
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Axially-chiral boramidine for detailed (chir)optical studies†
Nidal
Saleh
 *a,
Estefanía
Sucre-Rosales
b,
Francesco
Zinna
c,
Céline
Besnard
d,
Eric
Vauthey
*b and
Jérôme
Lacour
*a
*a,
Estefanía
Sucre-Rosales
b,
Francesco
Zinna
c,
Céline
Besnard
d,
Eric
Vauthey
*b and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: nidal.saleh@unige.ch; jerome.lacour@unige.ch
bDepartment of Physical Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: eric.vauthey@unige.ch
cDipartimento di Chimica e Chimica Industriale, University of Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy
dLaboratory of Crystallography, University of Geneva, Quai Ernest Ansermet 24, 1211 Geneva 4, Switzerland
First published on 2nd April 2024
Abstract
The inclusion of boron atoms into chiral π-conjugated systems is an effective strategy to unlock unique chiroptical properties. Herein, the preparation and characterization of a configurationally stable axially-chiral boramidine are reported, showcasing absorption in the UV domain, deep-blue fluorescence (Φ up to 94%), and ca. |10−3| gabs and glum values. Detailed photophysical studies and quantum-chemical calculations clearly elucidate the deactivation pathways of the emissive state to triplet excited states, involving increased spin–orbit coupling between the lowest singlet excited state and an upper triplet state.
Introduction
Small organic molecules exhibiting circularly polarized luminescence are attracting well deserved attention. These chiral chromophores, often abbreviated as SOM-CPL, are applied in cutting-edge research, ranging from 3D displays and optical sensors, to optical information storage or encryption.1 To address (chir)optical properties, main group elements can be incorporated into the π-conjugated systems. This strategy, often allied with efficient preparation routes, unlocks unique optoelectronic properties unattainable with traditional carbon- or metal-based materials.2 In this context, boron atoms are remarkably useful in either three or four-coordinated geometries3 and chiral B-containing derivatives can be readily generated with stereogenic elements of central, axial, planar or helical chirality.1,4,5 Recently, a new class of achiral boramidine chromophores 1 were prepared by simple treatment of α-amino pyridine with N-alkylnitriliumborane 2 in one step (Fig. 1A).6 Initial results by Yudin and coworkers indicated remarkable stability and high fluorescence quantum yields (up to 68%), making compounds 1 excellent candidates for SOM-CPL if transformed into chiral enantiopure moieties. Of all the previously mentioned chiral emitters,1,4,5 axially chiral derivatives are remarkable by the simplicity of their design, the ease of their synthesis, and the tuning of CPL emission by the control of the biaryl twist (dihedral angle) (Fig. 1B).5 Herein, the preparation, resolution and properties of axially-chiral boramidine 3 are reported (Fig. 1C). It displays absorption in the UV domain, narrow deep-blue fluorescence, large Stokes shifts, strong absorption and luminescence dissymmetric factors (gabs and glum ∼10−3) in electronic circular dichroism (ECD) and CPL. Of note, and contrary to the initial report,6 detailed time-resolved photophysical studies and quantum-chemical calculations unravel new deactivation pathways of the emissive state of 1 and 3, via triplet states in particular. The results further reveal that the introduction of axial chirality to the boramidine enhances spin–orbit coupling and facilitates intersystem crossing from the emissive state to the triplet manifold; a constraint not yet identified but of key importance in the design of biaryl-type fluorophores.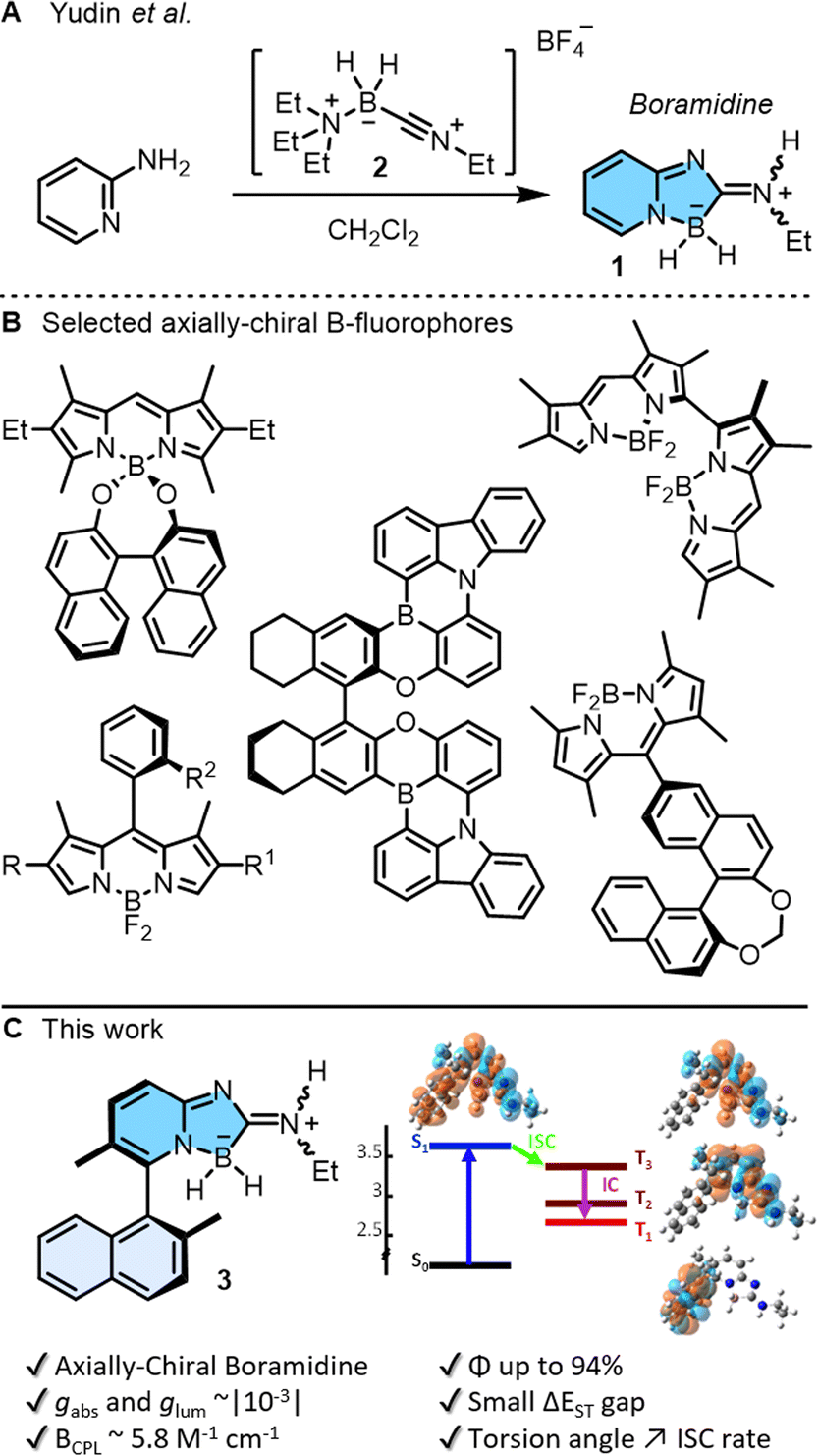 | ||
