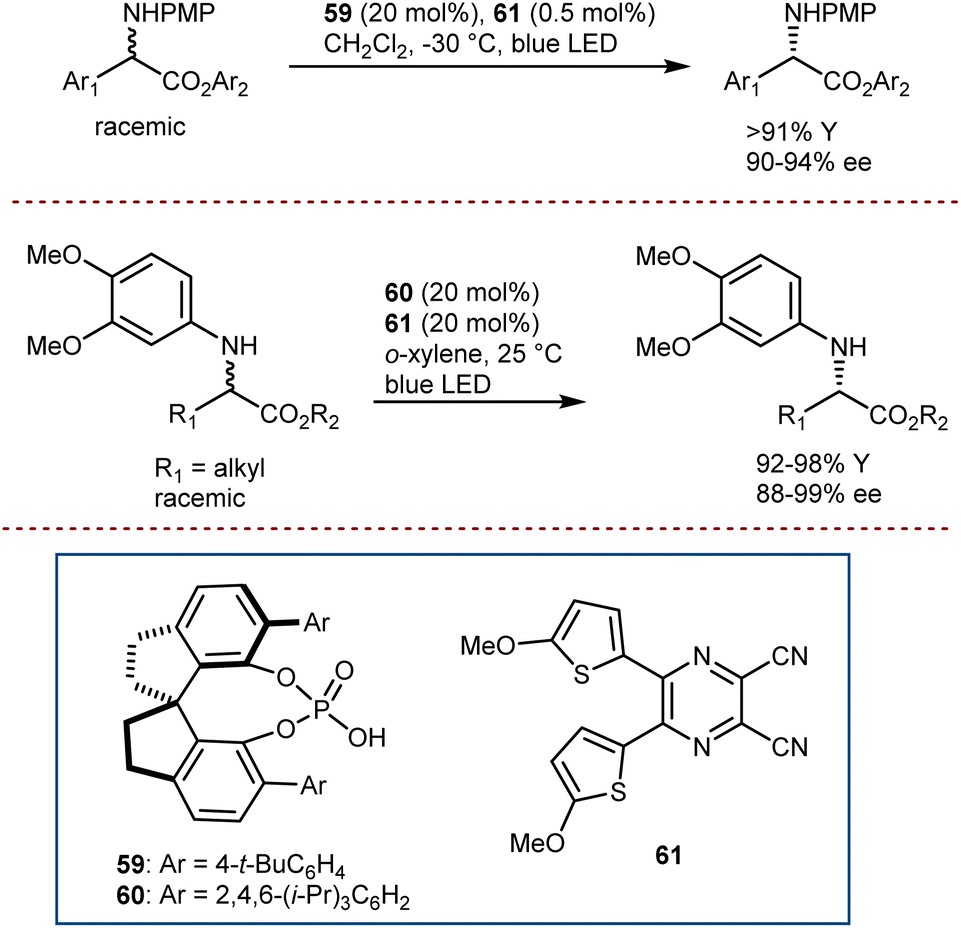 | ||
| Scheme 53 | ||
In this reaction, a critical enol intermediate is formed from both enantiomers through a cascade electron–proton–back electron transfer through the action of the photoexcited catalyst 61 (Scheme 54). This process, occurring at the same rate on both enantiomers, is slow compared to the fast (S)-selective tautomerisation (enol → (S)-product) driven by the CPA catalyst 59 or 60. Since the tautomerisation leading to the mirror image (R)-product is slower than the (S)-selective process, the overall result is the accumulation of the (S)-enantiomeric product over time.
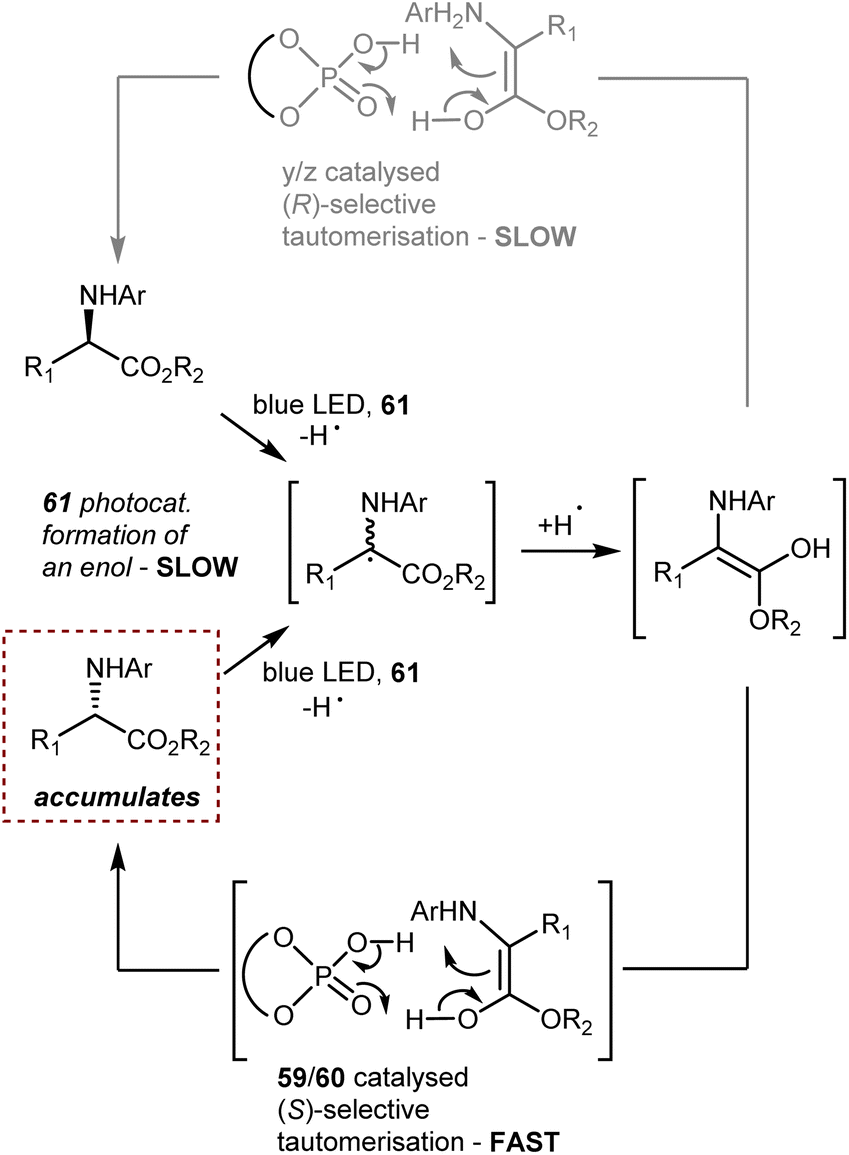 | ||
| Scheme 54 | ||
Examples of mechanistically distinct deracemization of AA derivatives – hydantoins and 2,5-diketopiperazines (DKP) – have been recently reported by Bach.157 As shown for DKP in Scheme 55, the stereochemistry is controlled by the chiral organophotocatalyst 62, operating under light irradiation at 366 nm, and the DKP can be readily taken to the enantioenriched N-substituted AAs by acidic hydrolysis. The reaction is remarkably general tolerating a wide range of aryl, heteroaryl and (cyclo)alkyl substituents, including functionalized ones, at the C6 and N1 of the DKP.
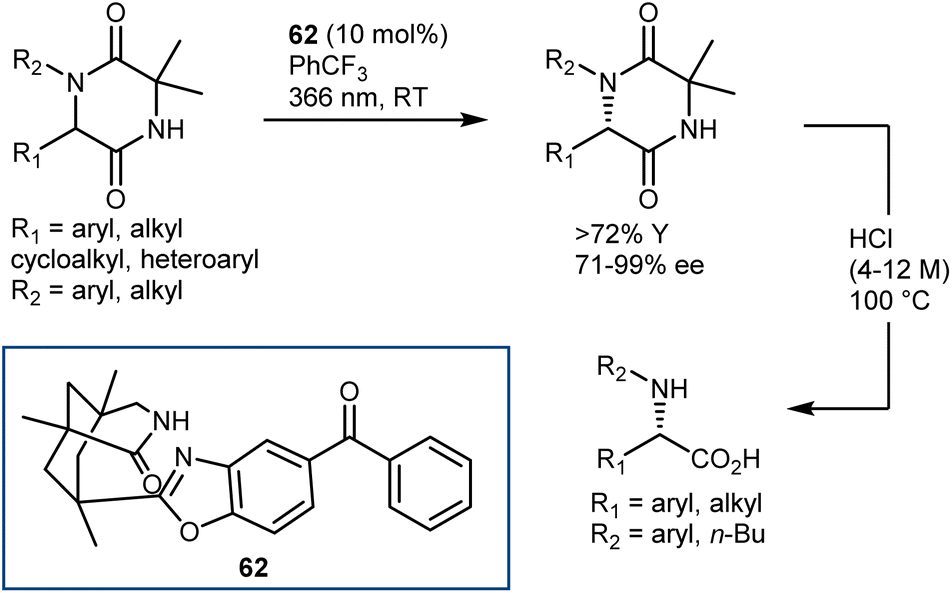 | ||
| Scheme 55 | ||
In simplified terms, the organophotocatalyst 62 racemizes selectively the (R)-DKP, exploiting the energy provided by light irradiation, making possible the accumulation of the (S)-DKP enantiomer (Scheme 56). The catalyst 62 was found to have a strong preference for the (R)-DKP, and there has been evidence gathered to support the theory that the photoexcited benzophenone moiety of 62 promotes a selective HAT from this DKP, followed by retro-HAT to the oxygen atom resulting in the formation and release of the DKP enol. Non-selective tautomerization delivers the racemic mixture of DKP, where only the (R)-enantiomer returns to the catalytic cycle, ultimately leading to the accumulation of the non-reacting (S)-DKP over time. A remarkable aspect about benzophenone catalyst 62 is that it can even function in a setting where the first HAT produces a carbon-centered radical that is only conjugated to the lactam entity that forms a H-bond with the catalyst.
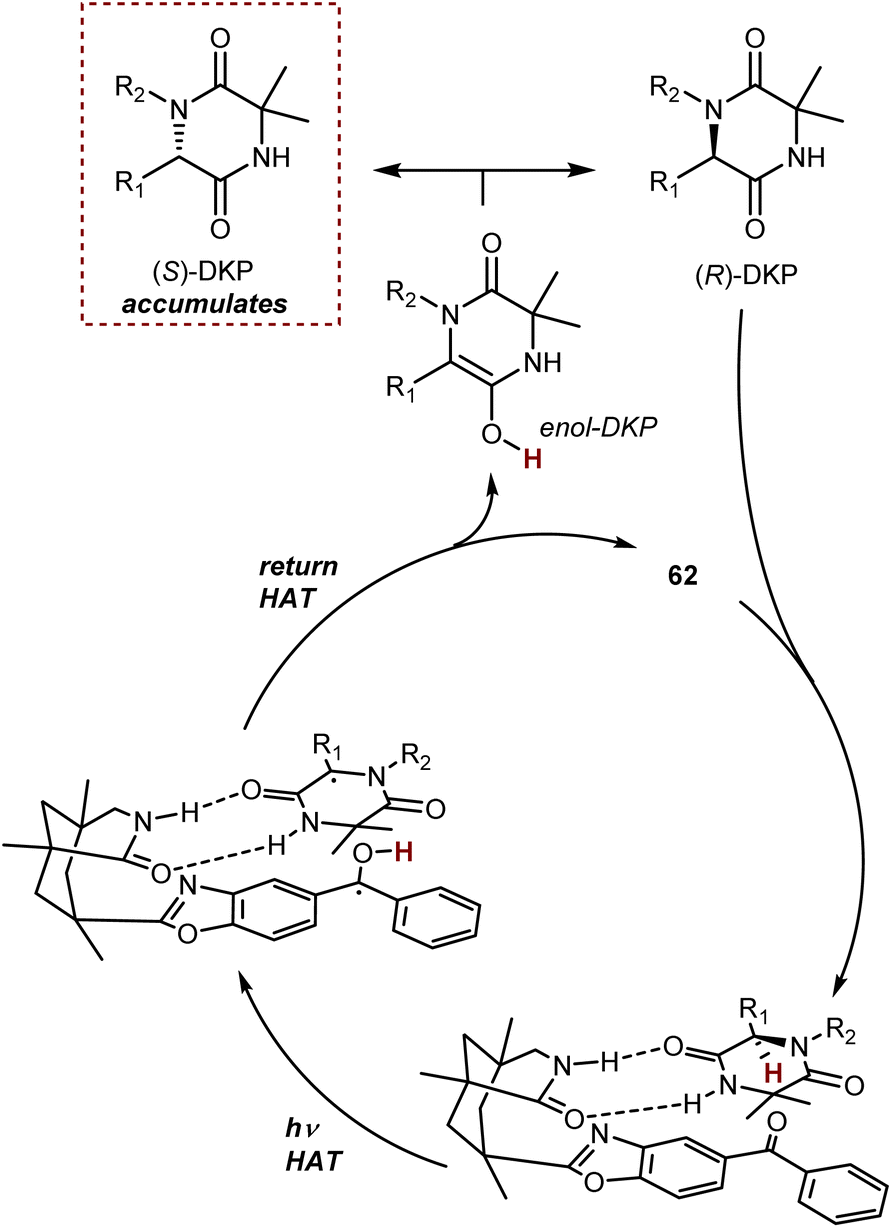 | ||
| Scheme 56 | ||
As demonstrated above, while other deracemisation methods have been documented, the photochemical method coupled with an organocatalytic process is especially desirable. Catalytic deracemization represents an attractive and fascinating strategy for stereochemical editing of organic molecules and AAs in particular.
