
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thiophene-fused γ-lactams inhibit the SARS-CoV-2 main protease via reversible covalent acylation†
Gayatri
a,
Lennart
Brewitz
 *a,
Lewis
Ibbotson
a,
Eidarus
Salah
a,
Shyam
Basak
a,
Hani
Choudhry
b and
Christopher J.
Schofield
*a
*a,
Lewis
Ibbotson
a,
Eidarus
Salah
a,
Shyam
Basak
a,
Hani
Choudhry
b and
Christopher J.
Schofield
*a
aChemistry Research Laboratory, Department of Chemistry and the Ineos Oxford Institute for Antimicrobial Research, University of Oxford, 12 Mansfield Road, OX1 3TA, Oxford, UK. E-mail: christopher.schofield@chem.ox.ac.uk; lennart.brewitz@chem.ox.ac.uk
bDepartment of Biochemistry, Center for Artificial Intelligence in Precision Medicines, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 16th April 2024
Abstract
Enzyme inhibitors working by O-acylation of nucleophilic serine residues are of immense medicinal importance, as exemplified by the β-lactam antibiotics. By contrast, inhibition of nucleophilic cysteine enzymes by S-acylation has not been widely exploited for medicinal applications. The SARS-CoV-2 main protease (Mpro) is a nucleophilic cysteine protease and a validated therapeutic target for COVID-19 treatment using small-molecule inhibitors. The clinically used Mpro inhibitors nirmatrelvir and simnotrelvir work via reversible covalent reaction of their electrophilic nitrile with the Mpro nucleophilic cysteine (Cys145). We report combined structure activity relationship and mass spectrometric studies revealing that appropriately functionalized γ-lactams can potently inhibit Mpro by reversible covalent reaction with Cys145 of Mpro. The results suggest that γ-lactams have potential as electrophilic warheads for development of covalently reacting small-molecule inhibitors of Mpro and, by implication, other nucleophilic cysteine enzymes.
Introduction
γ-Lactams are common in bioactive natural products,1 including e.g. in anantine and derivatives,2,3 monascuslactams A–D,4 the proteasome inhibitors lactacystin5–7 and salinosporamide A,8 and clausenamide.9 They are also present in clinically-used therapeutics, for example in the antiemetic rolapitant,10 the respiratory stimulant doxapram,11 piracetam, which is used to treat cortical myoclonus,12 the anti-cancer drug ivosidenib,13 and the antivirals nirmatrelvir (1)14 and simnotrelvir (2) (Fig. 1).15 The latter two inhibit the main protease (Mpro) of the severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2),16 which catalyses hydrolysis of the viral polyproteins pp1a/1ab into functional non-structural proteins; Mpro inhibition results in impaired viral replication.17–20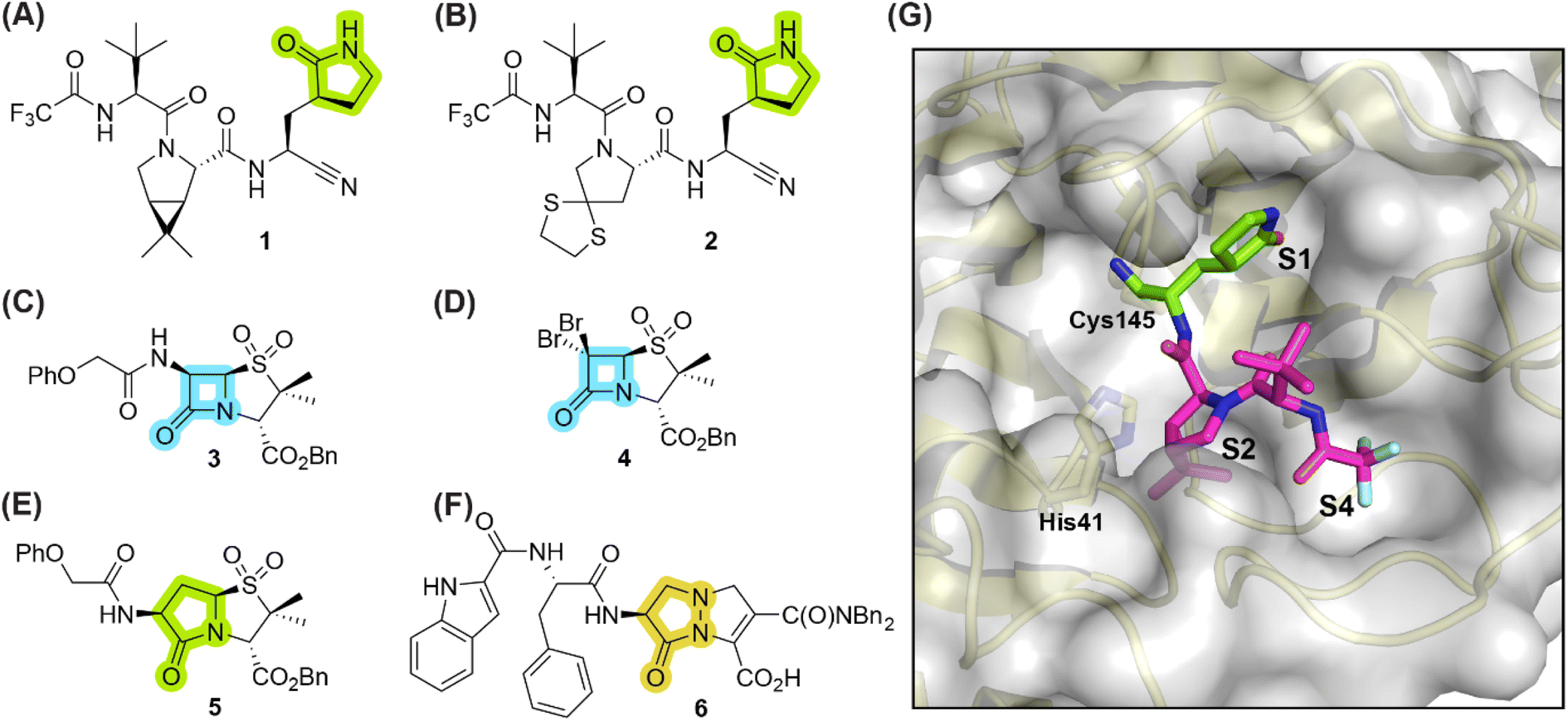 | ||
Fig. 1 Selected reported γ-lactam- and β-lactam-containing SARS-CoV-2 main protease (Mpro) inhibitors. (A) Nirmatrelvir (1);14 (B) simnotrelvir (2);15 (C) penicillin V sulfone benzyl ester 3;21 (D) β-lactam 4;21 (E) γ-lactam 5,22 derived from 3; (F) γ-lactam-derived pyrazolidinone 6;23 (G) view of the surface from the reported SARS-CoV-2 Mpro:1 complex structure active site (PDB ID: 7TE0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24), revealing that the γ-lactam group of 1 binds in the S1 subsite, whereas its bicyclic leucine mimic binds in the S2 subsite, its tert-butyl group is solvent exposed, and its trifluoroacetamide group binds in the S4 subsite. γ-Lactam, β-lactam, and pyrazolidinone groups are in green, blue, and ochre, respectively. Bn: benzyl; Ph: phenyl. 24), revealing that the γ-lactam group of 1 binds in the S1 subsite, whereas its bicyclic leucine mimic binds in the S2 subsite, its tert-butyl group is solvent exposed, and its trifluoroacetamide group binds in the S4 subsite. γ-Lactam, β-lactam, and pyrazolidinone groups are in green, blue, and ochre, respectively. Bn: benzyl; Ph: phenyl. | ||
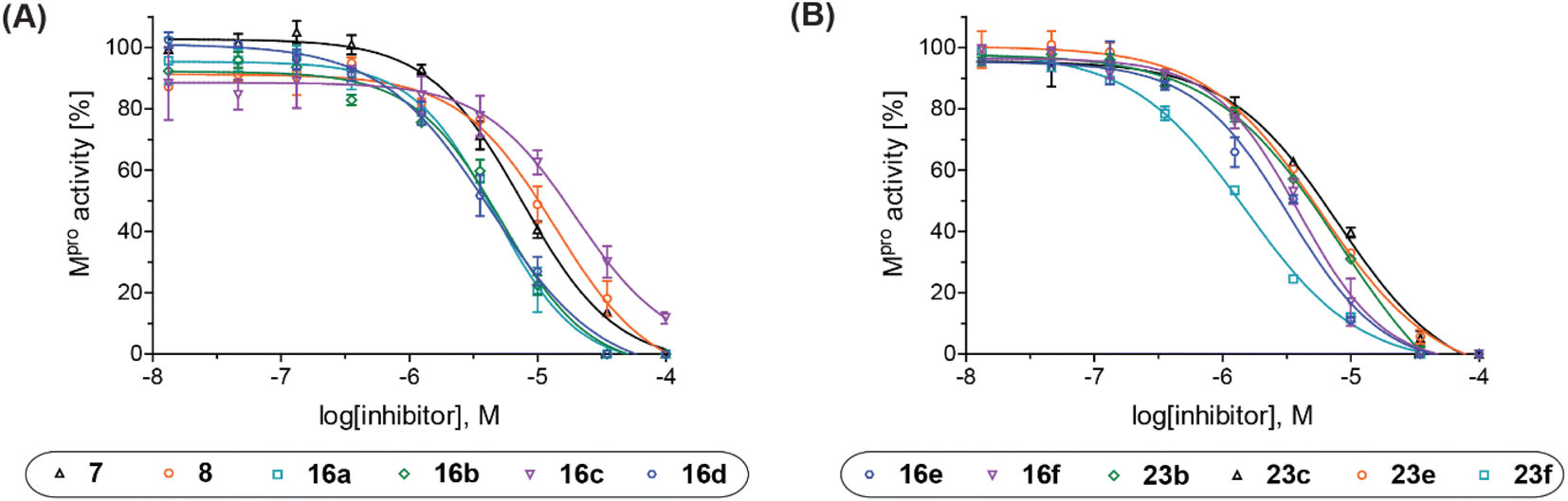 | ||
| Fig. 2 Representative dose–response curves of Mpro γ-lactam inhibitors used to determine IC50-values. (A) 7 (black triangles), 8 (orange circles), 16a (cyan boxes), 16b (green diamonds), 16c (violet inverse triangles), 16d (blue hexagons), and (B) 16e (blue hexagons), 16f (violet inverse triangles), 23b (green diamonds), 23c (black triangles), 23e (orange circles), 23f (cyan boxes). Two dose–response curves each composed of technical duplicates were independently determined using reported SPE-MS Mpro inhibition assays.42 | ||
Small-molecule inhibitors which acylate their target enzyme(s) via covalent reaction of a γ-lactam group with a nucleophilic residue have, to our knowledge, not yet been approved for therapeutic use. The lack of therapeutics which employ a γ-lactam as an electrophilic warhead for covalent reaction is remarkable, considering (i) the revived interest in the development of covalently reacting small-molecule therapeutics,25,26 (ii) recent advances in the synthesis of γ-lactams,1,27,28 (iii) the demonstrated safety of active pharmaceutical ingredients which contain a γ-lactam group that does not acylate the target enzyme, e.g., nirmatrelvir (1)14 and simnotrelvir (2),15 and, in particular, (iv) that many clinically used small-molecules employ a β-lactam as an electrophilic warhead to acylate their target enzyme(s), e.g., penicillin- and cephalosporin-based antibiotics.29 At least in part, this gap may reflect the reduced intrinsic reactivity of γ-lactams compared to more strained β-lactams based on (non-enzymatic) ring closure rates.30
The covalent reaction of γ-lactams with nucleophilic serine enzymes is reported;31–39 however, their analogous reactivity with nucleophilic cysteine enzymes, many of which are contemporary drug targets,25,26 is, to our knowledge, unknown. SARS-CoV-2 Mpro appears to be a suitable target to investigate the reactivity of γ-lactams with nucleophilic cysteine enzymes, because many small-molecule inhibitors are reported which employ an electrophilic group for reversible or irreversible covalent reaction with the nucleophilic thiolate of the catalytic cysteine residue of Mpro (i.e., Cys145, Fig. 1) and because of the important structural roles of a γ-lactam group in many reported substrate-derived Mpro inhibitors,40,41e.g., nirmatrelvir (1)14 and simnotrelvir (2).15 The γ-lactam group of these inhibitors binds in the S1 subsite of Mpro, that is proximate to Cys145 (Fig. 1).