| Fig. 1 (A) Synthesis of boramidine 1 by reaction of α-amino pyridine with N-alkylnitriliumborane 2.6 (B) Boron-containing axially chiral fluorophores. (C) Enantiospecific synthesis of axially-chiral 3 and subsequent (chir)optical properties; (−)-(M or R)-enantiomer shown arbitrarily. | ||
Results and discussion
Synthesis
Boramidines 1 have been first and only reported in 2020 as achiral materials.6 To create an asymmetric variant, the construction of axially-chiral 3 was considered knowing that metal-catalyzed cross-coupling reactions would offer efficient and direct synthetic routes to the core structure. 2-Amino-5-bromo-3-methylpyridine precursor 6 was prepared in two steps: firstly, the oxidation of commercially available 4 with m-CPBA to afford pyridine oxide 5 in 84% yield followed by a Zincke aminolysis to reach 6 in 74%; and secondly a Pd-catalyzed Suzuki–Miyaura cross-coupling reaction of 6 with (2-methylnaphthalen-1-yl)boronic acid which finally yielded rac-7 in essentially quantitative yield (97%) (Scheme 1).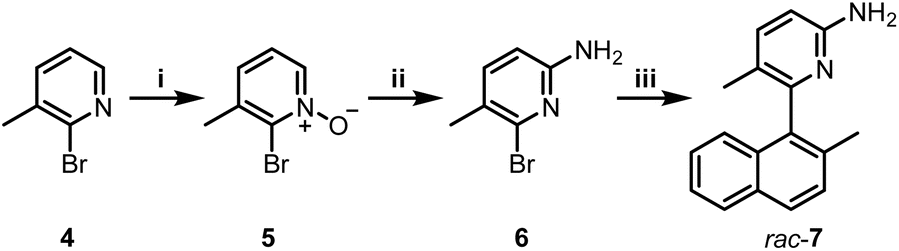 | ||
Scheme 1 (i) m-CPBA, CH2Cl2, 0 to 25 °C, 16 h, 84%; (ii) (a) trifluoroacetic anhydride, pyridine, CH2Cl2, 0 to 25 °C, 6 h; (b) ethanolamine, ACN, 16 h, 74%; (iii) (2-methylnaphthalen-1-yl)boronic acid, Pd(PPh3)4, Na2CO3, toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH:H2O, 100 °C, 20 h, 97%. :EtOH:H2O, 100 °C, 20 h, 97%. | ||
Resolution by chiral stationary phase (CSP) HPLC afforded rapidly and efficiently the single enantiomers (+) and (−)-7, which were obtained as first and second eluted fractions in excellent yields (>45%) and enantiomeric purity (>99% ee, Fig. S22 and S23†).7 Furthermore, samples of (−)-7 crystallized in chloroform, and their X-ray structural analysis revealed (R) or (M) absolute configurations for the chiral axis (Table S4 and Fig. S24†). With these (−)-(M or R) and (+)-(P or S) enantiomers of 7 in hand, by reaction with a slight excess of 2 (1.5 equiv.), (+)- and (−)-3 were prepared in 70% and 68% yields, respectively (Scheme 2). Of note, a 4:1 E:Z ratio is noticed for the exocyclic iminium appendage with either racemic and enantiopure samples, according to 1H and 13C NMR spectroscopy. In our hands, this ratio remained the same in various solvents and at different temperatures indicating that, for compound 3, an equilibrium between the E:Z configurations is unlikely.8 The geometrical isomers cannot be separated from one another and further studies have been performed with this mixture composition. Surprisingly, analytical CSP-HPLC conditions could not be found for enantiomers of 3 and the enantiospecificity (e.s. > 90%) of reaction 7 → 3 could only checked by 1H NMR spectroscopy using [Bu4N][Δ-BINPHAT] (Fig. S25 and S26†).9,10 Finally, care was taken to verify the configurational stability of compounds 3 at higher temperatures (vide infra). A series of ECD experiments were conducted with gradual temperature increase from 20 to 100 °C. The ECD spectra of (+)-3 (Fig. S10†), measured for periods of 30 minutes at the different increasing temperatures, remained unchanged. Compounds of type 3 are thus configurationally stable over a large range of temperatures and present hence a racemization barrier higher than 28 kcal mol−1.11 In view of the enantiospecificity and configurational stability of 3, absolute (P or S) and (M or R) configurations can be assigned with confidence for the (+)- and (−) enantiomers respectively.
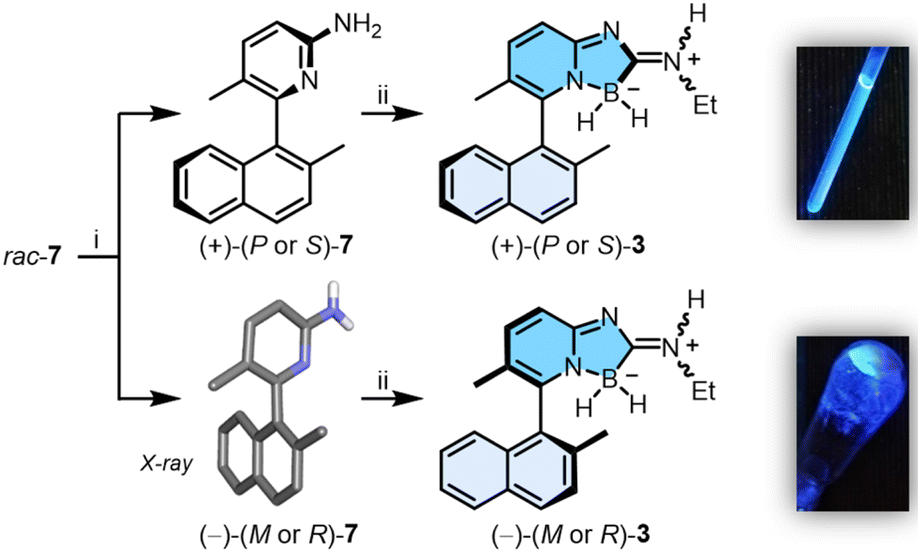 | ||
| Scheme 2 Synthesis of (+) and (−)-3: (i) CSP-HPLC resolution (CHIRALPAK® IC, iPrOH:hexane (+0.1% Et2NH), 5:95) and X-ray structure of (−)-(M or R)-7 (most H-atoms omitted); (ii) 2, CH2Cl2, 16 h, r.t., 68–70% yields. Right: CH2Cl2 solution and solid-state fluorescence (λexc = 366 nm). | ||
Photophysical and chiroptical measurements
A deep-blue fluorescence for rac, (+)- and (−)-3 was apparent both in solution and solid states (Scheme 2). The photophysical properties were first investigated by steady-state absorption and emission measurements in solution. In DCM (CH2Cl2), the absorption spectrum of 3 was compared to that of 7. As presented in Fig. 2, a new absorption band appears in the 300–400 nm domain, with a λmax at 357 nm and an extinction coefficient (ε) of 7720 M−1 cm−1. The absorption spectra of the chiral 3 and achiral 1 boramidines, used as a reference, present similar bands with the lower energy transition of 3 being slightly red-shifted (+7 nm, −560 cm−1) when compared to 1 (Fig. S1†).12 The intense absorption band centered at 225 nm in 3 corresponds to the contribution of the naphthalene moiety.13 A weak solvatochromism is noticed with blue and red shifts in MeOH (−7 nm, +560 cm−1) and DMF (+5 nm, −387 cm−1), respectively (Table 1 and Fig. 3, S2–S7†).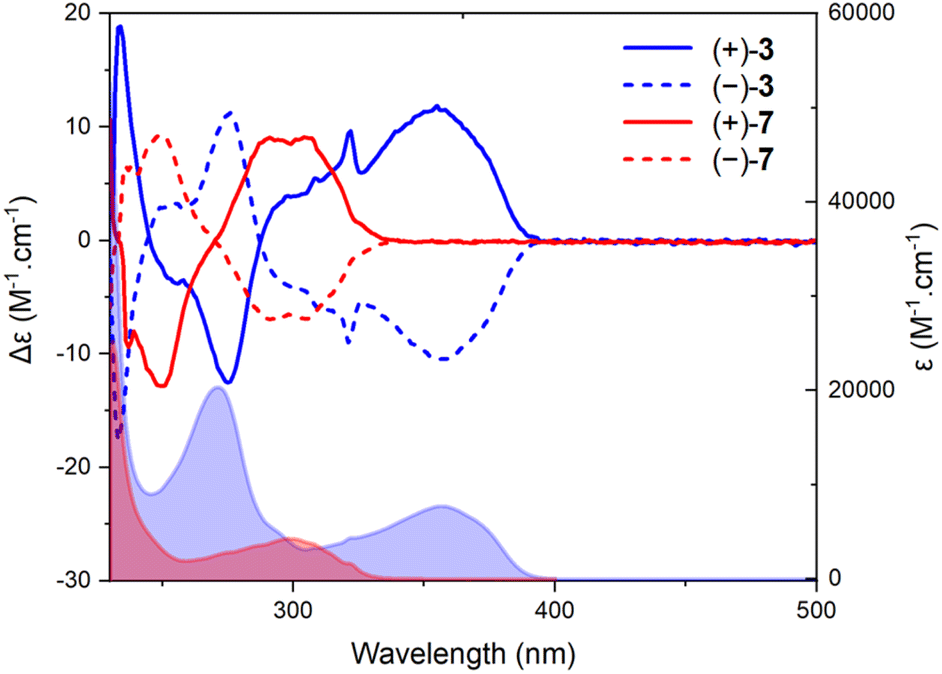 | ||
| Fig. 2 Electronic absorption and ECD spectra of 7 (red) and 3 (blue) in CH2Cl2 (2 × 10−5 M). Continuous (+)-(P) and dotted (−)-(M) lines. | ||