5. Introduction of the amino group
The AA structure can also be disconnected at the C–N bond. Using organocatalysis, the nitrogen functionality can be introduced according to two main approaches (Scheme 57): (i) amination of an enol-like intermediate with an electrophilic nitrogen source, and (ii) formal insertion of a carbenoid species into the N–H bond of a nucleophilic nitrogen source. To be more precise, the latter approach introduces the nitrogen and hydrogen atoms at the same time. We will discuss the realization of the first approach via the α-amination of aldehydes.158 The second approach will be exemplified with some recent advances in diazo compounds and sulfoxonium ylides as carbene precursors. While transamination and the less explored reductive amination also build the AA by introducing the amine, we prefer to treat these subjects in the previous Section 4 because the enantioselectivity is established through the addition of a hydrogen atom to an imine intermediate.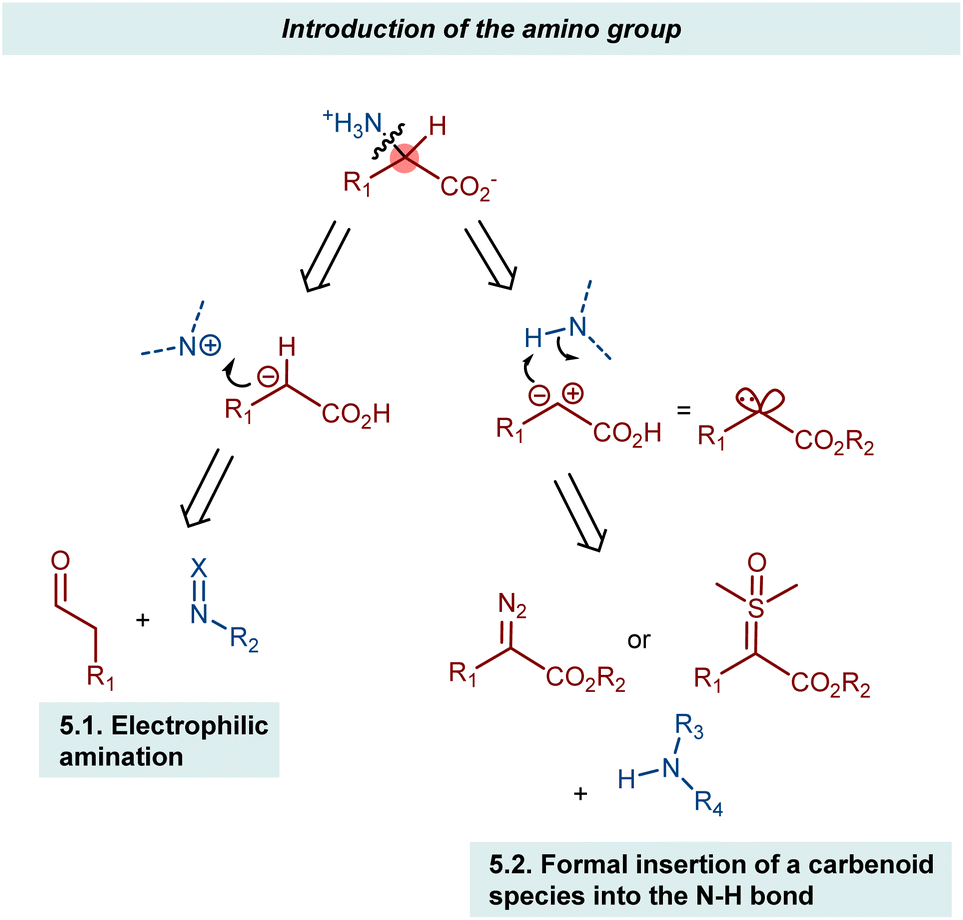 | ||
| Scheme 57 | ||
5.1. Electrophilic amination
Recognizing the analogy between previously successful π-acceptors (e.g., aldehydes and imines) and azodicarboxylates, List (Scheme 58, top) and Jørgensen (Scheme 58, bottom) independently published the L-proline (32)-catalyzed α-amination of aldehydes in 2002.159 These works highlighted the outstanding operational simplicity and efficiency of this reaction as well as some of its issues. The α-hydrazido aldehydes are configurationally unstable. Reduction to the alcohol, followed by cyclization to the oxazolidinone in one case, was preferred to enable product isolation. Furthermore, the cleavage of the N–N bond unveiling the amino function is not trivial, thus making the conversion of the catalytic adduct to the AA complicated.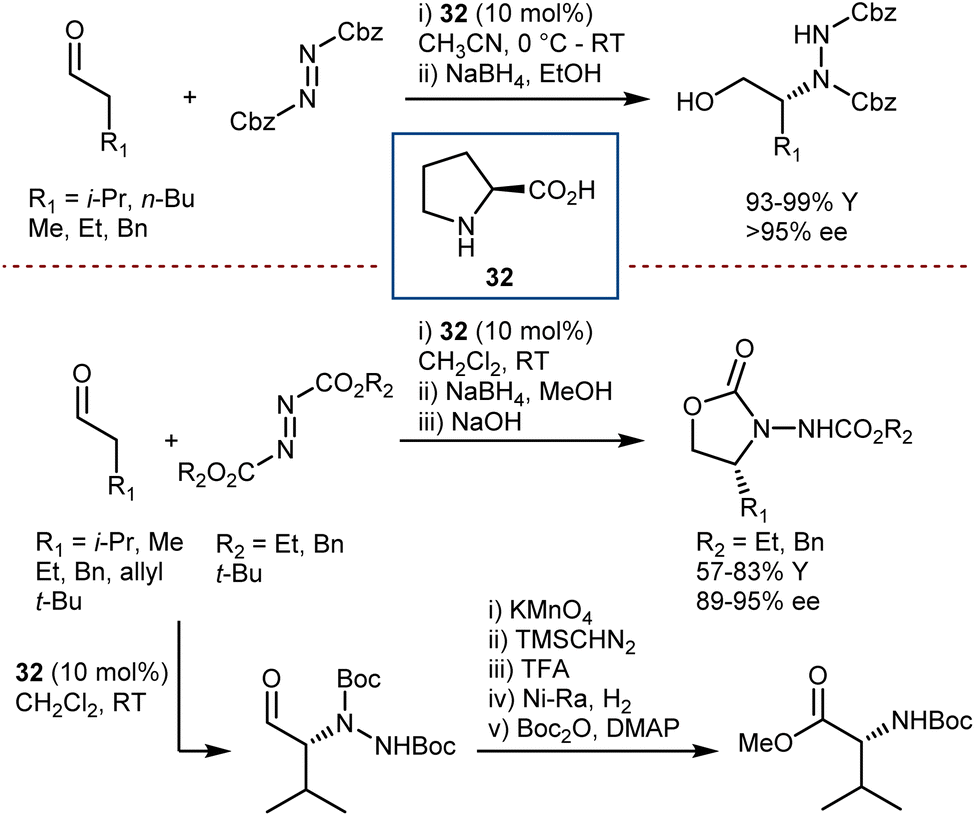 | ||
| Scheme 58 | ||
Nonetheless, the proline-catalyzed α-amination of aldehydes with azodicarboxylates has been repeatedly used for the asymmetric synthesis of target amine compounds of medium complexity.160 Lindel employed the closely related reaction with Ley's tetrazole catalyst 63 to obtain the challenging tetramethyl tryptophan unit of hemiasterlin, a cytotoxic marine natural product isolated from sponges (Scheme 59).161 In fact, Ley's catalyst 63 had been reported to be more efficient than proline (32) for highly hindered neohexyl aldehyde substrates such as the one required for the hemiasterlin unit.162 Furthermore, the reaction with another aminating agent (a nitrosocarbonyl compound, see below) did not work. The conversion of the catalytic product to the target AA derivative required substantial efforts to devise an efficient protocol for the N–N bond cleavage. Standard methods (e.g., SmI2, hydrogenation with Ni–Ra, Pd/C, or PtO2, and treatment with bromo acetate/Cs2CO3 to promote an E1cb reaction) failed. Ultimately, it was found that Pd(OH)2/C could catalyze the hydrogenolysis under high hydrogen pressure. Following a double oxidation and alkylation procedure, the target AA ester was obtained in a sufficient amount to eventually synthesize hemiasterlin.
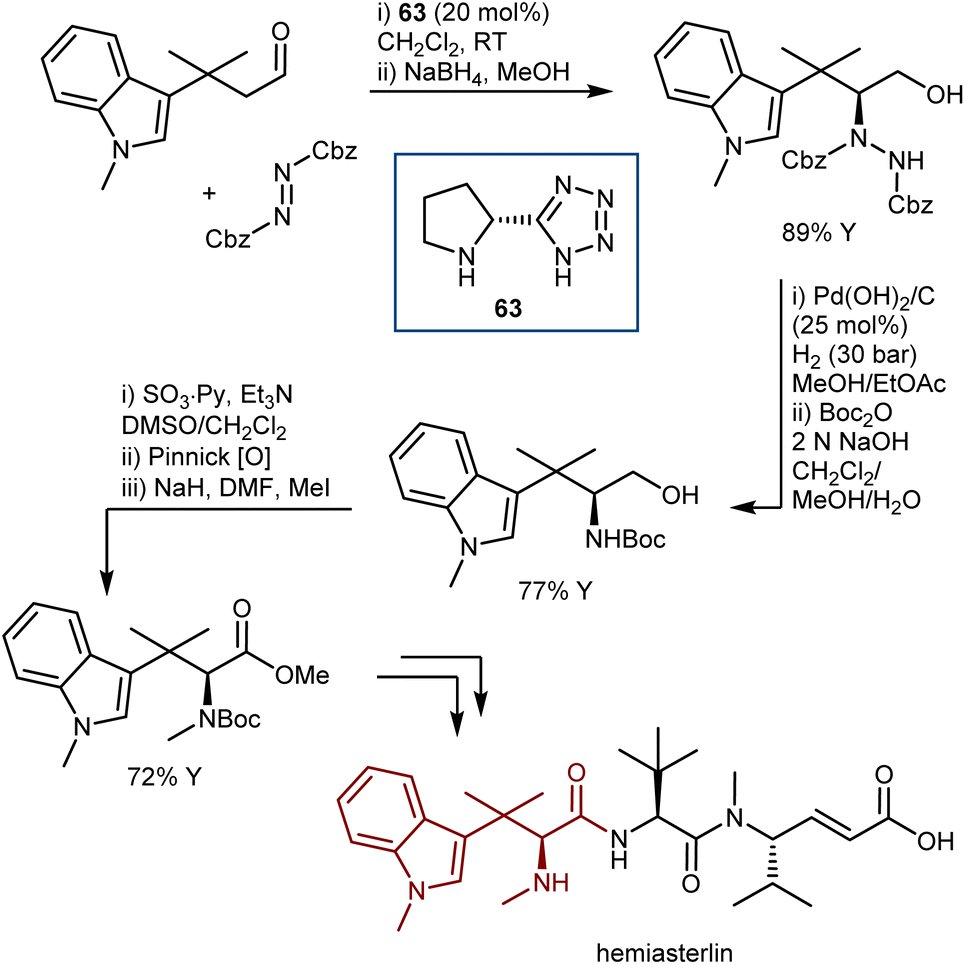 | ||
| Scheme 59 | ||
Moreover, the amination reaction has been inserted into domino reaction schemes, delivering α-hydrazido acid derivatives carrying an additional chiral center at the β-position,163 and, interestingly, the asymmetric α-amination of an α-branched aldehyde with dibenzyl azodicarboxylate has been recently applied on an industrial scale at AbbVie.164 However, the target of this process is a quaternary hydrazine-AA, eluding configurational lability issues and the requirement of N–N bond cleavage.
Alternatively, aryl nitroso compounds have been employed with great success in the α-amination of aldehydes. With these substrates, L-proline (32) promotes the α-aminoxylation, that is, the enamine attacks the acid-activated nitroso compound at its oxygen terminus. However, catalysts bearing functional groups that are less acidic than proline's carboxylic acid,165 or lacking them like 64,166 direct the reaction towards the oxyamination reaction, thus resulting in α-amination reactions (Scheme 60). The products were isolated as the corresponding alcohols upon reduction.
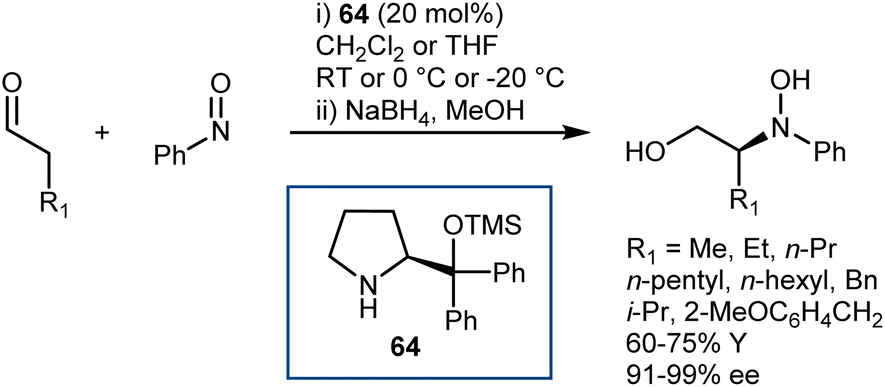 | ||
| Scheme 60 | ||
Aiming at an easier conversion of the catalytic product into a (protected) amine, focus was set on nitrosocarbonyl compounds.167 The poor stability of these electrophiles mandates their generation in situ, via oxidation of N-hydroxycarbamates.168 Three approaches to this reaction were reported within two years between 2013 and 2014 (Scheme 61).169 The first report by Maruoka employed the axially chiral catalyst 65 as well as the combination of TEMPO and benzoylperoxide as the oxidant (Scheme 61, top left).169a Yamamoto applied a more convenient oxidant (MnO2) with the tetrazole catalyst ent-63 (Scheme 61, top right).169b This oxidant was then adopted by Maruoka in the reaction with N-Boc-hydroxylamine and employing another catalyst 66 (Scheme 61, bottom left).169c As shown in the bottom right of Scheme 61, the unveiling of the amine functionality occurs under considerably milder conditions than for azodicarboxylates, using either catalytic hydrogenolysis or Mo(CO)6, although the isolation of the products as alcohols suggests a certain configurational instability of the aldehyde adducts.
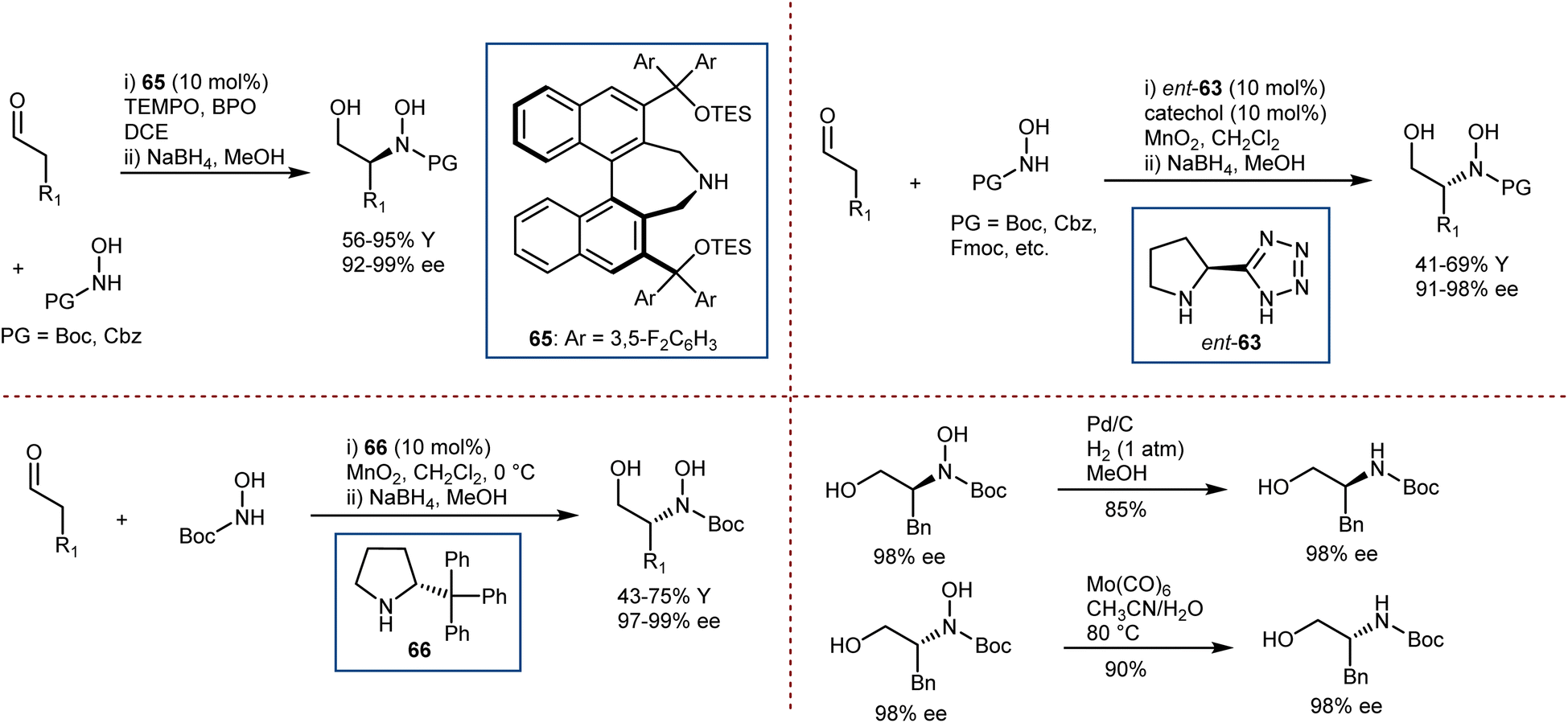 | ||
| Scheme 61 | ||
Conversely, aldehyde amination delivering directly protected amines was reported by Coombs, using sulfonyl azides as the electrophilic aminating agent.170 Elaborating an earlier investigation by Bräse with α-branched aldehydes and a stoichiometric amount of proline,171 Coombs' approach employed MacMillan's imidazolidinone catalyst 67 with linear aldehydes to afford the corresponding N-nosyl amino alcohols upon reduction of the aldehyde (Scheme 62). Oxidation and removal of the nosyl protecting group using the Fukuyama protocol led to the AA, avoiding the reductive steps typical of the use of azodicarboxylates and nitroso compounds.