Of Mpro inhibitors that react covalently, those that react reversibly may be preferred over those that react irreversibly, as the latter may also react with ‘off-targets’ in an irreversible manner. Indeed Cys145 of Mpro reacts reversibly with the nitrile group of the clinically-used drugs 1 and 2.14,15,42 Many investigational Mpro inhibitors, however, employ highly reactive electrophiles for covalent reaction with Cys145, including e.g., aldehydes, α-ketoamides, and Michael acceptors,19,43,44 which may potentially compromise safety, as reported for some clinically-used small-molecules bearing reactive electrophiles;26,45 The use of electrophilic groups with relatively low intrinsic reactivity is thus desirable. The observation that the γ-lactam of both 1 and 2 is stable in cells14,15 likely reflects its reduced reactivity compared to more reactive electrophiles, indicating that covalently reacting γ-lactams may have potential for development of safe COVID-19 therapeutics. However, by contrast with β-lactams,21,22 γ-lactams have, to our knowledge, not yet been considered as electrophilic warheads for covalent reaction with Mpro Cys145.
During the course of investigations aimed at developing penicillin-based Mpro inhibitors which acylate Cys145 via β-lactam ring opening (e.g., 3 and 4),21,22 we synthesized the γ-lactam analogue 5 to probe the effect of altering the lactam group on potency, including with respect to reversibility of acylation. Consistent with studies revealing that acylation of nucleophilic serine residues is more reversible with a γ-lactam compared to an analogous β-lactam,46,47 γ-lactam 5 inhibits isolated recombinant SARS-CoV-2 Mpro ∼4-fold less efficiently than the structurally-related β-lactam 3.22 Mass spectrometric analyses indicated that, by contrast with β-lactam 3, γ-lactam 5 did not react to form a stable acyl–enzyme complex, suggesting that it may bind to Mpro principally via non-covalent interactions.22
Here we report the synthesis of thiophene-fused γ-lactams which inhibit isolated recombinant SARS-CoV-2 Mpro more efficiently than β-lactam 3 and γ-lactam 5 (Fig. 1). Mass spectrometric evidence supports a mechanism involving reversible covalent reaction of the γ-lactam group with Cys145. The results reveal bicyclic γ-lactams are useful scaffolds for the inhibition of nucleophilic cysteine enzymes.
Results
Thiophene-fused γ-lactams inhibit SARS-CoV-2 Mpro
Natural product inspired trans-ring-fused γ-lactams can inhibit serine proteases via acylating their nucleophilic serine residue,31,32 as is also the case for thiophene-fused γ-lactams.33 In the latter case, it is proposed that, following acylation, the presence of the thiophene ring hinders deacylation by sequestering electron density on the γ-lactam-derived amine.33 Based on the proposal that a hydrophobic thiophene ring may bind in the hydrophobic S2 pocket of Mpro that is in proximity of Cys145 (Fig. 1), we synthesized an initial set of thiophene-fused γ-lactams (7–9) following modifications of reported procedures.33 The effect of these synthetic γ-lactams on catalysis of isolated recombinant SARS-CoV-2 Mpro was investigated using solid phase extraction coupled to mass spectrometry (SPE-MS) based assays, which directly monitor Mpro-catalysed hydrolysis of a pp1a/1ab-derived oligopeptide,21,22,42 and which we and others have used to characterise covalently and non-covalently binding Mpro inhibitors.21,22,42,48–56Analysis of the half-maximum inhibitory concentrations (IC50-values) revealed that both the regioisomeric thiophene-fused γ-lactams 7 and 8 moderately inhibited isolated recombinant SARS-CoV-2 Mpro with similar potencies (IC50 ∼ 8.5 and 8.4 μM, respectively; Table 1, entries i and ii). By contrast, the regioisomeric γ-lactam 9 did not inhibit Mpro over the tested concentration range (Table 1, entry iii), showing that the position of the thiophene sulfur atom with respect to the γ-lactam nitrogen atom affects inhibition potency. The substitution of the methylene group of 9 with an NSO2Me group to give 10 did not result in inhibition (Table 1, entry iv). To investigate the effect of the thiophene ring of 7 and 8, we synthesized the corresponding benzene-fused γ-lactam 11 using the route employed for synthesis of 7 and 8. 11 did not inhibit Mpro over the tested concentration range (Table 1, entry v), indicating that the size of the ring fused to the γ-lactam, nature of delocalization, and/or the presence of a sulfur atom in that ring are important for the inhibition manifested by 7 and 8.
| γ-Lactam | IC50 [μM] | |
|---|---|---|
| a Assays were performed as reported using SPE-MS, employing SARS-CoV-2 Mpro (0.05 μM) and substrate peptide (2.0 μM).42 Results are means of two independent runs each composed of technical duplicates (n = 2; mean ± standard deviation, SD). Representative dose–response curves of selected γ-lactams are shown in Fig. 2. | ||
| i |
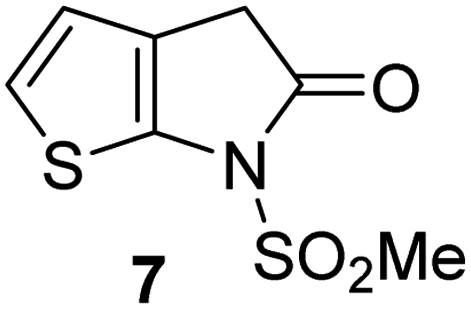
|
8.5 ± 1.4 |
| ii |

|
8.4 ± 0.5 |
| iii |

|
>100 |
| iv |
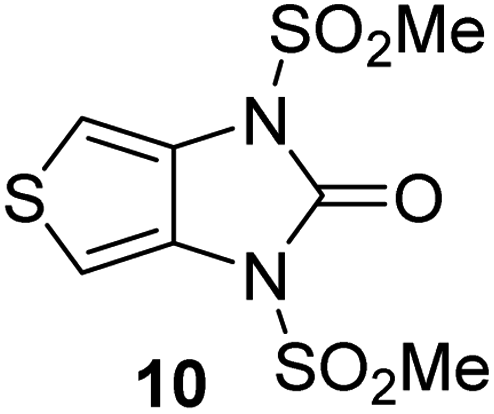
|
>100 |
| v |
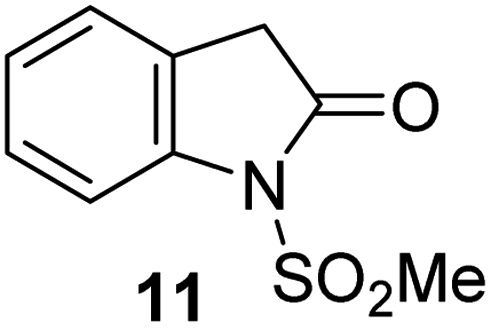
|
>100 |
The Mpro inhibition potency of γ-lactams 7 and 8 is in the range of that reported for penicillin V sulfone benzyl ester 3 (IC50 ∼ 6.6 μM;22Fig. 1), which inhibits Mprovia covalent reaction of its β-lactam with the active site Cys145.22 Notably, γ-lactams 7 and 8 inhibit Mpro ∼3-fold more efficiently than the reported γ-lactam 5 (IC50 ∼ 26.1 μM;22Fig. 1), which inhibits Mpro apparently via non-covalent interactions,22 and ∼5-fold more efficiently than the reported pyrazolidinone 6 (IC50 ∼ 45 μM;23Fig. 1).