| Solvent | λ max (nm) | ε (M−1 cm−1) | λ em (nm) | Stokes shift (cm−1) | Φ (ΦN2) % | g abs (10−3) | g lum (10−3) | B CPL (M−1 cm−1) |
|---|---|---|---|---|---|---|---|---|
| a Concentrations 2 × 10−5 M. b Calculated for the most red-shifted band 350–362 nm. c Diphenylanthracene (λexc = 373 nm, Φ = 97% in cyclohexane). d λ exc = 365 nm. e N2-saturated solution. | ||||||||
| MeOH | 350 | 9135 | 406 | 3940 | 66 (87) | +1.1/−1 | +1/−1.3 | 5.1e |
| DCM | 357 | 7720 | 406 | 3381 | 59 | +1.5/−1.4 | +1/−1.2 | 2.7 |
| ACN | 357 | 8820 | 407 | 3441 | 64 (89) | +1.2/−1.2 | +1/−1 | 3.9e |
| Toluene | 360 | 7650 | 404 | 3025 | 74 | +1.1/−1 | +1/−1.3 | 3.6 |
| DMF | 362 | 10360 |
414 | 3470 | 79 (94) | +1/−1.1 | +1.1/−1.2 | 5.8e |
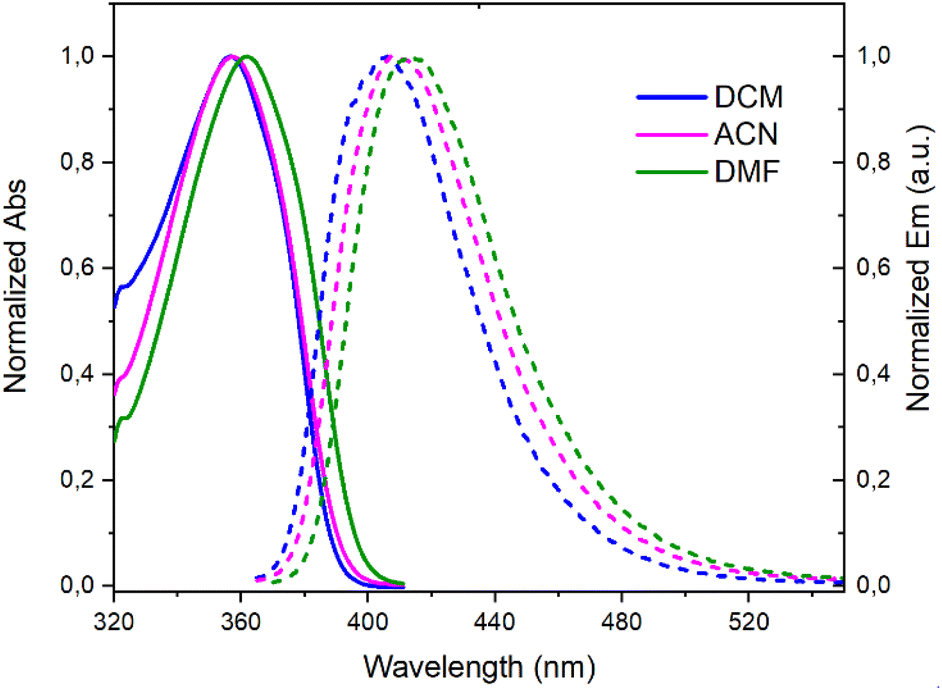 | ||
| Fig. 3 Normalized absorption (solid lines) and emission (dashed lines) spectra of 3 in CH2Cl2 (DCM), CH3CN (ACN), and DMF. | ||
The emission spectra spanned the 370–520 nm range, peaking at approximately 404–407 nm in toluene, MeOH, DCM, and ACN (CH3CN), and 414 nm in DMF (Table 1 and Fig. S8†). These spectra displayed a single narrow band with a full width at half maximum (FWHM) around 3000 cm−1 (Table S1†), accompanied by Stokes shifts within the 3400–3900 cm−1 range.
These features are consistent with high fluorescence quantum yields (Φ) of 64% and 79% in ACN and DMF, respectively. However, the emission intensity was dependent on the presence of dioxygen in solution; Φ increasing significantly to 89% and 94% under N2 saturation (ΦN2, Table 1 and Fig. S9†).
Then, the corresponding chiroptical spectroscopies were investigated using (+)- and (−)-3 (4:1 E:Z mixture). The ECD spectra were recorded for each enantiomer in the UV domain and, as expected, mirror-image Cotton effects were obtained (Fig. 2). A comparison with precursors (+)- and (−)-7 demonstrates relatively large differences in spectral shape with (same sign) lower energy signals around 300 nm for 7 and at 357 nm for 3, with |Δε| ca. 9 and 11 M−1 cm−1, respectively. However, the ECD measurements of 3 in MeOH, ACN, toluene and DMF did not reveal any significant change (Fig. S4–S7†). Overall, |gabs| values between 1.0 and 1.5 × 10−3 were obtained in various solvents (Table 1). All spectra present a feature around 320 nm which is possibly the result of a partial cancellation of two close-in-energy bands with opposite rotatory strength (Fig. 2 and S4–S7).† Interestingly, in CPL, the dextro and levoratory enantiomers of 3 displayed significant positive and negative signals in the visible domain (Fig. 4 and S8†), with luminescence dissymmetry factors (|glum|) of 10−3. Of note, changing solvents or performing measurements in either aerobic or N2-saturated conditions did not modify the glum values. Finally, comparable gabs and glum were hence found in the case of (+)- and (−)-3. This similarity indicates that only minor differences are present between the ground and excited state geometries, and it is consistent with previous correlations.14 The calculated CPL brightness (BCPL = ½ εΦ|glum|) magnitude is of approximately 2.7–5.8 M−1 cm−1 depending on the solvent, and it is similar to values reported for helicenes.4n
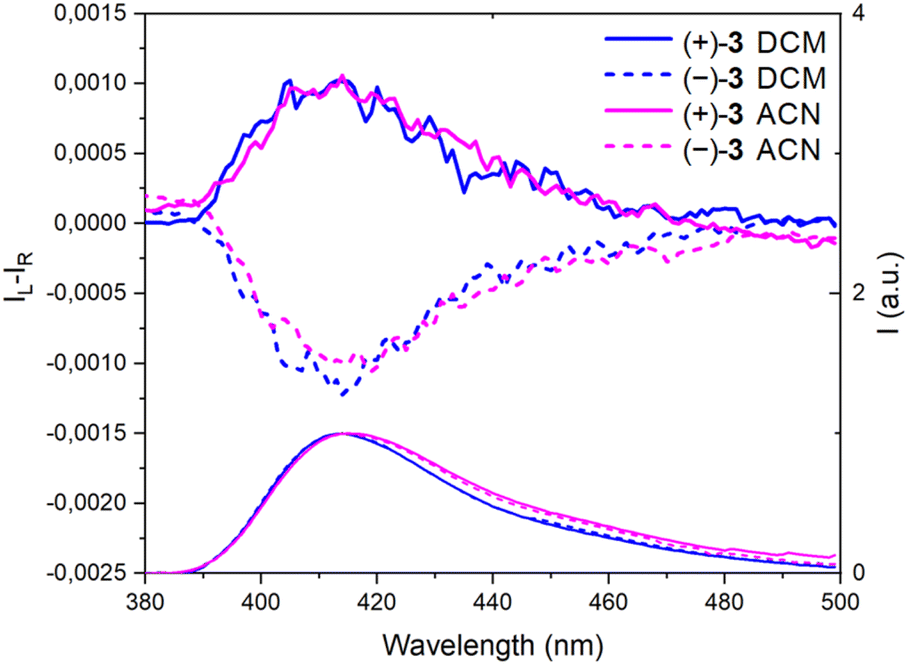 | ||
| Fig. 4 CPL (Top) and emission spectra (bottom) of (+)-(P) and (−)-(M)-3, continuous and dotted lines respectively, in CH2Cl2 (DCM) and CH3CN (ACN). | ||
To better understand the role of O2 on the fluorescence and look for a possible involvement of triplet excited states in the deactivation pathways, transient absorption (TA) spectroscopy measurements combined with quantum chemical calculations at the TD-DFT level were carried out. To this end, 3 was compared with the unsubstituted 1.
The evolution-associated difference absorption spectra (EADS) obtained from a global analysis of the TA data in ACN assuming two successive exponential steps (A → B →) are depicted in Fig. 5, whereas the TA spectra are shown in Fig. S12 and S13.†
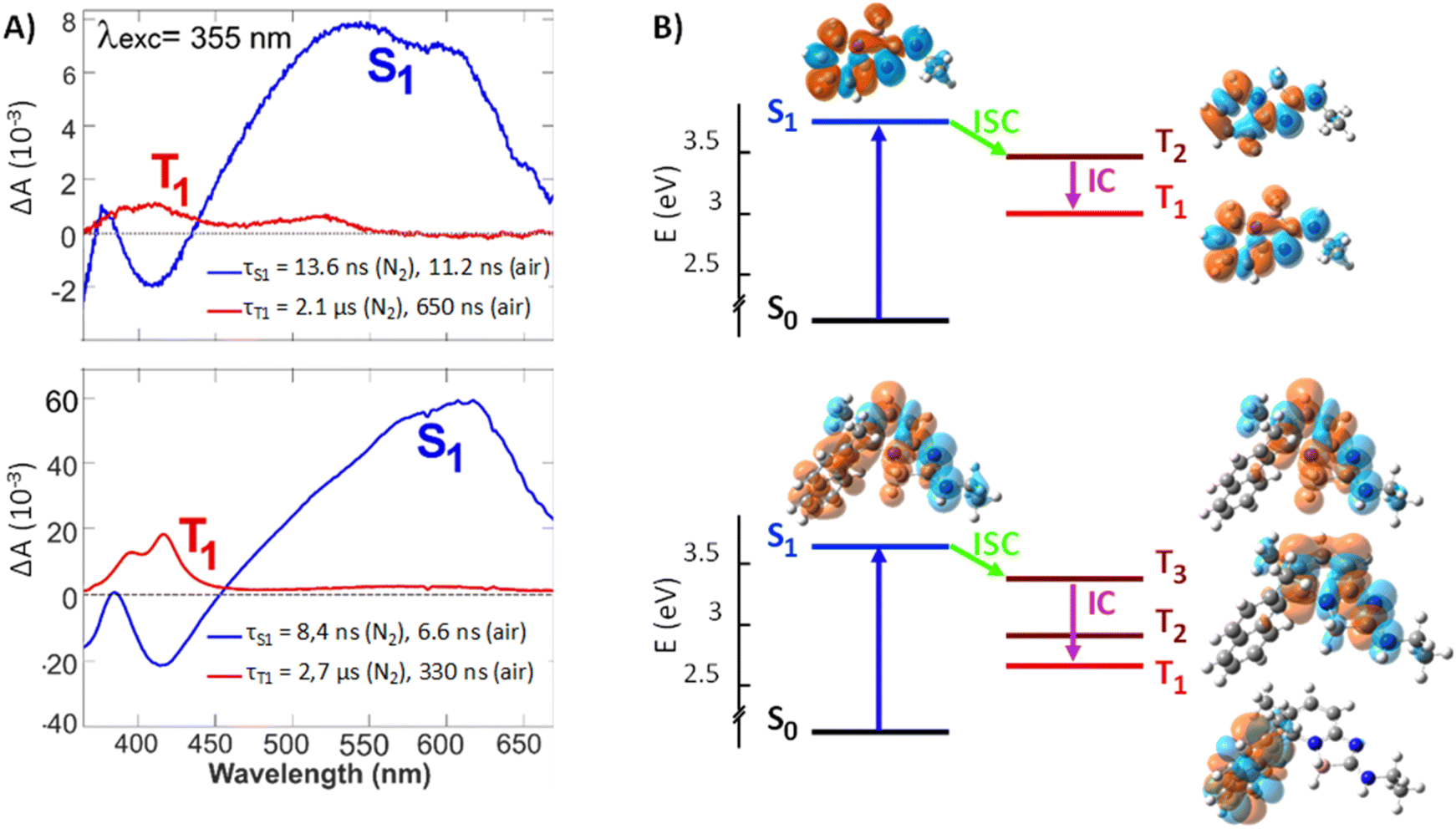 | ||
| Fig. 5 (A) Evolution-associated difference spectra resulting from a global analysis of the transient absorption data measured with 1 (top) and 3 (bottom) in ACN upon 355 nm excitation assuming two successive exponential steps (A → B →). (B) Mechanism for population of the lowest triplet state, together with the charge density difference isosurfaces (plotted at |Δρ = 0.0004|, orange and cyan colors represent, respectively, the increase and decrease of electronic density upon transition from the ground state). | ||
The spectra measured within the first ten ns are similar for both compounds and exhibit two negative features, which can be assigned to the ground state bleach (<370 nm) and the stimulated S1 → S0 emission (SE, around 420 nm), and a broad positive band above 500 nm. This band can be attributed to a Sn>1 ← S1 absorption, as it decays in parallel to the SE with a similar time constant as the fluorescence lifetime measured by time-correlated single photon counting (TCSPC, Table S2 and Fig. S11†). Concurrently to the decay of the spectral features of the S1 state, new positive bands appear, the most intense being around 420 nm. These later spectra can be attributed to the lowest triplet state, T1, of both compounds as their decay accelerates in the presence of oxygen. The triplet spectrum of 3 with its vibronic structure differs significantly from that of 1 and resembles closely that of naphthalene.15
Gas-phase quantum-chemical calculations were performed to better understand the transition from the boramidine-centered S1 state to the naphthalene-centered T1 state of 3. The optimized equilibrium geometries of 1 in the ground (S0) and excited (S1) states point to minor structural changes upon photoexcitation. This is consistent with the chiroptical data shown above. For 3, the main difference concerns the dihedral angle between the aromatic planes, which decreases from 86° to 68° upon going to the S1 state (Fig. S14†). The energy-level diagrams obtained from the TD-DFT calculations are presented in Fig. 5. For both molecules, the calculated S1 energy is larger by 0.4 eV from that measured in ACN. These calculations predict the presence of two or three triplet states below the S1 state of 1 and 3, respectively. The charge density difference (CDD) isosurfaces, which reflect the changes of electronic density upon transition from the ground to a given state, confirm that the S1 and T1 states of 3 are mostly localized on the boramidine and naphthalene sub-units, respectively, as inferred from the TA spectra. Furthermore, the T2 and T3 states of 3 are mostly localized on the boramidine and resemble the T1 and T2 states of 1. For boramidine 1, calculations predict the spin–orbit coupling (SOC) constant for the S1→ T2 transition to be four times as large as that for the S1 → T1 transition (Fig. S17†). Given that the ISC rate constant scales with the square of the SOC constant and that the small S1 – T2 gap favors a large Franck–Condon factor, the transition to the triplet manifold should mostly occur via S1 → T2 ISC. The subsequent internal conversion to the T1 state can be expected to occur within a few ps or less.16 Therefore, the T2 state is too short lived to be observable in a TA experiment. In the case of 3, SOC calculations predict S1 → T3 ISC to be the dominant pathway to the triplet manifold (Fig. S19†). This implies that, upon ISC, the excitation remains mostly localized on the boramidine sub-unit before migrating to the naphthalene group upon internal conversion to the T1 state. Direct S1 → T1 ISC can be expected to be relatively inefficient, because of the large S1 – T1 gap and the relatively weaker SOC. The shorter fluorescence lifetime of 3 relatively to 1 can be accounted by a faster ISC, which itself can be explained by a S1 – T3 SOC that is larger than the corresponding S1 – T2 SOC of boramidine 1. This difference most probably arises from a partial delocalization of the S1 excitation on the naphthalene substituent, conferring some twisted character to the excited state.17 According to quantum-chemical calculations (Table S3 and Fig. S20†), the S1 – T3 SOC of 3 depends critically on the dihedral angle between the aromatic planes of the boramidine and naphthalene subunits and decreases upon decreasing this angle below its equilibrium value (Fig. S21†). To conclude this section, these results reveal that the helicity introduced to confer an axial chirality to 3 has a detrimental effect on the fluorescence quantum yield as it favors conversion to the triplet state; ΦN2 values remaining still above 59% for 3.
Conclusions
In this study, through the introduction of atropisomeric conformations, enantiopure boramidines were prepared as configurationally-stable fluorophores. Absorption in the UV domain and deep-blue fluorescence (Φ up to 94% in N2 saturated solutions) are noted and corresponding |gabs| and |glum| values around 10−3 are further obtained in ECD and CPL for the excitation and emission dissymmetry factors. Detailed photophysical studies and quantum-chemical calculations helped elucidate deactivation pathways of the emissive state to triplet excited states of achiral and chiral boramidines. Of note, with chiral derivative 3, the necessary torsion (80–90°) between boramidine and naphthyl subunits favors spin–orbit coupling and accelerates intersystem crossing from the emissive state to the triplet manifold. In future studies, finding the right torsional balance to induce sufficient axial chirality without being detrimental to the fluorescence quantum yield should be considered in the design of biaryl-type fluorophores.Data availability
The data that support the findings of this study are openly available in https://yareta.unige.ch at https://doi.org/10.26037/yareta:xthmovjv45ccxghfhkdo7oq2ai. It will be preserved for 10 years.Author contributions
N. S. and J. L. conceived the original idea. N. S. designed the research approach, synthesized compounds, performed experiments and conducted data analysis. E. S.-R. and E. V. performed transient absorption and quantum-chemical calculations. F. Z. and C. B. completed CPL and X-ray structural analyses, respectively. J. L. supervised the project. N. S. and J. L. wrote the final manuscript with input and approval from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support (Grants 200020-184843, 200020-207539, and 200020-184607). We acknowledge the contribution of Mrs Bibiana Fabri to the CPL measurement. The computations were performed at University of Geneva using Baobab HPC service. Open access funding was provided by the University de Geneva.References
- T. Mori, Circularly polarized luminescence of isolated small organic molecules, Springer, 2020 Search PubMed.
- (a) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed; (b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (c) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed; (d) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- (a) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (b) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed; (c) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC; (d) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef CAS.
- (a) T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, J. Am. Chem. Soc., 2012, 134, 19600–19603 CrossRef CAS PubMed; (b) E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. d. l. Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef PubMed; (c) H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed; (d) T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef CAS PubMed; (e) X.-Y. Wang, X.-C. Wang, A. Narita, M. Wagner, X.-Y. Cao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 12783–12786 CrossRef CAS PubMed; (f) C. Shen, M. Srebro-Hooper, M. Jean, N. Vanthuyne, L. Toupet, J. A. G. Williams, A. R. Torres, A. J. Riives, G. Muller, J. Autschbach and J. Crassous, Chem.–Eur. J., 2017, 23, 407–418 CrossRef CAS PubMed; (g) X.-Y. Wang, T. Dienel, M. Di Giovannantonio, G. B. Barin, N. Kharche, O. Deniz, J. I. Urgel, R. Widmer, S. Stolz, L. H. De Lima, M. Muntwiler, M. Tommasini, V. Meunier, P. Ruffieux, X. Feng, R. Fasel, K. Müllen and A. Narita, J. Am. Chem. Soc., 2017, 139, 4671–4674 CrossRef CAS PubMed; (h) Z. Domínguez, R. López-Rodríguez, E. Álvarez, S. Abbate, G. Longhi, U. Pischel and A. Ros, Chem.–Eur. J., 2018, 24, 12660–12668 CrossRef PubMed; (i) C. Maeda, K. Nagahata, T. Shirakawa and T. Ema, Angew. Chem., Int. Ed., 2020, 59, 7813–7817 CrossRef CAS PubMed; (j) C. Maeda, S. Nomoto, K. Takaishi and T. Ema, Chem.–Eur. J., 2020, 26, 13016–13021 CrossRef CAS PubMed; (k) P. Moneva Lorente, A. Wallabregue, F. Zinna, C. Besnard, L. Di Bari and J. Lacour, Org. Biomol. Chem., 2020, 18, 7677–7684 RSC; (l) J. Full, S. P. Panchal, J. Götz, A.-M. Krause and A. Nowak-Król, Angew. Chem., Int. Ed., 2021, 60, 4350–4357 CrossRef CAS PubMed; (m) A. Macé, K. Hamrouni, E. S. Gauthier, M. Jean, N. Vanthuyne, L. Frédéric, G. Pieters, E. Caytan, T. Roisnel, F. Aloui, M. Srebro-Hooper, B. Carboni, F. Berrée and J. Crassous, Chem.–Eur. J., 2021, 27, 7959–7967 CrossRef PubMed; (n) L. Arrico, L. Di Bari and F. Zinna, Chem.–Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed; (o) J. Jiménez, F. Moreno, T. Arbeloa, T. A. Cabreros, G. Muller, J. Bañuelos, I. García-Moreno, B. L. Maroto and S. de la Moya, Org. Chem. Front., 2021, 8, 4752–4757 RSC; (p) L.-Y. Wang, Z.-F. Liu, K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Chem. Commun., 2022, 58, 3807–3810 RSC; (q) F. Zhang, F. Rauch, A. Swain, T. B. Marder and P. Ravat, Angew. Chem., Int. Ed., 2023, 62, e202218965 CrossRef CAS PubMed; (r) A. Macé, K. Hamrouni, P. Matozzo, M. Coehlo, J. Firlej, F. Aloui, N. Vanthuyne, E. Caytan, M. Cordier, G. Pieters, M. Srebro-Hooper, F. Berrée, B. Carboni and J. Crassous, Chirality, 2023, 35, 227–246 CrossRef PubMed; (s) D. Pecorari, E. Giuliani, A. Mazzanti, S. Stagni, V. Fiorini, G. Vigarani, F. Zinna, G. Pescitelli and M. Mancinelli, J. Org. Chem., 2023, 88, 871–881 CrossRef CAS PubMed; (t) C. Ray, E. Avellanal-Zaballa, M. Muñoz-Úbeda, J. Colligan, F. Moreno, G. Muller, I. López-Montero, J. Bañuelos, B. L. Maroto and S. de la Moya, Org. Chem. Front., 2023, 10, 5834–5842 RSC; (u) M. Ikeshita, S. Watanabe, S. Suzuki, M. Kitahara, Y. Imai and T. Tsuno, Chem.–Asian J., 2023, e202301024 Search PubMed; (v) Z. Ye, H. Wu, Y. Xu, T. Hua, G. Chen, Z. Chen, X. Yin, M. Huang, K. Xu, X. Song, Z. Huang, X. Lv, J. Miao, X. Cao and C. Yang, Adv. Mater., 2024, 36, 2308314 CrossRef CAS PubMed.
- (a) G. Beer, C. Niederalt, S. Grimme and J. Daub, Angew. Chem., Int. Ed., 2000, 39, 3252–3255 CrossRef CAS; (b) G. Beer, K. Rurack and J. Daub, Chem. Commun., 2001, 1138–1139 RSC; (c) A. Gossauer, F. Nydegger, T. Kiss, R. Sleziak and H. Stoeckli-Evans, J. Am. Chem. Soc., 2004, 126, 1772–1780 CrossRef CAS PubMed; (d) T. Bruhn, G. Pescitelli, S. Jurinovich, A. Schaumlöffel, F. Witterauf, J. Ahrens, M. Bröring and G. Bringmann, Angew. Chem., Int. Ed., 2014, 53, 14592–14595 CrossRef CAS PubMed; (e) S. Kolemen, Y. Cakmak, Z. Kostereli and E. U. Akkaya, Org. Lett., 2014, 16, 660–663 CrossRef CAS PubMed; (f) R. I. Lerrick, T. P. L. Winstanley, K. Haggerty, C. Wills, W. Clegg, R. W. Harrington, P. Bultinck, W. Herrebout, A. C. Benniston and M. J. Hall, Chem. Commun., 2014, 50, 4714–4716 RSC; (g) C. Wang, Z.-B. Sun, Q.-W. Xu and C.-H. Zhao, Chem.–Eur. J., 2016, 22, 16750–16754 CrossRef CAS PubMed; (h) F. Zinna, T. Bruhn, C. A. Guido, J. Ahrens, M. Bröring, L. Di Bari and G. Pescitelli, Chem.–Eur. J., 2016, 22, 16089–16098 CrossRef CAS PubMed; (i) Z. Jiang, T. Gao, H. Liu, M. S. S. Shaibani and Z. Liu, Dyes Pigm., 2020, 175, 108168 CrossRef CAS; (j) S.-P. Wan, H.-Y. Lu, M. Li and C.-F. Chen, J. Photochem. Photobiol., C, 2022, 50, 100500 CrossRef CAS; (k) Z.-P. Yan, L. Yuan, Y. Zhang, M.-X. Mao, X.-J. Liao, H.-X. Ni, Z.-H. Wang, Z. An, Y.-X. Zheng and J.-L. Zuo, Adv. Mater., 2022, 34, 2204253 CrossRef CAS PubMed.
- Y. Lebedev, C. Apte, S. Cheng, C. Lavigne, A. Lough, A. Aspuru-Guzik, D. S. Seferos and A. K. Yudin, J. Am. Chem. Soc., 2020, 142, 13544–13549 CrossRef CAS PubMed.
- Using semi-preparative conditions over a CHIRALPAK® IC column (250 mm × 20 mm, 5 μm) around 20 mg of rac-7 can be separated in 60 minutes into single enantiomers.
- The variable temperature ECD spectra of 3 display a complete lack of variations from 20 to 100 °C in DMF (Fig. S10†).
- Salt [Bu4N][Δ-BINPHAT] can act as NMR chiral solvating reagent with cationic but also neutral moieties.
- (a) J.-P. Djukic, A. Berger, M. Duquenne, M. Pfeffer, A. de Cian, N. Kyritsakas-Gruber, J. Vachon and J. Lacour, Organometallics, 2004, 23, 5757–5767 CrossRef CAS; (b) J. Lacour, L. Vial and C. Herse, Org. Lett., 2002, 4, 1351–1354 CrossRef CAS PubMed.
- At 100 °C, such a free energy of activation would entice a half-life in the range of 38 minutes for a first-order racemization. The total lack of ECD evolution indicates that the racemization barrier is clearly above 28 kcal·mol−1.
- In all spectroscopic experiments, both chiral 3 and achiral 1 boramidines were used as mixtures of geometrical isomers and presented a 4:1 E:Z ratio.
- R. Masrat, M. Maswal and A. A. Dar, J. Hazard. Mater., 2013, 244–245, 662–670 CrossRef CAS PubMed.
- (a) H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS; (b) M. Cei, L. Di Bari and F. Zinna, Chirality, 2023, 35, 192–210 CrossRef CAS PubMed.
- X. Wang, W. G. Kofron, S. Kong, C. S. Rajesh, D. A. Modarelli and E. C. Lim, J. Phys. Chem. A, 2000, 104, 1461–1465 CrossRef CAS.
- O. Yushchenko, G. Licari, S. Mosquera-Vazquez, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. Lett., 2015, 6, 2096–2100 CrossRef CAS PubMed.
- (a) S. S. Murali, J. K. Gallaher, C. Janiseck, E. J. Tay, I. Wagner, K. E. Thorn, A. Ilina, R. R. Tamming, J. Wang, C. Sester, J. J. Sutton, M. B. Price, K. C. Gordon, K. Chen, X. Zhan, J. M. Hodgkiss and P. A. Hume, J. Am. Chem. Soc., 2023, 145, 732–744 CrossRef CAS PubMed; (b) R. Letrun, B. Lang, O. Yushchenko, R. Wilcken, D. Svechkarev, D. Kolodieznyi, E. Riedle and E. Vauthey, Phys. Chem. Chem. Phys., 2018, 20, 30219–30230 RSC; (c) K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC; (d) B. F. Habenicht and O. V. Prezhdo, J. Am. Chem. Soc., 2012, 134, 15648–15651 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2321027. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00870g |
| This journal is © The Royal Society of Chemistry 2024 |