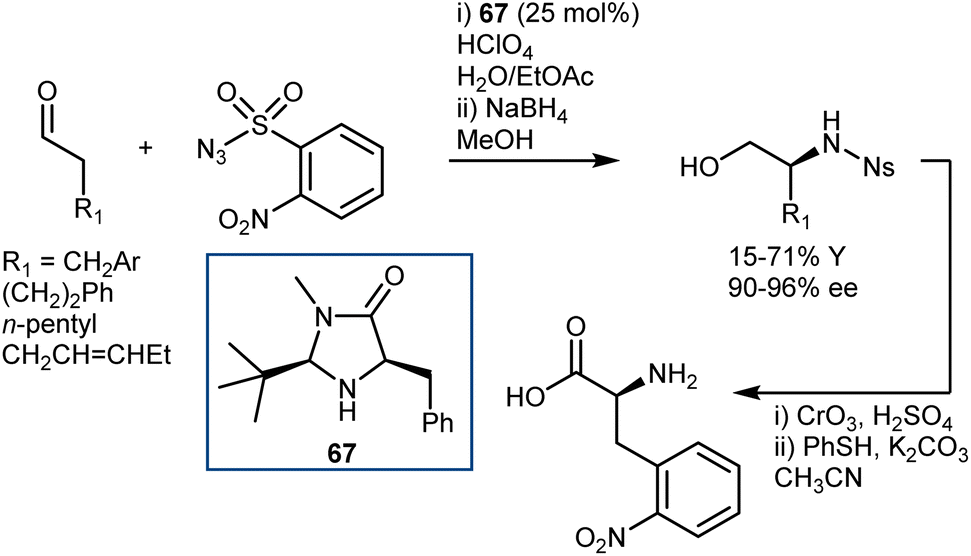 | ||
| Scheme 62 | ||
A conceptually distinct amination of aldehydes was reported by MacMillan by combining the organocatalytic activation of aldehydes with visible light photochemistry (Scheme 63).172 Using a 2,4-dinitrophenylsulfonyloxy precursor, the formation of a carbamyl radical occurs upon visible light irradiation followed by reduction. Thus, the formed radical is trapped by the enamine, resulting in the stereocontrolled formation of the C–N bond. Catalyst 68, featuring a peculiar quaternary center and a meta-ethyl substituent, is used in the reaction, enabling precise control over the enamine geometry and selective attack to one of its faces. Ultimately, N-alkyl, N-carbamoyl α-amino aldehydes were isolated with very good results. Oxidation of the aldehyde functionality provided the corresponding N-alkyl N-protected AA.
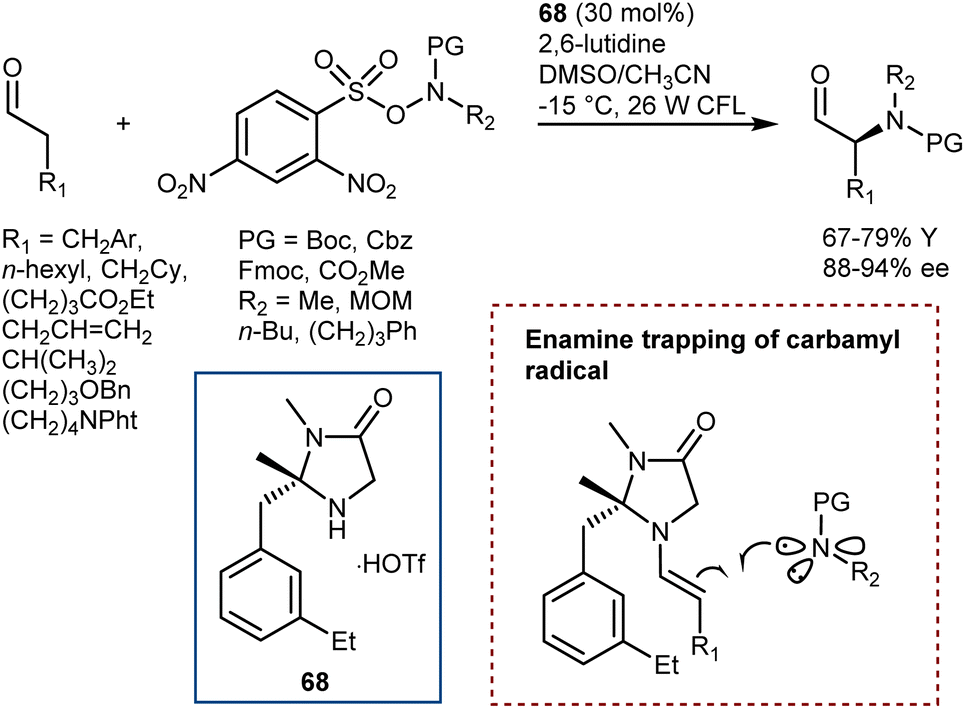 | ||
| Scheme 63 | ||
5.2. Formal insertion of N–H into a carbenoid species
This reaction often involves metal carbenes, and the role of the organocatalyst is to control the stereochemistry in a cooperative catalytic fashion. Historically, it has always been difficult to obtain enantioselectivity in this reaction. The first examples focused on the use of transition metals equipped with chiral ligands. Enantioselective N–H insertions were pioneered by McKervey in 1996 with Rh(II) carboxylates173 and Jørgensen in 2004 with Cu(I) and Ag(I) bisoxazolines,174 but the results presented were not satisfactory. A turning point was reported in 2007 by Zhou.175 The use of a spiro-bisoxazoline ligand in combination with Cu(I) as the catalyst led, for the first time, to excellent enantioselectivities (up to 98% ee). In the next few years, several other catalytic systems were proposed,176 many of which exploited cooperative catalysis between a metal and an organocatalyst. In this context, an important contribution was presented in 2011 by Zhu and Zhou, who demonstrated the enantioselective reaction between carbamates and α-aryldiazoacetates that was catalyzed by a combination of achiral tetra(triphenylacetate) dirhodium 69 and spirocyclic CPA 70 (Scheme 64).177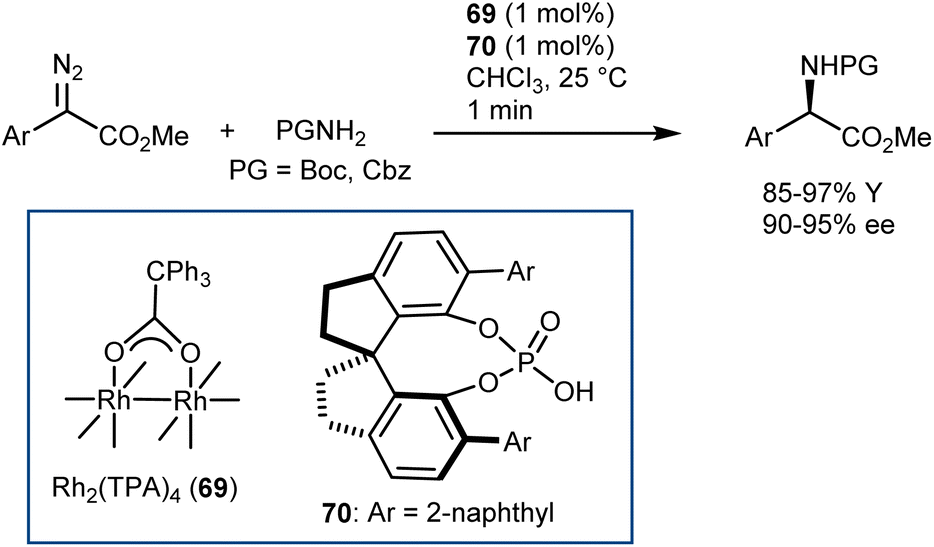 | ||
| Scheme 64 | ||
Besides the high appeal and efficiency of the reaction (production of N-Boc-protected AA derivatives, 1 min reaction time, 1 mol% loading, turnover frequency of 1000 min−1, and high enantioselectivities), the main highlight of this paper is the rationalization of the enantioselective step of the reaction as demonstrated by experimental evidence, including the following: (i) the ee decreased dramatically when the phosphoric acid was added as a sodium salt (from 98% to 7%); (ii) the 31P NMR signal of 70 did not change when 0.5 equiv. of [Rh2(TPA)4] 69 was introduced (thus ruling out a dirhodium phosphate catalyst); and (iii) no coordination between the carbamate and 70 was observed by 13C NMR or FT-IR. Overall, it was proposed that after the nucleophilic attack of the carbamate to the electrophilic rhodium carbene, the bifunctional catalyst could provide a proton and accept a proton synchronously from a zwitterionic intermediate through a cyclic transition state, acting as an enantioselective proton shuttle (Scheme 65). While a dirhodium enolate was considered as an intermediate in this reaction, the proton shuttle mechanism was proposed to occur on enol species in subsequent research on related systems (inset in Scheme 65).178
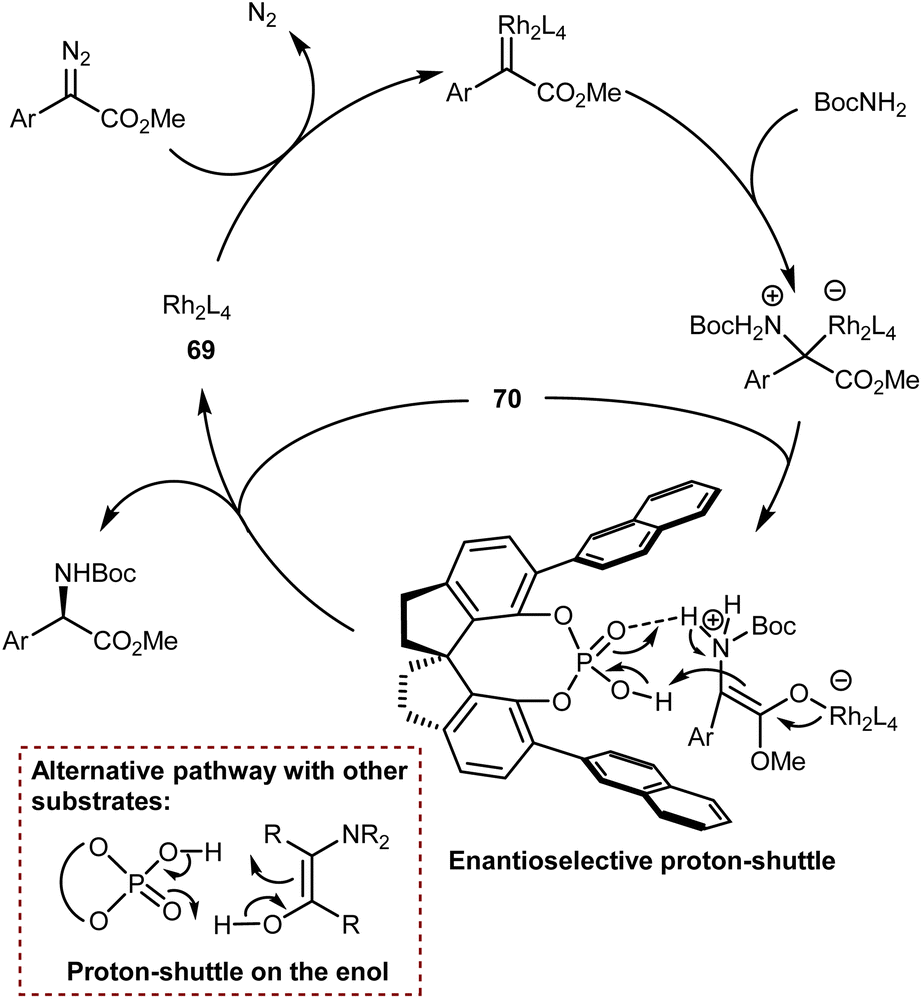 | ||
| Scheme 65 | ||
This approach, in which the protonation by an organocatalyst is recognized as the key enantioselective step, has been successful in several other reactions that encompass different chiral organocatalysts in combination with achiral metal catalysts, thus enabling the N–H insertion reaction of a range of diazo and nucleophilic nitrogen substrates (anilines, carbamates, amides, hydrazones, carbazoles, indoles, etc.).176 Recently, work by Zhu and Zhou extended this reaction to highly challenging aliphatic amines and ammonia as reaction partners. These nitrogen compounds are comparatively stronger Lewis bases; therefore, they are prone to poisoning the metal catalyst by strong coordination, interfering with the generation of the metal carbene intermediate. Subsequently, a new catalytic system was developed, a cooperative one between an achiral homoscorpionate copper complex (71) and chiral thioureas, in which the ligand coordination protects the copper center from the basicity of aliphatic amines or ammonia, permitting the formation of the metal carbene. The first paper published with this type of catalysis focused on the use of aliphatic amines.179 After a few years, the same group was able to provide remarkable results in the reaction with ammonia (Scheme 66).180 This process directly provides unprotected amines, which are convertible to the free AAs upon ester hydrolysis. The enantioinducing element is the relatively simple thiourea 72. Thus, by changing a single stereocenter, it is possible to obtain either L- or D-AAs in very good yields and ee values. A remarkable functional group tolerance was demonstrated with numerous examples, some of which include commercial drugs.