Substitution affects the inhibition potency of thiophene-fused γ-lactams
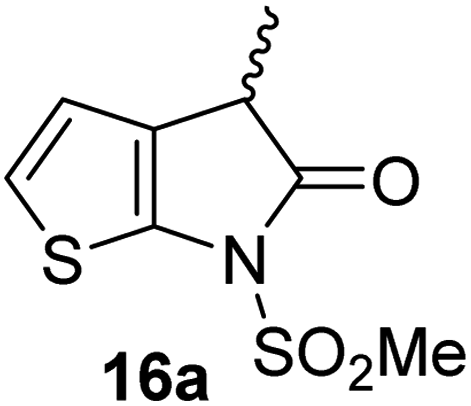
Structure activity relationship studies were performed to investigate whether the lactam nitrogen substituent and substituents α to the lactam carbonyl affect inhibition potency. Derivatives of γ-lactam 7 with a single α substituent were synthesized from commercially-sourced 2-nitrothiophene (12) in 5 steps following modification of reported procedures (Scheme 1A).33 Initially, 12 was efficiently alkylated with ethyl chloroacetate to give thiophene 13 as a single regioisomer. 13 was alkylated using an alkyl halide with Cs2CO3 as a base to give thiophenes 14a–d and 14f; 14e was synthesized from 13 using catalytic amounts of 1,1,3,3-tetramethylguanidine as a base and acrylonitrile as a Michael acceptor, as reported for related nitriles.57 Nitrothiophenes 14a–f were converted to the corresponding sulfonamides 15a–f following nitro-reduction, sulfonylation, and saponification. γ-Lactams 16a–f were obtained from 15a–fvia HATU58-mediated amide bond formation. Derivatives of γ-lactam 7 which bear two identical α substituents, i.e., 17a and 17b, were directly synthesized from 7via an alkylation reaction (Scheme 1B). | ||
| Scheme 1 Synthesis of α-substituted γ-lactam derivatives of 7. Reagents and conditions: (a) ethyl chloroacetate, KOtBu, THF, −50 °C to rt, 89%; (b) Cs2CO3, alkyl halide, DMF, rt, 52–94%, or for 14e: 1,1,3,3-tetramethylguanidine (20 mol%), acrylonitrile, THF, rt, 55%; (c) Fe(0), FeSO4 (8 mol%), dioxane:H2O (4:1), reflux; then: methylsulfonyl chloride, NEt3, 4-(N,N-dimethylamino)pyridine (10 mol%), CH2Cl2, rt, 10–30%; (d) LiOH, THF:H2O:EtOH (2:1:1), rt, 73–94%; (e) 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU),58iPr2NEt,59 MeCN:CH2Cl2 (1:1), rt, 46–76%; (f) Cs2CO3 (2.5 equiv.), alkyl halide (2.2 equiv.), DMF, rt, 9–90%. See Table 2 for structures of 16a–f and 17a–b. | ||
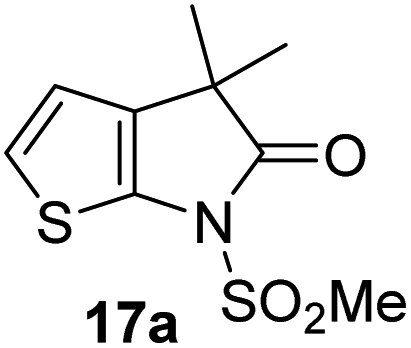
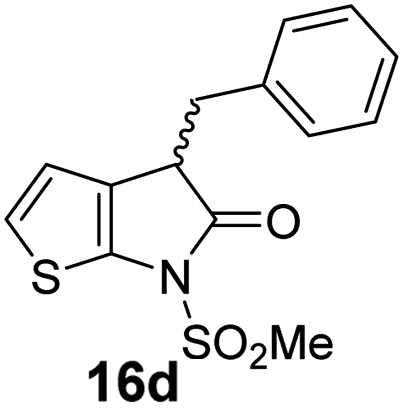
The Mpro inhibition results reveal that the addition of a methyl group α to the carbonyl of γ-lactam 7 increases inhibition potency by ∼2-fold, whereas the addition of a second methyl group ablates inhibition (Table 2, entries i and ii). The length of the alkyl substituent α to the γ-lactam carbonyl did not appear to substantially affect potency: 16b, which bears a propyl substituent α to the lactam carbonyl, inhibited isolated recombinant SARS-CoV-2 Mpro with similar potency as 16a which bears a methyl group at the same position (Table 2, entries i and iii). By contrast, isomeric isopropyl-substituted γ-lactam 16c inhibited Mpro ∼5-fold less efficiently than propyl-substituted 16b (IC50 ∼ 16 μM; Table 2, entry iv). However, sterically bulky substituents are not necessarily detrimental for efficient inhibition, since benzyl substituted γ-lactam 16d inhibited with similar potency as 16a and 16b (IC50 ∼ 4.0 μM; Table 2, entry v).
| γ-Lactam | IC50 [μM] | γ-Lactam | IC50 [μM] | ||
|---|---|---|---|---|---|
| a Assays were performed as reported using SPE-MS, employing SARS-CoV-2 Mpro (0.05 μM) and substrate peptide (2.0 μM).42 Results are means of two independent runs each composed of technical duplicates (n = 2; mean ± SD). Representative dose–response curves of selected γ-lactams are shown in Fig. 2. | |||||
| i |
|
3.9 ± 0.1 | vi |

|
2.7 ± 0.1 |
| ii |
|
>100 | vii |
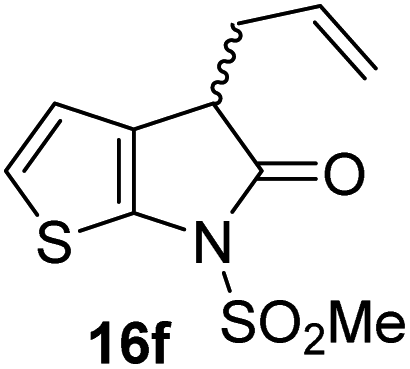
|
3.5 ± 0.2 |
| iii |
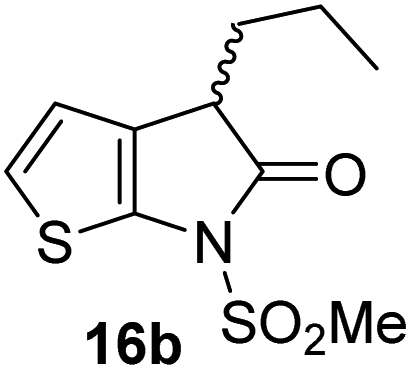
|
3.2 ± 0.8 | viii |
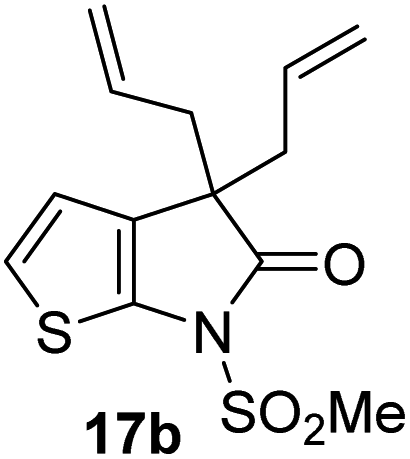
|
>100 |
| iv |
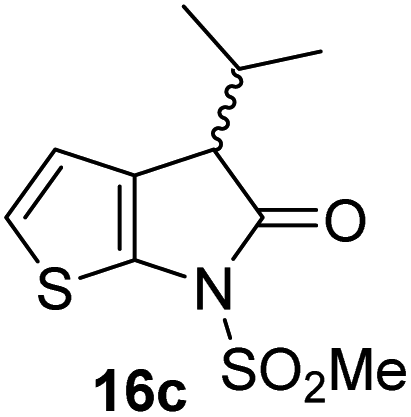
|
16 ± 2 | ix |
|
61 ± 4 |
| v |
|
4.0 ± 0.4 | x |
|
>100 |
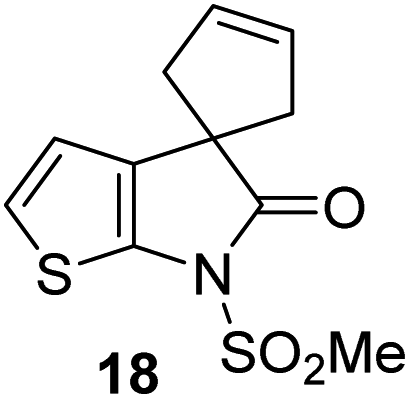
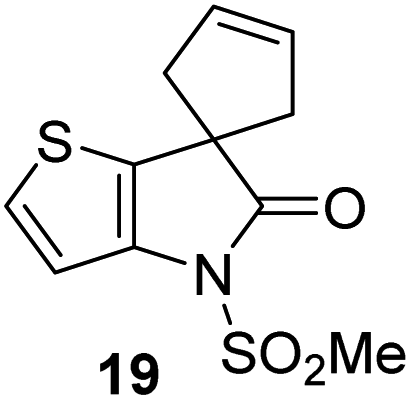
γ-Lactams 16e and 16f which are both derived from propyl-substituted γ-lactam 16b, but which contain a nitrile or olefin, respectively, in their alkyl substituent, inhibited Mpro with similar potency to 16b (IC50 ∼ 2.7 and 3.5 μM, respectively; Table 2, entries vi and vii). Similar to α-disubstituted γ-lactam 17a, 17b which bears two allyl substituents α to its lactam carbonyl did not inhibit Mpro (Table 2, entry viii). Both the spiro γ-lactam 18, which was synthesized from 17bvia a ring-closing metathesis,60 and the isomeric γ-lactam 19, which was synthesized from γ-lactam 8 following an analogous synthesis route (ESI†), did not efficiently inhibit Mpro (Table 2, entries ix and x).
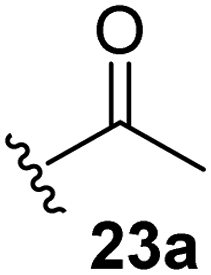
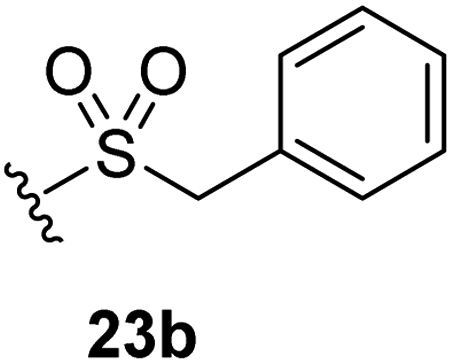
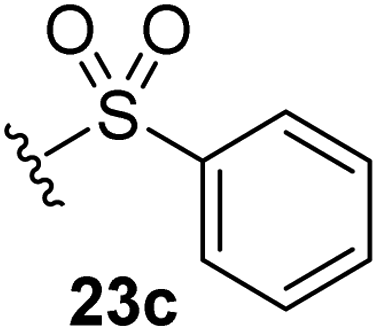
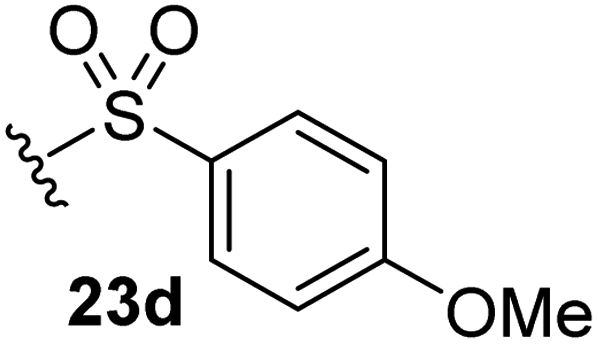
The effect of varying the γ-lactam nitrogen substituent of 7 on inhibition potency was investigated (Table 3). Derivatives of 7, i.e., 23a and 23c–f, were synthesized from commercially-sourced 2-(thiophen-3-yl)acetic acid (20) in 3 steps, employing copper-catalysed reaction of thiophene bromide 21 with activated amines (Scheme 2).61,62 γ-Lactam 23b was synthesized from thiophene 13 in a similar manner to which 7 was prepared (ESI†). Substituting the methylsulfonyl group of γ-lactam 7 for an acetyl group ablated Mpro inhibition (Table 3, entry ii), whereas use of benzylsulfonyl or phenylsulfonyl groups apparently increased potency by ∼2-fold (Table 3, entries iii and iv). In some cases, the addition of substituents on the phenyl ring of 23cpara to the sulfonyl group further increased potency (Table 3, entries v–vii); the CF3-substituted γ-lactam 23f appeared to be a particularly potent Mpro inhibitor, being ∼3- and ∼6-fold more potent than γ-lactams 23c and 7, respectively (IC50 ∼ 1.3 μM; Table 3, entry vii). Notably, γ-lactam 23f inhibits Mpro ∼20-fold more efficiently than our reported non-covalently reacting γ-lactam Mpro inhibitor 5 and ∼6-fold more efficiently than our reported covalently reacting β-lactam Mpro inhibitor 3 (Fig. 1).22
|
|
IC50 [μM] | |
|---|---|---|
| a Assays were performed as reported using SPE-MS, employing SARS-CoV-2 Mpro (0.05 μM) and substrate peptide (2.0 μM).42 Results are means of two independent runs each composed of technical duplicates (n = 2; mean ± SD). Representative dose response curves of selected γ-lactams are shown in Fig. 2. | ||
| i |
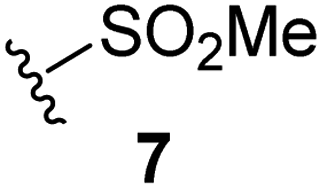
|
8.5 ± 1.4 |
| ii |
|
>100 |
| iii |
|
4.3 ± 0.1 |
| iv |
|
4.7 ± 1.1 |
| v |
|
2.7 ± 0.2 |
| vi |
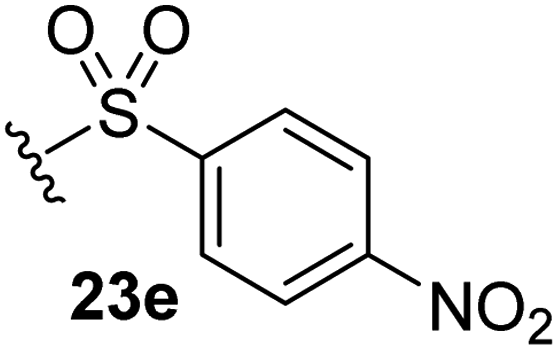
|
4.4 ± 0.7 |
| vii |
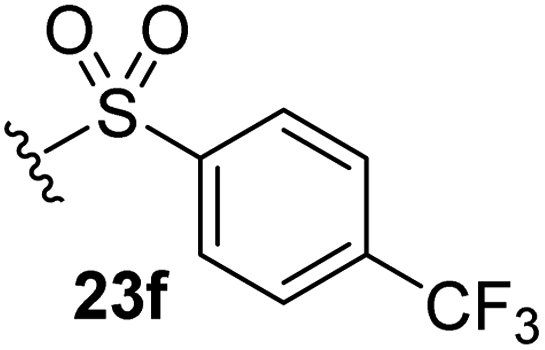
|
1.3 ± 0.1 |
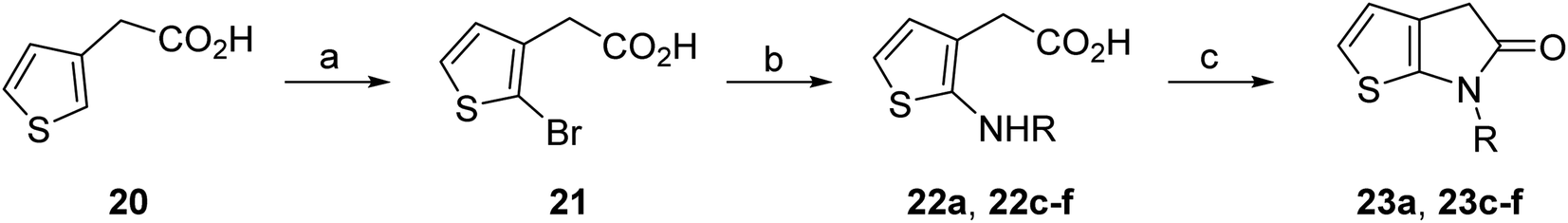 | ||
| Scheme 2 Synthesis of γ-lactams bearing different lactam nitrogen substituents. Reagents and conditions: (a) N-bromosuccinimide, THF, 0 °C to rt, 80%; (b) amine, K2CO3, tBuOH, CuI (10 mol%), N,N′-dimethylethylenediamine, 100 °C; or: amine, K2CO3, Cu(0), pyridine, 120 °C, 5–44%; (c) HATU,58iPr2NEt,59 MeCN:CH2Cl2 (1:1), rt, 22–67%. See Table 3 for structures of 23a and 23c–f. | ||
Thiophene-fused γ-lactams inhibit Mprovia reversible covalent reaction
Protein-observed MS studies under denaturing conditions were performed with selected synthetic γ-lactams to investigate whether they inhibit isolated recombinant SARS-CoV-2 Mprovia non-covalent interactions, as for γ-lactam 5,22 or via covalent reaction. The results reveal that some of the tested γ-lactams covalently react with Mpro during the tested time period (i.e., 4 h), as shown by the anticipated mass shifts (Fig. 3); the stoichiometry of the covalent reaction appears to be, at least predominantly, 1:1, suggesting that γ-lactams react selectively with a single nucleophilic Mpro residue, likely Cys145. Nonetheless, the MS studies imply that some of the γ-lactams, i.e., 16a, 16b, and 23d, may have capacity to covalently react with Mpro residues other than Cys145, albeit at substantially lower levels even when being used in excess; note that Mpro has eleven cysteine residues in addition to Cys145, all of which can covalently react with non-specific inhibitors such as ebselen.49
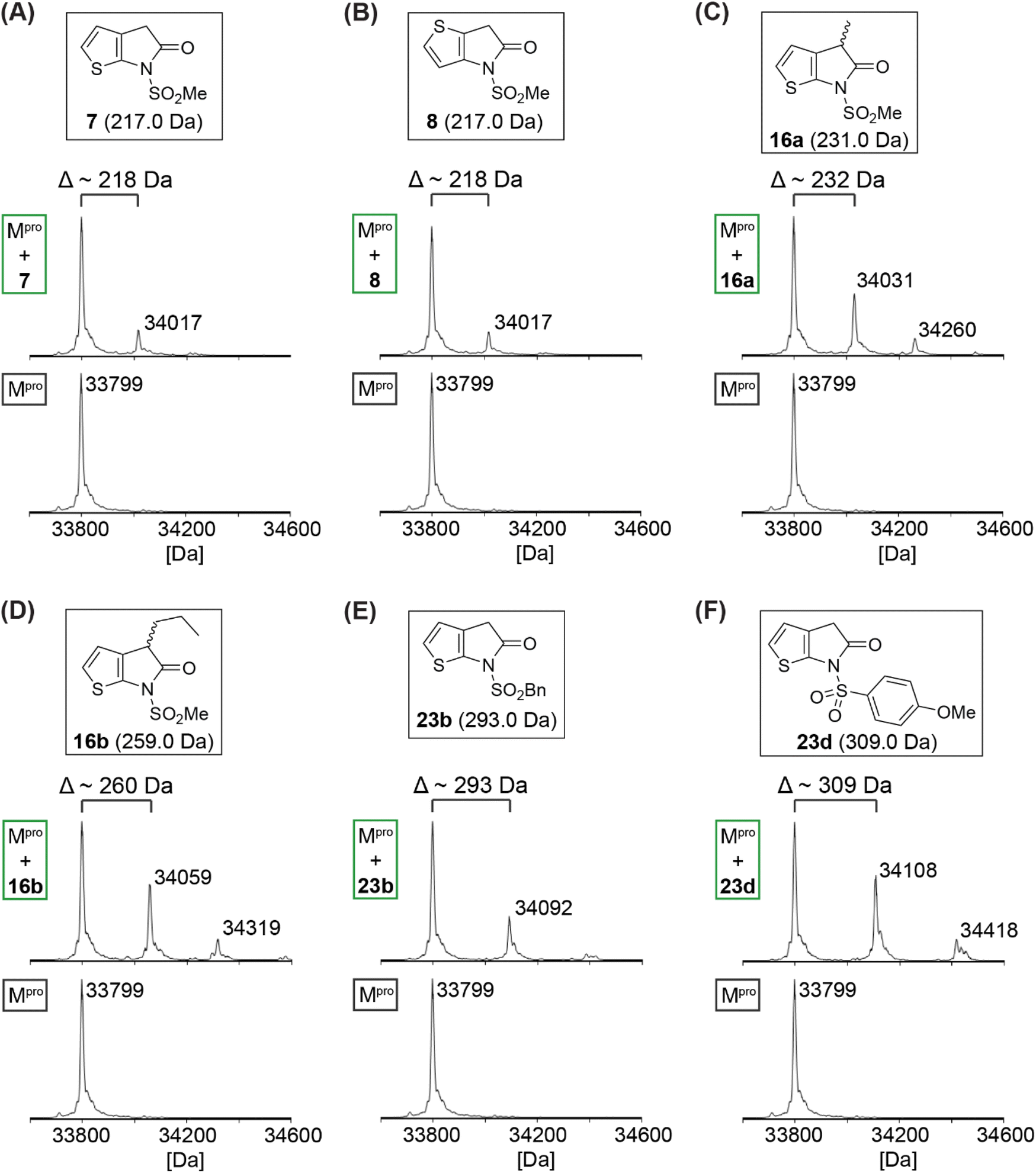 | ||
| Fig. 3 γ-Lactams react covalently with isolated recombinant Mpro. Analysis of a reaction mixture of Mpro and γ-lactams (A) 7, (B) 8, (C) 16a, (D) 16b, (E) 23b, and (F) 23d prior (bottom) and 4 h post (top) incubation with the respective γ-lactam. Assays were performed using SPE-MS as described in the Experimental section employing SARS-CoV-2 Mpro (3.0 μM) and, if appropriate, a γ-lactam (15 μM) in buffer (20 mM HEPES, pH 7.5). Representative spectra of technical duplicates are shown. | ||
Variable levels of Mpro acylation were observed depending on the γ-lactam employed, suggesting that initial binding constants, reaction rates, and/or stabilities of the acyl–enzyme complex differ depending on the substitution pattern. Notably, complete Mpro acylation was not observed under the tested conditions, an observation which may reflect the reversibility of the reaction and the comparatively low enzyme to γ-lactam ratio employed in the assay (i.e., 1:5); this ratio was chosen to avoid γ-lactam-induced ionization suppression of Mpro observed at higher γ-lactam concentrations, thus perturbing data analysis.
To localize the site of covalent modification and to probe whether covalent reaction occurs with Cys145, 16a was incubated with Mpro that had been previously reacted with a small-molecule inhibitor42 that selectively and irreversibly reacts with Cys145 (Fig. 4); 16a was used for this study because its levels of Mpro acylation were apparently higher than those for the unsubstituted 7 and 8, and because it has the least bulky substituent amongst those γ-lactams that covalently react with Mpro, a property which may favour more efficient covalent reaction. We have reported that an alkyne derivative of nirmatrelvir (24; Fig. 4), in which the electrophilic nitrile is substituted for an alkyne, reacts selectively and irreversibly with Cys145.42 Thus, isolated recombinant SARS-CoV-2 Mpro was incubated with a ∼16-fold excess of the alkyne derivative of 1 (i.e., 24) to block the thiol of Cys145 by stoichiometric thioenol ether formation (Fig. 4A).42 The excess of 24 was removed by washing and the resultant covalent Mpro:24 complex was incubated with 16a. The results reveal that 16a does not covalently react with the covalent Mpro:nirmatrelvir alkyne (24)42 complex within the tested time (i.e., 2 h). γ-Lactam 16a thus likely reacts selectively under the tested conditions with Cys145, but not, at least substantially, with other surface-exposed cysteine residues of Mpro.
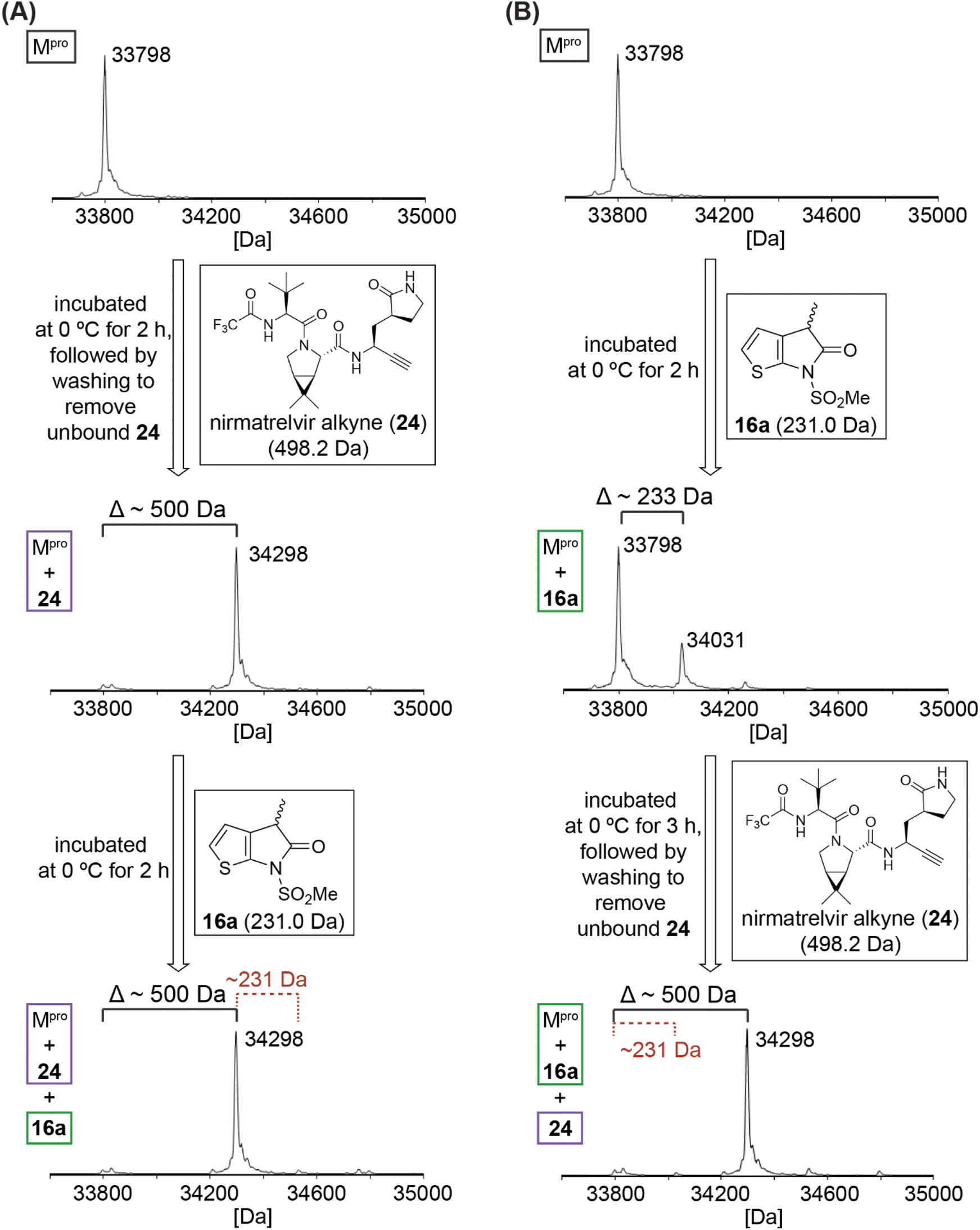 | ||
| Fig. 4 Evidence that γ-lactam 16a inhibits Mpro by selective reversible covalent reaction with Cys145. (A) γ-Lactam 16a does not covalently react with the covalent Mpro:nirmatrelvir alkyne derivative 2442 complex obtained via irreversible covalent reaction of Mpro Cys145 with 24,42 indicating that γ-lactams selectively react with the nucleophilic thiolate of Cys145 under the tested conditions; (B) addition of an excess of 2442 to a mixture containing the covalent Mpro:16a complex results in stoichiometric formation of the corresponding covalent Mpro:2442 complex, substantial levels of the Mpro:16a complex were not detected using SPE-MS implying that the reaction of γ-lactams with Mpro is reversible and/or that the acyl–enzyme complex is not stable towards hydrolysis. Assays were performed using SPE-MS, as described in the Experimental section, employing SARS-CoV-2 Mpro (3.0 μM), and, if appropriate, γ-lactam 16a (15 μM) and/or nirmatrelvir alkyne 2442 (50 μM) in buffer (20 mM HEPES, pH 7.5). Representative spectra of technical duplicates are shown. | ||
It was of interest to investigate whether the synthetic γ-lactams react reversibly with Cys145. Thus, γ-lactam 16a was incubated with Mpro at 0 °C for 2 h (enzyme/16a ratio: 1:5), before the nirmatrelvir alkyne derivative 24 was added to the reaction mixture. The resultant mixture was incubated for 3 h at 0 °C, followed by washing to remove excess 24 and analysis using SPE-MS (Fig. 4B). Stoichiometric formation of the covalent Mpro:24 (ref. 42) complex was observed, whereas substantial levels of the Mpro:16a complex were not detected (Fig. 4B). The results imply that the reaction of γ-lactams with Mpro is reversible and/or that the resultant acyl–enzyme complex is not stable towards hydrolysis. This proposal is precedented by the reported hydrolytic γ-lactamase activity of other nucleophilic serine63,64 and cysteine enzymes.65–67 The combined results indicate that γ-lactams have potential as electrophilic warheads for development of covalently reacting small-molecule inhibitors of Mpro and, consequently, other nucleophilic cysteine enzymes.
Discussion
Penicillins and related antibiotics inhibit bacterial cell wall biosynthesis via covalent reaction of their electrophilic β-lactam ring with the nucleophilic serine residue of transpeptidases to give stable acyl-enzyme complexes.68 Efforts to substitute the β-lactam ring of penicillins began in the 1940s, ultimately leading to the identification of suitably activated γ-lactams and related compounds, including the natural product lactivicin and derivatives, as antibiotics.69–75 Subsequently, 1,6-diazabicyclo[3.2.1]octane-based compounds (DBOs),76–79 including avibactam, have been developed for clinical use as reversibly reacting covalent inhibitors of nucleophilic serine β-lactamases.80–82γ-Lactams have been developed as inhibitors of both human and viral serine proteases including e.g., human neutrophil elastase,31–33 the hepatitis C virus (HCV) ns3/4a serine protease,34–37 and the human cytomegalovirus (HCMV) serine protease.38,39 They inhibit via acylation of the nucleophilic serine residue, at least in some cases in a reversible manner.38,46 By contrast with β-lactams,21,22,83–89 the reaction of γ-lactams with nucleophilic cysteine enzymes has not, to our knowledge, been well explored. Several γ-lactamases have been proposed to catalyse γ-lactam hydrolysis via nucleophilic attack by a cysteine residue,65–67 however, the intermediate acyl-enzyme complexes have not yet been structurally characterized.
Our combined results imply that γ-lactams have potential to be useful covalently reacting inhibitors of nucleophilic cysteine enzymes, in particular SARS-CoV-2 Mpro. They reveal that thiophene-fused γ-lactams can efficiently inhibit Mproin vitro (Tables 1–3), in accord with the proposal that the thiophene ring helps to sequester electron density of the γ-lactam-derived amine following acyl–enzyme complex formation.33 The γ-lactam thiophene ring appears to be important for efficient Mpro inhibition, since its substitution by a benzene ring ablates inhibition and, interestingly, the regioisomeric positioning of the sulfur atom within the thiophene ring also affects inhibition potency (Table 1). The reasons for this observation, including precisely how sequestration of the lone pair(s) on the γ-lactam-derived amine N atom affects the extent of inhibition, are under investigation. The results also show that substitution both α to the γ-lactam carbonyl and on the γ-lactam N atom affect inhibition potency (Tables 2 and 3). Thus, there is considerable scope for further optimization of the identified γ-lactam Mpro inhibitors, in particular with respect to optimal binding in the S1 or S2 pocket. The knowledge that γ-lactams can bind in the S1 pocket of Mprovia non-covalent interactions14,24,42 suggests that derivatives of 7 and 8 possessing a second γ-lactam binding in the S2 or S1 pocket are of interest.
Our MS studies reveal that γ-lactams acylate the nucleophilic thiolate of Cys145 in a reversible manner (Fig. 3–5). The results thus indicate that lactam rings other than β-lactams have potential for development as covalently reacting inhibitors of Mpro and, by implication, of other nucleophilic cysteine enzymes. However, it should be noted that γ-lactams do not necessarily have to covalently react with Mpro for efficient inhibition, because e.g., γ-lactam 5 (Fig. 1) is reported to likely inhibit Mpro without covalently reacting.22 The formation of a γ-lactam from an ester, including an acyl–enzyme complex, is intrinsically more favourable than that of a β-lactam, as indicated by studies on the reactivity of γ-lactams with a nucleophilic serine enzyme.30,46 The more reversible nature of γ-lactam versus β-lactam reaction with nucleophilic residues may, in some circumstances, be an advantage with respect to limiting (irreversible) off-target reactivity. Reversibility may, however, be an unfavourable property with respect to the relative potency of inhibition for analogous γ- and β-lactams.
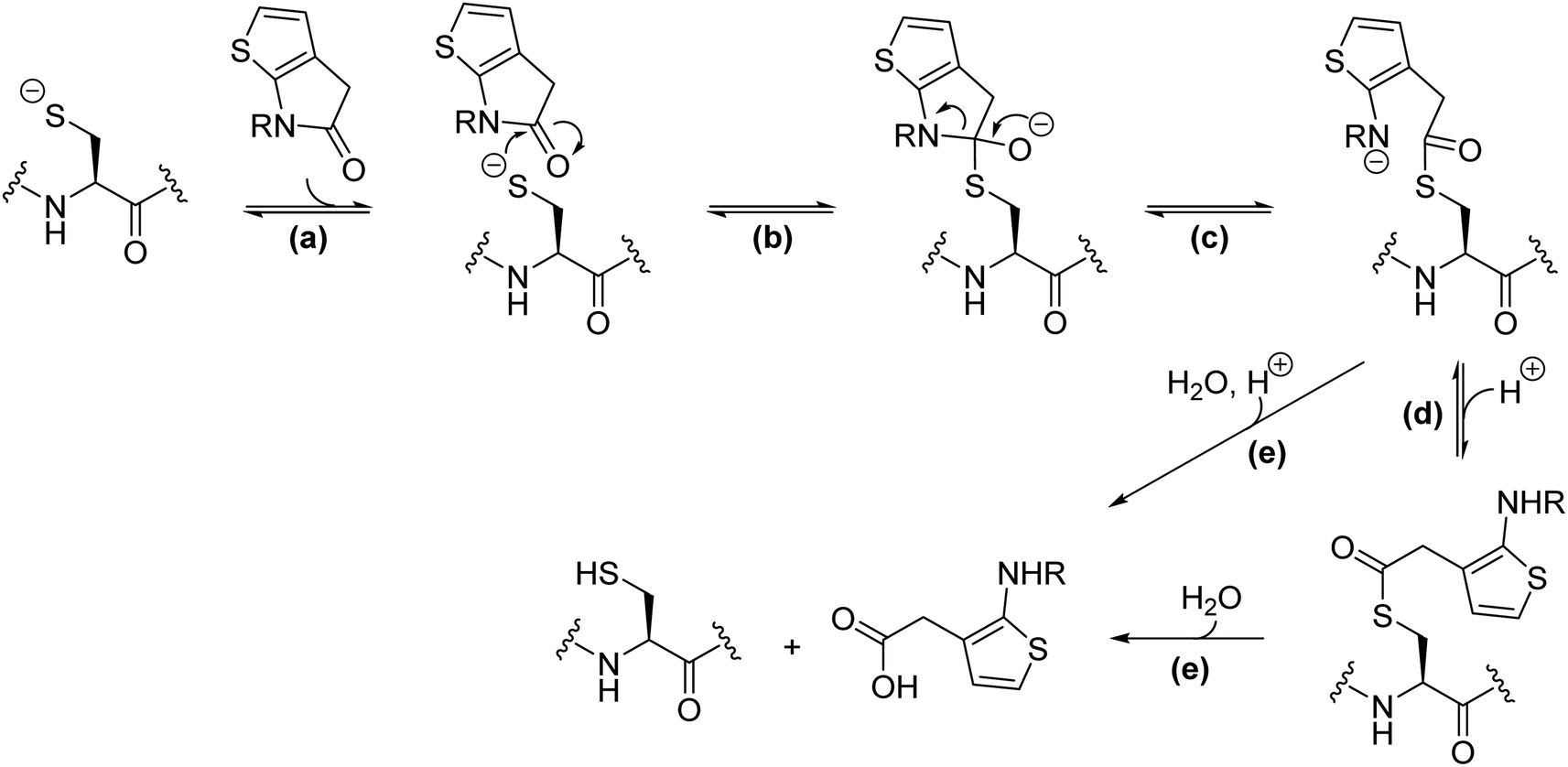 | ||
| Fig. 5 Proposed scheme for reaction of thiophene-fused γ-lactams with nucleophilic cysteine enzymes. Reaction steps include: (a) the reversible non-covalent binding of the thiophene-fused γ-lactam to the enzyme active site; (b) reversible nucleophilic attack of the cysteine thiolate to the proximate γ-lactam carbonyl forming a tetrahedral intermediate; (c) reversible γ-lactam fission and formation of an acyl–enzyme complex; (d) (reversible) protonation of the resultant amine, which may or may not be associated with a conformational change to form a hydrolytically more stable acyl–enzyme complex; (e) irreversible hydrolysis of the acyl–enzyme complex. Note there is variation in the general acid base machinery and oxy-anion stabilising mechanisms employed by nucleophilic cysteine enzymes. | ||
It is possible that the γ-lactam-derived acyl–enzyme complex can either react to reform the initial γ-lactam and/or be hydrolysed (Fig. 5), as proposed for other nucleophilic cysteine enzymes with reported γ-lactamase reactivity.65–67 We did not observe evidence for γ-lactam hydrolysis in our MS studies; this potential problem can be limited by steric extrusion of hydrolytic water from the active site, as precedented with work on inhibiting nucleophilic serine enzymes.33,46,47 The potential γ-lactam liability concerning reversibility in acylation can be overcome if the amine lone pair derived from the γ-lactam is sequestered in the acyl–enzyme complex; indeed, this concept enabled the initial development of thiophene-fused γ-lactams as serine protease inhibitors.33 It should be noted that the acylation ability of 5,5-trans-fused bicyclic pyrrolidine lactams31 and related γ-lactams is likely not, at least principally, a result of their ring strained structure.90 Indeed, the normally efficient natural substrates of proteases are themselves unstrained amides. Hence, empirical optimization remains of importance in inhibitor development.
We have not yet obtained a crystal structure of the acyl–enzyme complex formed by covalent reaction of Cys145 with a γ-lactam, something that may reflect reversible binding and/or the labile nature of this intermediate. In addition to inhibiting by covalent reaction with Cys145, our combined mass spectrometric evidence imply that γ-lactams can also inhibit Mprovia non-covalent binding.22 It is thus possible that γ-lactams can bind to the Mpro active site in different conformations, i.e., one that enables covalent reaction of the γ-lactam group with Cys145 or one that enables non-covalent binding of the γ-lactam group, for example in the S1 pocket, as crystallographically observed for the γ-lactam of nirmatrelvir (1).14,24 Our current work is thus focused on substituents α to the γ-lactam carbonyl which engage with residues at the Mpro active site to promote formation of a stable acyl–enzyme complex.
Conclusions
Our results expand the repertoire of covalently reacting groups for efficient SARS-CoV-2 Mpro inhibition to γ-lactams and suggest that γ-lactams may also covalently react with other disease-relevant nucleophilic cysteine enzymes. In this regard, it will be of interest to investigate γ-lactams as inhibitors of nucleophilic cysteine enzymes from different mechanistic sub-families, including the SARS-CoV-2 papain-like protease (PLpro), which employs a catalytic triad for catalysis, rather than a dyad as Mpro.91–93 Considering that both β-lactams21,22 and, as we now report, γ-lactams can covalently react with Cys145, other related ring systems also have potential for Mpro inhibition. Such ring systems include lactone derivatives, as precedented by work on β-lactone inhibitors of hepatitis A virus and plant nucleophilic cysteine enzymes.94–97 The corresponding β- and γ-sultam derivatives and δ-lactams, which are reported to inhibit human neutrophil elastase,98–100 may also be suited to covalent reaction with nucleophilic cysteine enzymes, including Mpro.Experimental section
γ-Lactam synthesis
Thiophene-fused γ-lactams were synthesized following modifications of reported procedures.33SARS-CoV-2 Mpro inhibition assays
Solid phase extraction coupled to mass spectrometry (SPE-MS) inhibition assays were performed using isolated recombinant SARS-CoV-2 Mpro (0.05 μM), which was based on the Wuhan-Hu-1 genome101 (National Center for Biotechnology Information (NCBI) reference sequence: NC_045512.2) and which was prepared according to established procedures,21 a 37mer oligopeptide (ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2), which was based on the on the sequence of the N-terminal SARS-CoV-2 Mpro self-cleavage site and synthesized as a C-terminal amide and purified by GL Biochem (Shanghai) Ltd (Shanghai, China), as a substrate (2.0 μM), and the N-terminally acetylated C-terminal product peptide (Ac-SGFRKMAFPS-NH2) as an internal standard (0.4 μM) in buffer (20 mM HEPES, pH 7.5, 50 mM NaCl, 20 °C), as reported.42Protein-observed Mpro assays
Assays were performed as reported using recombinant isolated SARS-CoV-2 Mpro (3.0 μM) and, if appropriate, the indicated γ-lactam (15 μM) and/or nirmatrelvir alkyne 2442 (50 μM) in buffer (20 mM HEPES, pH 7.5, 20 °C); reaction mixtures were analysed using SPE-MS.21,22,42Data availability
The synthetic procedures and the characterization of all products and NMR spectra of the lead inhibitors are given in the ESI.†Author contributions
G. and L. I. synthesised the γ-lactams with assistance from S. B.; L. B. performed assays; E. S. and H. C. provided resources; L. B. and C. J. S. supervised and conceived the research and wrote the manuscript with help from G.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The investigators acknowledge the philanthropic support of the donors to the University of Oxford's COVID-19 Research Response Fund and King Abdulaziz University, Saudi Arabia, for funding. This research was funded in part by the Wellcome Trust (106244/Z/14/Z). For the purpose of open access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. We thank Cancer Research UK (C8717/A18245) and the Biotechnology and Biological Sciences Research Council (BB/J003018/1 and BB/R000344/1) for funding.References
- J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC.
- F. Khuong-Huu, X. Monseur, G. Ratle, G. Lukacs and R. Goutarel, Tetrahedron Lett., 1973, 14, 1757–1760 CrossRef.
- P. G. Waterman and D. F. Faulkner, Phytochemistry, 1981, 20, 2765–2767 CrossRef CAS.
- Q. Guo, X.-M. Dai, W.-J. Lan, L.-P. Chen, C.-K. Lam, G.-K. Feng, R. Deng, X.-F. Zhu and H.-J. Li, Nat. Prod. Res., 2022, 36, 2534–2541 CrossRef CAS PubMed.
- S. Omura, T. Fujimoto, K. Otoguro, K. Matsuzaki, R. Moriguchi, H. Tanaka and Y. Sasaki, J. Antibiot., 1991, 44, 113–116 CrossRef CAS PubMed.
- S. Omura, K. Matsuzaki, T. Fujimoto, K. Kosuge, T. Furuya, S. Fujita and A. Nakagawa, J. Antibiot., 1991, 44, 117–118 CrossRef CAS.
- G. Fenteany and S. L. Schreiber, J. Biol. Chem., 1998, 273, 8545–8548 CrossRef CAS.
- R. H. Feling, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2003, 42, 355–357 CrossRef CAS.
- S. Chu, S. Liu, W. Duan, Y. Cheng, X. Jiang, C. Zhu, K. Tang, R. Wang, L. Xu, X. Wang, X. Yu, K. Wu, Y. Wang, M. Wang, H. Huang and J. Zhang, Pharmacol. Ther., 2016, 162, 179–187 CrossRef CAS.
- R. A. Duffy, C. Morgan, R. Naylor, G. A. Higgins, G. B. Varty, J. E. Lachowicz and E. M. Parker, Pharmacol., Biochem. Behav., 2012, 102, 95–100 CrossRef CAS PubMed.
- A. J. Wasserman and D. W. Richardson, Clin. Pharmacol. Therapeut., 1963, 4, 321–325 CrossRef CAS PubMed.
- B. Winblad, CNS Drug Rev., 2005, 11, 169–182 CrossRef CAS PubMed.
- J. Popovici-Muller, R. M. Lemieux, E. Artin, J. O. Saunders, F. G. Salituro, J. Travins, G. Cianchetta, Z. Cai, D. Zhou, D. Cui, P. Chen, K. Straley, E. Tobin, F. Wang, M. D. David, V. Penard-Lacronique, C. Quivoron, V. Saada, S. de Botton, S. Gross, L. Dang, H. Yang, L. Utley, Y. Chen, H. Kim, S. Jin, Z. Gu, G. Yao, Z. Luo, X. Lv, C. Fang, L. Yan, A. Olaharski, L. Silverman, S. Biller, S.-S. M. Su and K. Yen, ACS Med. Chem. Lett., 2018, 9, 300–305 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- X. Jiang, H. Su, W. Shang, F. Zhou, Y. Zhang, W. Zhao, Q. Zhang, H. Xie, L. Jiang, T. Nie, F. Yang, M. Xiong, X. Huang, M. Li, P. Chen, S. Peng, G. Xiao, H. Jiang, R. Tang, L. Zhang, J. Shen and Y. Xu, Nat. Commun., 2023, 14, 6463 CrossRef CAS PubMed.
- A. E. Gorbalenya, S. C. Baker, R. S. Baric, R. J. de Groot, C. Drosten, A. A. Gulyaeva, B. L. Haagmans, C. Lauber, A. M. Leontovich, B. W. Neuman, D. Penzar, S. Perlman, L. L. M. Poon, D. V. Samborskiy, I. A. Sidorov, I. Sola and J. Ziebuhr, Nat. Microbiol., 2020, 5, 536–544 CrossRef.
- P. V’kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Nat. Rev. Microbiol., 2021, 19, 155–170 CrossRef PubMed.
- L. V. Yevsieieva, K. O. Lohachova, A. Kyrychenko, S. M. Kovalenko, V. V. Ivanov and O. N. Kalugin, RSC Adv., 2023, 13, 35500–35524 RSC.
- G. Li, R. Hilgenfeld, R. Whitley and E. De Clercq, Nat. Rev. Drug Discovery, 2023, 22, 449–475 CrossRef CAS PubMed.
- Y. Duan, H. Wang, Z. Yuan and H. Yang, Curr. Opin. Struct. Biol., 2023, 82, 102667 CrossRef CAS PubMed.
- T. R. Malla, A. Tumber, T. John, L. Brewitz, C. Strain-Damerell, C. D. Owen, P. Lukacik, H. T. H. Chan, P. Maheswaran, E. Salah, F. Duarte, H. Yang, Z. Rao, M. A. Walsh and C. J. Schofield, Chem. Commun., 2021, 57, 1430–1433 RSC.
- T. R. Malla, L. Brewitz, D.-G. Muntean, H. Aslam, C. D. Owen, E. Salah, A. Tumber, P. Lukacik, C. Strain-Damerell, H. Mikolajek, M. A. Walsh and C. J. Schofield, J. Med. Chem., 2022, 65, 7682–7696 CrossRef CAS PubMed.
- D. Jelisejevs, A. L. Bula and L. Kinena, Bioorg. Med. Chem. Lett., 2023, 96, 129530 CrossRef CAS PubMed.
- K. S. Yang, S. Z. Leeuwon, S. Xu and W. R. Liu, J. Med. Chem., 2022, 65, 8686–8698 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- F. Rivas and T. Ling, Org. Prep. Proced. Int., 2016, 48, 254–295 CrossRef CAS.
- L.-W. Ye, C. Shu and F. Gagosz, Org. Biomol. Chem., 2014, 12, 1833–1845 RSC.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef PubMed.
- P. Imming, B. Klar and D. Dix, J. Med. Chem., 2000, 43, 4328–4331 CrossRef CAS PubMed.
- S. J. F. Macdonald, D. J. Belton, D. M. Buckley, J. E. Spooner, M. S. Anson, L. A. Harrison, K. Mills, R. J. Upton, M. D. Dowle, R. A. Smith, C. R. Molloy and C. Risley, J. Med. Chem., 1998, 41, 3919–3922 CrossRef CAS PubMed.
- S. J. F. Macdonald, M. D. Dowle, L. A. Harrison, P. Shah, M. R. Johnson, G. G. A. Inglis, G. D. E. Clarke, R. A. Smith, D. Humphreys, C. R. Molloy, A. Amour, M. Dixon, G. Murkitt, R. E. Godward, T. Padfield, T. Skarzynski, O. M. P. Singh, K. A. Kumar, G. Fleetwood, S. T. Hodgson, G. W. Hardy and H. Finch, Bioorg. Med. Chem. Lett., 2001, 11, 895–898 CrossRef CAS PubMed.
- M. E. Migaud, R. C. Wilmouth, G. I. Mills, G. J. Wayne, C. Risley, C. Chambers, S. J. F. Macdonald and C. J. Schofield, Chem. Commun., 2002, 1274–1275 RSC.
- V. Chung, A. R. Carroll, N. M. Gray, N. R. Parry, P. A. Thommes, K. C. Viner and E. A. D'Souza, Antimicrob. Agents Chemother., 2005, 49, 1381–1390 CrossRef CAS PubMed.
- M. J. Slater, D. M. Andrews, G. Baker, S. S. Bethell, S. Carey, H. Chaignot, B. Clarke, B. Coomber, M. Ellis, A. Good, N. Gray, G. Hardy, P. Jones, G. Mills and E. Robinson, Bioorg. Med. Chem. Lett., 2002, 12, 3359–3362 CrossRef CAS PubMed.
- D. M. Andrews, P. S. Jones, G. Mills, S. L. Hind, M. J. Slater, N. Trivedi and K. J. Wareing, Bioorg. Med. Chem. Lett., 2003, 13, 1657–1660 CrossRef CAS PubMed.
- D. M. Andrews, M. C. Barnes, M. D. Dowle, S. L. Hind, M. R. Johnson, P. S. Jones, G. Mills, A. Patikis, T. J. Pateman, T. J. Redfern, J. E. Robinson, M. J. Slater and N. Trivedi, Org. Lett., 2003, 5, 4631–4634 CrossRef CAS PubMed.
- A. D. Borthwick, S. J. Angier, A. J. Crame, A. M. Exall, T. M. Haley, G. J. Hart, A. M. Mason, A. M. K. Pennell and G. G. Weingarten, J. Med. Chem., 2000, 43, 4452–4464 CrossRef CAS PubMed.
- A. D. Borthwick, D. E. Davies, P. F. Ertl, A. M. Exall, T. M. Haley, G. J. Hart, D. L. Jackson, N. R. Parry, A. Patikis, N. Trivedi, G. G. Weingarten and J. M. Woolven, J. Med. Chem., 2003, 46, 4428–4449 CrossRef CAS PubMed.
- K. A. Scheidt, W. R. Roush, J. H. McKerrow, P. M. Selzer, E. Hansell and P. J. Rosenthal, Bioorg. Med. Chem., 1998, 6, 2477–2494 CrossRef CAS PubMed.
- J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- L. Brewitz, L. Dumjahn, Y. Zhao, C. D. Owen, S. M. Laidlaw, T. R. Malla, D. Nguyen, P. Lukacik, E. Salah, A. D. Crawshaw, A. J. Warren, J. Trincao, C. Strain-Damerell, M. W. Carroll, M. A. Walsh and C. J. Schofield, J. Med. Chem., 2023, 66, 2663–2680 CrossRef CAS PubMed.
- X. Li and Y. Song, Eur. J. Med. Chem., 2023, 260, 115772 CrossRef CAS PubMed.
- Y. L. Janin, RSC Med. Chem., 2024, 15, 81–118 RSC.
- S. H. Watterson, Q. Liu, M. Beaudoin Bertrand, D. G. Batt, L. Li, M. A. Pattoli, S. Skala, L. Cheng, M. T. Obermeier, R. Moore, Z. Yang, R. Vickery, P. A. Elzinga, L. Discenza, C. D'Arienzo, K. M. Gillooly, T. L. Taylor, C. Pulicicchio, Y. Zhang, E. Heimrich, K. W. McIntyre, Q. Ruan, R. A. Westhouse, I. M. Catlett, N. Zheng, C. Chaudhry, J. Dai, M. A. Galella, A. J. Tebben, M. Pokross, J. Li, R. Zhao, D. Smith, R. Rampulla, A. Allentoff, M. A. Wallace, A. Mathur, L. Salter-Cid, J. E. Macor, P. H. Carter, A. Fura, J. R. Burke and J. A. Tino, J. Med. Chem., 2019, 62, 3228–3250 CrossRef CAS PubMed.
- R. C. Wilmouth, S. Kassamally, N. J. Westwood, R. J. Sheppard, T. D. W. Claridge, R. T. Aplin, P. A. Wright, G. J. Pritchard and C. J. Schofield, Biochemistry, 1999, 38, 7989–7998 CrossRef CAS PubMed.
- R. C. Wilmouth, N. J. Westwood, K. Anderson, W. Brownlee, T. D. W. Claridge, I. J. Clifton, G. J. Pritchard, R. T. Aplin and C. J. Schofield, Biochemistry, 1998, 37, 17506–17513 CrossRef CAS PubMed.
- M. A. Redhead, C. D. Owen, L. Brewitz, A. H. Collette, P. Lukacik, C. Strain-Damerell, S. W. Robinson, P. M. Collins, P. Schäfer, M. Swindells, C. J. Radoux, I. N. Hopkins, D. Fearon, A. Douangamath, F. von Delft, T. R. Malla, L. Vangeel, T. Vercruysse, J. Thibaut, P. Leyssen, T.-T. Nguyen, M. Hull, A. Tumber, D. J. Hallett, C. J. Schofield, D. I. Stuart, A. L. Hopkins and M. A. Walsh, Sci. Rep., 2021, 11, 13208 CrossRef CAS PubMed.
- S. T. D. Thun-Hohenstein, T. F. Suits, T. R. Malla, A. Tumber, L. Brewitz, H. Choudhry, E. Salah and C. J. Schofield, ChemMedChem, 2022, 17, e202100582 CrossRef CAS PubMed.
