
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRestructuring multi-phase interfaces from Cu-based metal–organic frameworks for selective electroreduction of CO2 to C2H4†
Jiye
Feng
a,
Wenbiao
Zhang
ab,
Danni
Shi
a,
Yingshuai
Jia
b,
Yi
Tang
 b,
Yuying
Meng
a and
Qingsheng
Gao
*a
b,
Yuying
Meng
a and
Qingsheng
Gao
*a
aCollege of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, P. R. China. E-mail: tqsgao@jnu.edu.cn
bDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Laboratory of Advanced Materials, Collaborative Innovation Centre of Chemistry for Energy Materials, Fudan University, Shanghai 200433, P. R. China
First published on 13th May 2024
Abstract
Multi-phase interfaces are promising for surmounting the energy barriers of electrochemical CO2 reduction involving multiple electron transfer steps, but challenges still remain in constructing interfacial micro-structures and unraveling their dynamic changes and working mechanism. Herein, highly active Ag/Cu/Cu2O heterostructures are in situ electrochemically restructured from Ag-incorporating HKUST-1, a Cu-based metal–organic framework (MOF), and accomplish efficient CO2-to-C2H4 conversion with a high faradaic efficiency (57.2% at −1.3 V vs. RHE) and satisfactory stability in flow cells, performing among the best of recently reported MOFs and their derivatives. The combination of in/ex situ characterizations and theoretical calculations reveals that Ag plays a crucial role in stabilizing Cu(I) and increasing the CO surface coverage, while the active Cu/Cu2O interfaces significantly reduce the energy barrier of C–C coupling toward the boosted ethylene production. This work not only proves MOFs as feasible precursors to derive efficient electrocatalysts on site, but also provides in-depth understanding on the working interfaces at an atomic level.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) powered by renewable electricity provides a sustainable process toward carbon neutrality.1,2 Compared with C1 products (e.g., CO, HCOOH, and CH4), C2+ ones (e.g., C2H4 and C2H5OH) feature higher energy density and market value; however, their productivity is seriously limited by the multi-step hydrogenation competing with a series of side-reactions.3–5 So far, Cu is the only metal that can selectively convert CO2 to C2+ thanks to the benign bonding of *CO intermediates,6,7 and its interfaces with tailored valence states, grain boundaries and unsaturated sites are evidenced to surmount the energy barriers of such multiple electron transfer steps.8–10 Although progress has been made in C2+ production, it's still challenging to facilely construct active interfaces and effectively stabilize them during electrolysis.11Metal–organic frameworks (MOFs) have attracted extensive interest for the electrochemical CO2RR associated with the high surface area, controllable pore size/shape, and open metal sites,12–14 but as for the catalytic mechanism debates remain due to the dynamic change of frameworks during the tough electrolysis.15 In particular, Cu-based MOFs containing frangible Cu–O4 nodes would undergo in situ reconstruction since the electron transport along networks via the copper-ion redox demolishes the coordination extensively.16 The newly formed surface/interfaces, rather than the initial frameworks, should be brought into sharp focus for the mechanism study.17 Following the progressive insights, such electrochemical reconstruction was proved feasible to produce active and selective catalysts on site,18–22 avoiding the time and energy-consuming preparation of catalysts and working electrodes (e.g., thermally driven epitaxial growth of multi-phase interfaces23,24 and ultrasonic spraying for loading powdery electrocatalysts onto gas-diffusion electrodes25). The in situ derived electrocatalysts inherit the structural merits of MOFs, and more importantly generate plenteous interfaces abundant with coordinatively unsaturated sites under the manipulated electrochemical conditions.26 For example, the single-type Cu2O sites of Cu2O@CuHHTP and coordinatively unsaturated Cu paddle wheel clusters (Cu2(HCOO)3), partially reduced from CuHHTP and HKUST-1 at mild potentials, promoted the selective CO2RR to CH4 in H-type reactors, while C2+ production was still restricted.17,19 Moreover, the structure–activity relationship under working conditions was inconclusive in the context of the further reconfiguration at more negative potentials and higher current densities adopted for the CO2RR, especially in flow cells. As recently highlighted, the deep reconstruction of sulfur-doped HKUST-1 to ligand-free Cu/CuxSy interfaces could enable efficient ethylene production;18 however, such effort was hampered by the difficult control over the exchange of benzenetricarboxylic linkers by thioacetamide ligands in frangible frameworks. In other words, constructing, promoting and stabilizing interfacial active species, e.g., Cu(0)–Cu(I) ensembles and defective sites, via elaborative MOF reconstruction are promising for robust C2+ production, but still an arduous task under the harsh CO2RR conditions.27,28
Thanks to the thermodynamic merits of Ag in comparison with Cu, e.g., the relatively higher standard redox potential (EAg+/Ag = +0.80 V vs. ECu+/Cu = +0.52 V) and the less negative formation enthalpy of oxides (−31.1 kJ mol−1 Ag2O vs. −169 kJ mol−1 Cu2O), introducing foreign Ag atoms is anticipated to stabilize neighboring Cu(I) active for the CO2RR.29 Moreover, it's also expected to empower tandem electrolysis on the expanded interfaces via available *CO spillover from weakly bonding Ag to Cu(0)–Cu(I) sites capable of C–C coupling.30–32 However, the relevant research is still absent for directional MOF reconstruction. Here, Ag-incorporating HKUST-1 frameworks (Agn/HKUST-1, n denotes the molar ratio of Ag/Cu) were for the first time introduced to in situ restructure Ag/Cu/Cu2O heterostructures under practical electrolysis conditions, which were highly active for CO2-to-C2H4 conversion due to the rich Cu(I) species and Ag/Cu/Cu2O interfaces. With an optimal loading of Ag (nAg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nCu = 0.1), the in situ derived electrocatalysts afforded a high C2H4 faradaic efficiency (FE) of 57.2% at −1.3 V vs. RHE, superior to those of HKUST-1 derivatives (17.4%), electro-deposited Ag/Cu/Cu2O (16.7%), and their recently reported MOF-related counterparts, and maintained satisfactory long-term durability. The combination of in/ex situ characterizations and theoretical calculations confirmed that Ag played a crucial role in stabilizing Cu(I) and increasing CO surface coverage, while the resulting rich Cu/Cu2O interface significantly reduces the energy barrier of C–C coupling toward boosted ethylene production.
:nCu = 0.1), the in situ derived electrocatalysts afforded a high C2H4 faradaic efficiency (FE) of 57.2% at −1.3 V vs. RHE, superior to those of HKUST-1 derivatives (17.4%), electro-deposited Ag/Cu/Cu2O (16.7%), and their recently reported MOF-related counterparts, and maintained satisfactory long-term durability. The combination of in/ex situ characterizations and theoretical calculations confirmed that Ag played a crucial role in stabilizing Cu(I) and increasing CO surface coverage, while the resulting rich Cu/Cu2O interface significantly reduces the energy barrier of C–C coupling toward boosted ethylene production.
Experimental section
Chemicals
Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, >99.0%), polyvinylpyrrolidone (PVP, MW 58000) and N,N-dimethylformamide (DMF, 99.8%) were provided by Macklin Co., Ltd (Shanghai, China). 1,3,5-Benzenetricarboxylic acid (H3BTC, 98%), potassium hydroxide (KOH, 85%) and silver nitrate (AgNO3, 99.8%) were purchased from Aladdin Chemistry Co., Ltd (Shanghai, China). Nafion (5 wt%) solution was purchased from Sigma-Aldrich. Ethanol (99.7%) was bought from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All aqueous solutions were prepared using ultrapure water (>18 MΩ).
Material synthesis
:1 by volume) was mixed with 2.0 g of PVP under continuous stirring. After complete dispersion of the reactants, the two solutions were mixed directly. Subsequently, the mixed solution was stirred for 15 min and transferred into a 50 mL Teflon-lined stainless-steel autoclave. The autoclave was heated at 100 °C for 10 h. After naturally cooling down to room temperature, the product was washed with ethanol three times by centrifugation at 8000 rpm for 5 min. Finally, the blue powder of HKUST-1 was obtained after drying under vacuum at 60 °C overnight.
:VH2O = 9:1), and then 70 mg (0.32 mmol) of the as-prepared HKUST-1 was added into the above solution with continuous stirring for 20 h at 85 °C. The precipitate was centrifuged, washed with ethanol and dried in a vacuum oven at 60 °C overnight. Furthermore, varied amounts of AgNO3 were used to prepare Agn/HKUST-1 with different amounts of Ag incorporated.