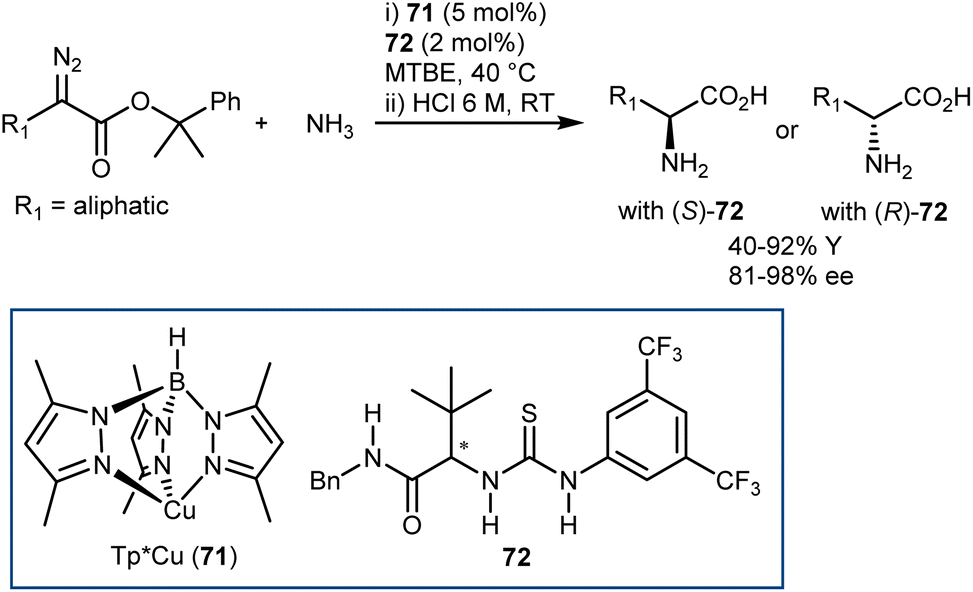 | ||
| Scheme 66 | ||
A crucial aspect investigated regarding the mechanism is the pKa of the thiourea catalyst 72, which deeply influences the enantioselectivity. The proton donation is directly linked to the pKa, which changes considerably between the free thiourea 72 and its complex with Tp*Cu 71 (Scheme 67, top). In the 1H NMR spectrum, the signal of the hydrogen of the aromatic amine (thiourea N–H) shifts downfield and then gradually disappears when the catalyst 72 is mixed with Tp*Cu 71, indicating an increase in acidity. Experimentally, the newly formed complex was found to be much more acidic (ΔpKa = −4.29). Such an increase in acidity enables the thiourea to behave like a Brønsted acid catalyst, an unusual behavior for thioureas. In fact, using a series of thioureas with different pKa values, a linear relationship between acidity and enantioselectivity was found. On the other hand, the yield is controlled by the first step of the reaction, i.e., by the formation of the metal carbene. This step does not significantly affect the ee, and the enantiodetermining step does not interfere with the yield, leading to the mechanistic hypothesis shown in the bottom of Scheme 67, which was confirmed by DFT calculations.
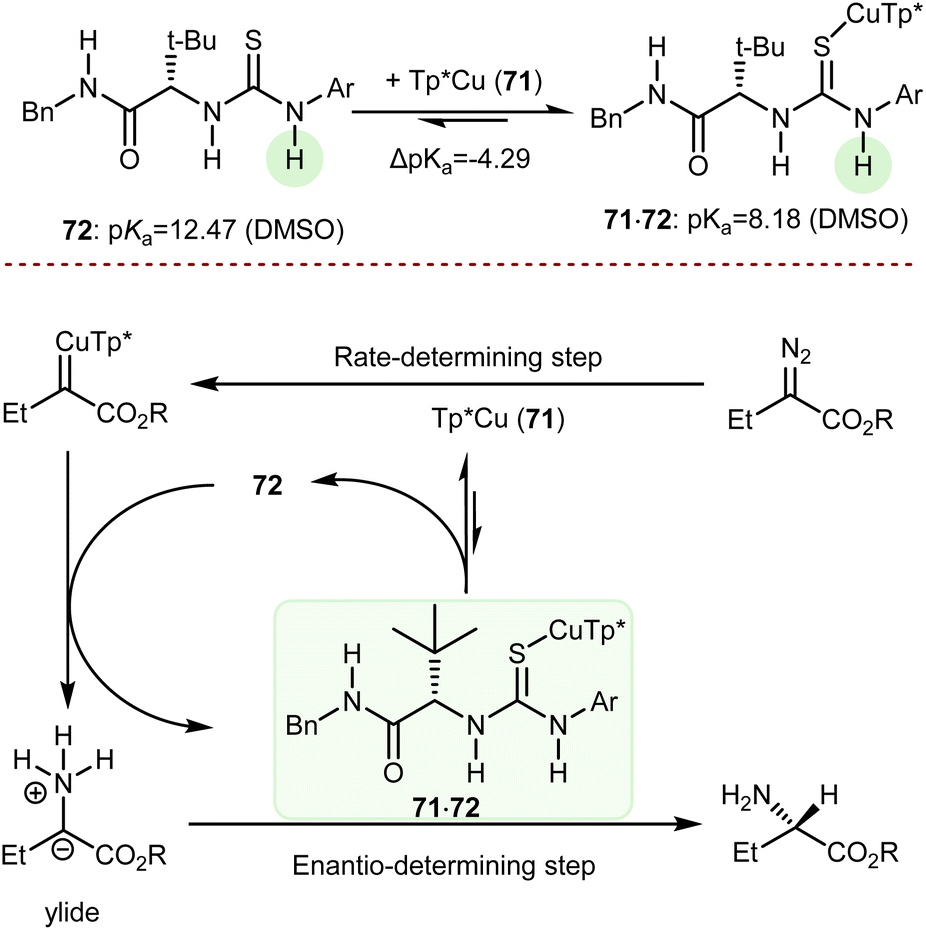 | ||
| Scheme 67 | ||
Alternative approaches to the N–H insertion reaction with diazo compounds that do not require metals and involve free carbenes have been reported as well. An early attempt by Miyairi used thermally generated carbenes, formed by heating α-aryldiazoacetates in toluene at 110 °C, in combination with a squaramide organocatalyst.181 The products were obtained with moderate enantioselectivities, and the yields were compromised by side reactions such as carbene dimerization. In fact, the reactivity of free carbenes is difficult to control and, although anilines can intercept their less abundant singlet state, many side reactions can occur.
Recently, using photochemical irradiation (visible region, 440 nm) of α-aryldiazoacetates to trigger the formation of a free carbene species under mild conditions, Li and Zhou reported a more efficient N–H insertion procedure (Scheme 68).182 This work shows an interesting parallelism with previous studies from the same laboratory180 for the influence of the catalyst's pKa on the enantioselection obtained, which is controlled during the proton transfer step. To investigate this aspect, a library of catalysts with different pKa values was synthesized. According to their analysis, two important points were understood: the pKa of the chiral catalyst must be less than that of the product so that the proton of the catalyst can be effectively transferred to the ylide intermediate. Second, the pKa of the catalyst must be greater than that of the protonated amine so that the proton of the ylide intermediate can be accepted by the catalyst at the end of the catalytic cycle. In practice, the product has a pKa of 23, while the intermediate's pKa varies between 13 and 4 (in DMSO). Ultimately, to induce enantioselectivity, a new class of catalysts with tailored acidity was specifically designed and synthesized—phosphamides; of the compounds studied, the spirocyclic derivative 73 proved optimal. In contrast to the previous work, the enantiodetermining step matches with the rate-determining step, since both the generation of the free carbene by visible light and its trapping by aniline should be fast processes. The optimization of this photo/organocatalytic system permitted the preparation of a wide range of molecules, with high ee values for the arylamines. It is remarkable that ammonia and benzylic amines work as well, but with lower yields and ee values. Considering the very rich chemistry of photochemically generated free carbenes, this methodology may open many possibilities for enantioselective metal-free X–H insertion reactions.
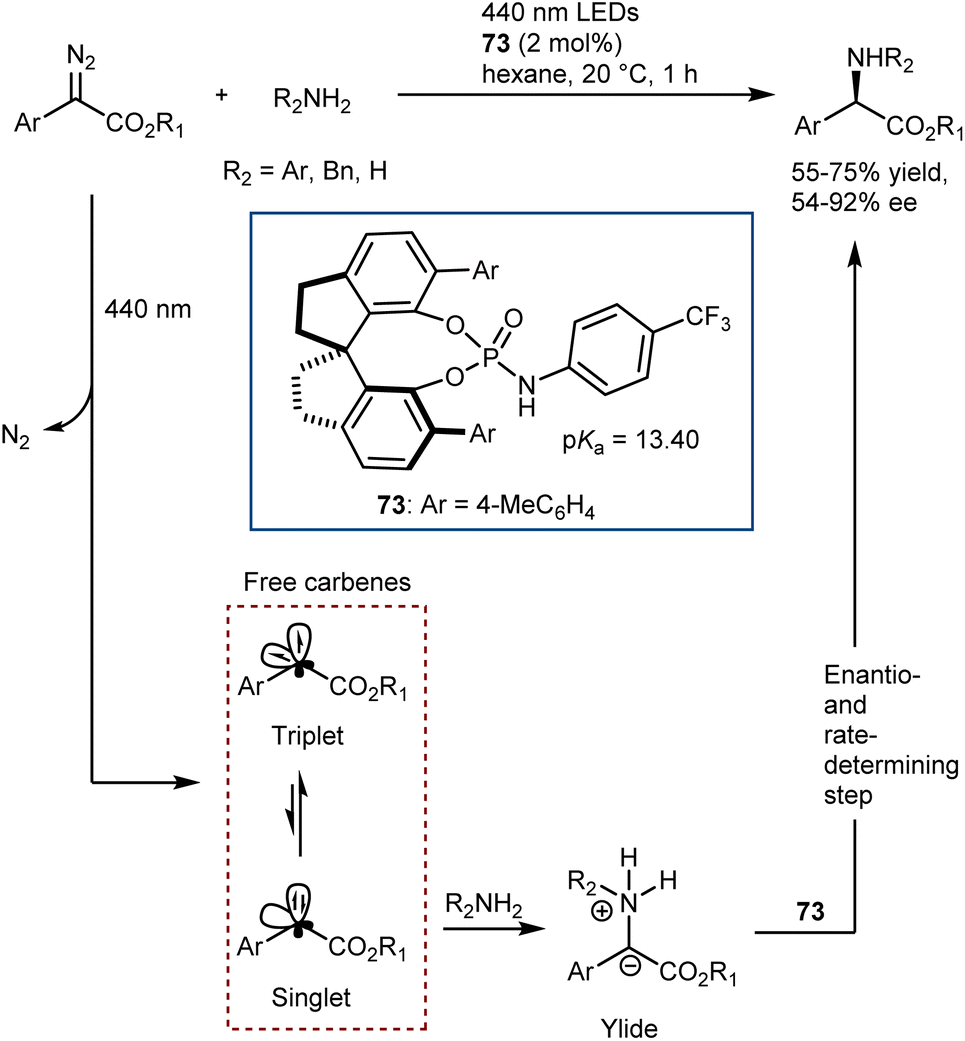 | ||
| Scheme 68 | ||
In fact, another reaction involving a free carbene species generated by visible light irradiation of α-aryldiazoacetates was recently reported in 2023 by Guo and Sun (Scheme 69). The reaction employs CPA 74 as the catalyst for the N–H insertion of anilines.183
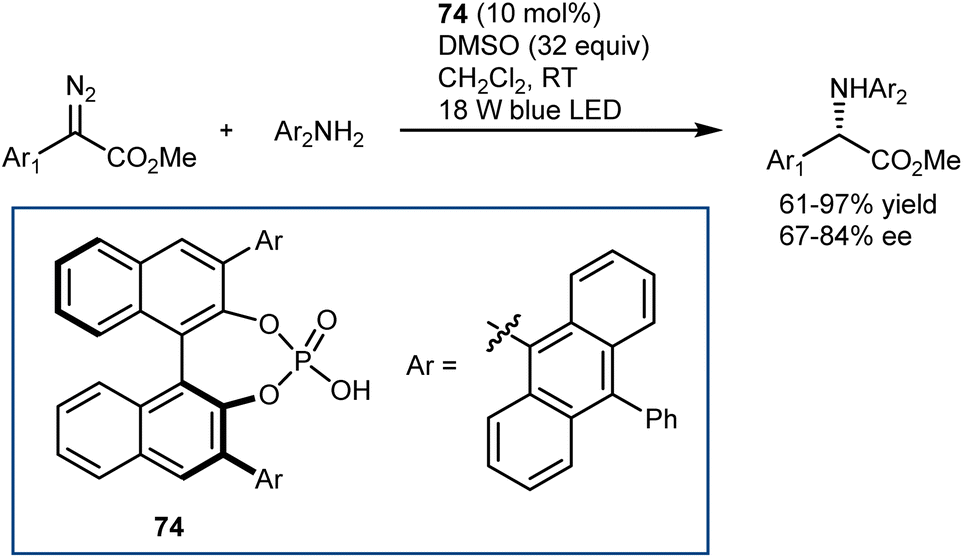 | ||
| Scheme 69 | ||
While Li and Zhou tackled the challenging reactivity of free carbenes by modulating the catalyst to perfectly fit the substrates, Guo and Sun adopted the strategy to lower the reactivity of the carbene by trapping it with a suitable additive, DMSO (Scheme 70). The result is a sulfoxonium ylide, which can undergo a formal enantioselective insertion reaction under the catalysis of CPA 74. The carbene capture must be rapid to avoid side reactions, and the additive must be reactive enough to outcompete the amine (k2 ≫ k1), which would result in a racemic N–H insertion reaction. The scope reported did not include basic amines such as ammonia or alkyl amines, and only aromatic amines rendered satisfactory results. The limitations of this methodology include not very high ee values; however, the novelty of the photo-activation with carbene trapping is remarkable and opens a new approach for this field.