- T. Miura, T. R. Malla, C. D. Owen, A. Tumber, L. Brewitz, M. A. McDonough, E. Salah, N. Terasaka, T. Katoh, P. Lukacik, C. Strain-Damerell, H. Mikolajek, M. A. Walsh, A. Kawamura, C. J. Schofield and H. Suga, Nat. Chem., 2023, 15, 998–1005 CrossRef CAS PubMed.
- M. de Munnik, J. Lithgow, L. Brewitz, K. E. Christensen, R. H. Bates, B. Rodriguez-Miquel and C. J. Schofield, Chem. Commun., 2023, 59, 12859–12862 RSC.
- B. Mahjour, R. Zhang, Y. Shen, A. McGrath, R. Zhao, O. G. Mohamed, Y. Lin, Z. Zhang, J. L. Douthwaite, A. Tripathi and T. Cernak, Nat. Commun., 2023, 14, 3924 CrossRef CAS PubMed.
- Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu, T. Suto, K. Kouki, A. Nakahashi, S. Kawashima, T. Sanaki, S. Toba, K. Uemura, T. Mizutare, S. Ando, M. Sasaki, Y. Orba, H. Sawa, A. Sato, T. Sato, T. Kato and Y. Tachibana, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed.
- M. L. Boby, D. Fearon, M. Ferla, M. Filep, L. Koekemoer, M. C. Robinson, The Covid Moonshot Consortium, J. D. Chodera, A. A. Lee, N. London, A. von Delft and F. von Delft, Science, 2023, 382, eabo7201 CrossRef CAS PubMed.
- T. Miura, T. R. Malla, L. Brewitz, A. Tumber, E. Salah, K. J. Lee, N. Terasaka, C. D. Owen, C. Strain-Damerell, P. Lukacik, M. A. Walsh, A. Kawamura, C. J. Schofield, T. Katoh and H. Suga, Bull. Chem. Soc. Jpn., 2024 DOI:10.1093/bulcsj/uoae018.
- L. Brewitz, J. J. A. G. Kamps, P. Lukacik, C. Strain-Damerell, Y. Zhao, A. Tumber, T. R. Malla, A. M. Orville, M. A. Walsh and C. J. Schofield, ChemMedChem, 2022, 17, e202200016 CrossRef CAS PubMed.
- J. P. Michael, C. B. de Koning, C. W. van der Westhuyzen and M. A. Fernandes, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 2055–2062 RSC.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS.
- S. Hünig and M. Kiessel, Chem. Ber., 1958, 91, 380–392 CrossRef.
- A. J. S. Alves, N. G. Alves, M. I. L. Soares and T. M. V. D. Pinho e Melo, Org. Chem. Front., 2021, 8, 3543–3593 RSC.
- I. G. C. Coutts and M. Hamblin, J. Chem. Soc., Perkin Trans. 1, 1975, 1, 2445–2446 RSC.
- A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS PubMed.
- H. Li, S. Zhu and G. Zheng, Bioorg. Med. Chem. Lett., 2018, 28, 1071–1076 CrossRef CAS PubMed.
- K. Line, M. N. Isupov and J. A. Littlechild, J. Mol. Biol., 2004, 338, 519–532 CrossRef CAS PubMed.
- S. Gao, Y. Zhou, W. Zhang, W. Wang, Y. Yu, Y. Mu, H. Wang, X. Gong, G. Zheng and Y. Feng, Sci. Rep., 2017, 7, 44542 CrossRef PubMed.
- Y. Su, S. Gao, H. Li and G. Zheng, Process Biochem., 2018, 72, 96–104 CrossRef CAS.
- S. Zhu and G. Zheng, J. Ind. Microbiol. Biotechnol., 2018, 45, 1017–1031 CrossRef CAS PubMed.
- A. Zapun, C. Contreras-Martel and T. Vernet, FEMS Microbiol. Rev., 2008, 32, 361–385 CrossRef CAS PubMed.
- Y. Nozaki, N. Katayama, H. Ono, S. Tsubotani, S. Harada, H. Okazaki and Y. Nakao, Nature, 1987, 325, 179–180 CrossRef CAS PubMed.
- Y. Nozaki, N. Katayama, S. Harada, H. Ono and H. Okazaki, J. Antibiot., 1989, 42, 84–93 CrossRef CAS PubMed.
- J. E. Baldwin, C. Lowe and C. J. Schofield, Tetrahedron Lett., 1990, 31, 2211–2212 CrossRef CAS.
- J. E. Baldwin, C. Lowe, C. J. Schofield and E. Lee, Tetrahedron Lett., 1986, 27, 3461–3464 CrossRef CAS.
- D. B. Boyd, T. K. Elzey, L. D. Hatfield, M. D. Kinnick and J. M. Morin, Tetrahedron Lett., 1986, 27, 3453–3456 CrossRef CAS.
- D. B. Boyd, B. J. Foster, L. D. Hatfield, W. J. Hornback, N. D. Jones, J. E. Munroe and J. K. Swartzendruber, Tetrahedron Lett., 1986, 27, 3457–3460 CrossRef CAS.
- J. E. Baldwin, G. P. Lynch and J. Pitlik, J. Antibiot., 1991, 44, 1–24 CrossRef CAS PubMed.
- T. F. Durand-Reville, A. A. Miller, J. P. O'Donnell, X. Wu, M. A. Sylvester, S. Guler, R. Iyer, A. B. Shapiro, N. M. Carter, C. Velez-Vega, S. H. Moussa, S. M. McLeod, A. Chen, A. M. Tanudra, J. Zhang, J. Comita-Prevoir, J. A. Romero, H. Huynh, A. D. Ferguson, P. S. Horanyi, S. J. Mayclin, H. S. Heine, G. L. Drusano, J. E. Cummings, R. A. Slayden and R. A. Tommasi, Nature, 2021, 597, 698–702 CrossRef CAS PubMed.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063–1084 CrossRef CAS PubMed.
- T. A. Blizzard, H. Chen, S. Kim, J. Wu, R. Bodner, C. Gude, J. Imbriglio, K. Young, Y.-W. Park, A. Ogawa, S. Raghoobar, N. Hairston, R. E. Painter, D. Wisniewski, G. Scapin, P. Fitzgerald, N. Sharma, J. Lu, S. Ha, J. Hermes and M. L. Hammond, Bioorg. Med. Chem. Lett., 2014, 24, 780–785 CrossRef CAS PubMed.
- M. Fujiu, K. Yokoo, T. Aoki, S. Shibuya, J. Sato, K. Komano, H. Kusano, S. Sato, M. Ogawa and K. Yamawaki, J. Org. Chem., 2020, 85, 9650–9660 CrossRef CAS PubMed.
- C. González-Bello, D. Rodríguez, M. Pernas, Á. Rodríguez and E. Colchón, J. Med. Chem., 2020, 63, 1859–1881 CrossRef PubMed.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS PubMed.
- J. A. Goldberg, V. Kumar, E. J. Spencer, D. Hoyer, S. H. Marshall, A. M. Hujer, K. M. Hujer, C. R. Bethel, K. M. Papp-Wallace, F. Perez, M. R. Jacobs, D. van Duin, B. N. Kreiswirth, F. van den Akker, M. S. Plummer and R. A. Bonomo, Eur. J. Med. Chem., 2021, 220, 113436 CrossRef CAS PubMed.
- E. L. Setti, D. Davis, T. Chung and J. McCarter, Bioorg. Med. Chem. Lett., 2003, 13, 2051–2053 CrossRef CAS PubMed.
- N. E. Zhou, D. Guo, G. Thomas, A. V. N. Reddy, J. Kaleta, E. Purisima, R. Menard, R. G. Micetich and R. Singh, Bioorg. Med. Chem. Lett., 2003, 13, 139–141 CrossRef CAS PubMed.
- J. W. Skiles and D. McNeil, Tetrahedron Lett., 1990, 31, 7277–7280 CrossRef CAS.
- M. Cordillot, V. Dubée, S. Triboulet, L. Dubost, A. Marie, J.-E. Hugonnet, M. Arthur and J.-L. Mainardi, Antimicrob. Agents Chemother., 2013, 57, 5940–5945 CrossRef CAS PubMed.
- P. Kumar, A. Kaushik, E. P. Lloyd, S.-G. Li, R. Mattoo, N. C. Ammerman, D. T. Bell, A. L. Perryman, T. A. Zandi, S. Ekins, S. L. Ginell, C. A. Townsend, J. S. Freundlich and G. Lamichhane, Nat. Chem. Biol., 2017, 13, 54–61 CrossRef CAS PubMed.
- M. de Munnik, C. T. Lohans, G. W. Langley, C. Bon, J. Brem and C. J. Schofield, ChemBioChem, 2020, 21, 368–372 CrossRef CAS PubMed.
- E. M. Steiner, G. Schneider and R. Schnell, FEBS J., 2017, 284, 725–741 CrossRef CAS PubMed.
- N. O. Sykes, S. J. F. Macdonald and M. I. Page, J. Med. Chem., 2002, 45, 2850–2856 CrossRef CAS PubMed.
- A.-T. Ton, M. Pandey, J. R. Smith, F. Ban, M. Fernandez and A. Cherkasov, Trends Pharmacol. Sci., 2022, 43, 906–919 CrossRef CAS PubMed.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K.-P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Nature, 2020, 587, 657–662 CrossRef CAS PubMed.
- J. Lei, Y. Kusov and R. Hilgenfeld, Antiviral Res., 2018, 149, 58–74 CrossRef CAS PubMed.
- M. S. Lall, C. J. Karvellas and J. C. Vederas, Org. Lett., 1999, 1, 803–806 CrossRef CAS PubMed.
- M. S. Lall, Y. K. Ramtohul, M. N. G. James and J. C. Vederas, J. Org. Chem., 2002, 67, 1536–1547 CrossRef CAS PubMed.
- J. Yin, E. M. Bergmann, M. M. Cherney, M. S. Lall, R. P. Jain, J. C. Vederas and M. N. G. James, J. Mol. Biol., 2005, 354, 854–871 CrossRef CAS PubMed.
- Z. Wang, C. Gu, T. Colby, T. Shindo, R. Balamurugan, H. Waldmann, M. Kaiser and R. A. L. van der Hoorn, Nat. Chem. Biol., 2008, 4, 557–563 CrossRef CAS PubMed.
- P. S. Hinchliffe, J. M. Wood, A. M. Davis, R. P. Austin, R. P. Beckett and M. I. Page, Org. Biomol. Chem., 2003, 1, 67–80 RSC.
- M. I. Page, Acc. Chem. Res., 2004, 37, 297–303 CrossRef CAS PubMed.
- J. Seibel, S. J. Macdonald and C. J. Schofield, J. Chem. Res., 2005, 826–832 CrossRef CAS.
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes and Y.-Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01027b |
| This journal is © The Royal Society of Chemistry 2024 |