:VH2O = 9:1) with continuous stirring for 20 h at 85 °C. The precipitate was centrifuged, washed with ethanol and dried in a vacuum oven at 60 °C overnight.
Physical characterization
Transmission electron microscopy (TEM), energy dispersive spectroscopy (EDS) and the corresponding elemental mapping were performed on a JEOL 2100F. Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) were conducted on a ZEISS ULTRA55. X-ray diffraction (XRD) analysis was performed on a Bruker D8 diffractometer using Cu Kα radiation (λ = 1.54056 Å). X-ray photoelectron and Auger electron spectroscopies (XPS and AES) were performed on a Thermo Scientific Escalab 250Xi. Fourier transform infrared (FT-IR) spectroscopy was carried out on a PerkinElmer spectrometer with the spectral range of 4000–400 cm−1. Raman investigation was performed on a laser confocal Raman microspectrometer (Horiba HR-800) with an excitation laser wavelength of 532 nm. The electron paramagnetic resonance (EPR) spectra were recorded on a Bruker A300. Inductively coupled plasma-optical emission spectrometry (ICP-OES) analysis was conducted on an OPTIMA 2000DV. X-ray absorption spectroscopy (XAS) was performed at beamline 12-BM of the Advanced Photon Source beamline at Argonne National Laboratory in Illinois, USA.Preparation of cathode electrodes
5 mg of electrocatalyst was ground to powder and then dispersed into anhydrous ethanol (450 μL) followed by adding 50 μL Nafion. After continuous ultrasonication for at least 30 min, a homogeneous ink was achieved. 100 μL of the ink was pipetted onto a carbon paper electrode (0.5 cm × 2 cm) with a loading of 1 mg cm−2. The electrode was then dried at room temperature naturally for the subsequent electrochemical tests.Electrochemical test
CO2 electrolysis was performed in a flow cell. The prepared gas diffusion electrode (GDE) was the working electrode. An electrolyte (1.0 M KOH) was circulated through the flow cell at a rate of 15 mL min−1 under the pressure applied by a peristaltic pump. Anode and cathode chambers were separated by an anion exchange membrane. A platinum electrode and a solid Ag/AgCl electrode served as the counter electrode and reference electrode, respectively. Electrode potentials in the study were converted to the reversible hydrogen electrode (RHE) scale according to the following equation:| E(vs. RHE) = E(vs. Ag/AgCl) + 0.059 × pH + 0.197 V |
All electrochemical measurements were carried out in a three-electrode system with a CHI660E potentiostat. Linear sweep voltammetry (LSV) curves were obtained to choose the appropriate potential range for the catalysts. The sweeping range was from 0 to −1.7 V (vs. RHE) at a scan rate of 100 mV s−1 in 1 M KOH solution with a CO2 flow.
Products analysis
Gaseous products were analyzed using a gas chromatograph (GC, FULI-9790 II) equipped with a flame ionization detector (FID for CO and hydrocarbons) and a thermal conductivity detector (TCD for H2). Gas-phase products were sampled every 20 min using high-purity nitrogen (N2, 99.999%) as the carrier gas. The column effluent (separated gas mixtures) was first passed through the TCD where hydrogen was quantified, then CO, CH4 and C2H4 were subsequently quantified by the FID. According to the peak areas in the GC, the partial current densities and FEs of CO, CH4, C2H4 and H2 were calculated using the following equations: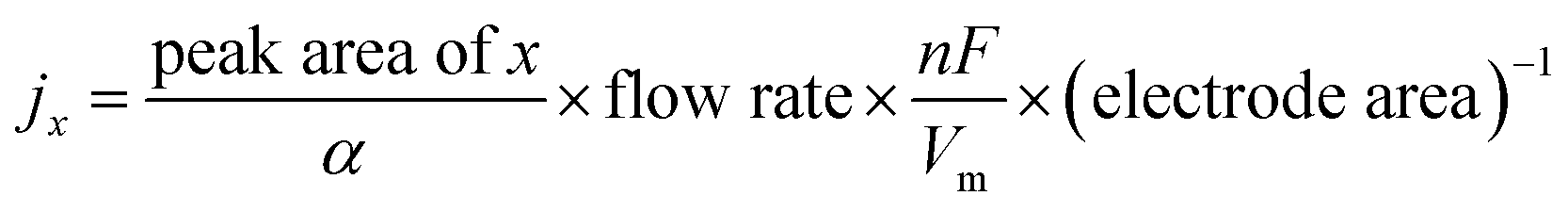
where x represents one of CO, CH4, C2H4 and H2, and n represents the number of electrons to be transferred to form the products, which are 2, 8, 12 and 2, respectively; α is the conversion factor for CO, CH4, C2H4 and H2, respectively, based on the calibration of standard samples; F is the Faraday constant (F = 96
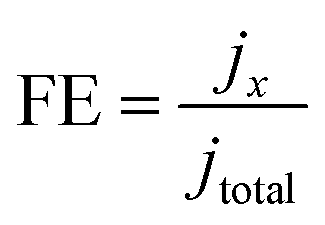 485 C mol−1), and Vm = 22.4 L mol−1.
485 C mol−1), and Vm = 22.4 L mol−1.
The liquid products were quantified using 1H nuclear magnetic resonance (NMR) (AVANCE III HD 400 MHz), in which 0.1 mL of the electrolyte was mixed with 0.5 mL of D2O, and DMSO was used as an internal standard.
Theoretical calculations
DFT calculations were performed at the GGA level within the Perdew–Burke–Ernzerhof (PBE) functional using the CASTEP software implemented in Materials Studio. The total energy calculation was performed using a kinetic energy cutoff of 450.0 eV assigned to the plane-wave basis set for calculating the density of states. The self-consistent field (SCF) tolerance was 1 × 10−6 eV. The Brillouin zone was sampled using 5 × 5 × 1 k-points. The core electrons were replaced with ultrasoft pseudopotentials. An fcc Cu model was used to further construct Cu–Ag and Cu–Cu2O. The fcc Cu–Ag was constructed by replacing a Cu atom with Ag. Then, we selected a 3 × 3 relaxed rhombus Cu2O(001) bilayer on top of a relaxed 3 × 3 Cu(001) surface to model Cu/Cu2O heterojunctions, in which O atoms were introduced to saturate the Cu atoms on the Cu(001) thus the surface atoms are well passivated. The Cu(100), Cu–Ag(100) and Cu–Cu2O(100) surfaces were modeled using five layer slabs to study the CO2 reduction activity. A vacuum region of 15 Å between any two repeated slabs was used to avoid interactions between repeated slabs along the z-direction.The binding energy (BE) of an adsorbate was calculated as:
| BE(adsorbate) = E(slab+adsorbate) − E(slab) − E(adsorbate) |
The Gibbs free energy (G) of a species was calculated as:
| G = E + ZPE − TS |
| ΔG = ΔE + ΔZPE − TΔS |
The transition state search used complete LST/QST in 0.25 eV Å−1 RMS convergence, with the optimized reactant and product geometries as starting points. CASTEP employed algorithms such as the nudged elastic band (NEB) or the dimer method to explore the potential energy surface and locate the transition state structure. The transition state was obtained between reactants and products while minimizing the energy in 10 QST steps.
Results and discussion
An HKUST-1 framework is built up of dimeric Cu units connected by benzene-1,3,5-tricarboxylate linkers. It was fabricated via a previously reported method,33 and then was incorporated with Ag after reacting with AgNO3 in a hydroalcoholic solution (VEtOH:VH2O = 9:1), where EtOH was the reducing agent that reduced Ag(I) to metallic Ag and partially reduced Cu(II) to Cu(I).34 Accordingly, we also prepared a reference sample via a similar process in the absence of AgNO3, which was named EtOH reduced HKUST-1 (ER-HKUST-1).
The crystal structure of the as-prepared samples was analyzed by XRD (Fig. 1a). Ag0.1/HKUST-1 presented a characteristic pattern consistent with those of HKUST-1 and ER-HKUST-1, confirming the well-retained framework after Ag loading and partial reduction. This is also supported by their same absorption bands in FT-IR spectra (Fig. S1, ESI†). A new peak of Ag(111) appearing in Ag0.1/HKUST-1 indicated the successful loading of Ag. An identical octahedral structure was observed in the three samples by SEM (insets of Fig. 1a), and the small nanoparticles (marked by red arrows) on the surface of Ag0.1/HKUST-1 should be assigned to the incorporated Ag. Accordingly, the elemental mapping performed with both SEM and TEM revealed the uniform distribution of O, Cu and Ag elements over Ag0.1/HKUST-1 octahedrons (Fig. S2, ESI†). In the Raman spectra (Fig. 1b), two new bands at 218 and 930 cm−1, responsible for the Cu(I)–O vibrations,34 appeared in ER-HKUST-1 and Ag0.1/HKUST-1, which indicated the partial reduction of Cu(II) to Cu(I).