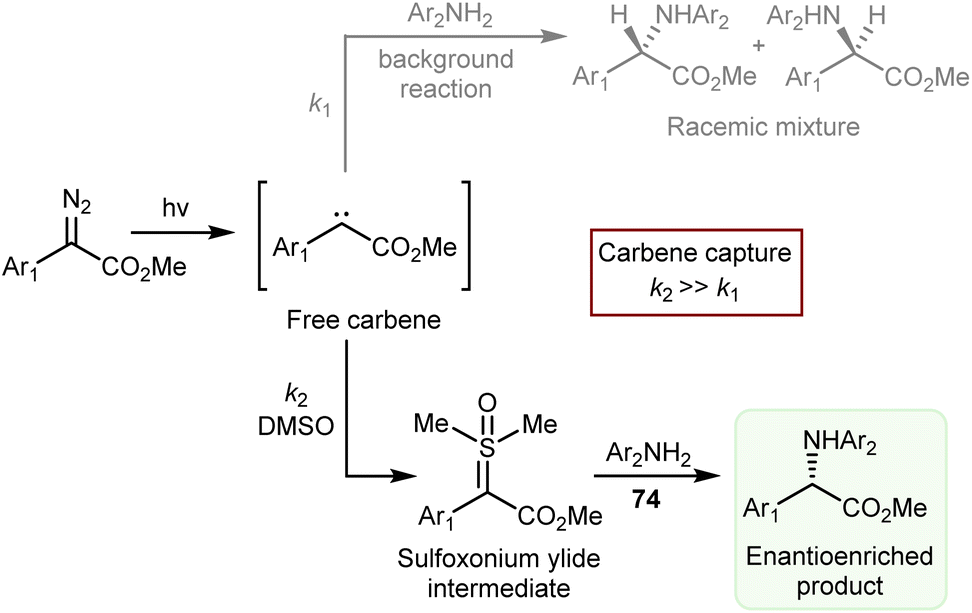 | ||
| Scheme 70 | ||
In effect, this work is related to a previous disclosure by Huang and Sun, from the same research group, in which sulfoxonium ylides were used as carbenoid species in a formal N–H insertion reaction catalyzed by the same CPA 74 (Scheme 71).184
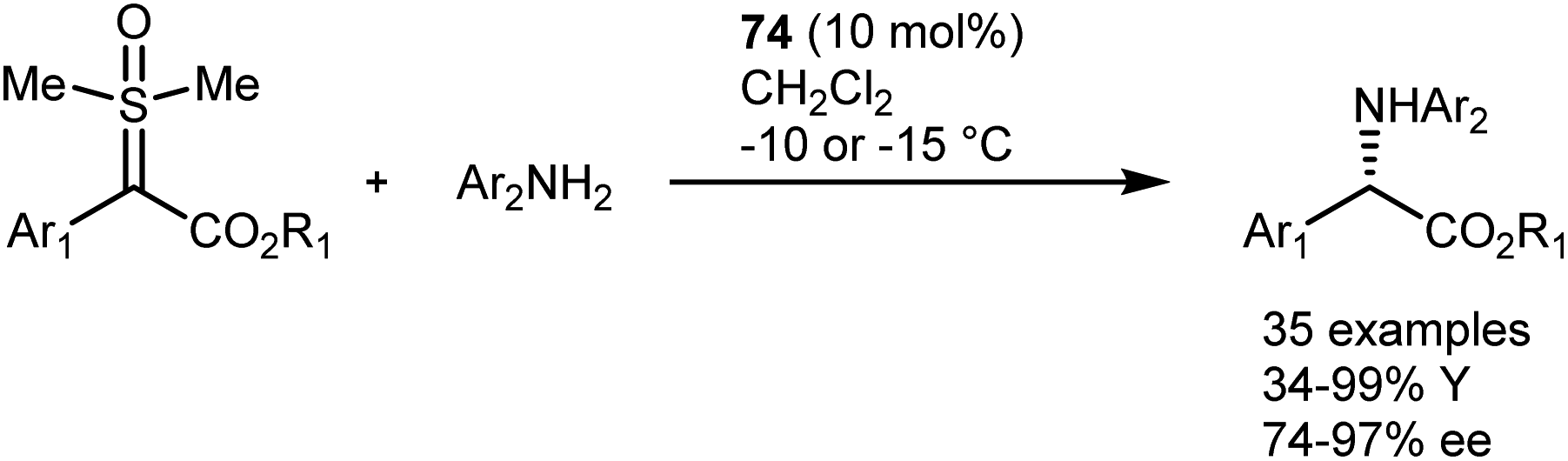 | ||
| Scheme 71 | ||
The mechanism of this purely organocatalytic reaction was investigated by 31P NMR to follow the catalytic species and by 19F NMR using anilines carrying fluorine substituents. These and other experiments pointed to the reaction pathway shown in Scheme 72, depicting that a rapid and reversible protonation is followed by the rate- and enantiodetermining nucleophilic displacement of DMSO by the aniline reaction partner, in an overall DKR process.
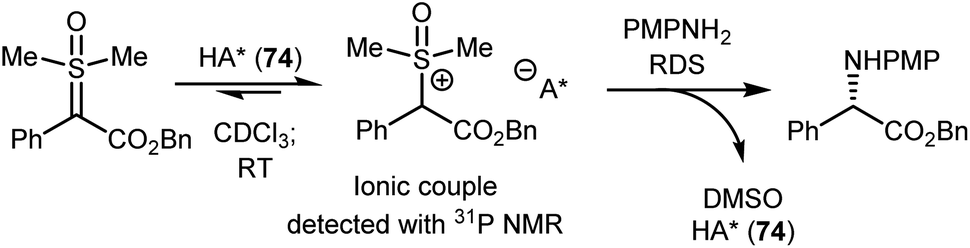 | ||
| Scheme 72 | ||
Although this methodology suffers from being limited to aniline derivatives, it presents several appealing aspects. For example, it employs only an organocatalyst, with no other activation sources, under mild conditions. Furthermore, sulfoxonium ylides, which present a more attractive safety profile compared to diazo compounds, are used as the carbenoid species. In fact, stabilized sulfoxonium ylides have been intensively studied in catalytic asymmetric settings in recent years.185 Another example related to AA synthesis was reported by Burtoloso (Scheme 73).186 This reaction recalls the previously discussed N–H insertion reactions of diazo compounds (see Schemes 66 and 67), rather than the CPA 74-catalyzed reactions: it employs the cooperative catalysis of a copper salt, which is capable of forming a copper carbene intermediate, and a squaramide organic catalyst (75) for the enantioselective proton-transfer step. The scope of the reaction encompasses aryl sulfoxonium ylides, primary anilines, and their N-methyl counterparts.
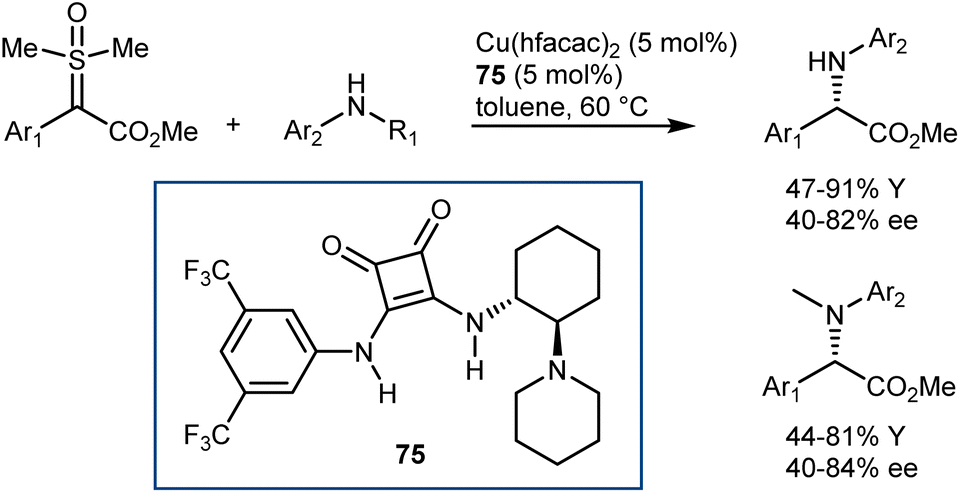 | ||
| Scheme 73 | ||
To conclude this section, we depart from the N–H insertion reactions by discussing a related MCR process with diazoacetates, sulfonamides, and imines, running under the cooperative catalytic action of Rh2(OAc)4 and spirocyclic CPA 76, reported by Zhang and Kang (Scheme 74).187 Their results were excellent, especially in terms of ee values and diastereomeric ratios. The removal of the tosyl protecting group was possible by treatment with triflic acid.
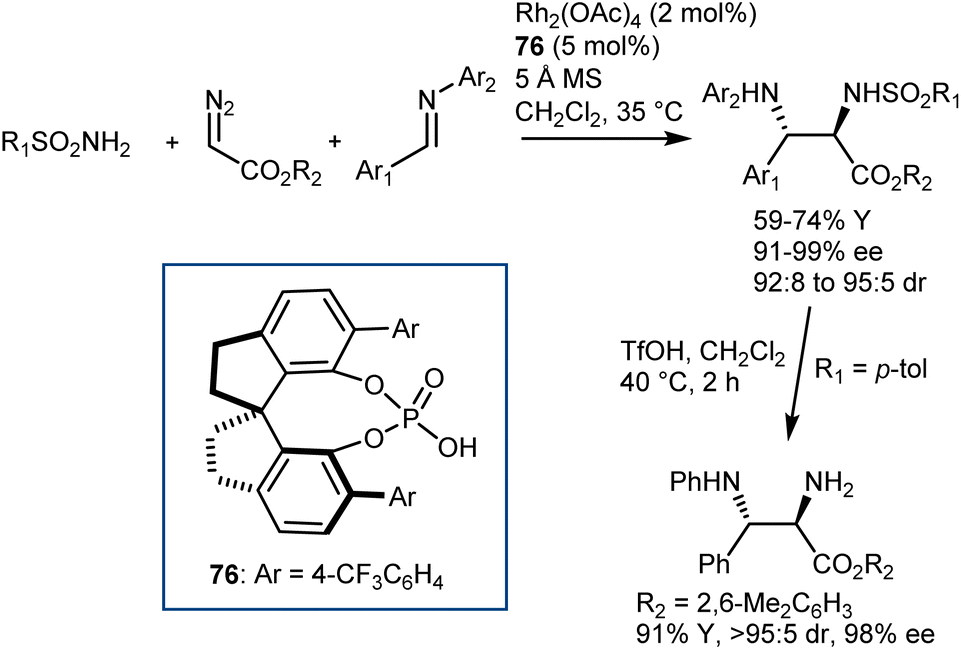 | ||
| Scheme 74 | ||
This reaction is part of the impressive series of MCRs with stabilized diazo compounds, electrophiles, and nucleophiles, under the cooperative catalysis of a metal and a CPA derivative, reported by Hu's laboratory.107 In this recent work, Zhang and Kang were able to use simple α-H diazoacetates instead of the α-substituted substrates typical of this reaction platform. In line with the previous MCRs, the pathway involves the interception of an enol intermediate, itself generated by the nucleophilic attack of the nucleophile (e.g., the sulphonamide) to a metal carbene (Scheme 75). The enol trapping by the imine occurs thanks to the metal/CPA cooperative catalysis. This and the N–H insertion pathway are intertwined. In the N–H insertion reaction (see also Scheme 65), the enol intermediate or an analogous zwitterionic species undergoes a proton-transfer step. In this MCR, the enol attacks the imine in a Mannich-type reaction, under the bifunctional coordination and activation of the CPA catalyst.
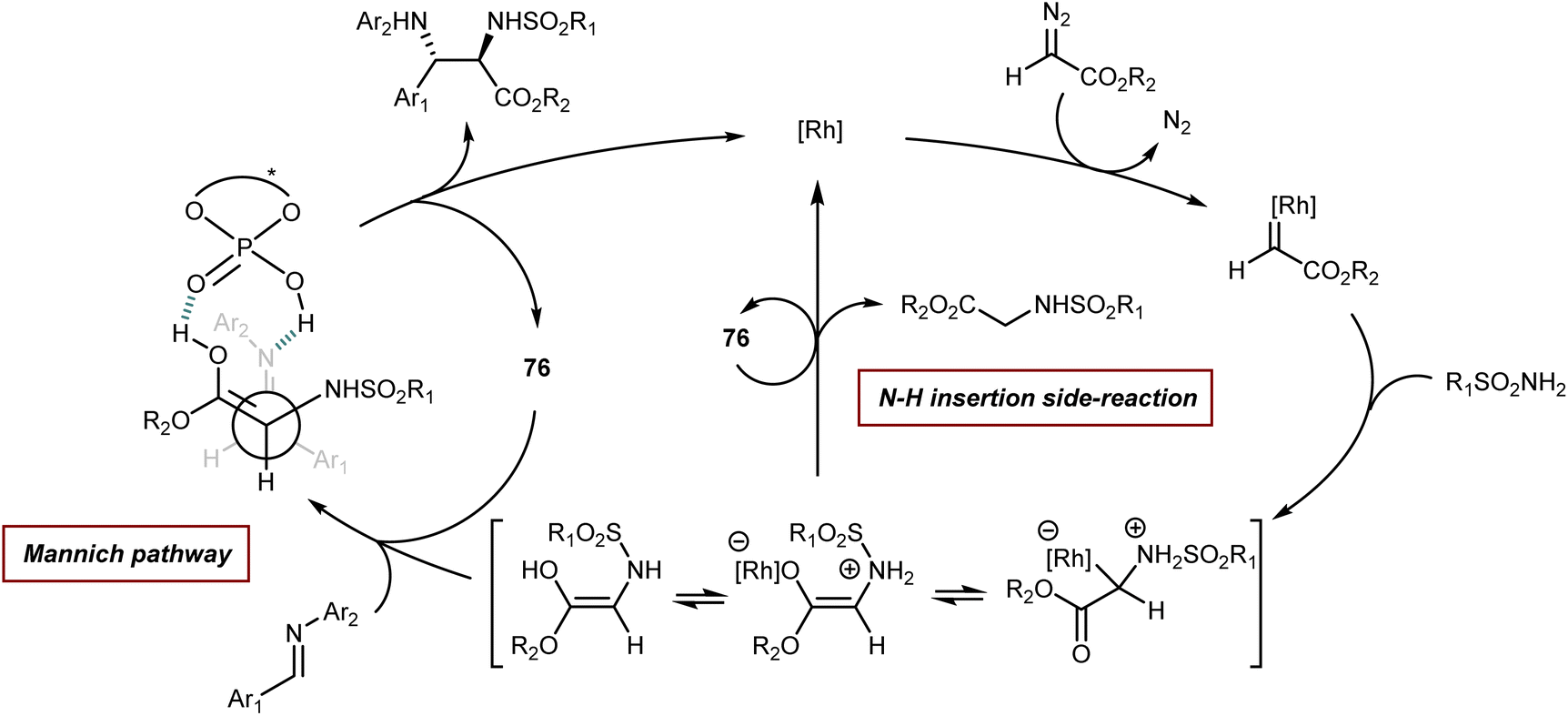 | ||
| Scheme 75 | ||
6. Conclusions
The roles played by enantiopure natural and unnatural AAs in different scientific fields cannot be overestimated. As an emerging and attractive tool to prepare enantioenriched compounds, organocatalysis has provided a number of pathways to AA derivatives in optically active form. Limiting the coverage to tertiary ncAAs, this perspective has discussed many of these reactions, providing a general overview of asymmetric organocatalysis. As outlined below, the information can be grouped into four main and closely intertwined themes. We expect that some of the next developments in asymmetric organocatalysis will follow these general themes.(i) Organocatalytic asymmetric versions of name and classical reactions, such as the Strecker synthesis, Mannich reaction, Ugi reaction, Michael addition, O'Donnell alkylation, and electrophilic amination, were disclosed several years ago—with the exception of the Ugi reaction—and have progressed dramatically over the years. Besides improved efficiency of the catalytic processes, the use of nitrogen protecting groups such as Boc boosts their synthetic appeal. In fact, in general terms, we expect that target-oriented applications of classic organocatalytic reactions will continue to grow, making this technology a solid platform for the synthesis of enantioenriched compounds at different scales, even in industrial settings.188
(ii) Biomimetic concepts explicitly appear in the asymmetric reduction as well as in the more recent disclosures on aldehyde catalysis and transamination reactions. On the other hand, organocatalysts and enzymes “speak the same language,” i.e., they use the same type of interactions with the substrates, thus accounting for the pervasiveness of biomimetic models in asymmetric organocatalysis.189 This relationship will likely last and evolve in different directions. Organocatalysis has been recently used in reaction networks approaching the complexity of living systems.190 Some (modified) enzymes have an “organocatalytic promiscuity,”191 bridging the gap between the worlds of small-molecule catalysis and biocatalysis.
