
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-cation-induced shifts in thiolate redox and reduced sulfur speciation†
W. T. Michael
Seo
 ,
Madeline N.
Riffel
,
Allen G.
Oliver
and
Emily Y.
Tsui
*
,
Madeline N.
Riffel
,
Allen G.
Oliver
and
Emily Y.
Tsui
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN, USA. E-mail: etsui@nd.edu
First published on 17th April 2024
Abstract
Sulfur-containing anions (e.g. thiolates, polysulfides) readily exchange in solution, making control over their solution speciation and distribution challenging. Here, we demonstrate that different redox-inactive alkali, alkaline earth, and transition metals (Li+, Na+, K+, Mg2+, Ca2+, Zn2+, and Cd2+) shift the equilibria of sulfur catenation or sulfur reduction/oxidation between thiolate, polysulfanide, and polysulfide anions in acetonitrile solution. The thermodynamic factors that govern these equilibria are examined by identification of intermediate metal thiolate and metal polysulfide species using a combination of NMR spectroscopy, electronic absorption spectroscopy, and mass spectrometry. Electrochemical measurements demonstrate that the metal cation of the electrolyte modulates both sulfur reduction and thiolate oxidation potentials. DFT calculations suggest that the changes in equilibria are driven by stronger covalent interactions between polysulfide anions and more highly charged cations.
Introduction
The solution chemistry of sulfur and sulfur-containing organic compounds (e.g. thiols, organic disulfides, etc.) is critically important in several disparate areas. For example, in energy storage technologies like Li–S batteries, the exchange and diffusion of soluble polysulfide anions formed upon S8 reduction at the cathode has been a major challenge.1 Polysulfide anions have also been invoked in C–S bond-forming reactions in the synthesis of pharmacologically-relevant sulfur-containing organic molecules.2–5 Lastly, reduction of elemental sulfur by amines has been demonstrated to form relevant precursors in the inorganic synthesis of metal chalcogenide semiconductor nanomaterials.6,7A major challenge for mechanistic understanding in each of these research areas is the complex nature of sulfur speciation. Chalcogens other than oxygen (e.g. sulfur, selenium, tellurium) readily form extended σ-bonded chains by catenation, an example of which is the reaction of S2− with zerovalent sulfur (S0) in the form of S8 to form a distribution of polysulfide dianions (eqn (1)).8 In a related reaction, disulfanide anions formed from S0 catenation with benzenethiolate and benzylthiolate have been characterized by single crystal X-ray diffraction (XRD) studies (eqn (2)).9–11
| S2− + S8 ⇌ Sx2− (x = 2–9) | (1) |
| RS− + S8 ⇌ RSx− (x ≥ 1, R = alkyl or aryl) | (2) |
At the same time, oxidation and reduction of sulfur compounds occur at relatively mild potentials. For example, S8 reduction forms polysulfide anions of different lengths (eqn (3) and (4)). Organosulfur compounds with electron-rich sulfur centers react similarly; thiolate anions are readily oxidized to the corresponding organic disulfide compounds (eqn (5)). Since sulfur catenation and redox reactions occur under similar conditions, it can be difficult to characterize or control the solution distribution of both inorganic and organic sulfur products, despite the importance of these equilibria in determining their final reactivity and outcome in their applications.
| S8 + 2e− → S82− | (3) |
| S82− + 2e− → 2S42− | (4) |
| 2RS− → RSSR + 2e− (R = alkyl, aryl) | (5) |
Metal ions can interact strongly with sulfur-containing anions, for example in transition-metal-stabilized polysulfide complexes.12–16 Polysulfide and disulfanide anions have also been crystallographically characterized with Li–S and K–S interactions, respectively.10,17 Lewis–acidic interactions between sulfur and metal cations or with B(C6F5)3 have also been demonstrated to shift the reduction potentials of elemental sulfur.18,19 Despite evidence for metal ion influence in reduced sulfur speciation provided by these reports, the factors governing the influence of metal ions on sulfur catenation vs. reduction to polysulfide anions remain unclear. Although metal ion effects on the electrochemistry of S8 have been investigated between the alkali metal ions18 and between Li+ and Mg2+,20,21 a systematic study of these effects with a wider range of metal cations under the same conditions has not yet been performed. Cation effects on polysulfanide anions has not yet been studied in detail.
Here, we combine NMR and electronic absorption spectroscopy with electrochemical studies and DFT calculations to investigate how redox-inactive metal cations affect the solution speciation of reduced sulfur compounds. Non-coordinating and alkali cations (Et4N+, tetrabutylammonium [TBA+], Li+, Na+, K+) favor sulfur catenation to thiolates to form disulfanide and polysulfanide anions. In contrast, alkaline earth (Mg2+, Ca2+) and redox-inert d-block metal cations (Zn2+, Cd2+) shift the equilibria of the same mixtures to favor S8 reduction to metal polysulfides and thiolate oxidation to organic disulfides. These processes appear to be dictated by stronger metal–sulfur covalency between the polysulfide anions and more highly charged metal ions, rather than by modulation of the electrochemical potentials. Implications for energy storage are discussed.
Results and analysis
Metal cations shift thiolate/polysulfanide/disulfide equilibria
Addition of S8 (1 S atom equiv.) to a CD3CN solution of [Et4N][p-tolS] ([Et4N][1Me]) forms a green mixture. The 1H NMR spectrum of this solution shows the formation of a new p-tolyl-containing major product ([2Me]−, 45% NMR conversion, Fig. 2 top). Based on a previous report by Chen and co-workers9 and in conjunction with negative ion mode electrospray ionization mass spectrometry (ESI-MS) data (Fig. S51†), [2Me]− is assigned as the p-tolyl disulfanide anion, p-tolSS− (Scheme 1). Resonances corresponding to higher order arylpolysulfanide anions (p-tolSx−, x ≥ 3) are also observed in the NMR spectrum (ca. 25% collectively). Addition of more S8 to the mixture results in greater conversion to [2Me]−, as well as formation of di(p-tolyl) disulfide (3Me). Electronic absorption spectroscopy of this mixture indicates the formation of S3˙− (λmax = 611 nm) and other polysulfide anions (Sx2−); these anions give rise to the green color of the mixture.22Scheme 1 shows the exchange of these reduced sulfur species.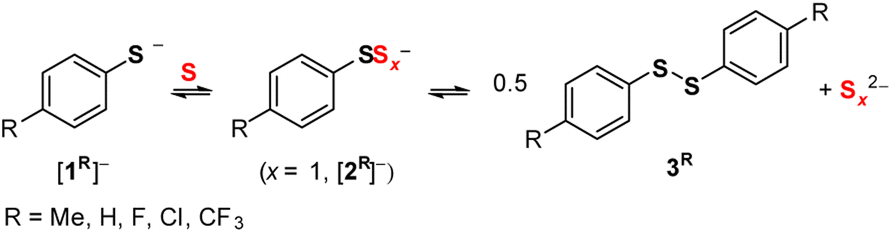 | ||
| Scheme 1 Substituted benzenethiolate anions ([1R]−) catenate with S8 (here abbreviated as S for 1/8 S8) to form disulfanide ([2R]−) anions, polysulfanide anions, and diaryl disulfides (3R). | ||
CD3CN mixtures of S8 (1 S atom equiv.) with tetraethylammonium salts of other p-substituted benzenethiolate anions ([Et4N][1R], R = Cl, F, CF3) exhibit substituent-dependent behavior. The 1H NMR spectra of mixtures prepared with [1F]− and [1Cl]− show a mixture of products with significantly broadened resonances, while the [1CF3]− reaction mixture shows only one broad set of signals that are shifted from those of the starting benzenethiolate anion (Fig. S21–S23†). We were unable to quantitatively compare the polysulfanide anion speciation of these mixtures; while basicity of the thiolate may affect S0 catenation to form the corresponding [2R]− anions, the broadness of the resonances may also indicate that more electron-poor thiolates exhibit different rates of sulfur exchange.
These acetonitrile solutions of [1R]− and S8 exhibit substituent-dependent colors; while the [1Me]− and S8 mixture is green, that of [1CF3]− and S8 is red. As such, although the 1H NMR data do not provide a quantitative assessment of relative benzenethiolate/polysulfanide concentrations, absorption spectroscopy quantified the concentration of S3˙− formed in these mixtures (Fig. 1A).23Fig. 1B plots [S3˙−] against the Hammett σ parameter of the para-substituent on the benzenethiolate anion. An increase in the σ parameter is correlated to decreasing benzenethiol pKa (4-CH3: 9.3, 4-F: 8.1, 4-Cl: 7.8 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone/water at 27 °C).24 This correlation indicates that more electron-rich thiolate anions more readily reduce zerovalent sulfur to polysulfide anions. Thus, the equilibrium reaction shown in Scheme 1 lies further to the right.
:1 acetone/water at 27 °C).24 This correlation indicates that more electron-rich thiolate anions more readily reduce zerovalent sulfur to polysulfide anions. Thus, the equilibrium reaction shown in Scheme 1 lies further to the right.
 | ||
| Fig. 1 (A) Electronic absorption spectra of CH3CN solutions of [1R]− (2.30 × 10−4 M) treated with S8 (1 S atom equiv.). (B) Negative log of [S3˙−] calculated from A613 nm (ref. 23) plotted against Hammett σ parameter of R. | ||
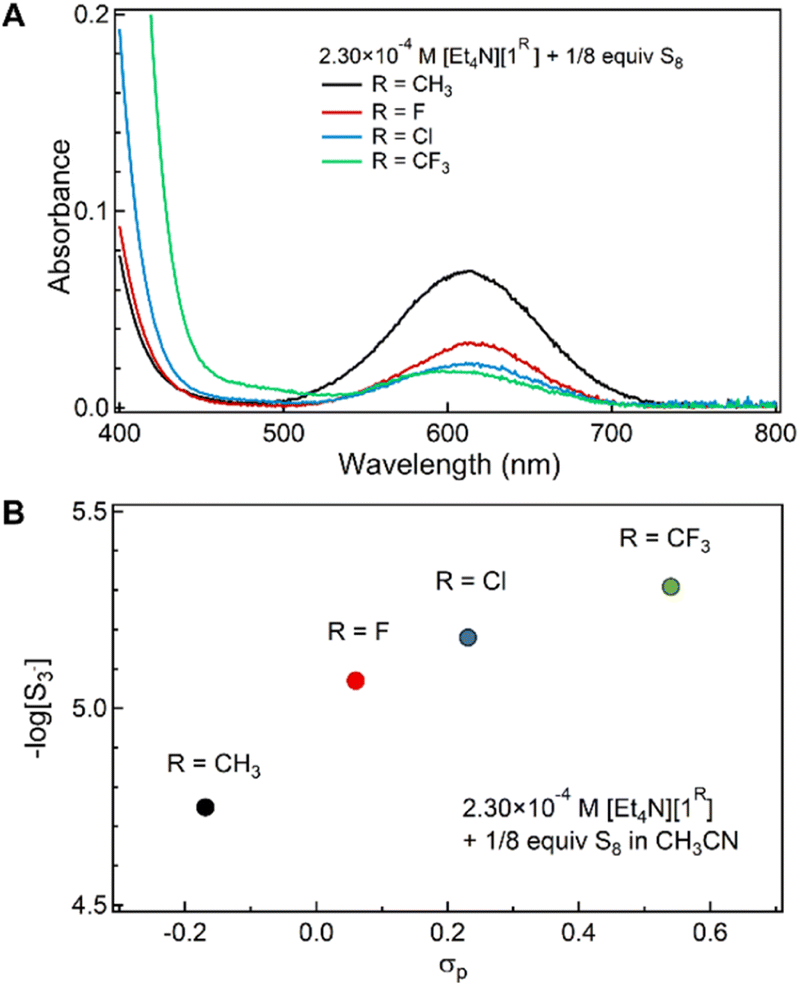
Fig. 2 shows the 1H NMR spectra of CD3CN mixtures of [Et4N][1Me] and S8 (1 S atom equiv.) upon addition of alkali metal triflate salts (MOTf, M+ = Li+, Na+, and K+). Compared with the mixtures with only Et4N+ as the counter-cation, the spectra for mixtures with added alkali metal ions showed no change in the distribution of [1Me]−, [2Me]−, and higher p-tolSx− species, although the 1H NMR resonances broadened with increasing metal triflate concentration (FWHM ∼ 12 to 36 Hz). This broadening may indicate faster exchange between reduced sulfur species in solution on the NMR time scale, although the chemical shifts of the resonances shift only minimally (<0.01 ppm). Increasing the amount of LiOTf added to 10 equiv. results only in slightly more broadening of the reaction mixture 1H NMR peaks (Fig. S33†). This broadening in the spectra is related to sulfur exchange, as addition of LiOTf (1 equiv.) to a solution of [1Me]− in CD3CN results in minor downfield shifts in the signals (0.02–0.08 ppm), which remain well-defined (Fig. S28†). 1H NMR spectroscopy of a solution of [Et4N][1Me], 1 equiv. S, and 0.5 equiv. LiOTf in CD3CN cooled to 260 K permits resolution of the broadened NMR features corresponding to p-tolS3− and p-tolS4− (Fig. S34†). ESI-MS of the mixture with added LiOTf (1 equiv.) shows a similar distribution of [1Me]−, [2Me]−, and higher order polysulfanide products (m/z = 123.0350, 155.0030, 186.9765, 218.9491, Fig. S52†).
 | ||
| Fig. 2 1H NMR spectra of CD3CN solutions of [Et4N][1Me] and S8 (1 S atom equiv.) with added alkali or alkaline earth metal triflates. Peak features corresponding to [1Me]− (blue), [2Me]− (red), and higher-order arylpolysulfanide species (green) are indicated. For Mg(OTf)2 and Ca(OTf)2, the broad chemical resonances match those of 3Me. | ||
Fig. 2 also shows that when the same CD3CN mixtures of [Et4N][1Me] of S8 are instead treated with alkaline earth metal triflate salts [M(OTf)2, M2+ = Mg2+, Ca2+], the 1H NMR resonances of the aromatic and benzylic protons gradually shift with increasing [M(OTf)2], approaching the chemical shifts of independently prepared di-p-tolyl disulfide [(p-tolS)2, 3Me] at higher metal ion concentrations (ca. 0.5 equiv.).
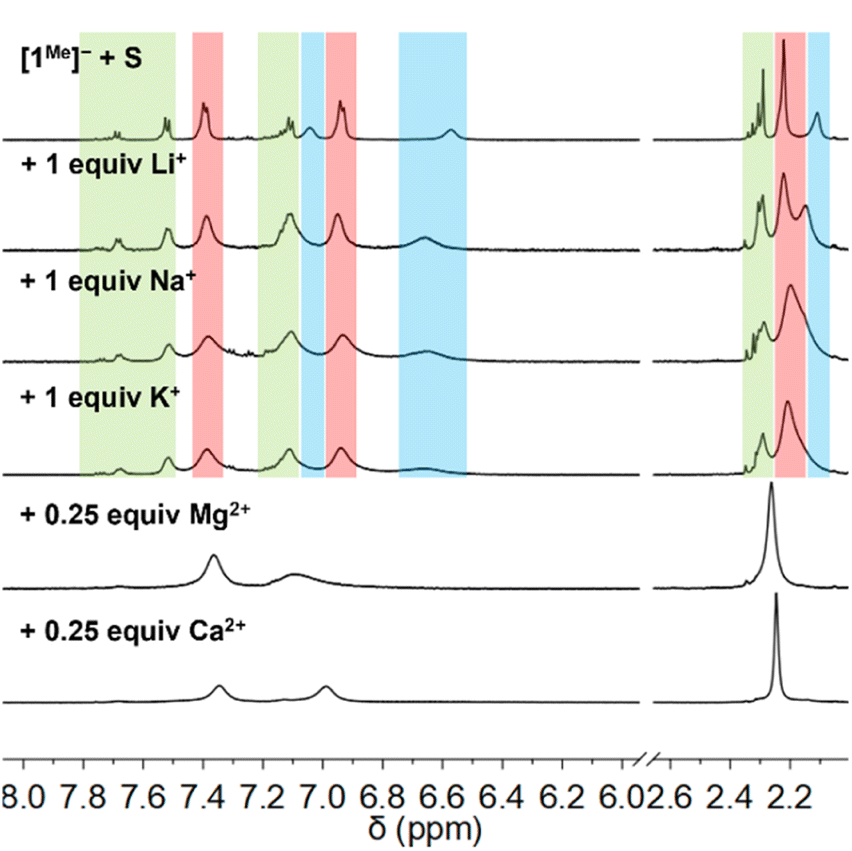
Fig. 3A plots the room temperature electronic absorption spectrum of a CH3CN solution of [Et4N][1Me] (77 μM) and S8 (1 S atom equiv.). As described above, a broad absorbance band is observed (λmax = 611 nm) that corresponds to S3˙−. The absorption feature at 304 nm is also slightly red-shifted from the absorption spectrum of only [1Me]− and exhibits a lower-energy shoulder (Fig. S58†), consistent with previously reported delocalization of the negative charge of the phenyldisulfanide anion and TD-DFT calculations (Fig. S89†).9 When increasing amounts of LiOTf (0 to 1 equiv.) are added to this solution, the band at 611 nm bleaches (Fig. 3B, ca. 44% at 1 equiv. LiOTf) and the band at 307 nm corresponding to [2Me]− also decreases in intensity (ca. 15% at 1 equiv. LiOTf). Similar absorption bleaches are observed when the same mixture of [1Me]− and S8 is instead treated with 1 equiv. NaOTf (40% S3˙−, 16% [2Me]−) or KOTf (27% S3˙−, 16% [2Me]−, Fig. S60 and S61†).
 | ||
| Fig. 3 Electronic absorption spectra of a CH3CN mixture of [Et4N][1Me] and S8 (1 S atom equiv.) treated with 1 equiv. LiOTf, showing absorbance decreases at (A) 304 nm (7.67 × 10−5 M [1Me]−) and (B) 611 nm (2.30 × 10−4 M [1Me]−). (C) Electronic absorption spectra of a CH3CN mixture of [Et4N][1Me] (7.67 × 10−5 M) and S8 (1 S atom equiv.) treated with Mg(OTf)2 (1 equiv.). (D) Absorbances at 304 nm of a CH3CN mixture of [Et4N][1Me] (7.67 × 10−5 M) and S8 (1 S atom equiv.) treated with 1 equiv. cation (group 1 as circles, where red: Li+, green: Na+, blue: K+; group 2 as squares, where red: Mg2+, green: Ca2+; group 12 as diamonds, where orange: Zn2+, purple: Cd2+). | ||
The electronic absorption spectra of acetonitrile mixtures of [1Me]− and S8 with addition of Mg(OTf)2 or Ca(OTf)2 show that the features corresponding to S3˙− (λ = 611 nm) and [2Me]− (λ = 304 nm) are bleached and a new absorbance at 243 nm is observed (Fig. 3C for M2+ = Mg2+). This new band corresponds to the formation of the disulfide compound 3Me (Fig. S57†). This bleaching occurs with an increased response to metal triflate concentration compared with the same experiments using the alkali metal triflate salts (Fig. 3D). At higher concentrations of Mg2+ or Ca2+ (>0.5 equiv.), solid material precipitates. Based on previous literature that reports the much lower solubility of Mg2+ polysulfides as compared with those of Li+,20 we initially hypothesized that these solids are Mg and Ca polysulfide species. However, powder X-ray diffraction (PXRD) analysis does not match any known MgS or CaS compounds, nor independently prepared mixtures of metal cation and thiolate (Fig. S86†).
As a control experiment, CD3CN solutions of [Et4N][1Me] were treated with the metal triflate salts with no added S8 in order to compare only metal–S(thiolate) interactions. Unlike LiOTf (described above), addition of Mg(OTf)2 or Ca(OTf)2 to solutions of [Et4N][1Me] results in significant broadening and downfield shifts (ca. 0.11–0.30 ppm for Mg2+) of the aromatic 1H NMR resonances (Fig. S27†). These shifts of the NMR resonances are consistent with stronger association of thiolate anion to these divalent cations. Absorbance spectroscopy data supports this hypothesis, as a solution of [1Me]2− in CH3CN treated with 0–1 equiv. Mg2+ shows consumption of starting thiolate features and growth of absorbances likely corresponding to Mg thiolate species (Fig. S62†).
In a second control experiment, CH3CN solutions of [TBA]2[S6] were treated with the same metal triflate salts, with no added benzenethiolate. S62− is in dynamic exchange with S3˙−, favoring the radical at dilute concentrations, and consumption of the polysulfide anions can be monitored by measurement of the absorption feature at 611 nm. Addition of LiOTf (1 equiv.) results in an 89% decrease in [S3˙−]. Addition of NaOTf and KOTf resulted in 64% and 71% consumption, respectively. As expected, treatment with Mg(OTf)2 and Ca(OTf)2 (0.5 equiv.) of the same solution results in near-quantitative consumption of polysulfide (Fig. S70 and S71†).
For the experiments discussed above and below, we chose acetonitrile as the solvent for its ability to solubilize each of the components, including the metal triflate salts, metal thiolate/polysulfide complexes, and the tetraethylammonium arenethiolate salts. The use of less coordinating solvents such as ethers was challenging due to the low solubility of non-lithium-containing metal salts. Halogenated solvents (e.g., chloroform) showed similar poor solubilization of the metal salts and also underwent nucleophilic substitution reactions with the thiolate anions.
Taken together, these data show that the interactions between metal cations and both thiolate or polysulfide anions for the divalent alkaline earth metals are strong, but are weaker for the alkali metals, with no apparent dependence on metal ion size. The stronger metal–polysulfide interactions of Mg2+ and Ca2+ drive the equilibria shown in Scheme 1 forward, favoring the formation of polysulfide anions and organic disulfides (sulfur redox) and disfavoring polysulfanide anions (sulfur catenation).
Transition metal effects on reduced sulfur species equilibria
The 1H NMR spectrum of a CD3CN solution of [Et4N][1Me] and S8 (1 S atom equiv.) with added Zn(OTf)2 (1 equiv.) shows two p-tolyl-containing species in addition to 3Me, but no [1Me]− or [2Me]−. These new species were identified as the tetra(4-methylbenzenethiolato)zincate dianion [(p-tolS)4Zn]2− ([4Me]2−) and the di(4-methylbenzenethiolato)zinc(II) polysulfide dianion, [(p-tolS)2ZnSx]2− (x = 4–6, [5Me]2−). Analogous benzenethiolate-supported zinc complexes [(PhS)2ZnS4]2− ([5H]2−) and [(PhS)2ZnS5]2− ([4H]2−) have been previously reported by Coucouvanis and co-workers and synthesized by treatment of [(PhS)4Zn]2− with BnSSSBn.12 This reaction was presumed to proceed via benzenethiolate oxidation to PhSSPh, along with sulfur reduction to polysulfide anions (Scheme 2). To confirm these assignments and further study their reactions with S8, a series of analogous tetraethylammonium zinc tetrathiolate salts ([Et4N]2[4R], R = Me, Cl, F, CF3) with para-substituted benzenethiolate ligands was synthesized and characterized by NMR spectroscopy, combustion analysis, and, in the case of [Et4N]2[4Me], ESI-MS and XRD.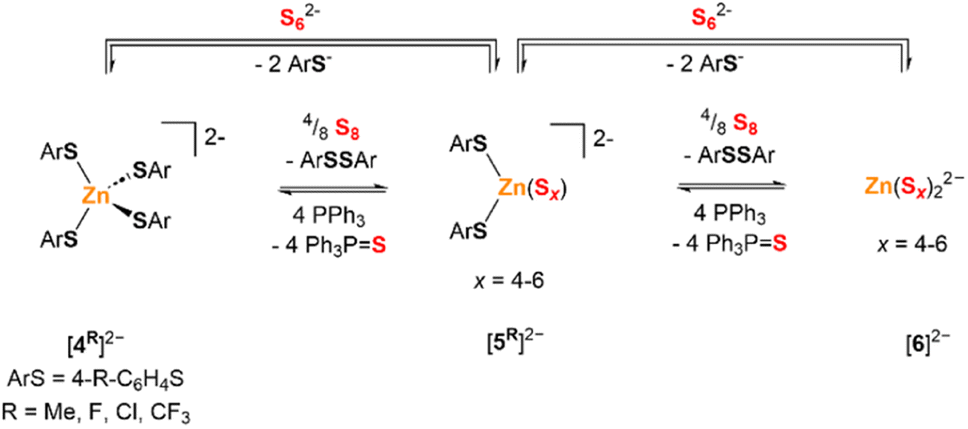 | ||
| Scheme 2 Formation of zinc polysulfide products [5R]2− and [6]2−via reduction of elemental sulfur by zinc thiolate or via substitution of thiolate by polysulfide anion. | ||
1H NMR spectroscopy of mixtures of [Et4N]2[4Me] with additional S8 (8 S atom equiv.) under the same conditions shows only NMR resonances corresponding to 3Me and Et4N+, consistent with complete oxidation of thiolate and formation of the bis(polysulfido)zincate species [6]2− (Scheme 2). The bis(tetrasulfido)zincate anion has been previously reported formation upon treatment of [4H]2− with BnSSSBn,12 while the bis(hexasulfido)zincate dianion has also been reported under other conditions.25 These different polysulfide nuclearities likely exchange in solution with free S8 under our reaction conditions. Fig. 4 shows the 1H NMR spectra of a CD3CN solution of [4Me]2− treated with increasing amounts of S8 to form [5Me]2−, [6]2−, and 3Me. ESI-MS of solutions of [4Me]2− treated with 6/8 and 1 equiv. S8 revealed the expected progression of starting material consumption to form increasing amounts of [5Me]2− and [6]2− (Fig. S54 and S55†).
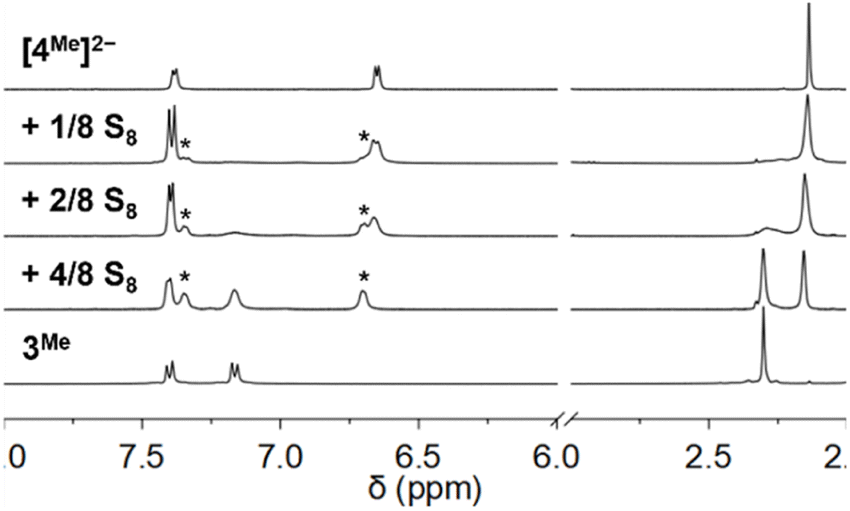 | ||
| Fig. 4 1H NMR spectra of a CD3CN solution of [4Me]2− with increasing added S8. Peaks assigned to [5Me]2− are marked with asterisks. | ||
The thiolate-supported zinc polysulfide complex [Et4N]2[5H] has previously been characterized only by combustion analysis and PXRD.12 While we were unable to grow X-ray-quality single crystals to confirm the assignment of [5Me]2−, ESI-MS data are consistent with the assignment of [5Me]2− as a dithiolate zinc complex with bound polysulfide dianion chelate. The mass spectrum of a CH3CN solution of [Et4N]2[4Me] shows signals at 123.0268 and 432.9922 m/z that correspond to p-tolS− and [Zn(p-tolS)3]− fragments. We note that the bimetallic, hexathiolate dimer [Zn2(SPh)6]2− has been reported26 and that the p-tolyl analogue may be forming under ionizing conditions and would be detected at the same m/z value. Similarly, the mass spectrum of a CH3CN solution of [Et4N]2[5Me] shows signals at 123.0334, 218.9330, 250.9061, 284.8675, and 314.8485 m/z, corresponding to [1Me]− and a mixture of [Zn(p-tolS)(Sx)]− (x = 4–5).
Diffusion ordered spectroscopy (DOSY) NMR experiments were performed to explore the metal ion-sulfur speciation in CD3CN. The diffusion coefficients (and corresponding hydrodynamic radius, RH) are similar for [Et4N][2Me] before and after added LiOTf (2.10 × 10−9 and 2.02 × 10−9 m2 s−1, respectively), supporting the idea of weak interactions and the monomeric nature of the disulfanide anion. In comparison, DOSY of the zinc complex anions [Et4N]2[5Me] and [Et4N]2[4Me] shows smaller diffusion constants (1.26 × 10−9 and 9.84 × 10−10 m2 s−1, respectively), consistent with the larger expected size of the metal complexes. Estimation of RH and of the formula weight using the Stokes–Einstein–Gierer–Wirtz model show sizes that are consistent with the monometallic zincate anions shown in Scheme 2 rather than larger multimetallic clusters (Table S1†).27,28 However, we are unable to use this method to assign the specific polysulfide nuclearity of [5Me]2−.
Our assignment of [5Me]2− and [6]2− featuring multiple polysulfide nuclearities is supported by reported 113Cd NMR spectra of DMF solutions of [PPh4]2[Cd(S6)2], in which several resonances are observed.29 We synthesized the Cd2+ congener of [4Me]2−, [Cd(p-tolS)4]2−. The 113Cd NMR spectrum of this complex in DMSO-d6 shows a resonance at δ 581 ppm, consistent with the previously reported chemical shift at δ 589 ppm for [Cd(SPh)4]2−.30 Similar to [4Me]2−, 1H NMR spectra of [Cd(p-tolS)4]2− treated with 6 and 12 S atom equiv. of S8 results in increasing formation of 3Me (Fig. S25†). Unlike the zinc congener, which forms an intermediate species [5Me]2−, downfield shifts of the p-tolyl signals are observed rather than a distinct intermediate. In the 113Cd NMR spectra, the S8-treated mixtures show complete consumption of starting [Cd(p-tolS)4]2− complex and formation of several species consistent with cadmium polysulfide complexes with multiple sulfur nuclearities (Fig. S26†). The addition of Cd(OTf)2 to mixtures of [Et4N][1Me] and S8 also forms similar mixtures to those observed with Zn(OTf)2, further highlighting the similarities in thiolate oxidation and sulfur reduction behavior between the two metals.
The formation of the zinc polysulfide complexes [5Me]2− and [6]2− by addition of S8 is reversible. A CD3CN solution of [4Me]2− was treated with S8 (4 S atom equiv.), followed by PPh3 (4 equiv.). The 1H NMR spectrum before PPh3 addition displayed the expected resonances corresponding to [5Me]2− and 3Me. After the addition of PPh3, the spectrum showed only resonances corresponding to [4Me]2− and triphenylphosphine sulfide (Fig. S42†).
Addition of S8 (4 S atom equiv.) to tetra(benzenethiolato)zincate anions with other para substituents ([4R]2−, R = F, Cl, CF3) in CD3CN forms mixtures whose 1H NMR spectra are also consistent with equilibria between [4R]2−, [6]2−, and the corresponding diaryl disulfide, 3R. Decreases in both conversion of [4R]2− and formation of 3R were observed with more electron-withdrawing substituents, where treatment of [4CF3]2− with S8 resulted in no 3CF3 formation at all (Fig. S29–S31†). No additional peaks corresponding to [5R]2− could be identified after treating [4Cl]2− or [4CF3]2− with S8. However, cooling these solutions to −20 °C reveals mixtures of several aromatic signals, in which [5R]2− may be included.
To deconvolute ligand binding stability from redox reactions with elemental sulfur, the exchange of anionic ligands coordinated to Zn2+ (Scheme 2, upper arrow) was studied. A CD3CN solution of [4Me]2− was treated with increasing amounts of [TBA]2[S6] (0.50–1.50 equiv.). 1H NMR spectroscopy of the mixture shows the formation of [5Me]−, [6]2−, and [2Me]−, along with higher order polysulfanide anions. These spectra are consistent with displacement of [1Me]− from [4Me]2− by S62− at Zn2+. The resulting S62− chelate can dissociate to extrude “S0” equivalents in the form of S8 to generate the distribution of other chelating Sx2− chain lengths (x = 4–6). The [1Me]− in solution then adds to the released S8 to form the disulfanide anion [2Me]− and higher order polysulfanide species. Consistent with Scheme 2, no disulfide 3Me or free thiolate [1Me]− are observed by 1H NMR spectroscopy. Unfortunately, we were unable to calculate accurate equilibrium constants for these processes due to some S8 precipitation in these mixtures.
Reaction mixtures of the thiolate/polysulfanide anions with other d-block metal salts are more complicated. Treatment of a CD3CN mixture of [Et4N][1Me] and S8 (1 S atom equiv.) with Sc(OTf)3 (>0.3 equiv.) causes the solution to become colorless and to precipitate a white solid material. The solution 1H NMR spectrum shows a distribution of (p-tolS)2Sx (x ≥ 2). We assign the insoluble solid as a scandium polysulfide species, leaving a stoichiometric excess of sulfur in solution that can insert into the S–S bonds of 3Me, forming the distribution we observe by NMR spectroscopy. However, PXRD of the solid is not consistent with reported patterns of Sc2S3. Similar results were obtained with redox-active metal cations—addition of 1 equiv. of (CH3CN)4CuPF6 or AgOTf to the same polysulfanide mixture in CD3CN results in immediate solution color change from dark green to colorless and rapid precipitation of brown and black insoluble materials, respectively. Again, 1H NMR spectra of the colorless filtrates shows a distribution of (p-tolS)2Sx (x ≥ 2). PXRD analysis of the Ag+ precipitate matches those of Ag2S; however, the PXRD pattern of the precipitate from the Cu+ reaction mixture could not be assigned to a reported compound. In all cases, we propose the oxidation of [1Me]− to 3Me and precipitation of an insoluble metal sulfide or polysulfide product through strong covalent metal–sulfur interactions and S8 reduction to S2−. The identification of relevant intermediates and of the final insoluble precipitate is ongoing.
Metal cation effects on electrochemical potentials of sulfur reduction and thiolate oxidation
The effect of electrolyte cations on the electrochemical reduction of S8 has been previously studied in the context of Li–S and Mg–S batteries.18,20,21 Lu and co-workers reported that larger monocations impart greater stabilization on polysulfide anions in DMSO due to weaker cation solvation energies. Stronger solvent–cation interactions and weaker cation–polysulfide interactions for the smaller cations results in polysulfide electrochemical oxidation potentials increasing from Rb+ > K+ > Na+ > Li+ ∼TBA+.18 Spectroelectrochemical measurements performed by Bieker and co-workers showed that the reduction of S8 in Mg2+-containing electrolytes forms less soluble polysulfide species compared to Li+-containing electrolytes, indicating stronger binding of the cations to the polysulfide anions.20,21 From these examples, it is clear that metal–sulfur interactions in solution can affect the electrochemical potentials or thermodynamic stabilities of their redox transformations.The cyclic voltammograms (CVs) of a saturated solution of S8 (ca. 0.1 mM) in CH3CN with different electrolytes [TBAPF6, MOTf (M+ = Li+, Na+, K+), 0.1 M] show two reductive events between −1 and −2 V vs. Fc+/Fc, as expected (Fig. S73†). These reduction events have been previously assigned to S8 reduction and formation of polysulfide anions (S82−, S42−, eqn (3) and (4)). In contrast to the reported CVs of S8 reduction in DMSO (see above),18 the first reduction wave is largely unaffected by monocation size. We attribute this difference to weaker solvation by CH3CN compared to DMSO on metal cations, as measured by Gutmann donor number (14.1 vs. 29.8 for CH3CN and DMSO, respectively).31 Additionally, the corresponding oxidation of the polysulfide anions is not observed for the CVs measured in CH3CN.
Fig. 5A shows the CVs of S8 performed in a mixture of TBAPF6 and M(OTf)2 (M2+ = Mg2+, Ca2+, 0.01 M) in CH3CN, as the CH3CN solubilities of these alkaline earth triflate salts are lower than those of the alkali cations. For these examples, the first reduction of S8 shifts to more positive potentials with the different cations added (+0.030 V with Mg2+; +0.060 mV with Ca2+vs. TBA+) and for the return oxidation event (+0.166 V with Mg2+; +0.335 V with Ca2+). The second cathodic wave corresponding to the S82−/2S42− reduction event is absent with added Mg(OTf)2 or Ca(OTf)2. This observation is consistent with spectroelectrochemical studies by Bieker and co-workers in which S82− in the presence of Mg2+ dissolved in CH3CN were found to disproportionate to shorter-chain polysulfide anions; computational evidence suggests these smaller polysulfides coordinate more strongly to Mg2+.21
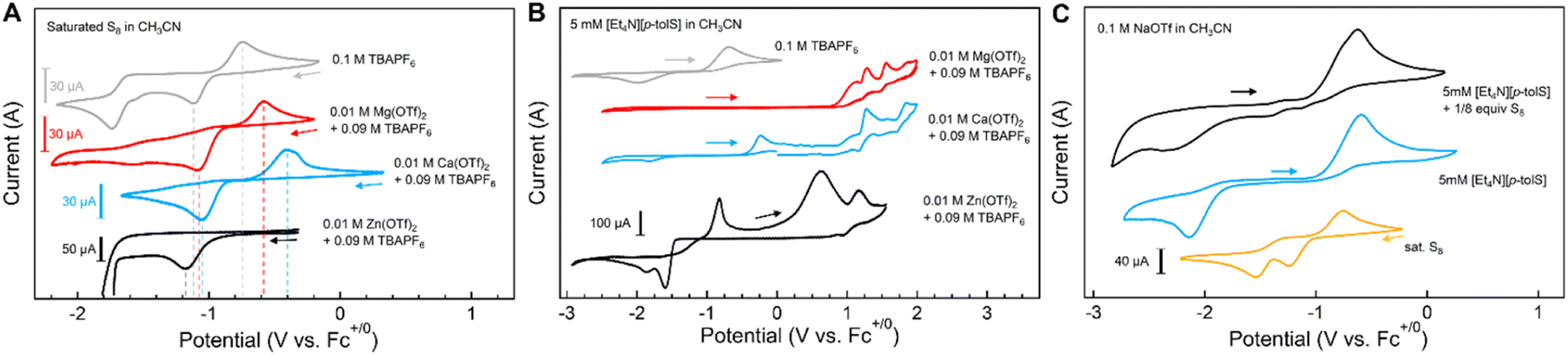 | ||
| Fig. 5 (A) CVs of saturated solutions of S8 in CH3CN (ca. 0.1 mM) in the presence of M(OTf)2 salts. (B) CVs of [Et4N][1Me] (5 mM) in CH3CN with different electrolytes. (C) CVs of S8 (ca. 0.1 mM), [Et4N][1Me] (5 mM), and [Et4N][1Me] (5 mM) + S8 (1 S atom equiv.) in CH3CN. All cyclic voltammograms were obtained under inert atmosphere with a glassy carbon working electrode, Ag+/0 reference electrode, and a Pt wire counter electrode at a scan rate of 0.1 V s−1. | ||
Fig. 5A also shows the CV of S8 with added Zn(OTf)2; when scanning to more negative potentials beyond the first S8 reduction event, a steep negative current is observed (Fig. S76†). Furthermore, the return oxidation is not observed, and subsequent cycles do not show the S8 reduction event while the large negative current persists. We tentatively assign this process to catalytic reduction of S82− to form [6]2− in the presence of Zn2+, though we are unsure of the mechanistic details of this system. In comparison, the voltammogram of isolated [6]2− is much more defined, but with onset potentials that match those of the S8 and Zn2+ salt mixture (Fig. S72†). Interestingly, we observe a group of oxidation events with one broad re-reduction, which we attribute to multiple possible zinc-bound polysulfide chain lengths, as reported in literature. A similar CV is observed when Cd(OTf)2 is added to S8 (Fig. S75†).
Next, the electrolyte effect on 3Me reduction and [1Me]− oxidation was studied. CVs of [Et4N][1Me] with different electrolytes (TBAPF6, LiOTf, NaOTf, and KOTf) show a large peak separation between the thiolate oxidation wave and the related reduction event (ΔEp ∼ 1.3 V for TBA+). The oxidation of the benzenethiolate anion and reduction of diaryl disulfide compounds have been previously shown to proceed by ECE mechanisms (eqn (6)–(8)).32
| p-tolS− → p-tolS˙ + e− | (6) |
| p-tolS˙ + p-tolS− ⇌ (p-tol)2S2˙− | (7) |
| (p-tol2)S2˙− → (p-tol2)S2 + e− | (8) |
From these assignments, the oxidation wave in the CV corresponds to thiolate oxidation to the thiyl radical, and the reduction event corresponds to disulfide reduction to the corresponding disulfide radical anion. The CVs conducted in electrolytes with alkali metal triflate salts (M+ = Li+, Na+, and K+) are all similar to the CV measured with TBAPF6 as the electrolyte. While this comparison is made qualitatively by comparing the onset potentials of oxidation and reduction, a more quantitative study of these potentials via scan rate dependence measurements also yielded minimal differences.32
In contrast, CVs of [Et4N][1Me] measured in electrolyte solutions of the triflate salts of dicationic metals (Mg2+, Ca2+) show oxidation waves that are shifted to much more positive potentials, with a broadened reduction wave (Fig. 5B). The CV performed in Mg(OTf)2 electrolyte also shows multiple oxidation waves. We attribute these additional oxidation events to the formation of different magnesium and calcium thiolate complexes that shift the oxidation potential of the thiolate sulfur center to more positive potentials. Similar, albeit less well-defined, positive shifts in oxidation features are observed with CVs of [1Me]− in the presence of Zn2+ (Fig. 5B), Cd2+, and Sc3+, corresponding to formation of more complex mixtures of metal thiolate species under these conditions.
CVs of [1Me]− + S8 (1 equiv. S) with 0.1 M MOTf (M+ = Li+, Na+, and K+) in CH3CN show features that resemble combinations of the disparate solution components ([1Me]− and S8 separately) with additional features that we assign to [2Me]− or to other polysulfanide anions (Fig. 5C). New oxidation events are observed at less positive potentials than that of thiolate oxidation or reduced sulfur re-oxidation, and the reduction waves from 3Me and S8 broaden significantly. CVs of the same mixture in 0.1 M LiOTf taken before and after addition of 0.01 M Mg(OTf)2 result in disappearance of the thiolate oxidation wave over successive scans, as well as the positive shifts in S8 reduction described with Mg2+ above, demonstrating the much stronger metal–sulfur interactions of Mg2+ over Li+ in solution despite much lower relative concentration.
Comparison of structures and interaction energies
Based on the hypothesis that the solution-phase stabilization of metal thiolate and metal polysulfide species dictate these reaction equilibria, we compared the structures of previously reported metal benzenethiolate and metal polysulfide complexes. Numerous transition metal polysulfide complexes have been synthesized and structurally characterized,13–16 including several examples of zinc polysulfide structures of different polysulfide chain lengths supported by nitrogen donor ancillary ligands (e.g.Fig. 6A, 9)13,15,33,34 or homoleptic bis(polysulfido)zincate anions (Fig. 6A, [6A]2− and [6B]2−).12,25 For the other cations, despite the widespread interest in lithium polysulfide species that are components of Li–S batteries, there are few examples of structurally-characterized lithium polysulfide complexes. One rare example is a hexasulfide-bridged dilithium complex supported by N,N-tetramethylenediamine (TMEDA) ancillary ligands (Fig. 6A, 7).35 In contrast, there are no crystallographically-characterized examples of magnesium or calcium polysulfide complexes that display metal–sulfur bonds, although examples of magnesium polysulfide salts in which the Mg2+ ion is coordinatively saturated by neutral donor ligands and the polysulfide species acts as an outer sphere counteranion have been isolated.20,36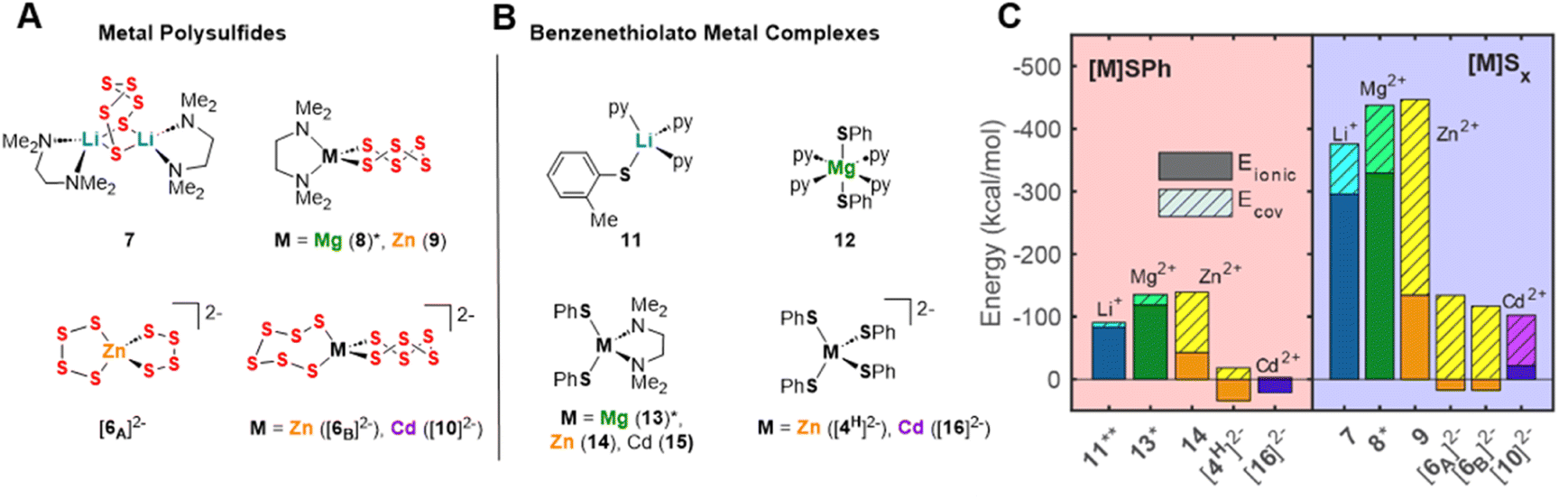 | ||
| Fig. 6 Selected examples of structurally characterized* (A) metal polysulfide and (B) terminal metal benzenethiolate complexes. (C) Comparison of Ecov and Eionic interaction energies for selected polysulfide and benzenethiolate complexes calculated by DFT. *Mg complexes 8 and 13 have not been structurally characterized and calculations are of optimized DFT structures. **DFT calculations were performed on the analogue of 11 with an unsubstituted benzenethiolate ligand. | ||
Thiolate complexes exhibit many different structural motifs, including terminally-bound thiolate moieties and multimetallic clusters with bridging thiolate ligands that include bimetallic, cubane-like, and adamantane-like motifs. Fig. 6B shows examples of terminally-bound benzenethiolate complexes of the same series of metal ions studied above. These structures include neutral pyridine-supported benzenethiolate complexes of Li+ and Mg2+ (11 and 12, respectively),37,38 TMEDA-supported di(benzenethiolato)zinc and cadmium complexes (14 for M = Zn2+),39,40 and tetra(benzenethiolato)metallate anions of Zn2+ and Cd2+ ([4H]2− and 162−, respectively).30
DFT calculations were used to compare the metal–sulfur interaction energies within these compounds and to distinguish covalent from electrostatic contributions to these energies. The compounds studied here were the crystallographically characterized polysulfide and benzenthiolate complexes of Li+, Zn2+, and Cd2+ (Fig. 6A and B), and the Mg2+ congeners of tetrahedral TMEDA-supported polysulfide and benzenethiolate complexes (8* and 13*, Fig. 6A and B), which have not been isolated. For these calculations, the benzenethiolate variants of 11 and 14 were used in place of the structurally characterized 2-methylthiolate and 4-amino-benzenthiolate complexes, respectively. These calculations follow a previously reported procedure used to study first-row transition metal thiolate interactions in tris(pyrazolyl)borate-supported metal thiolate complexes.41,42 First, the interaction energy Eint was calculated between the polysulfide or benzenethiolate fragments and the remaining metal complex fragment using the B3LYP functional with a counterpoise correction to account for the basis set superposition error (BSSE, see ESI†). This interaction energy was then divided into two components (eqn (9)) where Ecov is the covalent or orbital interaction energy, and Eionic is the electrostatic interaction energy. Eionic was estimated as a sum of the electrostatic interactions (eqn (10)) between the charges of the atoms of each fragment from Natural Population Analysis (NPA).
| Eint = Ecov + Eionic | (9) |
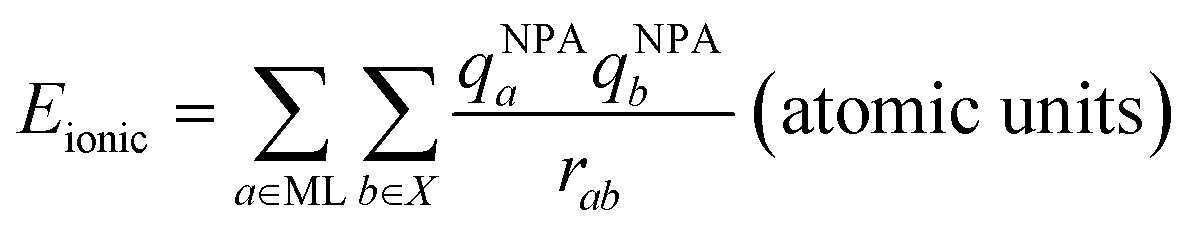 | (10) |
Fig. 6C plots these interaction energies for the polysulfide and benzenethiolate complexes shown in Fig. 6A and B. For all complexes studied, the interaction energies for the metal polysulfide complexes are more negative than those of the same benzenethiolate complexes. While some of this difference is due to the greater negative charge of polysulfide dianions compared to monoanionic thiolate moieties, the polysulfide complexes also exhibit greater covalent contributions to the interaction energies. The covalent contributions also significantly increase from Li+ to Mg2+ to Zn2+ and Cd2+. In fact, for the dianionic bis(polysulfido)zinc or bis(polysulfido)cadmium complexes, the interaction energies show a destabilizing positive ionic energetic contribution due to the negatively charged complex. In these cases, the covalent contribution result in the observed net stability of the dianionic complexes.
Discussion
Scheme 3 shows the relevant equilibria for the species in exchange in solution. In the absence of a metal ion that can be coordinated by sulfur donors (i.e. with Et4N+ or similar countercations), the reduced sulfur species follow the equilibria shown in the first row of the scheme. Addition of S8 to a thiolate anion forms a distribution of thiolate, disulfanide, and higher order polysulfanide ions. Excess S8 converts this assortment to a mixture of the corresponding organic disulfide and polysulfide anions, here shown as S3˙− but likely existing as a distribution of polysulfide nuclearities.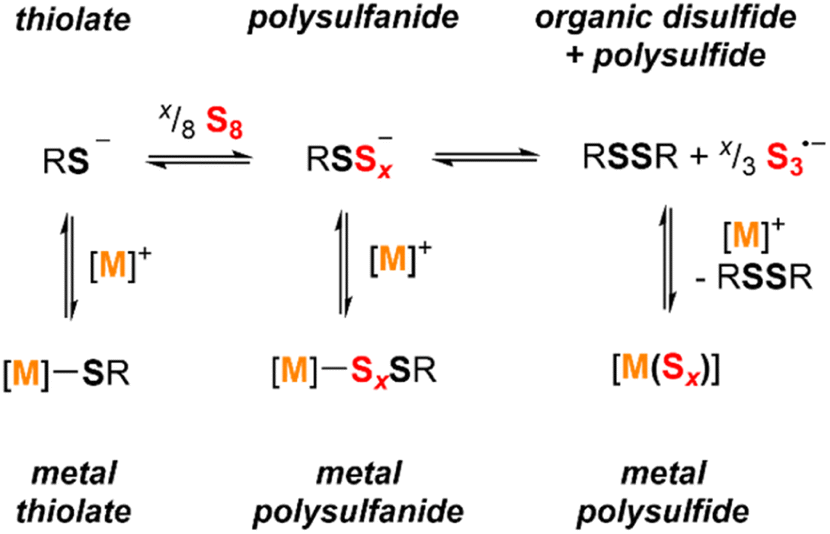 | ||
| Scheme 3 Reduced sulfur species (thiolates, polysulfanides, and polysulfides) dynamically exchange in solution, with equilibria driven by metal coordination and added sulfur content. | ||
The results above show that the addition of different metal ions can shift these equilibria due to stabilizing effects of the metal–sulfur interactions. The 1H NMR data and the electronic absorption spectra indicate that the alkali metals (Li+, Na+, and K+) do not shift these equilibria significantly. In contrast, the alkaline earth metals (Mg2+ and Ca2+) and the group 12 transition metals (Zn2+ and Cd2+) greatly favor formation metal polysulfide species over metal polysulfanide compounds. These observations suggest that while aryldisulfanide anions can be stabilized with respect to the corresponding thiolate anions and due to delocalization of the negative charge across the sulfur atoms and onto the aromatic ring,9 these energetic factors are outweighed by stronger metal–sulfur interactions in the metal thiolate and metal polysulfide complexes in comparison with the metal polysulfanide complexes for the dicationic metal ions.
We originally considered the possibility that these metal–sulfur interactions shift the redox potentials of the sulfur centers of the corresponding species. As shown above, the alkali metal ions do not significantly shift the reduction potentials of S8 or the oxidation potential of the substituted benzenethiolate anions. However, the addition of the alkali earth metal ions Mg2+ and Ca2+ as well as Zn2+ and Cd2+ result in a more positive potential of benzenethiolate oxidation due to stronger metal-thiolate interactions. As the relevant half-reactions for thiolate oxidation (eqn (6)–(8)) would be expected to couple with S8 reduction during this process, these results would be expected to disfavor reduction of elemental sulfur by metal thiolates from an electrochemical standpoint. As such, the electrochemical data show metal-ion-induced shifting of the redox potentials of sulfur-centered oxidation or reduction is not the primary factor that dictates the final speciation of the reduced sulfur species.
Scheme 4 shows the thermodynamic square scheme of the relevant exchanging species, while disregarding the polysulfanide complexes. In this scheme, the reaction described by K1 shows the exchange between metal thiolate and metal polysulfide complexes. As discussed above, this equilibrium lies further to the left for the alkali metal ions or when the counter-cation cannot be coordinated by sulfur.
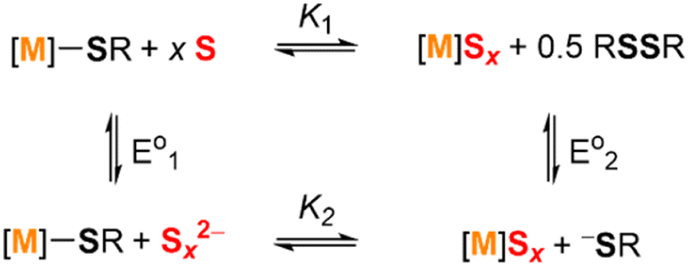 | ||
| Scheme 4 Thermodynamic square scheme of metal thiolate species treated with zerovalent sulfur to form metal polysulfide species. | ||
The equilibrium constant K1 is related to the free energy term ΔG1, which could therefore be expressed as a sum of free energy terms derived from 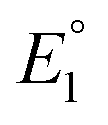 , K2, and
, K2, and 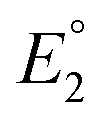 .
. 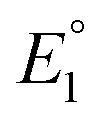 and
and 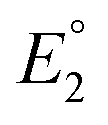 do not change upon variation of the metal ion. As such, the differences in behavior between the alkali metals, the alkali earth metal ions, and Zn2+ or Cd2+ are related to K2, which compares the relative stabilities of the metal thiolate complexes to the metal polysulfide species. We note that this reaction equation does not take into account entropic effects or rearrangement of the relevant species into multimetallic ions, complexes, or clusters. Based upon both experimental results and upon computation, this equilibrium process is nearly thermoneutral for the alkali metals, but lies further toward the right for Cd2+ and Zn2+, likely due to increased covalency.
do not change upon variation of the metal ion. As such, the differences in behavior between the alkali metals, the alkali earth metal ions, and Zn2+ or Cd2+ are related to K2, which compares the relative stabilities of the metal thiolate complexes to the metal polysulfide species. We note that this reaction equation does not take into account entropic effects or rearrangement of the relevant species into multimetallic ions, complexes, or clusters. Based upon both experimental results and upon computation, this equilibrium process is nearly thermoneutral for the alkali metals, but lies further toward the right for Cd2+ and Zn2+, likely due to increased covalency.
In attempts to recognize trends in the effects of metal cations on reduced sulfur speciation and to quantitively correlate metal–sulfur interactions, we note that these Lewis acidic metal cations have been demonstrated to modulate transition metal redox potentials or electron transfer rates.43–45 In these studies, the effects of these metal ions has been correlated primarily to the Lewis acidity of the metal ions, often reported as the pKa of the corresponding metal aqua complex in water. From our studies, RSS equilibria appear to be more complicated and do not correlate as readily to metal ion Lewis acidity (Fig. S40†), and may be better understood in terms of metal “thiophilicity.”46
These results provide salient considerations in future metal–sulfur studies in the context of energy storage applications. Although many efforts in recent years have been applied toward the Mg–S battery,47–49 it should be clear that strategies toward mitigating polysulfide ions in these systems should be considered separately from existing Li–S battery sulfur sequestration methods. In short, although Mg2+ and Li+ are often related due to the diagonal relationship of similar charge density and other electronic properties,50–52 their relative degrees of covalency with sulfur and resulting influence on sulfur redox chemistry are demonstrably distinct. Such considerations should also be made in the development of batteries with other metals, including zinc-based batteries.53–56
Conclusions
We have demonstrated that metal ions perturb the equilibria between sulfur species in solution. Broadly speaking, more charged metal ions favor sulfur-centered redox reactions; the benzenethiolate moieties reduce S8 to form the corresponding metal polysulfide complexes and organic disulfides. This reaction appears to be driven by stronger metal–sulfur interactions rather than by metal-induced perturbation of the sulfur or thiolate electrochemical potentials, with significant contribution from metal–sulfur covalency. For alkali metal ions or for non-coordinating countercations, “S0” from elemental sulfur appears to act as a better stabilizing Lewis acid compared to the countercation, therefore favoring S–S catenation and the formation of polysulfanide anions instead of sulfur redox. These results stress the need for understanding the differences in solution metal–sulfur interactions when comparing alkali, alkaline earth, and transition metal cations in fields such as sulfur-based energy storage.Data availability
Experimental procedures and characterization data can be found in the ESI.†Author contributions
W. T. M. S. synthesized zinc and cadmium complexes and performed NMR spectroscopy, absorption spectroscopy, and voltammetry experiments. M. N. R. performed mass spectrometry experiments. A. G. O. performed crystallographic structure refinement. E. Y. T. ran the DFT calculations. W. T. M. S. and E. Y. T. designed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF (CHE-2047045). We thank Dr Evgenii Kovrigin for assistance with 113Cd NMR spectroscopy.Notes and references
- R. Steudel and T. Chivers, Chem. Soc. Rev., 2019, 48, 3279–3319 RSC.
- P. Song, W. Rao, T. Chivers and S.-Y. Wang, Org. Chem. Front., 2023, 10, 3378–3400 RSC.
- G. Zhang, H. Yi, H. Chen, C. Bian, C. Liu and A. Lei, Org. Lett., 2014, 16, 6156–6159 CrossRef CAS PubMed.
- Z.-Y. Gu, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067–4072 RSC.
- J.-H. Li, Q. Huang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2018, 20, 4704–4708 CrossRef CAS PubMed.
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036–5041 CrossRef CAS PubMed.
- J. M. Rhodes, C. A. Jones, L. B. Thal and J. E. Macdonald, Chem. Mater., 2017, 29, 8521–8530 CrossRef CAS.
- D. L. Pringle, The Nature of the Polysulfide Anion, Iowa State University, 1967 Search PubMed.
- S. Jungen, E. Paenurk and P. Chen, Inorg. Chem., 2020, 59, 12322–12336 CrossRef CAS PubMed.
- K. Li, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2023, 145, 13435–13443 CrossRef CAS PubMed.
- U. Krautscheid, S. Dev, H. Krautscheid, P. P. Paul, S. R. Wilson and T. B. Rauchfuss, Z. Naturforsch., B, 1993, 48, 653–658 CrossRef CAS.
- D. Coucouvanis, P. R. Patil, M. G. Kanatzidis, B. Detering and N. C. Baenziger, Inorg. Chem., 1985, 24, 24–31 CrossRef CAS.
- R. J. Pafford and T. B. Rauchfuss, Inorg. Chem., 1998, 37, 1974–1980 CrossRef CAS.
- M. Draganjac and T. B. Rauchfuss, Angew Chem. Int. Ed. Engl., 1985, 24, 742–757 CrossRef.
- A. K. Verma, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 1995, 34, 3072–3078 CrossRef CAS.
- T. B. Rauchfuss, Inorg. Chem., 2004, 43, 14–26 CrossRef CAS PubMed.
- K. Tatsumi, H. Kawaguchi, K. Inoue, K. Tani and R. E. Cramer, Inorg. Chem., 1993, 32, 4317–4323 CrossRef CAS.
- Q. Zou, Z. Liang, G.-Y. Du, C.-Y. Liu, E. Y. Li and Y.-C. Lu, J. Am. Chem. Soc., 2018, 140, 10740–10748 CrossRef CAS PubMed.
- L. L. Liu, L. L. Cao, Y. Shao and D. W. Stephan, J. Am. Chem. Soc., 2017, 139, 10062–10071 CrossRef CAS PubMed.
- G. Bieker, J. Wellmann, M. Kolek, K. Jalkanen, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2017, 19, 11152–11162 RSC.
- G. Bieker, D. Diddens, M. Kolek, O. Borodin, M. Winter, P. Bieker and K. Jalkanen, J. Phys. Chem. C, 2018, 122, 21770–21783 CrossRef CAS.
- T. Chivers and I. Drummond, Inorg. Chem., 1972, 11, 2525–2527 CrossRef CAS.
- F. Taitiro, K. Tooru, O. Satoshi and H. Masashi, Bull. Chem. Soc. Jpn., 1980, 53, 2851–2855 CrossRef.
- B. Dmuchovsky, F. B. Zienty and W. A. Vredenburgh, J. Org. Chem., 1966, 31, 865–869 CrossRef CAS.
- A. Muller, J. Schimanski, U. Schimanski and H. Bogge, Z. Naturforsch., B, 1985, 40, 1277–1288 CrossRef.
- I. L. Abrahams, C. D. Garner and W. Clegg, J. Chem. Soc., Dalton Trans., 1987, 1577–1579 RSC.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 3987–3994 CrossRef CAS PubMed.
- R. M. H. Banda, I. G. Dance, T. D. Bailey, D. C. Craig and M. L. Scudder, Inorg. Chem., 1989, 28, 1862–1871 CrossRef CAS.
- D. Swenson, N. C. Baenziger and D. Coucouvanis, J. Am. Chem. Soc., 1978, 100, 1932–1934 CrossRef CAS.
- V. Gutmann, Electrochim. Acta, 1976, 21, 661–670 CrossRef CAS.
- Q. Zhu, C. Costentin, J. Stubbe and D. G. Nocera, Chem. Sci., 2023, 14, 6876–6881 RSC.
- S. Dev, E. Ramli, T. B. Rauchfuss and C. L. Stern, J. Am. Chem. Soc., 1990, 112, 6385–6386 CrossRef CAS.
- H. Li, S. Du and X. Wu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 498–500 CrossRef.
- A. J. Banister, D. Barr, A. T. Brooker, W. Clegg, M. J. Cunnington, M. J. Doyle, S. R. Drake, W. R. Gill, K. Manning, P. R. Raithby, R. Snaith, K. Wade and D. S. Wright, J. Chem. Soc., Chem. Commun., 1990, 105–107 RSC.
- S. Dev, E. Ramli, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 1991, 30, 2514–2519 CrossRef CAS.
- A. J. Banister, W. Clegg and W. R. Gill, J. Chem. Soc., Chem. Commun., 1987, 850–852 RSC.
- S. Chadwick, U. Englich, M. O. Senge, B. C. Noll and K. Ruhlandt-Senge, Organometallics, 1998, 17, 3077–3086 CrossRef CAS.
- K. S. Anjali, J. T. Sampanthar and J. J. Vittal, Inorg. Chim. Acta, 1999, 295, 9–17 CrossRef CAS.
- M. Yosef, A. K. Schaper, M. Fröba and S. Schlecht, Inorg. Chem., 2005, 44, 5890–5896 CrossRef CAS PubMed.
- S. I. Gorelsky, L. Basumallick, J. Vura-Weis, R. Sarangi, K. O. Hodgson, B. Hedman, K. Fujisawa and E. I. Solomon, Inorg. Chem., 2005, 44, 4947–4960 CrossRef CAS PubMed.
- E. I. Solomon, S. I. Gorelsky and A. Dey, J. Comput. Chem., 2006, 27, 1415–1428 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS PubMed.
- H. Yoon, Y.-M. Lee, X. Wu, K.-B. Cho, R. Sarangi, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 9186–9194 CrossRef CAS PubMed.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- Q. Zou, Y. Sun, Z. Liang, W. Wang and Y.-C. Lu, Adv. Energy Mater., 2021, 11, 2101552 CrossRef CAS.
- P. He and J. L. Schaefer, ACS Energy Lett., 2022, 7, 4352–4361 CrossRef CAS.
- M. D. Qian, F. A. L. Laskowski, S. D. Ware and K. A. See, ACS Appl. Mater. Interfaces, 2023, 15, 9193–9202 CrossRef CAS PubMed.
- G. Rayner-Canham, Found. Chem., 2011, 13, 121–129 CrossRef CAS.
- T. Chlupatý, Z. Růžičková, H. Kampová, J. Merna and A. Růžička, Inorg. Chem., 2022, 61, 9392–9404 CrossRef PubMed.
- X. Sun, X. Wang, Y. Wan, Y. Guo, T. Deng and X. Yu, Chem. Eng. J., 2023, 452, 139610 CrossRef CAS.
- T. A. Bendikov, C. Yarnitzky and S. Licht, J. Phys. Chem. B, 2002, 106, 2989–2995 CrossRef CAS.
- Y. Zhao, D. Wang, X. Li, Q. Yang, Y. Guo, F. Mo, Q. Li, C. Peng, H. Li and C. Zhi, Adv. Mater., 2020, 32, 2003070 CrossRef CAS PubMed.
- M. M. Gross and A. Manthiram, ACS Appl. Mater. Interfaces, 2018, 10, 10612–10617 CrossRef CAS PubMed.
- M. R. Tuttle, C. Walter, E. Brackman, C. E. Moore, M. Espe, C. Rasik, P. Adams and S. Zhang, Chem. Sci., 2021, 12, 15253–15262 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332488. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01025f |
| This journal is © The Royal Society of Chemistry 2024 |