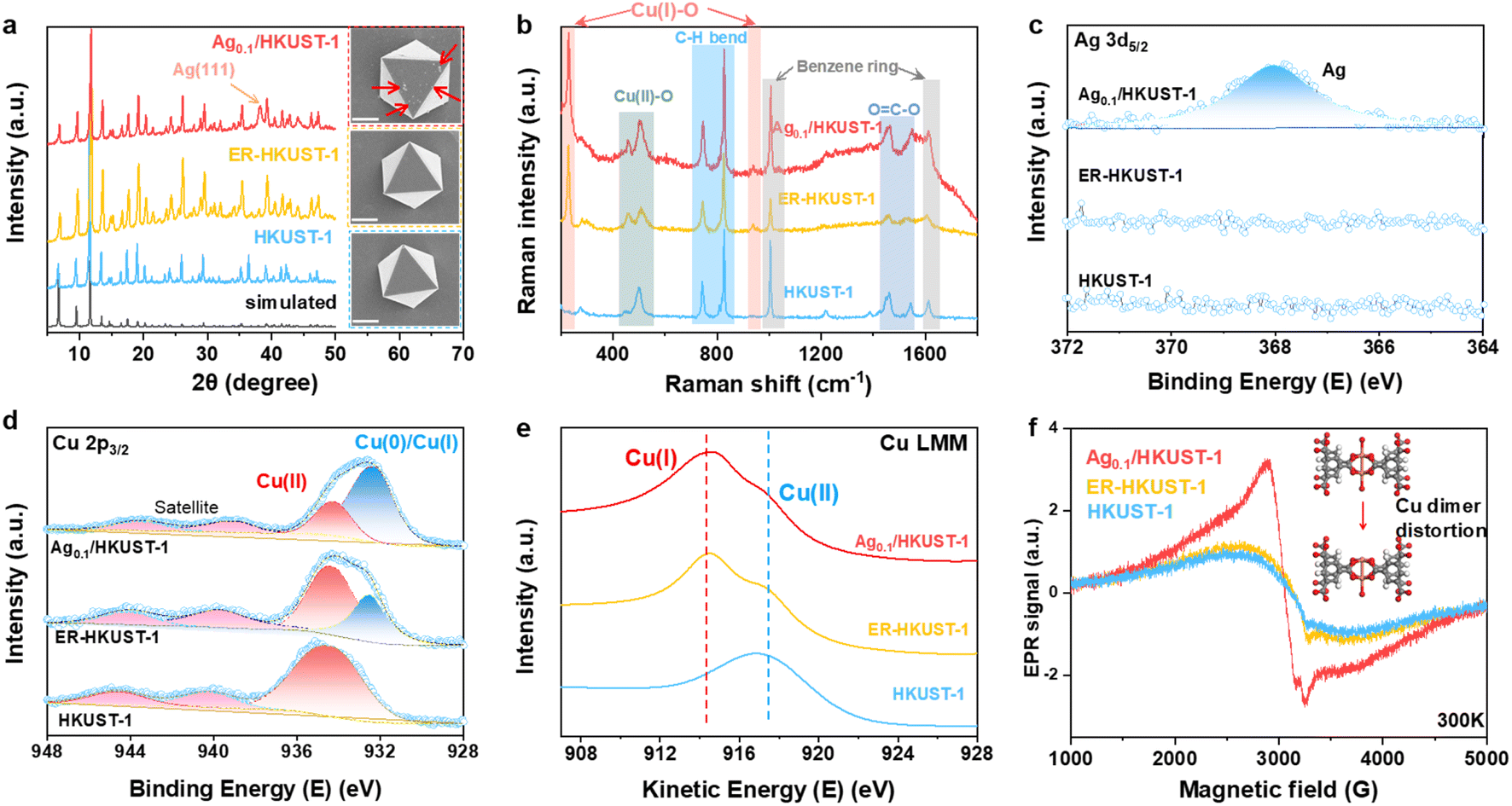 | ||
| Fig. 1 Structural characterization of HKUST-1, ER-HKUST-1 and Ag0.1/HKUST-1: (a) XRD patterns, (insets of a) SEM images with the scale bar of 1 μm, (b) Raman spectra, (c) high-resolution Ag 3d5/2 XPS, (d) high-resolution Cu 2p3/2 XPS, (e) Cu LMM AES and (f) EPR spectra. | ||
The chemical composition and element valence state were further studied by XPS and AES. Different from HKUST-1 and ER-HKUST-1, Ag0.1/HKUST-1 displayed a broad Ag 3d peak at 368.1 eV in XPS (Fig. 1c), confirming the successful incorporation of Ag. In the profile of Cu 2p3/2, the peak at 934.5 eV was attributed to Cu(II) species, and that at 932.4 eV could be assigned to Cu(0) or Cu(I) (Fig. 1d).18 AES with the good resolution of Cu(0)/Cu(I) further identified the co-presence of Cu(I) and Cu(II) in both Ag0.1/HKUST-1 and ER-HKUST-1, in comparison with the dominant Cu(II) in HKUST-1 (Fig. 1e). Therefore, the peak at 932.4 eV in the Cu 2p3/2 XPS profile should be ascribed to the Cu(I) species in Ag0.1/HKUST-1 and ER-HKUST-1, which was derived from the partial reduction of Cu(II) by EtOH. It's noteworthy that Ag0.1/HKUST-1 showed a relatively higher intensity of Cu(I) in both XPS and AES profiles as compared with ER-HKUST-1, suggesting the stabilization of Cu(I) by additive Ag. The presence of Cu(I) would cause the distortion of the Cu–Cu dimer, as confirmed by the high signal intensity of Ag0.1/HKUST-1 in EPR (Fig. 1f).35 In addition, a series of Agn/HKUST-1 frameworks with an identical structure but different Ag/Cu molar ratios could be obtained after varying the feeding ratio (Fig. S3–S7, ESI†), which showed a volcano relationship between Cu(I)/Cu(II) and Ag/Cu ratios (Fig. S8, ESI†), indicating the enriched Cu(I) at a moderate Ag loading.
We conducted in situ Raman analysis combined with a CO2RR test at various potentials to assess the reconstruction of such Cu-based MOFs (Fig. S9, ESI†). The bands corresponding to Cu(II)–O and C–H disappeared quickly at the applied bias, indicating the collapse of the metal–organic framework. The emerging band of Cu–CO further suggested the formation of *CO on the in situ formed metallic surface.36 Of particular interest is the appearance of Cu2O bands at 520 and 610 cm−1 in Ag0.1/HKUST-1 and ER-HKUST-1 compared to HKUST-1,37 whose intensity increased with the negative potentials and reached a maximum at −1.3 V vs. RHE. Thus, the in situ reconstruction of the three samples was further analyzed at −1.3 V vs. RHE in a flow cell. As the model sample, Ag0.1/HKUST-1 afforded a fluctuating current density initially in chronoamperometry (Fig. 2a), which afterwards became more stable after 1 h electrolysis, suggesting that the reconfiguration can be completed within the first hour. To observe the reconstruction process more directly, the reconstituted samples were subjected to SEM (Fig. 2b–d). Obviously, the octahedral structure collapsed after only 1 min, and the final morphology of HKUST-1 and ER-HKUST-1 was granular, whereas Ag0.1/HKUST-1 exhibited dendritic nanostructures that can obviously enhance surface hydrophobicity (Fig. S10, ESI†). As previously evidenced,38,39 electrocatalysts with a hydrophobic surface would be favorable for the CO2RR because of the inhibited hydrogen evolution competition. Interestingly, the reconfigured samples after 1 h and 5 h of electrolysis presented identical nanostructures, further confirming the rapid reconstruction finished within 1 h. Accordingly, the disappearance of the characteristic peaks of the benzene ring in FT-IR indicated that the organometallic framework has been drastically destroyed (Fig. S11, ESI†). This is distinct from the previous reports of ligand-retaining Cu surfaces (e.g., Cu2O@CuHHTP,17 Cu2(HCOO)3 clusters,19 PPy/HKUST-1,21 and PANI/HKUST-1 (ref. 21)), and guarantees a reliable platform to unravel the structure–activity relationship of in situ formed interfaces.
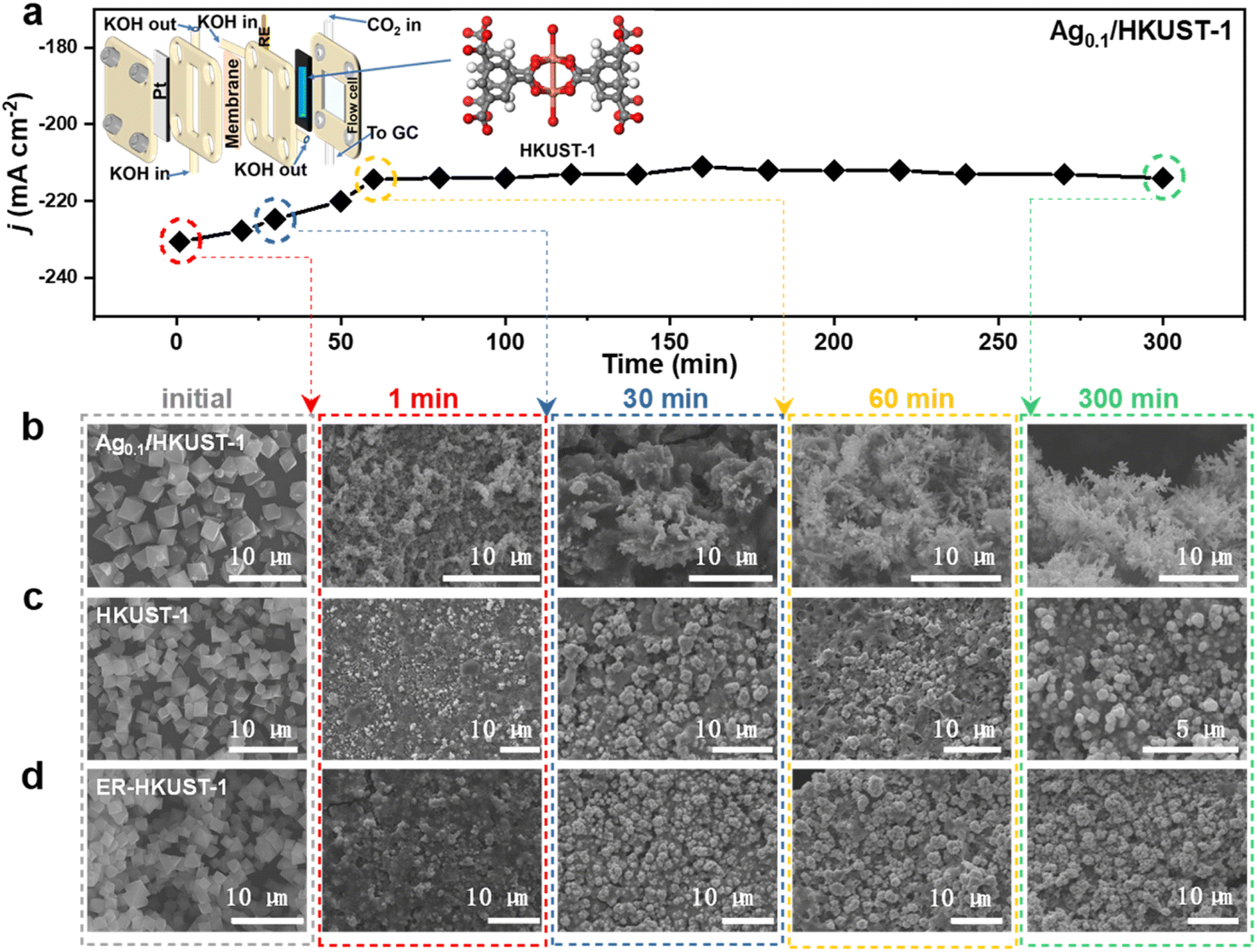 | ||
| Fig. 2 (a) Chronoamperometry curve of Ag0.1/HKUST-1 during in situ electrochemical reconstruction at −1.3 V vs. RHE, and SEM images of (b) Ag0.1/HKUST-1, (c) HKUST-1 and (d) ER-HKUST-1 collected at 0, 1, 30, 60, and 300 min. | ||
The restructured electrocatalysts from HKUST-1, ER-HKUST-1 and Ag0.1/HKUST-1 after 1 h of CO2RR were denoted as CuHKUST-1, Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1, respectively. To minimize the impact of oxidation, the samples were immediately transferred to an argon-filled glovebox for further characterization. As depicted in their XRD patterns (Fig. 3a), the diffraction peaks of the original framework disappeared. Instead, typical peaks of Cu(111) and (200) were detected at 2θ = 43.3° and 50.4°, respectively. Besides, the visible Cu2O(111) and (200) in both Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1, and the Ag(111) and (200) in Ag/Cu/Cu2OAg0.1/HKUST-1 confirmed the co-presence of multiple phases, i.e., Cu/Cu2O and Ag/Cu/Cu2O. In the Cu 2p XPS profiles (Fig. 3b), unlike the pre-catalysts, the peak of Cu(II) was absent in the reconstructed samples, and only the peak at 932.4 eV associated with Cu(0)/Cu(I) was observed, which indicated the reduction of Cu(II) at the applied potential. Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1 showed peaks at 914.3 and 917.3 eV in the AES, corresponding to Cu(I) and Cu(0) species,40 while only Cu(0) was present on CuHKUST-1 (Fig. 3c), which were consistent with the XRD results. Compared to the pre-catalysts, the peak of Ag 3d on Ag/Cu/Cu2OAg0.1/HKUST-1 shifted toward a lower binding energy (Fig. S12, ESI†), probably due to Ag aggregation along with the electrochemical reconstruction.41
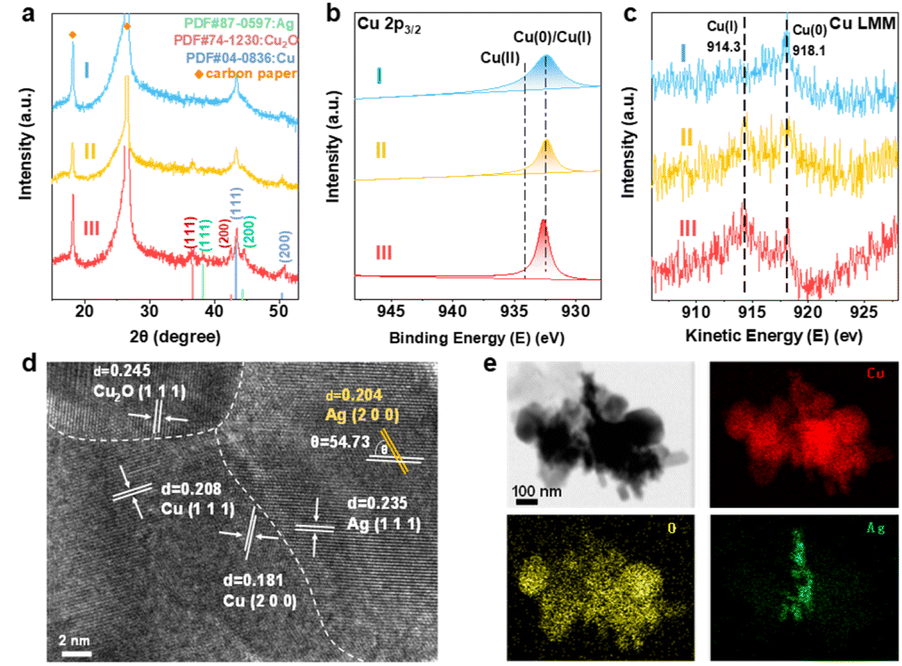 | ||
| Fig. 3 (a) XRD patterns, (b) high-resolution Cu 2p3/2 XPS and (c) Cu LMM AES of (I) CuHKUST-1, (II) Cu/Cu2OER-HKUST-1 and (III) Ag/Cu/Cu2OAg0.1/HKUST-1. (d) HR-TEM image and (e) elemental mapping of Ag/Cu/Cu2OAg0.1/HKUST-1. | ||
The high-resolution TEM (HR-TEM) image of Ag/Cu/Cu2OAg0.1/HKUST-1 clearly showed the interfaces of Ag/Cu/Cu2O (Fig. 3d). The lattice fringes of 0.181, 0.204 and 0.245 nm could be indexed to Cu(200), Cu(111) and Cu2O(111), respectively, and those of 0.204 and 0.235 nm were assigned to Ag(200) and Ag(111). In comparison, the lattice planes of Cu(111) and Cu2O(111) were visible in Cu/Cu2OER-HKUST-1, and only Cu(111) was observed in CuHKUST-1 (Fig. S13, ESI†). In Fig. 3e, the elements of Cu and O were uniformly distributed throughout the nanostructures, while the concentrated distribution of Ag suggested the formation of an Ag/Cu/Cu2O multi-phase with abundant interfaces, rather than Ag–Cu alloys.
Furthermore, the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) of Ag/Cu/Cu2OAg0.1HKUST-1 were analysed to understand the local environments of Ag and Cu (Fig. S14, ESI†). The Ag K-edge overlapped with that of Ag foil, indicating a close-to-zero valence state of Ag in Ag/Cu/Cu2OAg0.1HKUST-1. But the white line peak was slightly lower, consistent with the XPS data (Fig. S12, ESI†), which was probably due to the electron transfer from Cu to Ag. The electron transfer between Ag and Cu may lead to the formation of Ag–Cu bonds, as well as the existence of an Ag–Cu interface.42 Accordingly, the Cu K-edge analysis presented a slightly higher valence state of Cu than that of the Cu2O reference. Moreover, the k2-weighted Ag K-edge EXAFS showed that the bond length and coordination number of Ag in Ag/Cu/Cu2OAg0.1HKUST-1 were slightly reduced, suggesting the formation of a heterostructure rather than alloys. Analogously, the increased Cu–O signal in the Cu K-edge EXAFS as compared with that of a Cu reference indicated the considerable amount of Cu2O in the restructured interfaces, and the lower Cu–O coordination number and the accordingly longer Cu–O bond in comparison with those of a Cu2O reference might be due to the presence of heterogeneous interfaces. In addition, wavelet transform analysis highlighted the dominance of Ag–Ag and Cu–O bonds in the heterostructures, while Ag–O and Cu–Cu bonds are negligible, matching well with the EXAFS fitting results (Fig. S15 and Table S1, ESI†).
The electrochemical CO2RR performance of CuHKUST-1, Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1 was evaluated in a flow cell with 1.0 M KOH as the electrolyte. The polarization curves showed the highest current densities of Ag/Cu/Cu2OAg0.1/HKUST-1 among the three samples (Fig. 4a), indicative of the high activity of the restructured Ag/Cu/Cu2O. The electrolyte after reaction was analyzed by using 1H nuclear magnetic resonance (NMR) spectroscopy, which suggested the negligible formation of liquid products (Fig. S16, ESI†). As evidenced by recent reports,38,43 the higher coverage and transport efficiency of chemisorbed *H derived from abundant H2O on gas–liquid–solid interfaces would benefit the generation of alcohols, the typical liquid products of the CO2RR. Herein, the super-hydrophobic surface of the restructured Ag/Cu/Cu2O (Fig. S10, ESI†) probably prohibits the formation of alcohol products. With further regard to the same key step of *CO coupling shared by ethylene and ethanol formation, we thereby focused on the gas products quantified by on-line GC for the understanding of the restructured interfaces, to avoid the superposition of instrumental errors (e.g., GC and NMR). Potentiostatic measurements at −1.0 to −1.5 V (vs. RHE) showed that Ag/Cu/Cu2OAg0.1/HKUST-1 produced mainly C2H4 with the maximum FE of 57.2% at −1.3 V vs. RHE, while this value was only 17.4% on CuHKUST-1 (Fig. 4b). The reaction profiles of CuHKUST-1, Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1, including product FEs and current densities, are depicted in Fig. S17, ESI.† Accordingly, Ag/Cu/Cu2OAg0.1/HKUST-1 afforded a higher partial current density of C2H4 (jC2H4) than CuHKUST-1 and CuER-HKUST-1 (Fig. 4c), confirming the promoted ethylene production over Ag/Cu/Cu2O heterostructures. For comparison, either the reference sample consisting of physically mixed Cu, Ag and Cu2O nanoparticles or the electro-deposited one delivered a lower FE of C2H4 than Ag/Cu/Cu2OAg0.1/HKUST-1 under the same conditions (Fig. S18, ESI†), which highlighted the key contribution of MOF precursors to construct active Ag/Cu/Cu2O multi-interfaces. To further unravel the effect of Ag, the CO2RR performance of a series of Ag/Cu/Cu2O derived from Agn/HKUST-1 frameworks was measured (Fig. S19, ESI†). With the increase of Ag content, the FE of C2H4 gradually increased and then tended to decrease, which indicated the benefited synergy of active components on the in situ generated Ag/Cu/Cu2O interfaces with an optimal composition.
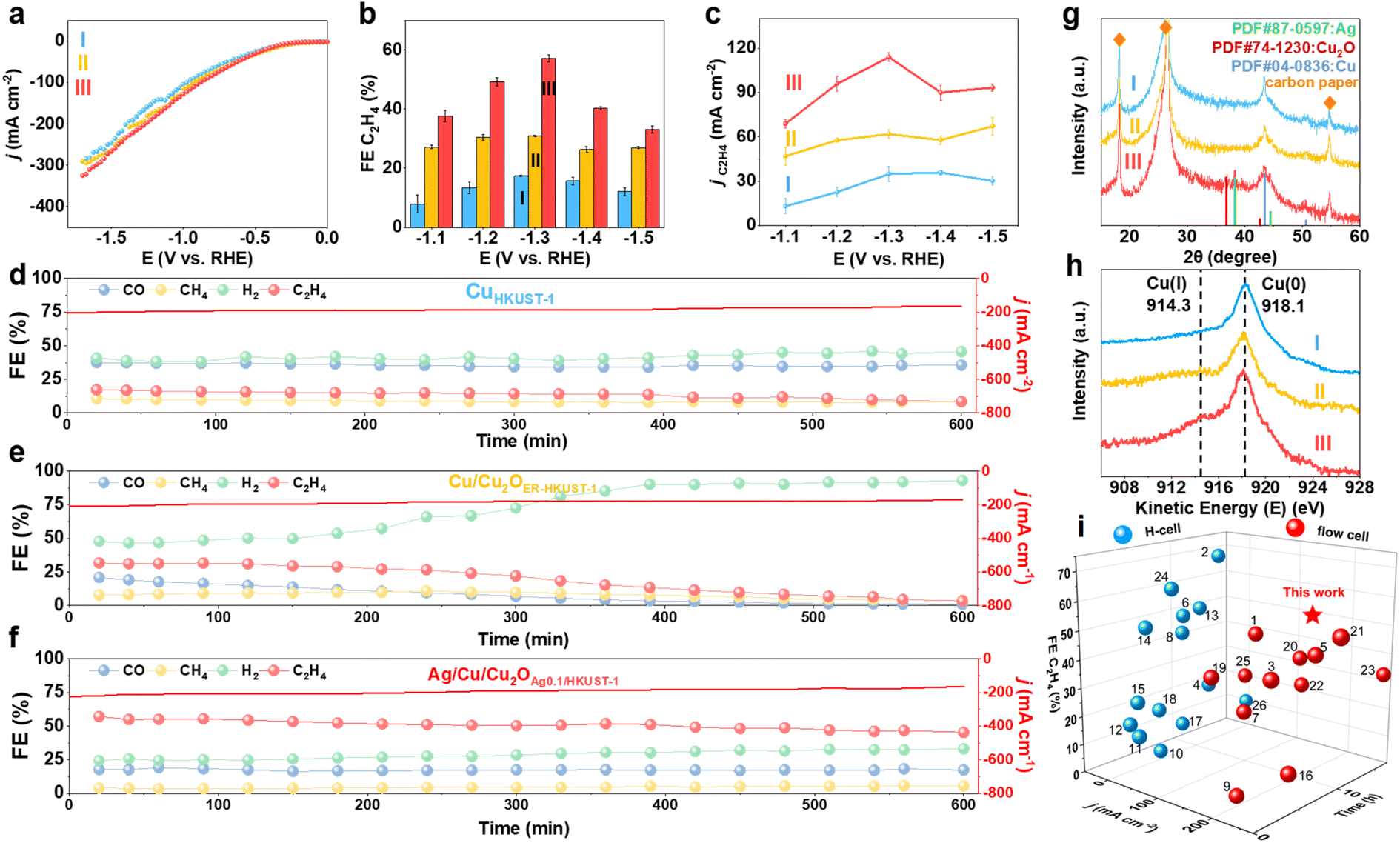 | ||
| Fig. 4 (a) Polarization curves, (b) FE and (c) partial current density of C2H4 at different applied potentials of (I) CuHKUST-1, (II) Cu/Cu2OER-HKUST-1 and (III) Ag/Cu/Cu2OAg0.1/HKUST-1 in CO2-saturated 1.0 M KOH. Chronoamperometric stability tests of (d) CuHKUST-1, (e) Cu/Cu2OER-HKUST-1 and (f) Ag/Cu/Cu2OAg0.1/HKUST-1. (g) XRD patterns and (h) Cu LMM AES of CuHKUST-1, Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1 after the CO2RR (600 min). (i) Performance of Ag/Cu/Cu2OAg0.1/HKUST-1 compared with the recently reported Cu-based electrocatalysts (the numbers of reference samples are obtained from Table S2, ESI†).16,18–20,35,44–64 | ||
The electrochemically active surface area (ECSA) of the catalysts could be evaluated in terms of double-layer capacitance (Cdl, Fig. S20, ESI†), on the basis of their proportional correlation. Ag/Cu/Cu2OAg0.1/HKUST-1 presented a Cdl value of 3.24 mF cm−2, exceeding those of CuER-HKUST-1 (2.68 mF cm−2) and Cu/Cu2OHKUST-1 (2.04 mF cm−2), which indicated enriched active-sites after restructuring Ag/Cu/Cu2O multi-interfaces. Meanwhile, electrochemical impedance spectroscopy (EIS) pointed out the relatively smaller electron-transfer resistance (Rct) on Ag/Cu/Cu2OAg0.1/HKUST-1 (Fig. S21, ESI†), in good accordance with its higher activity.
The long-term stability was evaluated by conducting a chronoamperometric test (Fig. 4d–f). On CuHKUST-1, the main products were H2 and CO with FEs of ∼40% and ∼35%, respectively, and the FE of C2H4 was lower than 18%. Although the C2H4 production was impressively enhanced on CuER-HKUST-1 (FEC2H4 ∼30%) during the initial reaction, it suffered from a drastic decrease to <20% after 300 min, concomitant with the increase of H2 production. In sharp comparison, Ag/Cu/Cu2OAg0.1/HKUST-1 manifested a remarkably stable FE of C2H4 (∼50%) within 600 min, which should be ascribed to the Ag additives. In addition, compared to CuHKUST-1, Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1 showed a significantly lower CO yield and higher C2H4 yield during the initial reactions, which was accounted for by the effective consumption of *CO to produce C2H4. Afterwards, the CO production suffered a continuous decrease on Cu/Cu2OER-HKUST-1, whereas it was almost constant on Ag/Cu/Cu2OAg0.1/HKUST-1, suggesting a dynamic balance of *CO formation and self-coupling on Ag/Cu/Cu2O interfaces. In other words, *CO generated on Ag sites might spill over onto the Cu/Cu2O interface to increase the surface coverage of *CO, thereby promoting C–C coupling toward C2H4 and retaining the balance with CO formation and consumption.30,32 Furthermore, we characterized the three catalysts after the long-term CO2RR test. The spent Ag/Cu/Cu2OAg0.1/HKUST-1 showed a well-retained phase composition (Ag/Cu/Cu2O) in XRD (Fig. 4g). The HR-TEM images and the elemental mapping of Ag/Cu/Cu2OAg0.1/HKUST-1 clearly presented the interfaces of Ag/Cu/Cu2O (Fig. S22, ESI†), indicating the good structural stability. By contrast, the Cu2O(111) and (200) disappeared on the used Cu/Cu2OER-HKUST-1, consistent with the drastic decrease of C2H4 FE. Accordingly, the AES results (Fig. 4h) identified the total disappearance of Cu(I) on Cu/Cu2OER-HKUST-1, but the signal on Ag/Cu/Cu2OAg0.1/HKUST-1 was still visible after the CO2RR, which confirmed the promoted stability of Cu(I) assisted by Ag additives. There are two related thermodynamic factors taken into consideration. First, the substantial disparity in the formation enthalpies of Cu2O (−169 kJ mol−1) and Ag2O (−31.1 kJ mol−1) suggests that any Ag oxide resulting from oxygen exposure will be rapidly reduced by the adjacent Cu.32 Second, Ag, with its higher redox potential relative to Cu, can accept electrons from Cu. In addition, the negligible change of the Ag 3d signal in XPS further underscored the durability of the Ag/Cu/Cu2O ternary interfaces in Ag/Cu/Cu2OAg0.1/HKUST-1 (Fig. S23, ESI†).
In further comparison with recently reported Cu-based electrocatalysts (Fig. 4i and Table S2, ESI†), Ag/Cu/Cu2OAg0.1/HKUST-1 presented an outstanding overall performance with excellent C2H4 FE, current output, and service life. Although some previous reports demonstrated the high FE of C2H4, they did not gain the high levels of current density and stability synchronously. For example, CuPz2-Act-30 exhibited a high C2H4 FE of 67% in an H-cell, but the performance under high-current conditions such as in flow cells is still unclear.20 S-HKUST-1 displayed a C2H4 FE of 57.2% at a current density of −400 mA cm−2 in a flow cell, but the stability test (480 min) was only conducted at −150 mA cm−2.18 Similarly, Cu–PzH yielded a C2H4 FE of 60% at −1.0 V (vs. RHE) with a large partial current density of 346.46 mA cm−2 while its stability remained for only 242 min.59 More counterparts with rich Cu(I) species but prepared via other methods were further taken for comparison. For example, Cu2O–BN and Cu2O@SiO2–NH2 demonstrated stability exceeding 600 min,61,64 whereas the C2H4 FEs of 35% and 15% were deemed suboptimal. An AgCu single-atom alloy blended with Ag nanoparticles showed a 94 ± 4% FE towards multi-carbon products at a high current density of ∼720 mA cm−2 in a flow cell, but the FE of C2H4 was below 40%.42 In contrast, Ag/Cu/Cu2OAg0.1/HKUST-1 can achieve the superiority of C2H4 FE (∼50%), current output (>180 mA cm−2), and stability (600 min) at the same time, underscoring its promise for practical use. When a membrane electrode assembly (MEA) reactor is rationally designed for such restructured catalysts, we believe greater advances in performance, in particular the greater current density and long-term stability, can be accomplished.65
In situ Raman spectra were obtained and the CO2RR test was performed to understand the dynamic changes of the restructured interfaces. Different from CuHKUST-1, Cu/Cu2OER-HKUST-1 and Ag/Cu/Cu2OAg0.1/HKUST-1 showed the band assigned to Cu(I) species at 520 and 610 cm−1 (Fig. 5a and b).37 This band was well retained on Ag/Cu/Cu2OAg0.1/HKUST-1 for 300 min, while it became weaker and disappeared on Cu/Cu2OER-HKUST-1. In the meantime, this evolution was confirmed by the in situ Raman spectra collected at various potentials (Fig. S24, ESI†). Such in situ monitoring, highly consistent with the post-CO2RR AES analysis (Fig. 4h), identified again the stabilization of the Cu(I) species by Ag. Furthermore, the peaks at 280 and 364.3 cm−1, corresponding to the restricted rotating (P1) and stretching (P2) models of adsorbed Cu–CO, respectively, could be detected on the surfaces, and the intensity ratio of P2/P1 peaks was a valid measure of the surface coverage of *CO.36 The P2 band was the main peak while the P1 was very weak on CuHKUST-1 (Fig. 5c and d), and their large ratio of 8.5–9.0 indicated the extremely high *CO coverage on the Cu(0)-dominant surface,66 consistent with the main distribution of CO in the products (Fig. 4d). In comparison with Cu/Cu2OER-HKUST-1, Ag/Cu/Cu2OAg0.1/HKUST-1 delivered a larger P2/P1 ratio associated with the higher *CO coverage. More importantly, this ratio was kept at a high level above 2.0 on Ag/Cu/Cu2OAg0.1/HKUST-1 during the CO2RR (0–300 min), but it drastically decreased on Cu/Cu2OER-HKUST-1 (Fig. 5d). According to the recently evidenced tandem electrocatalysis on Ag/Cu interfaces,67,68 we reasonably assumed that the CO spillover from Ag sites could increase the *CO coverage on Cu(0)–Cu(I) ensembles, leading to the well-maintained P2/P1 ratio on Ag/Cu/Cu2OAg0.1/HKUST-1 for the further C–C coupling.
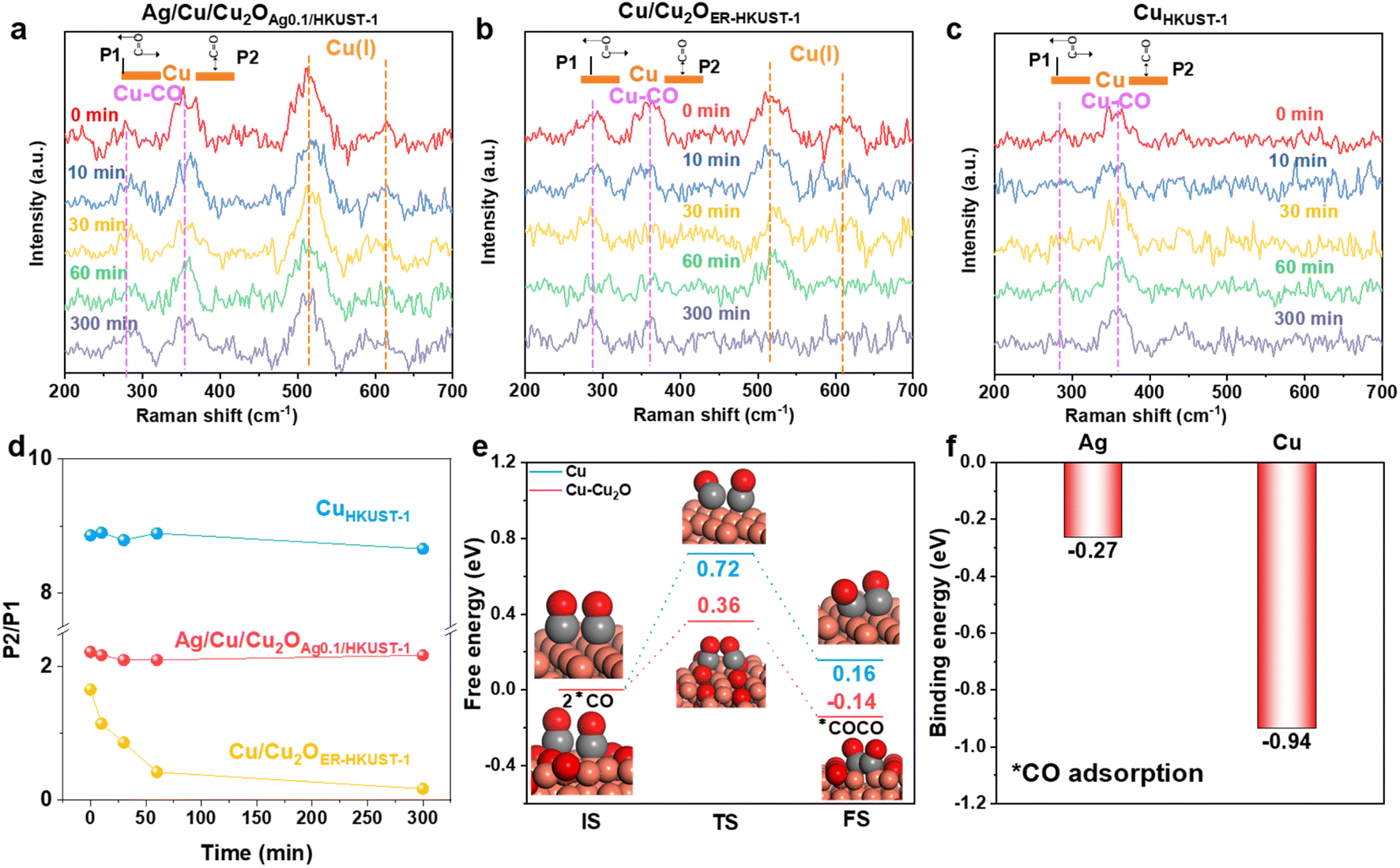 | ||
| Fig. 5 In situ Raman spectra during the CO2RR under different reaction times of (a) Ag/Cu/Cu2OAg0.1/HKUST-1, (b) Cu/Cu2OER-HKUST-1 and (c) CuHKUST-1, and (d) the corresponding evolution of P2/P1 ratios. (e) Energy profiles for the initial states (ISs), transition states (TSs) and final states (FSs) of *CO–*CO coupling on Cu and Cu–Cu2O models. (f) Energy profiles of *CO adsorption on Ag and Cu. | ||
To further understand the contribution of Ag and Cu2O on the C–C coupling and ethylene production, the model structures of Cu and Cu–Cu2O were taken for DFT calculations. As a critical step toward C2+ products, the coupling of two *CO was brought into focus. As shown in Fig. 5e, the kinetic barrier (0.36 eV) and enthalpy change (−0.14 eV) for the *CO coupling on Cu–Cu2O are lower than those on Cu (0.72 and 0.16 eV), suggesting that the interfaces of Cu(0)–Cu(I) can effectively boost C2 production. The coupling of two adjacent *CO on Cu–Ag should be difficult because of the high energy barrier (1.44 eV) (Fig. S25, ESI†), wherein the spillover of *CO from Ag to Cu would be preferred. Numerous studies have found that Ag has a high selectivity for CO in the CO2RR which can provide adequate *CO for the tandem catalysis toward C2 products.30–32,67,68 As indicated by the more negative *CO binding energy on Cu (−0.94 eV) than that on Ag (−0.27 eV) (Fig. 5f), the spillover of the as-formed *CO from Ag to Cu and even Cu(0)–Cu(I) interfaces is thermodynamically favorable. Therefore, a tandem mechanism is proposed, in which the CO produced by Ag sites can diffuse to the reactive interface of Cu–Cu2O with a low kinetic barrier for further coupling toward C2H4. In addition, Ag can stabilize the Cu(I) species to enable high ethylene production for a long time, which has been well validated by in situ Raman and ex situ XRD and AES.
Finally, we adopted other Cu-MOFs with the varied coordination as the pre-catalysts towards Ag/Cu/Cu2O interfaces, to examine the applicability of this strategy. With a similar Cu–O4 structure but a layered crystalline structure, CuBDC (BDC = 1,4-benzenedicarboxylic acid) underwent extensive reconstruction from 2D crystals to smaller nanoparticles, which was beneficial for restructuring Ag/Cu/Cu2O interfaces when Ag was incorporated (Fig. S26, ESI†). Thanks to the rich interfaces of Ag/Cu/Cu2O, the reconstructed samples afforded the obviously higher FE of C2H4 in comparison with that free from Ag. By contrast, CuPz2 (Pz = pyrazol) with a Cu–N4 structure cannot undergo the reconstruction under these working conditions due to the strong coordination of Cu–N4 (Fig. S27, ESI†). When Ag was incorporated into CuPz2, the resulting samples unfortunately increased the FE of CO, probably due to the preferred generation of CO on the loaded Ag nanoparticles. The accordingly reduced FE of C2H4 also indicated the severely competitive reduction of CO2 on Cu and Ag sites because of the absence of Ag/Cu interfaces. This is indirect proof for the tandem electrolysis towards C2H4 over Ag/Cu/Cu2OAg0.1/HKUST-1via *CO spillover. It's clear that the coordination of Cu-based MOFs is important for restructuring Ag/Cu/Cu2O interfaces, although the ligands make negligible influences on the electrocatalytic performance indeed because they have already been removed during the reconstruction (Fig. S11, ESI†). The frangible Cu–O4 nodes are the prerequisite for the reconstruction, while the strong Cu–N4 requires more tough conditions (e.g., more negative potentials) to drive the reconfiguration.20
Conclusions
In summary, the directional in situ reconfiguration of Ag incorporating HKUST-1 frameworks was successfully introduced to restructure multi-phase Ag/Cu/Cu2O electrocatalysts highly active for the CO2RR. The reconstituted Ag/Cu/Cu2O exhibited outstanding performance for the selective electro-reduction of CO2 to C2H4, with superior FE, current output, and service life to the most of recently reported counterparts. The combination of in/ex situ characterizations (Raman, XRD, XPS and AES) and theoretical calculations demonstrated that Ag plays a crucial role in stabilizing Cu(I) and increasing CO surface coverage, while the resulting Cu/Cu2O interfaces significantly reduced the C–C coupling energy barrier toward the boosted ethylene production. This work not only offered a rational method to prepare highly active catalysts via electrochemical activation, but also deepened the understanding of the working state and mechanism on multi-phase interfaces. It's envisioned to gain further improvement via utilizing booming MOF pre-catalysts with more precise control over the coordination chemistry and dynamic evolution.Data availability
Data supporting the findings of this study are available within the article ESI.†Author contributions
Jiye Feng: synthesis, investigations, formal analysis, writing of the original draft, data curation. Wenbiao Zhang: DFT calculation, supervision. Danni Shi: experimental analysis, image polishing. Yingshuai Jia: proofreading of the original draft. Yi Tang: conceptualization, supervision, funding acquisition. Yuying Meng: conceptualization. Qingsheng Gao: conceptualization, supervision, writing – review and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (No. 22175077), Innovation Team Project in Guangdong Colleges and Universities (No. 2021KCXTD009), Guangdong Basic and Applied Basic Research Foundation (2023A1515240081), Guangzhou Science and Technology Program (No. 202201020071) and Fundamental Research Funds for the Central Universities (No. 21623103).Notes and references
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2020, 367, eaav3506 Search PubMed.
- D. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- T. Yan, X. Chen and L. Kumari, et al. , Chem. Rev., 2023, 123, 10530–10583 CrossRef CAS PubMed.
- M. Kuang and G. Zheng, Chem Catal., 2023, 3, 100565 CrossRef CAS.
- Q. Chen, X. Wang, Y. Zhou, Y. Tan, H. Li, J. Fu and M. Liu, Adv. Mater., 2024, 36, 2303902 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen and S. B. Scott, et al. , Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, D. Cao-Thang, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- H. Yang, S. Li and Q. Xu, Chin. J. Catal., 2023, 48, 32–65 CrossRef CAS.
- R. M. Aran-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef CAS.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS PubMed.
- H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, ACS Cent. Sci., 2022, 8, 1506–1517 CrossRef CAS PubMed.
- X. Kang, Q. Zhu, X. Sun, J. Hu, J. Zhang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 266–273 RSC.
- D. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 18178–18184 CrossRef CAS PubMed.
- X. Xie, X. Zhang and M. Xie, et al. , Nat. Commun., 2022, 13, 63 CrossRef CAS PubMed.
- J. D. Yi, R. Xie, Z. L. Xie, G. L. Chai, T. F. Liu, R. P. Chen, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- C. F. Wen, M. Zhou and P. F. Liu, et al. , Angew. Chem., Int. Ed., 2022, 61, e202111700 CrossRef CAS PubMed.
- W. Zhang, C. Huang and J. Zhu, et al. , Angew. Chem., Int. Ed., 2022, 61, e202112116 CrossRef CAS PubMed.
- C. Liu, X.-D. Zhang, J.-M. Huang, M.-X. Guan, M. Xu and Z.-Y. Gu, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS.
- J. Cheng, L. Chen and X. Xie, et al. , Angew. Chem., Int. Ed., 2023, 62, e202312113 CrossRef CAS PubMed.
- J.-X. Wu, S.-Z. Hou, X.-D. Zhang, M. Xu, H.-F. Yang, P.-S. Cao and Z.-Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- J.-Y. Kim, D. Hong and J.-C. Lee, et al. , Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed.
- C. Hahn, T. Hatsukade, Y.-G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison and J. Wicks, et al. , Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- X.-D. Zhang, T. Liu and C. Liu, et al. , J. Am. Chem. Soc., 2023, 145, 2195–2206 CrossRef CAS PubMed.
- S.-C. Lin, C.-C. Chang, S.-Y. Chiu, H.-T. Pai, T.-Y. Liao, C.-S. Hsu, W.-H. Chiang, M.-K. Tsai and H. M. Chen, Nat. Commun., 2020, 11, 3525 CrossRef CAS PubMed.
- W. Zhang, Y. Yang, Y. Tang and Q. Gao, J. Energy Chem., 2022, 70, 414–436 CrossRef CAS.
- C.-J. Chang, S.-C. Lin, H.-C. Chen, J. Wang, K. J. Zheng, Y. Zhu and H. M. Chen, J. Am. Chem. Soc., 2020, 142, 12119–12132 CrossRef CAS PubMed.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- T. Yang, M. Kuang and J. Yang, Nano Res., 2023, 16, 8670–8683 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- Z. Song, Y. Liu, Y. Zhong, Q. Guo, J. Zeng and Z. Geng, Adv. Mater., 2022, 34, 2204306 CrossRef CAS PubMed.
- H. Sun, X. Han, K. Liu, B. Shen, J. Liu, D. Wu and X. Shi, Ind. Eng. Chem. Res., 2017, 56, 9541–9550 CrossRef CAS.
- N. Dae-Hyun, O. S. Bushuyev and J. Li, et al. , J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef PubMed.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kuhl, R. Garcia-Muelas, N. Lopez and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- Y. Lin, T. Wang and L. Zhang, et al. , Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed.
- Y. Wu, J. Feng, D. Shi, W. Zhang, Y. Tang and Q. Gao, Chem. Commun., 2023, 59, 10428–10431 RSC.
- Y. Guo, C. Feng and S. Wang, et al. , J. Mater. Chem. A, 2020, 8, 24477–24485 RSC.
- I. Lopez-Salido, D. C. Lim and Y. D. Kim, Surf. Sci., 2005, 588, 6–18 CrossRef CAS.
- C. Du, J. P. Mills and A. G. Yohannes, et al. , Nat. Commun., 2023, 14, 6142 CrossRef CAS PubMed.
- M. Luo, Z. Wang and Y. C. Li, et al. , Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- Z. Weng, J. Jiang and Y. Wu, et al. , J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- S. Kusama, T. Saito, H. Hashiba, A. Sakai and S. Yotsuhashi, ACS Catal., 2017, 7, 8382–8385 CrossRef CAS.
- Y.-S. Cheng, X.-P. Chu, M. Ling, N. Li, K.-L. Wu, F.-H. Wu, H. Li, G. Yuan and X.-W. Wei, Catal. Sci. Technol., 2019, 9, 5668–5675 RSC.
- F. Yang, A. Chen, P. L. Deng, Y. Zhou, Z. Shahid, H. Liu and B. Y. Xia, Chem. Sci., 2019, 10, 7975–7981 RSC.
- A. Guan, Z. Chen and Y. Quan, et al. , ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- J.-D. Yi, R. Xie, Z.-L. Xie, G.-L. Chai, T.-F. Liu, R.-P. Chen, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- Y. Zhou, S. Chen, S. Xi, Z. Wang, P. Deng, F. Yang, Y. Han, Y. Pang and B. Y. Xia, Cell Rep. Phys. Sci., 2020, 1, 100182 CrossRef.
- H. Huo, J. Wang, Q. Fan, Y. Hu and J. Yang, Adv. Energy Mater., 2021, 11, 2102447 CrossRef CAS.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- L. Zhang, X.-X. Li and Z.-L. Lang, et al. , J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS PubMed.
- Y. Zhang, L.-Z. Dong, S. Li, X. Huang, J.-N. Chang, J.-H. Wang, J. Zhou, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS PubMed.
- R. Chen, L. Cheng, J. Liu, Y. Wang, W. Ge, C. Xiao, H. Jiang, Y. Li and C. Li, Small, 2022, 18, 2200720 CrossRef CAS PubMed.
- D.-S. Huang, H.-L. Zhu, Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 8444–8450 CrossRef CAS.
- L.-L. Zhuo, P. Chen and K. Zheng, et al. , Angew. Chem., Int. Ed., 2022, 61, e202204967 CrossRef CAS PubMed.
- B. Kim, Y. C. Tan and Y. Ryu, et al. , ACS Energy Lett., 2023, 8, 3356–3364 CrossRef CAS.
- R. Wang, J. Liu and Q. Huang, et al. , Angew. Chem., Int. Ed., 2021, 60, 19829–19835 CrossRef CAS PubMed.
- P. P. Guo, Z. H. He, S. Y. Yang, W. T. Wang, K. Wang, C. C. Li, Y. Y. Wei, Z. T. Liu and B. X. Han, Green Chem., 2022, 24, 1527–1533 RSC.
- Y. Zhou, Y. Yao and R. Zhao, et al. , Angew. Chem., Int. Ed., 2022, 134, e202205832 CrossRef.
- X. Ma, X. Song and L. Zhang, et al. , Green Chem., 2023, 25, 7635–7641 RSC.
- F. Yu, X. Liu, L. Liao, G. Xia and H. Wang, Small, 2023, 19, 2301558 CrossRef CAS PubMed.
- Z.-Y. Zhang, H. Tian, H. Jiao, X. Wang, L. Bian, Y. Liu, N. Khaorapapong, Y. Yamauchi and Z.-L. Wang, J. Mater. Chem. A, 2024, 12, 1218–1232 RSC.
- Y. Liang, J. Zhao and Y. Yang, et al. , Nat. Commun., 2023, 14, 474 CrossRef CAS PubMed.
- T.-C. Chou, C.-C. Chang and H.-L. Yu, et al. , J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- Y. Yu, D. Wang, Y. Hong, T. Zhang, C. Liu, J. Chen, G. Qin and S. Li, Chem. Commun., 2022, 58, 11163–11166 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and data for CO2RR performance, EDS mapping, additional XRD, IR, XPS and CVs, NMR spectra, XANES and EXAFS. See DOI: https://doi.org/10.1039/d4sc00967c |
| This journal is © The Royal Society of Chemistry 2024 |