(iii) Diastereodivergency, which has arisen from the use of different catalysts and reaction conditions, or from (sequential) multi-catalytic reactions, has unfolded the full potential of some asymmetric transformations, giving access to all product stereoisomers. Examples will continue to emerge.
(iv) Organocatalysts have provided tremendous opportunities when combined with metal catalysts, as shown in the formal insertion reactions of carbenoid species in N–H bonds as well as with photochemistry (deracemization). In these reactions, the efficiency of organocatalysts in exerting stereocontrol in a highly challenging reaction, such as asymmetric protonation, exemplifies the level of sophistication reached by this technology. In addition, we expect that these synergies will continue to grow, thus providing novel reaction pathways enabling unprecedented synthetic transformations.192
Finally, we note the evolution of organocatalysts from simple catalysts (L-proline, Cinchona alkaloids, etc.), which can be used without any precaution to exclude moisture or air, in untreated solvents to complex structures requiring nontrivial, multi-step syntheses to be used under rigorous conditions and with nonstandard set-up procedures. In this respect, the original claim of an exceedingly user-friendly technology has been in part lost. Despite this downside, we expect that the future of asymmetric organocatalysis will provide exciting and unforeseen synthetic opportunities.193
Author contributions
The manuscript was written with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Italian Ministry for University and Research (MUR, PRIN 2020, 2020AEX4TA project), the University of Bologna (RFO program), and FIS (Fabbrica Italiana Sintetici – S.p.A.).Notes and references
- G. Wu, Amino Acids, 2009, 37, 1 CrossRef CAS PubMed.
- (a) D. A. Evans , G. Helmchen and M. Reuping, Asymmetric Synthesis: The Essentials, 2007, pp. 3–9 Search PubMed; (b) Y. Gnas and F. Glorius, Synthesis, 2006, 12, 1899 Search PubMed.
- (a) E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed. Engl., 1998, 37, 1986 CrossRef CAS; (b) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CrossRef CAS PubMed; (c) K. Sakthivel, W. Notz, T. Bui and C. F. Barbas, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS PubMed; (d) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed; (e) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed; (f) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (g) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed.
- Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833 CrossRef CAS PubMed.
- (a) A. Pinazo, R. Pons, L. Pérez and M. R. Infante, Ind. Eng. Chem. Res., 2011, 50, 4805 CrossRef CAS; (b) M. J. Rowland, E. A. Appel, R. J. Coulston and O. A. Scherman, J. Mater. Chem. B, 2013, 1, 2904 RSC.
- (a) M. T. Reetz, Angew. Chem. Int. Ed. Engl., 1991, 30, 1531 CrossRef; (b) P. Singh, K. Samanta, S. K. Das and G. Panda, Org. Biomol. Chem., 2014, 12, 6297 RSC.
- Although peptide-based therapeutics comprise a small portion of the global pharmaceutical market, their average rate as newly approved “lead compounds” has steadily increased over the last two decades. (a) K. Fosgerau and T. Hoffmann, Drug Discov. Today, 2015, 20, 122 CrossRef CAS; (b) J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700 CrossRef CAS PubMed; (c) M. Muttenthaler, G. E. King, D. J. Adams and P. E. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309 CrossRef CAS PubMed.
- Y. A. Haggag, Biomed. J. Sci. Tech. Res., 2018, 8, 6659 Search PubMed.
- (a) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS PubMed; (b) A. C.-L. Lee, J. L. Harris, K. K. Khanna and J.-H. Hong, Int. J. Mol. Sci., 2019, 20, 2383 CrossRef.
- (a) C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098 CrossRef CAS; (b) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764 CrossRef CAS; (c) C. D. Spicer and B. G. Davies, Nat. Commun., 2014, 5, 4740 CrossRef CAS; (d) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174 CrossRef CAS PubMed; (e) M. B. E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem, 2019, 3, 147 CrossRef.
- (a) M. J. O'Donnell, C. Zhou and W. L. Scott, J. Am. Chem. Soc., 1996, 118, 6070 CrossRef; (b) A. Abdildinova, M. J. Kurth and Y.-D. Gong, Asian J. Org. Chem., 2021, 10, 2300 CrossRef CAS.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980 CrossRef CAS PubMed.
- (a) P. M. England, Biochemistry, 2004, 43, 11623 CrossRef CAS PubMed ; ; (b) C. H. Kim, J. Y. Axup and P. G. Schultz, Curr. Opin. Chem. Biol., 2013, 412 CrossRef CAS PubMed; (c) W. Brown, J. Liu and A. Deiters, ACS Chem. Biol., 2018, 2375 CrossRef CAS PubMed; (d) E. N. Mirts, A. Bhagi-Damodaran and Y. Lu, Acc. Chem. Res., 2019, 935 CrossRef CAS; (e) T. J. Hallam, E. Wold, A. Wahl and V. V. Smider, Mol. Pharmaceut., 2015, 1848 CrossRef CAS PubMed; (f) Y. Yu, C. Hu, L. Xia and J. Wang, ACS Catal., 2018, 8, 1851 CrossRef CAS.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed; (b) N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952 CrossRef CAS PubMed.
- (a) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807 CrossRef CAS PubMed; (b) Y. Ding, J. P. Ting, J. Liu, S. Al-Azzam, P. Pandya and S. Afshar, Amino Acids, 2020, 52, 1207 CrossRef CAS PubMed.
- A. S. Bommarius, M. Schwarm and K. Drauz, J. Mol. Catal. B Enzym., 1998, 5, 1 CrossRef CAS.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654 CrossRef PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586 CrossRef CAS PubMed.
- A. J. Fowell and K. L. Nash, Adv. Ther., 2010, 27, 512 CrossRef CAS PubMed.
- M. L. Merz, S. Habeshian, B. Li, J.-A. G. L. David, A. L. Nielsen, X. Ji, K. I. Khwildy, M. M. D. Benitez, P. Phothirath and C. Heinis, Nat. Chem. Biol., 2023 DOI:10.1038/s41589-023-01496-y.
- (a) A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) L. Pollegioni and S. Servi, Unnatural Amino Acids, Springer, New York, 2012 CrossRef; (c) T. W. Muir and J. N. Abelson, Non-Natural Amino Acids, Elsevier: San Diego, CA, 2009 Search PubMed.
- (a) V. A. Soloshonok and K. Izawa, Asymmetric Synthesis and Application of α-Amino Acids, American Chemical Society, Washington DC, ACS Symposium Series, 2009 Search PubMed; (b) A. S. Saghyan and P. Langer, Asymmetric Synthesis of Non-Proteinogenic Amino Acids, Wiley-VCH, Weinheim, 2016 CrossRef; (c) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef.
- For some references, see: (a) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (b) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed; (c) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed; (d) R.-Y. Zhu, K. Tanaka, G.-C. Li, J. He, H.-Y. Fu, S.-H. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 7067 CrossRef CAS PubMed; (e) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS PubMed; (f) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700 CrossRef CAS; (g) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506 CrossRef CAS PubMed; (h) F. Yuan, Z.-L. Hou, P. K. Pramanick and B. Yao, Org. Lett., 2019, 21, 9381 CrossRef CAS; (i) A. Correa, Eur. J. Inorg. Chem., 2021, 2928 CrossRef CAS; (j) T. A. King, J. M. Kandemir, S. J. Walsh and D. R. Spring, Chem. Soc. Rev., 2021, 50, 39 RSC; (k) T. Brandhofer and O. G. Mancheño, Eur. J. Org Chem., 2018, 6050 CrossRef CAS.
- (a) P. J. Almhjell, C. E. Boville and F. H. Arnold, Chem. Soc. Rev., 2018, 47, 8980 RSC; (b) A. D. Pagar, M. D. Patil, D. T. Flood, T. H. Yoo, P. E. Dawson and H. Yun, Chem. Rev., 2021, 121, 6173 CrossRef CAS; (c) H. D. Biava, ChemBioChem, 2020, 21, 1265 CrossRef CAS PubMed; (d) Y.-P. Xue, C.-H. Cao and Y.-G. Zheng, Chem. Soc. Rev., 2018, 47, 1516 RSC.
- For other, less investigated, synthetic equivalents, see: (a) K. S. Yang and V. H. Rawal, J. Am. Chem. Soc., 2014, 136, 16148 CrossRef CAS PubMed; (b) R. A. Kovalevsky, A. S. Kucherenko and S. G. Zlotin, Chem. Commun., 2022, 58, 12827 RSC.
- K. Tanaka, A. Mori and S. Inoue, J. Org. Chem., 1990, 55, 181 CrossRef CAS.
- M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, J. Am. Chem. Soc., 1996, 118, 4910 CrossRef CAS . Retraction: M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, J. Am. Chem. Soc., 2023, 145, 15016..
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901 CrossRef CAS.
- E. J. Corey and M. J. Grogan, Org. Lett., 1999, 1, 157 CrossRef CAS PubMed.
- S. J. Zuend, M. P. Coughlin, M. P. Lalonde and E. N. Jacobsen, Nature, 2009, 461, 968 CrossRef CAS PubMed.
- S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 15358 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210 CrossRef CAS PubMed.
- R. P. Herrera, V. Sgarzani, L. Bernardi, F. Fini, D. Pettersen and A. Ricci, J. Org. Chem., 2006, 71, 9869 CrossRef CAS.
- H. Yan, J. S. Oh, J.-W. Lee and C. E. Song, Nat. Commun., 2012, 3, 1212 CrossRef PubMed.
- A. M. Nauth and T. Opatz, Org. Biomol. Chem., 2019, 17, 11 RSC.
- S. C. Pan, J. Zhou and B. List, Angew. Chem., Int. Ed., 2007, 46, 612 CrossRef CAS.
- S. C. Pan and B. List, Org. Lett., 2007, 9, 1149 CrossRef CAS PubMed.
- S. Saravanan, N.-u. H. Khan, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, ACS Catal., 2013, 3, 2873 CrossRef CAS.
- A. Sadhukhan, S. Saravanan, N.-u. H. Khan, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, J. Org. Chem., 2012, 77, 7076 CrossRef CAS PubMed.
- (a) P. Merino, E. Marqués-López, T. Tejero and R. P. Herrera, Tetrahedron, 2009, 65, 1219 CrossRef CAS; (b) N. Kurono and T. Ohkuma, ACS Catal., 2016, 6, 989 CrossRef CAS; (c) B. Ullah, N. K. Gupta, Q. Ke, N. Ullah, X. Cai and D. Liu, Catalysts, 2022, 12, 1149 CrossRef CAS.
- (a) T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625 CrossRef CAS PubMed; (b) B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418 CrossRef CAS PubMed; (c) T. P. Yoon and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 466 CrossRef CAS; (d) L. Bernardi, F. Fini, R. P. Herrera, A. Ricci and V. Sgarzani, Tetrahedron, 2006, 62, 375 CrossRef CAS; (e) X. Xu, T. Furukawa, T. Okino, H. Miyabe and Y. Takemoto, Chem.–Eur. J., 2006, 12, 466 CrossRef PubMed.
- (a) F. Fini, V. Sgarzani, D. Pettersen, R. P. Herrera, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 7975 CrossRef CAS PubMed; (b) C. Palomo, M. Oiarbide, A. Laso and R. López, J. Am. Chem. Soc., 2005, 127, 17622 CrossRef CAS PubMed.
- (a) K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nuñez, A. M. Goldys and D. J. Dixon, Org. Lett., 2012, 14, 2492 CrossRef CAS PubMed; (b) Y. Wei, W. He, Y. Liu, P. Liu and S. Zhang, Org. Lett., 2012, 14, 704 CrossRef CAS PubMed.
- E. Gomez-Bengoa, A. Linden, R. López, I. Múgica-Mendiola, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2008, 130, 7955 CrossRef CAS PubMed.
- (a) E. d. F. Martins and J. R. Pliego, ACS Catal., 2013, 3, 613 CrossRef; (b) C. Yang, W. Zhang, Y.-H. Li, X.-S. Xue, X. Li and J.-P. Cheng, J. Org. Chem., 2017, 82, 9321 CrossRef CAS; (c) F. Buttard and P. A. Champagne, ACS Catal., 2022, 12, 8185 CrossRef CAS; (d) C. Q. He, A. Simon, Y.-h. Lam, A. P. J. Brunskill, N. Yasuda, J. Tan, A. M. Hyde, E. C. Sherer and K. N. Houk, J. Org. Chem., 2017, 82, 8645 CrossRef CAS PubMed.
- C. Palomo, M. Oiarbide, R. Halder, A. Laso and R. López, Angew. Chem., Int. Ed., 2006, 45, 117 CrossRef CAS PubMed.
- E. Foresti, G. Palmieri, M. Petrini and R. Profeta, Org. Biomol. Chem., 2003, 1, 4275 RSC.
- J. Li, M. J. Lear, Y. Kawamoto, S. Umemiya, A. R. Wong, E. Kwon, I. Sato and Y. Hayashi, Angew. Chem., Int. Ed., 2015, 54, 12986 CrossRef CAS PubMed.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027 CrossRef CAS PubMed.
- K. E. Schwieter and J. N. Johnston, Chem. Sci., 2015, 6, 2590 RSC.
- K. E. Schwieter and J. N. Johnston, ACS Catal., 2015, 5, 6559 CrossRef CAS PubMed.
- For reviews, see: (a) E. Marqués-López, P. Merino, T. Tejero and R. P. Herrera, Eur. J. Org Chem., 2009, 2401 CrossRef; (b) A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887 CrossRef CAS PubMed.
- Enantioselective reduction of β-amino nitroalkenes with Hantzsch esters gives a conceptually distinct approach to the same compounds, see for example: (a) A. Ferraro, L. Bernardi and M. Fochi, Adv. Synth. Catal., 2016, 358, 1561 CrossRef CAS; (b) Z. Deng, M. A. Padalino, J. E. L. Jan, S. Park, M. W. Danneman and J. N. Johnston, J. Am. Chem. Soc., 2024, 146, 1269 CrossRef CAS PubMed.
- T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 6717 CrossRef CAS PubMed.
- (a) Y. Zhang, Y.-F. Ao, Z.-T. Huang, D.-X. Wang, M.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 5282 CrossRef CAS; (b) T. Hashimoto, H. Kimura, Y. Kawamata and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 7279 CrossRef CAS; (c) W. Zhao, L. Huang, Y. Guan and W. D. Wulff, Angew. Chem., Int. Ed., 2014, 53, 3436 CrossRef CAS PubMed.
- J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, 1087 CrossRef CAS.
- B.-B. Sun, K. Liu, Q. Gao, W. Fang, S. Lu, C.-R. Wang, C.-Z. Yao, H.-Q. Cao and J. Yu, Nat. Commun., 2022, 13, 7065 CrossRef CAS PubMed.
- For other glycine α-anion equivalents in organocatalysis, see: (a) L. Li, E. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2008, 130, 12248 CrossRef CAS PubMed; (b) R. Thayumanavan, F. Tanaka and C. F. Barbas, Org. Lett., 2004, 6, 3541 CrossRef CAS PubMed; (c) T. Kano, R. Sakamoto, M. Akakura and K. Maruoka, J. Am. Chem. Soc., 2012, 134, 7516 CrossRef CAS PubMed.
- M. J. O'Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353 CrossRef.
- M. J. O'Donnell, S. Wu and J. C. Huffman, Tetrahedron, 1994, 50, 4507 CrossRef.
- E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414 CrossRef CAS.
- B. Lygo and P. G. Wainwright, Tetrahedron Lett., 1997, 38, 8595 CrossRef CAS.
- B. Hu, M. W. Bezpalko, C. Fei, D. A. Dickie, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2018, 140, 13913 CrossRef CAS.
- (a) M. J. O'Donnell, Tetrahedron, 2019, 75, 3667 CrossRef; (b) T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222 CrossRef CAS PubMed; (c) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed; (d) S.-s. Jew and H.-g. Park, Chem. Commun., 2009, 7090 RSC; (e) M. Waser, M. Winter and C. Mairhofer, Chem. Rec., 2023, 23, e202200198 CrossRef CAS PubMed; (f) D. C. M. Albanese and M. Penso, Eur. J. Org Chem., 2023, 26, e202300224 CrossRef CAS.
- (a) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 6519 CrossRef CAS; (b) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 5139 CrossRef CAS PubMed; (c) M. Kitamura, S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2005, 44, 1549 CrossRef CAS.
- M. Mąkosza and M. Fedoryński, Catalysts, 2020, 10, 1436 CrossRef.
- (a) D. E. Patterson, S. Xie, L. A. Jones, M. H. Osterhout, C. G. Henry and T. D. Roper, Org. Process Res. Dev., 2007, 11, 624 CrossRef CAS; (b) D. E. Patterson, S. Xie, L. A. Jones, M. H. Osterhout, C. G. Henry and T. D. Roper, Large-Scale Application of Asymmetric Phase-Transfer Catalysis for Amino Acid Synthesis, in Asymmetric Catalysis on Industrial Scale, ed. H.-U. Blaser and H.-J. Federsel, Wiley-VCH, Weinheim, 2nd edn, 2010, ch. 26, p. 473 Search PubMed.
- S. Murayama, Z. Li, H. Liang, Y. Liu, H. Naka and K. Maruoka, Chem.–Eur. J., 2023, 29, e202301866 CrossRef CAS PubMed.
- (a) J. Tan and N. Yasuda, Org. Process Res. Dev., 2015, 19, 1731 CrossRef CAS; (b) D. L. Hughes, Org. Process Res. Dev., 2022, 26, 2224 CrossRef CAS.
- M. J. O'Donnell, W. D. Bennett, W. A. Bruder, W. N. Jacobsen, K. Knuth, B. LeClef, R. L. Polt, F. G. Bordwell, S. R. Mrozack and T. A. Cripe, J. Am. Chem. Soc., 1988, 110, 8520 CrossRef.
- (a) M. Ikunaka and K. Maruoka, Asymmetric Phase-Transfer Catalysis for the Production of Non-Proteinogenic α-Amino Acids In Asymmetric Catalysis on Industrial Scale, ed. H.-U. Blaser and H.-J. Federsel, Wiley-VCH, Weinheim, 2nd edn, 2010, ch. 9, p. 151. See also: Search PubMed; (b) J. Lu, L. Huang, H. Liang, Z. Wang, T. Kato, Y. Liu and K. Maruoka, Org. Lett., 2024 DOI:10.1021/acs.orglett.3c04290.
- M. Majdecki, P. Niedbala and J. Jurczak, Org. Lett., 2019, 21, 8085 CrossRef CAS PubMed.
- C. Xu, Y. Qi, X. Yang, X. Li, Z. Li and L. Bai, Org. Lett., 2021, 23, 2890 CrossRef CAS PubMed.
- S. Wen, X. Li and Y. Lu, Asian J. Org. Chem., 2016, 5, 1457 CrossRef CAS.
- R. Schettini, F. De Riccardis, G. Della Sala and I. Izzo, J. Org. Chem., 2016, 81, 2494 CrossRef CAS PubMed.
- A. Homberg, R. Hrdina, M. Vishe, L. Guénée and J. Lacour, Org. Biomol. Chem., 2019, 17, 6905 RSC.
- M. Kondo, K. Nakamura, C. G. Krishnan, S. Takizawa, T. Abe and H. Sasai, ACS Catal., 2021, 11, 1863 CrossRef CAS.
- See for example: (a) E. J. Corey, M. C. Noe and F. Xu, Tetrahedron Lett., 1998, 39, 5347 CrossRef CAS; (b) G. N. Gururaja, R. Herchl, A. Pichler, K. Gratzer and M. Waser, Molecules, 2013, 18, 4357 CrossRef CAS PubMed; (c) S. Shirakawa, Y. Liu, A. Usui and K. Maruoka, ChemCatChem, 2012, 4, 980 CrossRef CAS; (d) L. Bernardi, M. Fochi, R. Carbone, A. Martinelli, M. E. Fox, C. J. Cobley, B. Kandagatla, S. Oruganti, V. H. Dahanukar and A. Carlone, Chem.–Eur. J., 2015, 21, 19208 CrossRef CAS PubMed; (e) J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2012, 134, 5552 CrossRef CAS PubMed.
- S. E. Franz and J. D. Stewart, Adv. Appl. Microbiol., 2014, 88, 57 Search PubMed.
- H. Kuzuhara, N. Watanabe and M. Ando, J. Chem. Soc., Chem. Commun., 1987, 95 10.1039/C39870000095.
- J. T. Koh, L. Delaude and R. Breslow, J. Am. Chem. Soc., 1994, 116, 11234 CrossRef CAS.
- Y. Murakami, J.-i. Kicuchi, T. Miyajima and Y. Hisaeda, Chem. Lett., 1994, 23, 55 CrossRef.
- (a) W. Wen and Q.-X. Guo, Synthesis, 2023, 55, 719 CrossRef CAS; (b) W. Wen and Q.-X. Guo, Acc. Chem. Res., 2024, 57, 776 CrossRef CAS PubMed.
- X. Xiao and B. Zhao, Acc. Chem. Res., 2023, 56, 1097 CrossRef CAS PubMed.
- B. Xu, L.-L. Shi, Y.-Z. Zhang, Z.-J. Wu, L.-N. Fu, C.-Q. Luo, L.-X. Zhang, Y.-G. Peng and Q.-X. Guo, Chem. Sci., 2014, 5, 1988 RSC.
- W.-Z. Wang, H.-R. Shen, J. Liao, W. Wen and Q.-X. Guo, Org. Chem. Front., 2022, 9, 1422 RSC.
- J. Chen, X. Gong, J. Li, Y. Li, J. Ma, C. Hou, G. Zhao, W. Yuan and B. Zhao, Science, 2018, 360, 1438 CrossRef CAS PubMed.
- See for example: (a) W. Wen, L. Chen, M.-J. Luo, Y. Zhang, Y.-C. Chen, Q. Ouyang and Q.-X. Guo, J. Am. Chem. Soc., 2018, 140, 9774 CrossRef CAS PubMed; (b) C. Hou, B. Peng, S. Ye, Z. Yin, J. Cao, X. Xiao and B. Zhao, Nat. Catal., 2022, 5, 1061 CrossRef CAS; (c) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159 CrossRef CAS PubMed; (d) P. Ji, J. Li, Y. Tao, M. Li, W. Ling, J. Chen and B. Zhao, ACS Catal., 2023, 13, 9150 CrossRef CAS , and references cited therein..
- (a) E. Hagiwara, A. Fujii and M. Sodeoka, J. Am. Chem. Soc., 1998, 120, 2474 CrossRef CAS; (b) D. Ferraris, B. Young, T. Dudding and T. Lectka, J. Am. Chem. Soc., 1998, 120, 4548 CrossRef CAS; (c) W. J. Drury III, D. Ferraris, C. Cox, B. Young and T. Lectka, J. Am. Chem. Soc., 1998, 120, 11006 CrossRef; (d) S. Yao, X. Fang and K. A. Jørgensen, Chem. Commun., 1998, 2547 RSC; (e) A. E. Taggi, A. M. Hafez and T. Lectka, Acc. Chem. Res., 2003, 36, 10 CrossRef CAS PubMed.
- B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2017, 117, 8326 CrossRef CAS PubMed.
- For some reviews on organocatalytic Mannich reactions: (a) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC; (b) A. Ting and S. E. Schaus, Eur. J. Org Chem., 2007, 5797 CrossRef CAS; (c) Y. Hayashi, J. Synth. Org. Chem. Jpn., 2014, 72, 1228 CrossRef CAS; (d) I. Bagheri, L. Mohammadi, V. Zadsirjan and M. M. Heravi, ChemistrySelect, 2021, 6, 1008 CrossRef CAS.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS.
- (a) A. Córdova, W. Notz, G. Zhong, J. M. Betancort and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1842 CrossRef PubMed; (b) A. Córdova, S.-i. Watanabe, F. Tanaka, W. Notz and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1866 CrossRef PubMed.
- R. Martín-Rapún, X. Fan, S. Sayalero, M. Bahramnejad, F. Cuevas and M. A. Pericás, Chem.–Eur. J., 2011, 17, 8780 CrossRef PubMed.
- J. Franzén, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 18296 CrossRef PubMed.
- R. C. Simon, E. Busto, J. H. Schrittwieser, J. H. Sattler, J. Pietruszka, K. Faber and W. Kroutil, Chem. Commun., 2014, 50, 15669 RSC.
- S. K. Ghosh, B. Somanadhan, K. S.-W. Tan, M. S. Butler and M. J. Lear, Org. Lett., 2012, 14, 1560 CrossRef CAS PubMed.
- (a) B. T. Hahn, R. Fröhlich, K. Harms and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 9985 CrossRef CAS PubMed; (b) S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakayama and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187 CrossRef CAS PubMed; (c) Y. Zhang, J.-K. Li, F.-G. Zhang and J.-A. Ma, J. Org. Chem., 2020, 85, 5580 CrossRef CAS PubMed.
- Y. Nakamura, R. Matsubara, H. Kiyohara and S. Kobayashi, Org. Lett., 2003, 5, 2481 CrossRef CAS PubMed.
- For comprehensive references, see: S. P. Roche, Synthesis, 2021, 53, 2767 CrossRef CAS.
- C. Gianelli, L. Sambri, A. Carlone, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2008, 47, 8700 CrossRef CAS PubMed.
- Y. You, L. Zhang, L. Cui, X. Mi and S. Luo, Angew. Chem., Int. Ed., 2017, 56, 13814 CrossRef CAS PubMed.
- M. Wasa, R. Y. Liu, S. P. Roche and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 12872 CrossRef CAS PubMed.
- H. Xu, A. Nazli, C. Zou, Z.-P. Wang and Y. He, Chem. Commun., 2020, 56, 14243 RSC.
- (a) S. Saaby, X. Fang, N. Gathergood and K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 4114 CrossRef CAS; (b) D. Enders, M. Seppelt and T. Beck, Adv. Synth. Catal., 2010, 352, 1413 CrossRef CAS.
- S. Brunen, B. Mitschke, M. Leutzsch and B. List, J. Am. Chem. Soc., 2023, 145, 15708 CrossRef CAS PubMed.
- (a) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed; (b) D. Zhang and W. Hu, Chem. Rec., 2017, 17, 739 CrossRef CAS PubMed.
- (a) Y. Qian, C. Jing, S. Liua and W. Hu, Chem. Commun., 2013, 49, 2700 RSC; (b) C. Jing, D. Xing and W. Hu, Org. Lett., 2015, 17, 4336 CrossRef CAS PubMed; (c) S.-K. Jia, Y.-B. Lei, L.-L. Song, S.-Y. Liu and W.-H. Hu, Chin. Chem. Lett., 2017, 28, 213 CrossRef CAS.
- A. V. Malkov, S. Stoncius, K. N. MacDougall, A. Mariani, G. D. McGeoch and P. Kocovsky, Tetrahedron, 2006, 62, 264 CrossRef CAS.
- (a) J. W. Yang, M. T. Echavarria Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660 CrossRef PubMed; (b) J. W. Yang, M. T. Echavarria Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2005, 44, 108 CrossRef CAS PubMed; (c) S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32 CrossRef CAS PubMed; (d) M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781 CrossRef CAS PubMed.
- T. J. Stillman, P. J. Baker, K. L. Britton and D. W. Rice, J. Mol. Biol., 1993, 234, 1131 CrossRef CAS PubMed.
- L. Guilong, L. Yuxue and J. C. Antilla, J. Am. Chem. Soc., 2007, 129, 5830 CrossRef PubMed.
- Q. Kang, Z.-A. Zhao and S.-L. You, Adv. Synth. Catal., 2007, 349, 1657 CrossRef CAS . For an elegant extension of this catalytic system using β,γ-alkynyl-α-imino esters as a substrate to afford enantioenriched trans-alkenyl α-amino esters where both alkyne and imine functionalities are reduced, see: Q. Kang, Z.-A. Zhao and S.-L. You, Org. Lett., 2008, 10, 2031..
- Y. Qian, C. Jing, C. Zhai and W.-h. Hu, Adv. Synth. Catal., 2012, 354, 301 CrossRef CAS.
- J. Mazuela, T. Antonsson, M. J. Johansson, L. Knerr and S. P. Marsden, Org. Lett., 2017, 19, 5541 CrossRef CAS PubMed.
- V. N. Wakchaure, P. S. J. Kaib, M. Leutzsch and B. List, Angew. Chem., Int. Ed., 2015, 54, 11852 CrossRef CAS PubMed.
- (a) C. Zhu and T. Akiyama, Adv. Synth. Catal., 2010, 35, 1846 CrossRef; (b) T. Sakamoto, K. Mori and T. Akiyama, Org. Lett., 2012, 14, 3312 CrossRef CAS PubMed.
- (a) T. Li, Q. Zhou, F. Meng, W. Cui, Q. Li, J. Zhu and Y. Cao, Eur. J. Org Chem., 2023, 26, e202300507 CrossRef CAS; (b) A. M. Faisca Phillips and A. J. L. Pombeiro, Catalysts, 2023, 13, 419 CrossRef CAS; (c) S. Das, V. N. Wakchaure and B. List, in Asymmetric Hydrogenation and Transfer Hydrogenation, ed. V. Ratovelomanana-Vidal and P. Phansavath, Wiley-VCH, 2021, ch. 11, p. 339 Search PubMed.
- M. D. Toney and J. F. Kirsch, Biochemistry, 1993, 32, 1471 CrossRef CAS PubMed.
- A. Hjelmencrantz and U. Berg, J. Org. Chem., 2002, 67, 3585 CrossRef CAS PubMed.
- X. Xiao, Y. Xie, C. Su, M. Liu and Y. Shi, J. Am. Chem. Soc., 2011, 133, 12914 CrossRef CAS PubMed.
- (a) X. Xiao, M. Liu, C. Rong, F. Xue, S. Li, Y. Xie and Y. Shi, Org. Lett., 2012, 14, 5270 CrossRef CAS PubMed; (b) C. Su, Y. Xie, H. Pan, M. Liu, H. Tian and Y. Shi, Org. Biomol. Chem., 2014, 12, 5856 RSC.
- Q. Peng, B. Yan, F. Li, M. Lang, B. Zhang, D. Guo, D. Bierer and J. Wang, Commun. Chem., 2021, 4, 148 CrossRef CAS PubMed.
- Q.-K. Kang, S. Selvakumar and K. Maruoka, Org. Lett., 2019, 21, 2294 CrossRef CAS PubMed.
- Y. E. Liu, Z. Lu, B. Li, J. Tian, F. Liu, J. Zhao, C. Hou, Y. Li, L. Niu and B. Zhao, J. Am. Chem. Soc., 2016, 138, 10730 CrossRef CAS PubMed.
- R. Breslow, Acc. Chem. Res., 1995, 28, 146 CrossRef CAS.
- W. Cai, X. Qiao, H. Zhang, B. Li, J. Guo, L. Zhang, W. W. Chen and B. Zhao, Nat. Commun., 2021, 12, 5174 CrossRef CAS PubMed.
- Y. Xie, H. Pan, M. Liu, X. Xiao and Y. Shi, Chem. Soc. Rev., 2015, 44, 1740 RSC.
- J. W. Bogart and A. A. Bowers, Org. Biomol. Chem., 2019, 17, 3653 RSC.
- D. Leow, S. Lin, S. Chittimalla, X. Fu and C.-H. Tan, Angew. Chem., Int. Ed., 2008, 47, 5641 CrossRef CAS PubMed.
- T. Jousseaume, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1410 CrossRef CAS PubMed.
- (a) S.-W. Duan, J. An, J.-R. Chen and W.-J. Xiao, Org. Lett., 2011, 13, 2290 CrossRef CAS PubMed; (b) J. Gao, J.-R. Chen, S.-W. Duan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Asian J. Org. Chem., 2014, 3, 530 CrossRef CAS.
- M. Retini, S. Bartolucci, F. Bartoccini and G. Piersanti, Chem.–Eur. J., 2022, 28, e202201994 CrossRef CAS PubMed.
- (a) M. Righi, F. Bartoccini, S. Lucarini and G. Piersanti, Tetrahedron, 2011, 67, 7923 CrossRef CAS; (b) F. Bartoccini, M. Mari, M. Retini, S. Bartolucci and G. Piersanti, J. Org. Chem., 2018, 83, 12275 CrossRef CAS PubMed; (c) M. Retini, S. Bartolucci, F. Bartoccini, M. Mari and G. Piersanti, J. Org. Chem., 2019, 84, 12221 CrossRef CAS PubMed.
- For early examples with enzymatic and non-enzymatic catalysts, see: (a) R.-L. Gu, I.-S. Lee and C. J. Sih, Tetrahedron Lett., 1992, 33, 1953 CrossRef CAS; (b) J. Z. Crich, R. Brieva, P. Marquart, R.-L. Gu, S. Flemming and C. J. Sih, J. Org. Chem., 1993, 58, 3252 CrossRef CAS; (c) R. Noyori, M. Tokunaga and M. Kitamura, Bull. Chem. Soc. Jpn., 1995, 68(36), 3252 Search PubMed; (d) N. J. Turner, J. R. Winterman, R. McCague, J. S. Parratt and S. J. C. Taylor, Tetrahedron Lett., 1995, 36, 1113 CrossRef CAS; (e) D. Seebach, G. Jaeschke, K. Gottwald, K. Matsuda, R. Formisano, D. A. Chaplin, M. Breuning and G. Bringmann, Tetrahedron, 1997, 53, 7539 CrossRef CAS; (f) K. Gottwald and D. Seebach, Tetrahedron, 1999, 55, 723 CrossRef CAS; (g) S. A. Brown, M. C. Parker and N. J. Turner, Tetrahedron: Asymmetry, 2000, 11, 1687 CrossRef CAS.
- P. P. de Castro, G. M. F. Batista, H. F. dos Santos and G. W. Amarante, ACS Omega, 2018, 3, 3507 CrossRef CAS PubMed.
- For reviews, see: (a) P. P. de Castro, A. G. Carpanez and G. W. Amarante, Chem.–Eur. J., 2016, 22, 10294 CrossRef CAS PubMed; (b) I. F. S. Marra, P. P. de Castro and G. W. Amarante, Eur. J. Org Chem., 2019, 5830 CrossRef CAS.
- L. Xie, W. Hua, A. S. C. Chan and Y. C. Leung, Tetrahedron: Asymmetry, 1999, 10, 4715 CrossRef CAS.
- J. Liang, J. C. Ruble and G. C. Fu, J. Org. Chem., 1998, 63, 3154 CrossRef CAS.
- A. Berkessel, F. Cleemann, S. Mukherjee, T. N. Müller and J. Lex, Angew. Chem., Int. Ed., 2005, 44, 807 CrossRef CAS PubMed.
- A. Berkessel and K. Etzenbach-Effers, Computational Studies of Organocatalytic Processes Based on Hydrogen Bonding, in Hydrogen Bonding in Organic Synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009, ch. 3, pp. 15–42 Search PubMed.
- (a) A. Berkessel, S. Mukherjee, F. Cleemann, T. N. Müller and J. Lex, Chem. Commun., 2005, 1898 RSC; (b) A. Berkessel, S. Mukherjee, T. N. Mueller, F. Cleemann, K. Roland, M. Brandenburg, J.-M. Neudoerfl and J. Lex, Org. Biomol. Chem., 2006, 4, 4319 RSC.
- A. Peschiulli, C. Quigley, S. Tallon, Y. K. Gun'ko and S. J. Connon, J. Org. Chem., 2008, 73, 6409 CrossRef CAS PubMed.
- (a) Z. Rodríguez-Docampo, C. Quigley, S. Tallon and S. J. Connon, J. Org. Chem., 2012, 77, 2407 CrossRef PubMed; (b) C. Palacio and S. J. Connon, Eur. J. Org Chem., 2013, 5398 CrossRef CAS.
- (a) J.-W. Lee, T. H. Ryu, J.-S. Oh, H. Y. Bae, H. B. Jang and C. E. Song, Chem. Commun., 2009, 46, 7224 Search PubMed; (b) J.-S. Oh, J.-W. Lee, T. H. Ryu, J. H. Lee and C. E. Song, Org. Biomol. Chem., 2012, 10(5), 1052 RSC.
- V. Corti, R. Riccioli, A. Martinelli, S. Sandri, M. Fochi and L. Bernardi, Chem. Sci., 2021, 12, 10233 RSC.
- S. Tallon, F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2015, 54, 813 CrossRef CAS PubMed.
- (a) G. Lu and V. B. Birman, Org. Lett., 2011, 13, 356 CrossRef CAS PubMed; (b) S. Izumi, Y. Kobayashi and Y. Takemoto, Org. Lett., 2016, 18, 696 CrossRef CAS PubMed.
- (a) H. Mandai, K. Hongo, T. Fujiwara, K. Fujii, K. Mitsudo and S. Suga, Org. Lett., 2018, 20, 4811 CrossRef CAS PubMed; (b) M. S. Xie, B. Huang, N. Li, Y. Tian, X. X. Wu, Y. Deng, G. R. Qu and H. M. Guo, J. Am. Chem. Soc., 2020, 142, 19226 CrossRef CAS PubMed.
- A. J. Metrano and S. J. Miller, J. Org. Chem., 2014, 79, 1542 CrossRef CAS PubMed.
- K. Wakafuji, S. Iwasa, K. N. Ouchida, H. Cho, H. Dohi, E. Yamamoto, T. Kamachi and M. Tokunaga, ACS Catal., 2021, 11, 14067 CrossRef CAS and references therein..
- M. Huang, T. Pan, X. Jiang and S. Luo, J. Am. Chem. Soc., 2023, 145, 10917 CrossRef CAS PubMed.
- I. Chibata, T. Tosa and R. Sano, Appl. Microbiol., 1965, 13, 618 CrossRef CAS PubMed.
- F.-R. Alexandre, D. P. Pantaleone, P. P. Taylor, I. G. Fotheringham, D. J. Agerc and N. J. Turner, Tetrahedron Lett., 2002, 43, 707 CrossRef CAS.
- For a recent review, see: J. Großkopf and T. Bach, Angew. Chem., Int. Ed., 2023, 62, e202308241 CrossRef PubMed.
- Z. Gu, L. Zhang, H. Li, S. Cao, Y. Yin, X. Zhao, X. Ban and Z. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202211241 CrossRef CAS PubMed.
- (a) J. Großkopf, M. Plaza, A. Seitz, S. Breitenlechner, G. Storch and T. Bach, J. Am. Chem. Soc., 2021, 143, 21241 CrossRef PubMed; (b) R. J. Kutta, J. Großkopf, N. van Staalduinen, A. Seitz, P. Pracht, S. Breitenlechner, C. Bannwarth, P. Nuernberger and T. Bach, J. Am. Chem. Soc., 2023, 145, 2354 CrossRef CAS PubMed; (c) J. Großkopf, M. Plaza, R. J. Kutta, P. Nuernberger and T. Bach, Angew. Chem., Int. Ed., 2023, 62, e202313606 CrossRef PubMed.
- S. Bräse, H. Vogt and T. Baumann, Comprehensive Chirality, 2012, vol. 6, p. 374 Search PubMed.
- (a) A. Bøgevig, K. Juhl, N. Kumaragurubaran, W. Zhuang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 1790 CrossRef; (b) B. List, J. Am. Chem. Soc., 2002, 124, 5656 CrossRef CAS PubMed.
- B. B. Ahuja and A. Sudalai, Stud. Nat. Prod. Chem., 2020, 64, 418 Search PubMed.
- J. H. Lang, P. G. Jones and T. Lindel, Chem.–Eur. J., 2017, 23, 12714 CrossRef CAS PubMed.
- A. Shigenaga, J. Yamamoto, N. Nishioka and A. Otaka, Tetrahedron, 2010, 66, 7367 CrossRef CAS.
- B. Simmons, A. M. Walji and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 4349 CrossRef CAS PubMed.
- A. D. Huters, J. Stambuli, R. C. Klix, M. A. Matulenko, V. S. Chan, J. Simanis, D. R. Hill, R. E. Reddy, T. B. Towne, J. R. Bellettini, B. J. Kotecki, B. Cardinal-David, J. Ji, E. A. Voight, M. Shou, S. Balaraman, A. Ashok and S. Ghosh, J. Org. Chem., 2022, 87, 1986 CrossRef CAS PubMed.
- T. Kano, M. Ueda, J. Takai and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 6046 CrossRef CAS PubMed.
- C. Palomo, S. Vera, I. Velilla, A. Mielgo and E. Gómez-Bengoa, Angew. Chem., Int. Ed., 2007, 46, 8054 CrossRef CAS PubMed.
- B. Maji and H. Yamamoto, Bull. Chem. Soc. Jpn., 2015, 88, 753 CrossRef CAS.
- M. G. Memeo and P. Quadrelli, Chem. Rev., 2017, 117, 2108 CrossRef CAS PubMed.
- (a) T. Kano, F. Shirozu and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 18036 CrossRef CAS PubMed; (b) B. Maji and H. Yamamoto, Angew. Chem., Int. Ed., 2014, 53, 8714 CrossRef CAS PubMed; (c) T. Kano, F. Shirozu and K. Maruoka, Org. Lett., 2014, 16, 1530 CrossRef CAS PubMed.
- R. J. McGorry, S. K. Allen, M. D. Pitzen and T. C. Coombs, Tetrahedron Lett., 2017, 58, 4623 CrossRef CAS.
- H. Vogt, T. Baumann, M. Nieger and S. Bräse, Eur. J. Org Chem., 2006, 5315 CrossRef CAS.
- G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521 CrossRef CAS PubMed.
- C. F. Garcia, M. A. McKervey and T. Ye, Chem. Commun., 1996, 1465 RSC.
- S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044 RSC.
- B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834 CrossRef CAS PubMed.
- For a recent review, see: Y.-X. Su, M.-Y. Huang and S.-F. Zhu, ChemCatChem, 2023, 15, e202300539 CrossRef CAS.
- B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483 CrossRef CAS PubMed.
- B. Xu, S.-F. Zhu, X.-D. Zuo, Z.-C. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913 CrossRef CAS PubMed.
- M.-L. Li, J.-H. Han, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990 CrossRef CAS PubMed.
- M.-L. Li, J.-B. Pan and Q.-L. Zhou, Nat. Catal., 2022, 5, 571 CrossRef CAS.
- H. Saito, D. Morita, T. Uchiyama, M. Miyake and S. Miyairi, Tetrahedron Lett., 2012, 53, 6662 CrossRef CAS.
- J.-B. Pan, X.-G. Zhang, Y.-F. Shi, A.-C. Han, Y.-J. Chen, J. Ouyang, M.-L. Li and Q.-L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202300691 CrossRef CAS PubMed.
- W. Guo, Y. Zhou, H. Xie, X. Yue, F. Jiang, H. Huang, Z. Han and J. Sun, Chem. Sci., 2023, 14, 843 RSC.
- W. Guo, M. Wang, Z. Han, H. Huang and J. Sun, Chem. Sci., 2021, 12, 11191 RSC.
- C. A. D. Caiuby, L. G. Furniela and A. C. B. Burtoloso, Chem. Sci., 2022, 13, 1192 RSC.
- L. G. Furniel, R. Echemendìa and A. C. B. Burtoloso, Chem. Sci., 2021, 12, 7453 RSC.
- H. Sha, X. Xie, D. Zhang, W. Hu and Z. Kang, Adv. Synth. Catal., 2023, 365, 1 CrossRef.
- L. Bernardi, A. Carlone and F. Fini, in Methodologies in Amine Synthesis, Challenges and Applications, eds. A. Ricci and L. Bernardi, Wiley-VCH, 2021, ch. 6, p. 187 Search PubMed.
- L. Bernardi, M. Fochi, M. Comes Franchini and A. Ricci, Org. Biomol. Chem., 2012, 10, 2911 RSC.
- M. ter Harmsel, O. R. Maguire, S. A. Runikhina, A. S. Y. Wong, W. T. S. Huck and S. R. Harutyunyan, Nature, 2023, 621, 87 CrossRef CAS PubMed.
- H. Poddar, M. Rahimi, E. M. Geertsema, A.-M. W. H. Thunnissen and G. J. Poelarends, ChemBioChem, 2015, 16, 738 CrossRef CAS PubMed.
- M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed.
- (a) O. García Mancheño and M. Waser, Eur. J. Org Chem., 2023, 26, e202200950 CrossRef PubMed . For some recent advances in AAs that appeared during the writing of this perspective, see: ; (b) V. Battaglia, S. Meninno, A. Pellegrini, A. Mazzanti and A. Lattanzi, Org. Lett., 2023, 25, 5038 CrossRef CAS PubMed; (c) W.-F. Zheng, J. Chen, X. Qi and Z. Huang, Nat. Chem., 2023, 15, 1672 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |