DOI:
10.1039/D4SC00822G
(Edge Article)
Chem. Sci., 2024,
15, 7725-7731
Enantioselective synthesis of α-aryl α-hydrazino phosphonates†
Received
2nd February 2024
, Accepted 22nd April 2024
First published on 23rd April 2024
Abstract
Catalysts generated in situ by the combination of pyridine–hydrazone N,N-ligands and Pd(TFA)2 have been applied to the addition of arylboronic acids to formylphosphonate-derived hydrazones, yielding α-aryl α-hydrazino phosphonates in excellent enantioselectivities (96 → 99% ee). Subsequent removal of the benzyloxycarbonyl (Cbz) N-protecting group afforded key building blocks en route to appealing artificial peptides, herbicides and antitumoral derivatives. Experimental and computational data support a stereochemical model based on aryl-palladium intermediates in which the phosphono hydrazone coordinates in its Z-configuration, maximizing the interactions between the substrate and the pyridine–hydrazone ligand.
Introduction
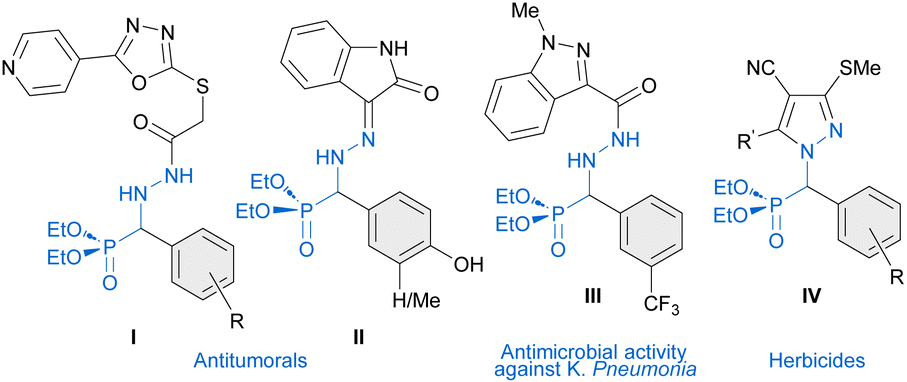
Phosphonic acids, phosphonates and their derivatives are involved in numerous biological processes.1 In drug design, the phosphonate group is often used as a bioisostere of carboxylate. For instance, various phosphonate analogues of known neuraminidase inhibitors exhibit improved pharmacokinetic properties to treat influenza infection.2 α-Amino phosphonic acid derivatives, acting as amino acid mimics, are known to exhibit important biological activities such as antifungal,3 antibacterial,4 antioxidant,5 anti-Alzheimer,6 antiviral,7 and anticancer activities,8 among others.9 Additionally, non-proteogenic α-hydrazino phosphonic acid derivatives, in which intramolecular H-bonds (hydrazino turns)10 might modify their biological activities, have emerged as promising targets in medicinal chemistry. In particular, α-hydrazinophosphonate oxadiazoles I,11 isatin derivatives II12 and hydrazide III4a have been reported to exhibit promising activities against different types of cancer (I and II) and antimicrobial activity against K. Pneumonia (III) (Fig. 1). Moreover, α-aryl α-hydrazino phosphonates are also the key precursors of pyrazoles IV13,14 which have been shown to have a potent herbicidal activity.
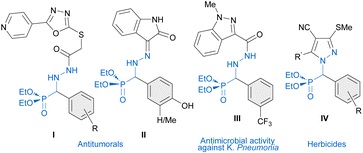 |
| | Fig. 1 Biologically active molecules derived from α-aryl α-hydrazino phosphonates. | |
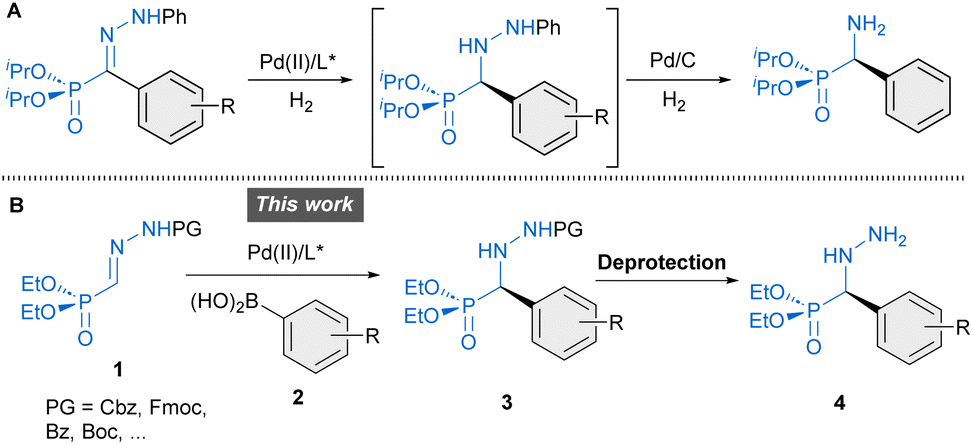
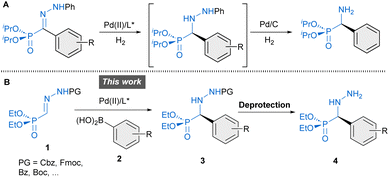
To the best of our knowledge, however, enantiomerically pure forms of these molecules have never been evaluated nor even synthesized. To date, existing routes for the synthesis of α-hydrazino phosphonates-related derivatives rely on various strategies such as metal-catalyzed electrophilic amination of β-keto phosphonates with azodicarboxylates (α-alkyl substitution),15 cycloaddition reactions employing α-(diazomethyl)-phosphonate for the construction of cyclic heterocycles16 and asymmetric hydrophosphonylation reaction of azomethine imines to afford cyclic hydrazides (C–P bond forming reaction).17 Other approaches based on enantioselective reactions involving phosphono hydrazones remain underdeveloped. For instance, PdII-catalyzed enantioselective hydrogenation of α-hydrazono phosphonates has been shown to provide α-aryl α-hydrazine phosphonates.18 However, the resulting products lack removable N-protecting groups necessary to access versatile free hydrazines. Hence, subsequent transformation into α-amino phosphonates by reductive N–N bond cleavage was performed instead (Scheme 1A). To overcome these limitations, we have developed diverse enantioselective arylation reactions of related hydrazones containing readily removable protecting groups (Cbz, Fmoc, etc.), which allowed the access to free hydrazino building blocks in enantiomerically enriched forms.19 In this article, we present a straightforward approach to α-aryl α-hydrazino phosphonates 4 based on PdII-catalyzed enantioselective addition of aryl boronic acids to α-hydrazono phosphonates 1 (C–C bond-forming reaction), enabling, for the first time, a convenient access to this type of molecules in enantioselective fashion (Scheme 1B). In addition, Density Functional Theory (DFT) calculations were carried out to understand the factors leading to the excellent enantioselectivity of the transformation.
 |
| | Scheme 1 Enantioselective transformations of phosphono hydrazones towards α-aryl α-amino phosphonates (A) and α-aryl α-hydrazino phosphonates (B, this work). | |
Results and discussion
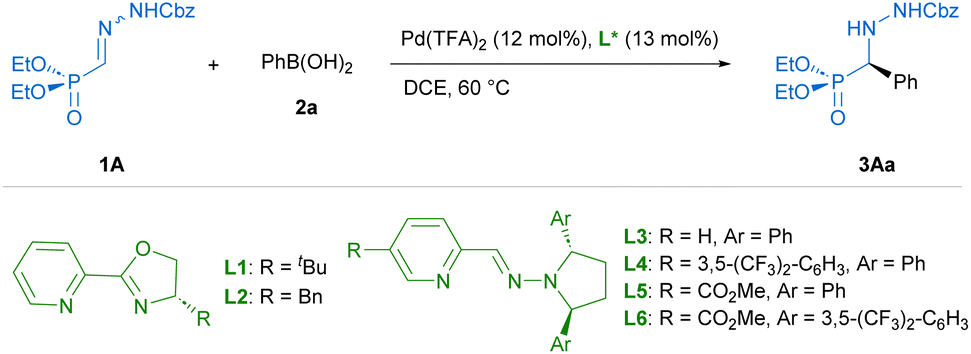
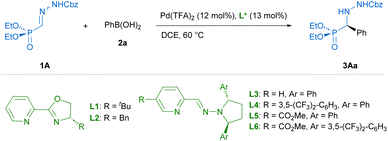
Preliminary experiments were conducted with benzyloxycarbonyl Cbz-protected hydrazone 1A and phenylboronic acid (2a) as model reagents. A preliminary screening of palladium complexes [prepared in situ from bipyridine (bipy) and different PdII sources] and conditions served to identify bipy/Pd(TFA)2 as the most efficient achiral catalyst and dichloroethane (DCE) at 60 °C as the best reaction medium.† Then, diverse chiral pyridine-oxazolines20 and pyridine–hydrazones21 were evaluated as chiral N,N′ ligands for the enantioselective version of the reaction (Table 1). To our delight, the use of pyridine–hydrazone L3 provided, after 24 hours, the desired adduct 3Aa with 70% conversion and 94% ee (entry 3), clearly outperforming the results obtained with commercially available pyridine-oxazolines L1 and L2 (entries 1 and 2). The (2S,5S)-2,5-diphenylpyrrolidine unit was retained as optimal chiral scaffold and the influence of the substituent attached to the C5 of the pyridine ring was also investigated. The presence of 3,5-bis-trifluoromethylphenyl group (R = ArF) led to a diminished catalytic activity (entry 4). On the contrary, ligand L5 bearing a methyl carboxylate group (R = CO2Me) afforded 3Aa in 84% conversion and excellent enantiocontrol (99% ee). Finally, the incorporation of (2S,5S)-2,5-di-(3,5-bis-trifluoromethylphenyl) pyrrolidine in ligand L6 led to a slightly lower enantioselectivity (93% ee) compared to ligands L3–L5, with the same level of conversion than L5 (entry 6). Interestingly, a similar result and the same stereochemical outcome were observed in an additional experiment employing the cis isomer (Z)-1A (entry 7), suggesting the participation of a common PdII-intermediate. During the scaling-up, we observed that the eventual presence of boronic acid anhydrides, normally present in commercial boronic acids, might be the origin of some erratic results. In fact, a control experiment revealed that reactivity dropped when 2,4,6-triphenylboroxine (2a′) was used instead of phenyl boronic acid (entry 8). Therefore, the remaining optimization was performed employing 2a after purification by filtration through a short pad of silica gel.† Under optimal conditions, the model reaction on a 0.2 mmol scale required a longer reaction time (36 hours), affording (R)-3Aa in 66% yield and 99% ee (Scheme 2). The scope and limitations of the reaction were then explored. First, modifications of hydrazone reagent 1 were investigated. Other N-protecting groups such as 9-fluorenylmethoxycarbonyl (Fmoc) and benzoyl (Bz) were well tolerated, affording (R)-3Ba and (R)-3Ca in good yields (68–69%) and 98% ee in both cases. Interestingly, a tert-butoxycarbonyl (Boc) group in hydrazone 1D hampered the process, giving 3Da with high enantioselectivity, albeit in low yield (17%). Overall, these results suggest that the aromatic scaffolds in N-carbamoyl/benzyl groups might stabilize the reactive intermediates through additional π⋯π interactions, although a more significant steric repulsion by the Boc group cannot be disregarded. Modification of the phosphonate moiety was also investigated. The dimethyl derivative (1E) afforded (R)-3Ea in 62% yield and slightly lower enantioselectivity (96% ee). Next, Cbz protected hydrazono phosphonate (E)-1A was made to react with a variety of arylboronic acids 2a–o to afford α-aryl α-hydrazide phosphonates 3 in variable yields (30–92%) and excellent enantioselectivities (96–99% ee) in all cases (Scheme 2). Mono-substituted electron-rich aryl boronic acids [2b: p-Me–C6H4–B(OH)2, 2c: p-MeO–C6H4–B(OH)2, 2d: p-BnO–C6H4–B(OH)2 and 2e: o-Me–C6H4–B(OH)2] were suitable reagents, whereas electron-poor boronic acids [2f: p-Cl–C6H4–B(OH)2, 2g: p-CF3–C6H4–B(OH)2] reacted more slowly, leading to side reactions such as homo-coupling and protodeboronation. Hence, portionwise addition of these reagents was required for the synthesis of (R)-3Af and (R)-3Ag in 57 and 30% yield, respectively. Di- [ortho/meta (2h), meta/para (2i and 2j), meta/meta (2k)] and tri-substituted (2l) aryl boronic acids afforded 3Ah–3Al in good yields (51–92%) and high enantioselectivities (97–99% ee). Additionally, aryl boronic acids bearing extended rings, such as 2m and naphthyl scaffolds (2n and 2o) gave essentially enantiopure α-hydrazide phosphonates 3Am–3Ao (98 → 99% ee). Finally, heteroaryl (3-furanyl and 3-thienyl) boronic acids and aryl boronic acids 2 containing nitrogenous functional groups (R = p-dimethylamino and R = p-acetamido) as well as a vinyl chain (R = CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) were unproductive reagents for this transformation. In a couple of representative examples (3Ai and 3Ao), the reaction could be scaled to 1 mmol without significant loss in yield or enantioselectivity. The absolute R configuration of products 3Aa, 3Ba and 3Da was assigned by chemical correlation.† Assuming a uniform reaction pathway, the absolute configurations of all other α-aryl α-hydrazide phosphonates 3 were assigned by analogy.
CH2) were unproductive reagents for this transformation. In a couple of representative examples (3Ai and 3Ao), the reaction could be scaled to 1 mmol without significant loss in yield or enantioselectivity. The absolute R configuration of products 3Aa, 3Ba and 3Da was assigned by chemical correlation.† Assuming a uniform reaction pathway, the absolute configurations of all other α-aryl α-hydrazide phosphonates 3 were assigned by analogy.
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
1 |
L* |
3 |
Conv. (%)b |
ee (%)c |
|
Reactions at 0.1 mmol scale. Reaction time: 24 hours.
Estimated by 31P-NMR.
Determined by HPLC on chiral stationary phases.
Performed with 2,4,6-triphenylboroxine (0.05 mmol).
|
| 1 |
(E)-1A |
L1
|
(S)-3Aa
|
20 |
33 |
| 2 |
(E)-1A |
L2
|
(S)-3Aa
|
46 |
rac
|
| 3 |
(E)-1A |
L3
|
(R)-3Aa
|
70 |
94 |
| 4 |
(E)-1A |
L4
|
(R)-3Aa
|
52 |
97 |
| 5 |
(E)-1A |
L5
|
(R)-3Aa
|
84 |
99 |
| 6 |
(E)-1A |
L6
|
(R)-3Aa
|
85 |
93 |
| 7 |
(Z)-1A |
L5
|
(R)-3Aa
|
86 |
98 |
| 8d |
(E)-1A |
L5
|
(R)-3Aa
|
68 |
98 |
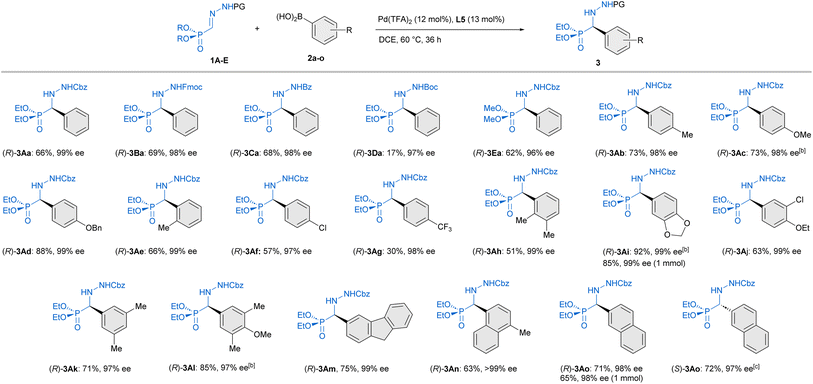 |
| | Scheme 2 Scope of the reaction. Yields of isolated products after column chromatography. Enantiomeric excesses were determined by HPLC on chiral stationary phases aReactions performed at 0.2 mmol scale. bReaction time: 20 h. c(S)-3Ao was synthesized employing ent-L5. | |
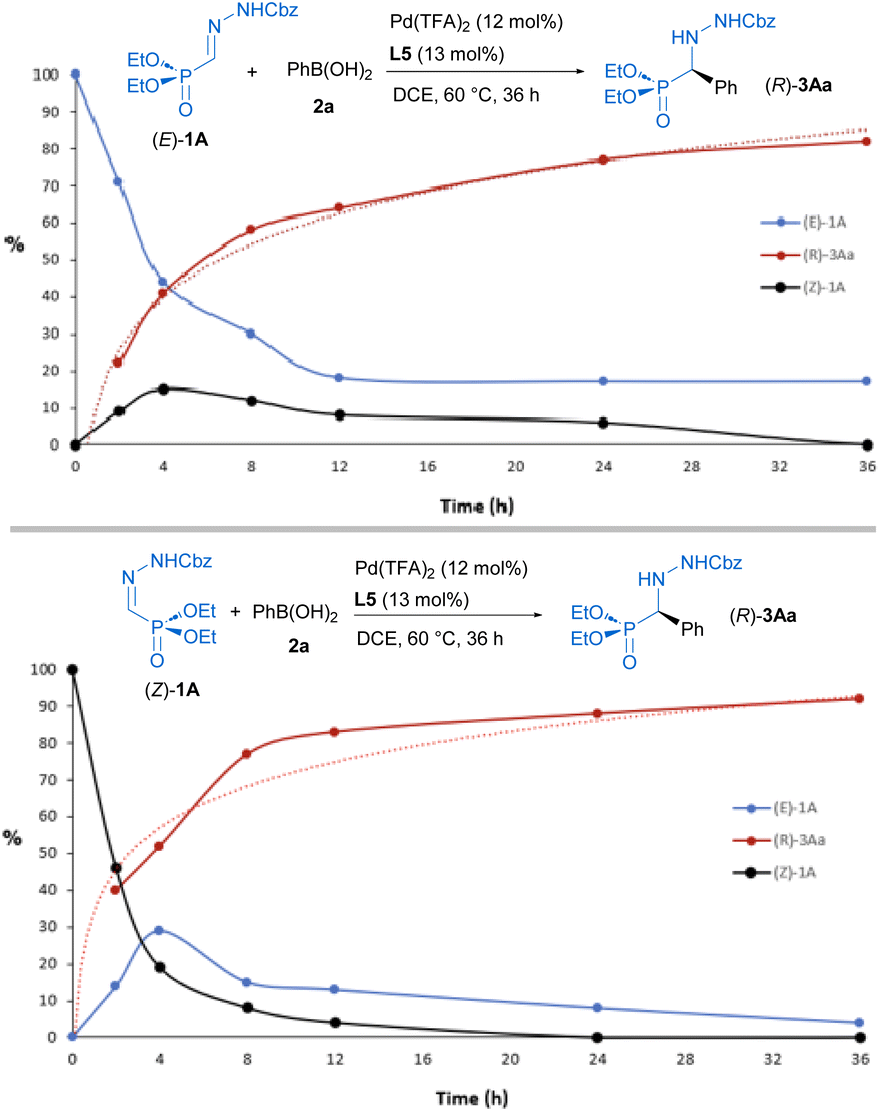
Experimental data, such as the observed stereoconvergence and the detection of both isomers in independent reactions starting from (E)-1A or (Z)-1A, suggest that the reaction proceeds under Curtin-Hammett control22 with a relatively fast interconversion of stereoisomers. It is known that depending on subtle structure variations and reaction conditions, such as heating in the presence of acids, these types of E and Z isomers might equilibrate.23 In order to shed light over reaction mechanism, 31P-NMR was used for monitoring the E/Z isomerization process at 60 °C, showing that the interconversion is facilitated either by the presence of phenyl boronic acid (1 equiv) (E/Z from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 75:25, after 1 hour) or catalytic amounts (12 mol%) of Pd(TFA)2 (E/Z from 100:0 to 75:25, after 5 h). Next, the reactions starting from (E)-1A or (Z)-1A were also independently monitored and their profiles over time comparatively analyzed (Fig. 2).
:0 to 75:25, after 1 hour) or catalytic amounts (12 mol%) of Pd(TFA)2 (E/Z from 100:0 to 75:25, after 5 h). Next, the reactions starting from (E)-1A or (Z)-1A were also independently monitored and their profiles over time comparatively analyzed (Fig. 2).
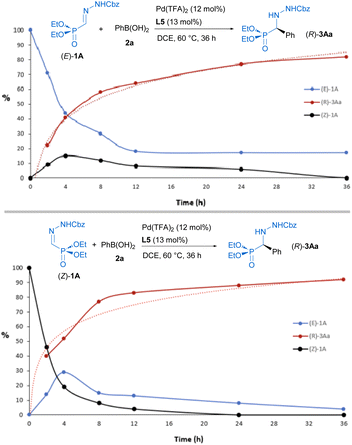 |
| | Fig. 2 Progression of ratios (%) of (E)-1A (in blue), (Z)-1A (in black) and (R)-3Aa (in red) under catalytic conditions (0.2 mmol). Estimated by 31P-NMR. (Top) starting from (E)-1A. (Bottom) starting from (Z)-1A. | |
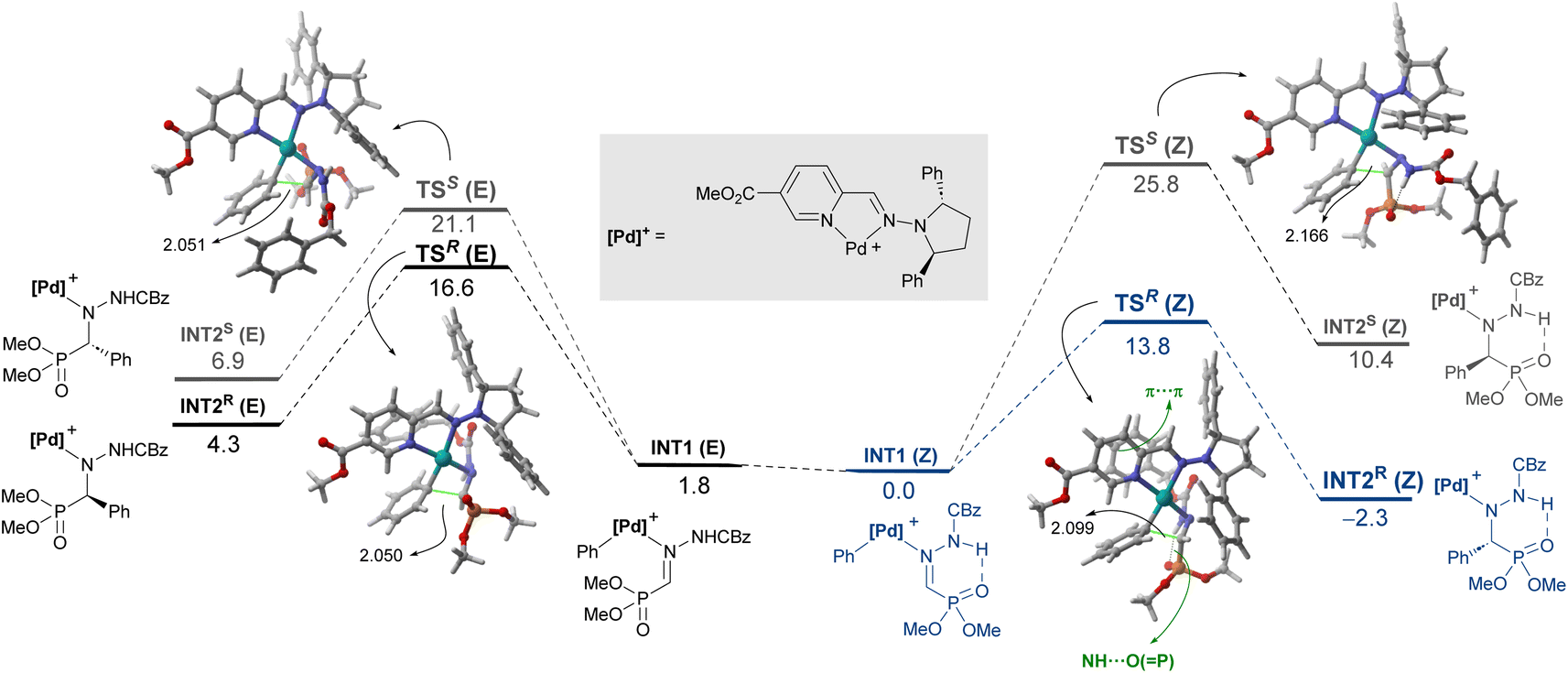
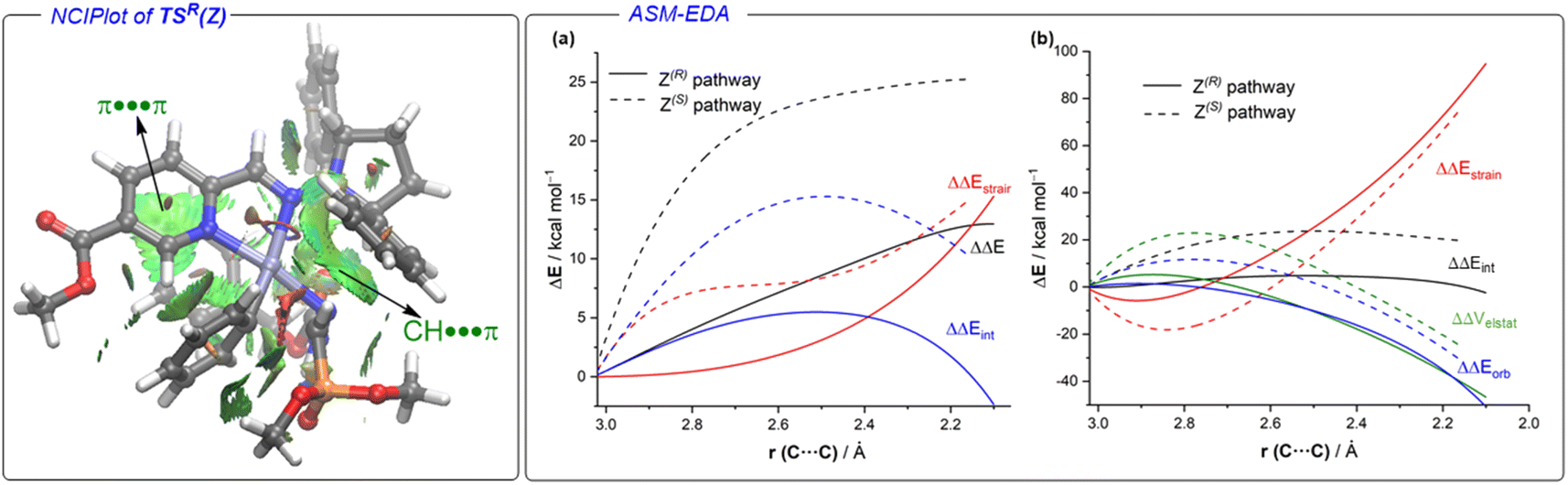
Coexistence of both isomers during the process suggests a relatively low E/Z interconversion barrier upon reaction conditions. Nevertheless, it is observed that conversion to product (R)-3Aa over time remains higher when starting from the (Z)-isomer, supporting a preferred pathway through palladium complexes involving (Z)-1A. Density Functional Theory (DFT) calculations were carried out to understand the origin of the observed complete enantioselectivity of the transformation. According to a previous report on related PdII-catalyzed asymmetric addition of arylboronic acids to cyclic N-sulfonyl ketimine esters,20e the transformation involves the initial phenyl transfer from the boronic acid to the active palladium(II) catalyst, coordination of the hydrazone followed by intramolecular C–C bond formation, protonation and concomitant regeneration of the catalyst (see Fig. S1 in the ESI† for the computed entire reaction profile). Therefore, the intramolecular C–C bond formation constitutes the enantiodetermining step of the transformation. We consequently focused on this particular reaction step for our system, starting from the initially formed intermediated INT1, where the reactive phenyl group and the hydrazone 1E (Z or E) are directly attached to the PdII-center bearing the chiral ligand L5. Our calculations indicate, not surprisingly, that INT1(Z) is 1.8 kcal mol−1 more stable than its isomer INT1(E), which is mainly due to the occurrence of a stabilizing PO⋯HN intramolecular hydrogen bond (see Fig. 3). Despite this, after a conformational search, we were able to locate two transition states for each isomer associated with the formation of the key C–C bond leading to the R (TSR) or S (TSE) enantiomers (with respect to the newly formed stereocenter, Fig. 3). Regardless of the initial intermediate, the formation of the R-enantiomer is favored from both kinetic and thermodynamical points of view, which agrees with the complete enantioselectivity towards the (R)-3 reaction product observed experimentally (see above). From the data in Fig. 3, it is clear that the pathway involving the transition state TSR(Z) becomes the preferred one, leading to intermediate INT2R(Z) in an exergonic transformation (ΔGR = −2.3 kcal mol−1 with an activation barrier of only 13.8 kcal mol−1), consistent with the reaction conditions used in the experiments. Remarkably, there is an energy gap of 7.3 kcal mol−1 with TSS(E), fully consistent with the almost complete enantioselectivity observed systematically in the experiments. The remarkable stability of TSR(Z) can be initially ascribed, according to the NCIPlot method,24 to the existence of stabilizing noncovalent π⋯π interactions between the phenyl group of CBz and the pyridine fragment of the chiral ligand, along with CH⋯π interactions involving the azomethine proton (NC)H of the hydrazone and a phenyl group of the pyrrolidine moiety in L5 (see Fig. 4, left). As these interactions are absent in the analogous transition state leading to the S-isomer, it is not surprising that TSR(Z) is by far the most stable saddle point. More quantitative insight into the factors favoring the pathway involving TSR(Z) can be gained by applying the Activation Strain Model (ASM) of reactivity.25 This analysis involves decomposing the electronic energy (ΔE) into two terms: the strain (ΔEstrain) resulting from the distortion of the individual reactants and the interaction (ΔEint) between the deformed reactants along the reaction coordinate, defined in this case by the formation of the key C⋯C bond. As this particular transformation occurs intramolecularly, the fragments, i.e., the hydrazone and the [Pd–Ph]+ complex, were referred to the geometry they adopt in the initial intermediate INT1(Z), constituting therefore the zero level of the different ASM terms. Fig. 4a (right) shows the corresponding activation strain diagrams (ASDs) computed for the R and S pathways starting from INT1(Z) up to the respective transition states. From the data in Fig. 4a, it becomes clear that the process leading to the R-isomer benefits from a less destabilizing distortion (measured by the ΔΔEstrain curve) and, particularly, from a much stronger interaction between the hydrazone and the palladium complex along the entire reaction coordinate. Indeed, whereas the ΔΔEint term becomes clearly stabilizing (i.e., negative) in the proximity of TSR(Z), the situation sharply contrasts in the S-pathway, where the change in the interaction from the initial intermediate INT1(Z) becomes destabilizing. The reasons behind the stronger interaction computed for the R-pathway were further analyzed with the help of the Energy Decomposition Analysis (EDA) method,26 which involves decomposing the ΔEint into three chemically meaningful terms: classical electrostatic interaction (ΔVelstat), Pauli repulsion between closed-shell orbitals, responsible for steric repulsion, and stabilizing orbital attractions (ΔΔEorb). As graphically depicted in Fig. 4b, which shows the evolution of the EDA terms along the reaction coordinate and referred again to the starting intermediate, it becomes clear that the stronger interaction computed for the R-pathway does not result from the ΔΔEPauli repulsion term, which is actually less destabilizing for the S-pathway (mainly due to the longer C⋯C bond-forming distance in TSS(Z) and the closer proximity of the CBz group to the chiral ligand in the Z-pathway), but exclusively from much stronger electrostatic and orbital interactions (in a nearly identical extent) between the palladium complex and the hydrazone fragments, once again along the entire reaction coordinate, and particularly, at the transition state region. Therefore, it can be concluded that the (practically exclusive) formation of the R-enantiomer finds its origin mainly in the electrostatic, orbital and non-covalent interactions between the palladium-complex and hydrazone substrate, which are maximized in the favored Z(R) reaction pathway.
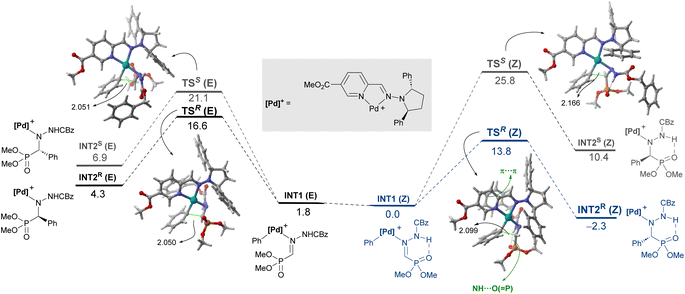 |
| | Fig. 3 Computed reaction profile for the key enantiodetermining step from intermediates INT1. Relative free energies (ΔG, at 333 K) and bond distances are given in kcal mol−1 and angstroms, respectively. | |
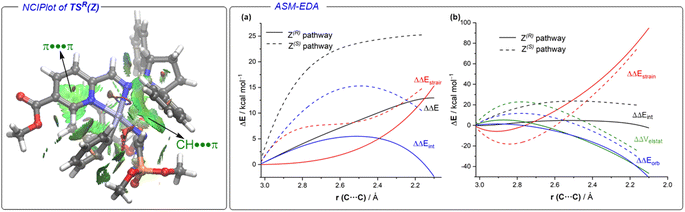 |
| | Fig. 4 (Left): Contour plots of the reduced density gradient isosurfaces (density cutoff of 0.04 a.u.) for TSR(Z). The greenish surfaces indicate attractive non-covalent interactions (computed at the PCM(dichloroethane)-M06L/def2-SVP level). (Right): (a) comparative activation strain analyses (PCM(dichloroethane)-M06L/def2-SVP level) and (b) energy decomposition analyses (ZORA-M06L/DZP//PCM(dichloroethane)-M06L/def2-SVP level) of the key enantiodetermining step projected onto the C⋯C bond-forming distance and involving the R (solid lines) and S (dotted lines) pathways. | |
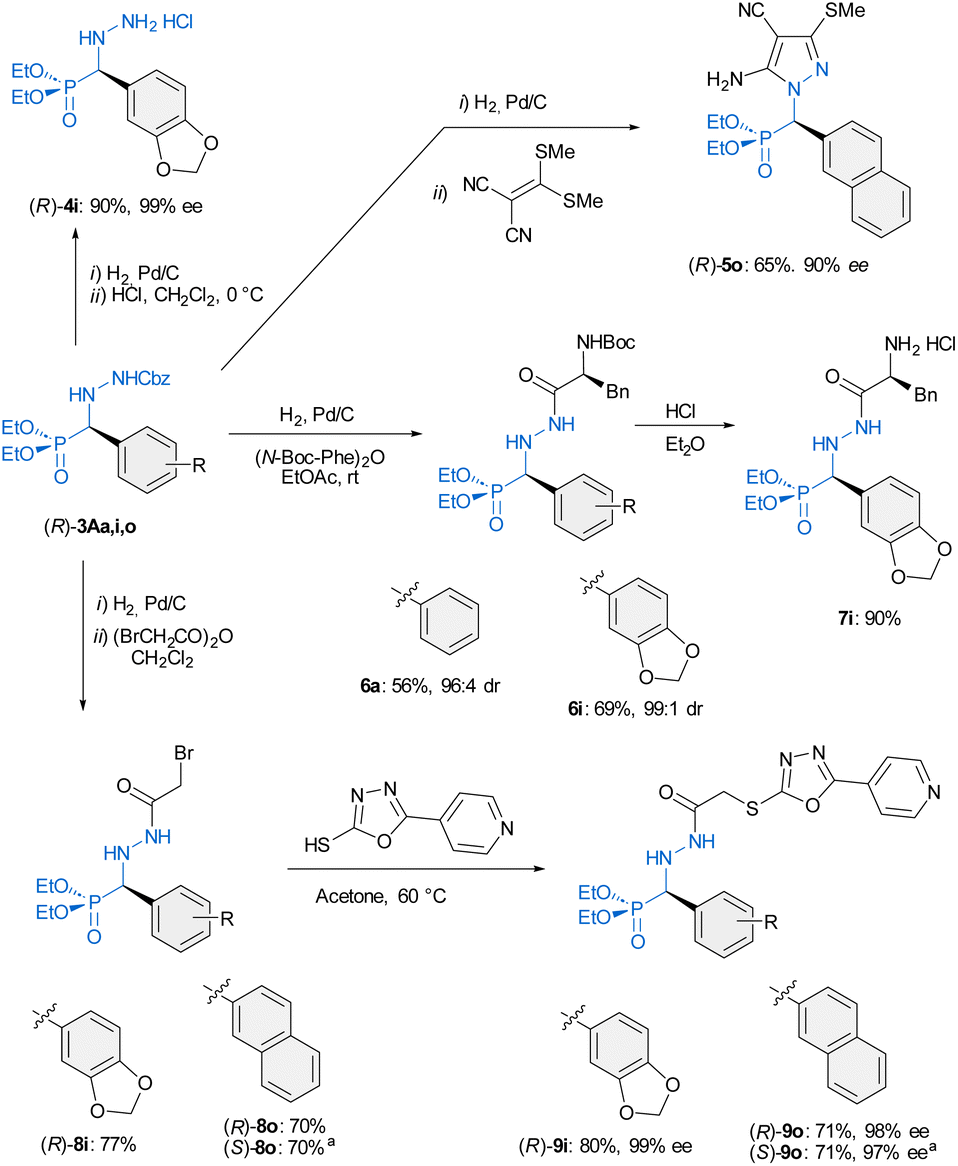
Finally, to further demonstrate the usefulness of this transformation, different derivatization reactions were carried out. N′-Cbz-protected α-aryl α-hydrazino phosphonates 3 are versatile building blocks for accessing compounds with potential applications in diverse fields (Scheme 3). Applying standard hydrogenolysis [Pd(C)/H2 (1 atm), rt], the benzyloxycarbonyl group (Cbz) of (R)-3A was efficiently removed to afford the corresponding hydrazino phosphonates, as exemplified in the synthesis of (R)-4i which was isolated as its hydrochloride salt in 90% yield and 99% ee. Additionally, the 2-nathphyl derivative (R)-3Ao reacted with 2-[bis(methylthio)-methylene]malononitrile to afford pyrazole (R)-5o (a representative of products III in Fig. 1) in good yield. Moreover, the introduction of amino acids by N(2) couplings from (R)-3Aa and (R)-3Ai was performed in one-pot fashion. Thus, hydrogenolysis in the presence of the N-Boc-L-phenyl alanine anhydride [(N-Boc-Phe)2O] afforded N(2)-hydrazides (R)-6a and (R)-6i in good overall yields (56–69%, two steps, only one chromatographic purification), without erosion of the enantioselectivity (96:4–99:1 dr).
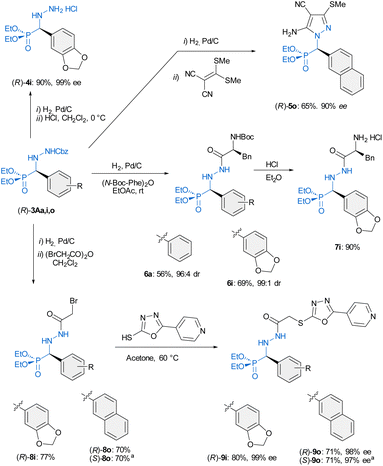 |
| | Scheme 3 Transformations of adducts 3 into appealing targets. aFrom (S)-3Ao. | |
Subsequent deprotection of the Boc group in (R)-6i allowed the isolation of (R)-7i in 90% yield, emphasizing the potential of hybrid amino acid/hydrazide phosphonates as versatile intermediates for the synthesis of artificial peptides. Finally, the synthesis of enantiopure anti-cancer α-aryl α-hydrazino phosphonates (compounds I in Fig. 1) was accomplished by reaction of deprotected hydrazines with bromoacetic anhydride to yield key intermediates (R)-8i and (R)-8o followed by reaction with commercially available 5-(pyridine-4-yl)-1,3,4-oxadiazole-2-thiol in acetone at 60 °C. In this way, the target compounds (R)-9i and (R)-9o were obtained in 80 and 71% yield, respectively, and without erosion of enantiomeric purity. The synthesis of the enantiomer (S)-9o was also successfully accomplished starting from the adduct (S)-3Ao.
Conclusions
In summary, catalysts generated by combinations of Pd(TFA)2 and pyridine–hydrazone ligands provide excellent enantioselectivities in the 1,2-addition of aryl boronic acids to formylphosphonate-derived hydrazones, yielding α-aryl α-hydrazino phosphonates in high enantioselectivities (96 → 99% ee). Experimental evidence, DFT calculations and ASM-EDA analyses support a stereochemical model in which the hydrazone in its Z-configuration is coordinated to the aryl-palladium complex. The preferred transition state is not only stabilized by key noncovalent π⋯π and CH⋯π interactions between the hydrazone and the ligand, but also by significant electrostatic and orbital interactions, resulting in a remarkable enantiocontrol of the process. Moreover, the presence of a readily removable protecting group, such as Cbz in adducts 3, has been exploited for targeting pyrazoles, phosphonopeptides and antitumoral hydrazide phosphonate oxadiazoles in enantiomerically pure forms, illustrating the utility of the methodology.
Author contributions
D. M, J. M. L. and R. F. designed the project. S. A and J. R.-P. carried out experiments. I. F. performed computational studies and data analysis. D. M, J. M. L and I. F. wrote the manuscript – original draft. R. F: review and editing. All authors discussed the experimental results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Spanish MICINN (grants PID2019-106358GB-C21, PID2019-106358GB-C22, PID2019-106184GB-I00, PID2022-137888NB-I00, PID2022-143230NB-I00, PID2022-139318NB-I00 and RED2022-134287-T), European FEDER funds and the Junta de Andalucía (grants P18-FR-3531, P18-FR-644 and US-1262867) for financial support.
Notes and references
- G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704 CrossRef CAS PubMed.
-
(a) J.-J. Shie, J.-M. Fang, P.-T. Lai, W.-H. Wen, S.-Y. Wang, Y.-S. E. Cheng, K.-C. Tsai, A.-S. Yang and C.-H. Wong, J. Am. Chem. Soc., 2011, 133, 17959 CrossRef CAS PubMed;
(b) J.-J. Shie, J.-M. Fang, S.-Y. Wang, K.-C. Tsai, Y.-S. E. Cheng, A.-S. Yang, S.-C. Hsiao, C.-Y. Su and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 11892 CrossRef CAS;
(c) P.-C. Wang, J.-M. Fang, K.-C. Tsai, S.-Y. Wang, W.-I. Huang, Y.-C. Tseng, Y.-S. E. Cheng, T.-J. R. Cheng and C.-H. Wong, J. Med. Chem., 2016, 59, 5297 CrossRef CAS.
- Y. Quin, R. Xing, S. Liu, H. Yu, K. Li, L. Hu and P. Li, Int. J. Biol. Macromol., 2014, 63, 83 CrossRef.
-
(a) D. Koszelewski, P. Kowalczyk, P. Śmigielski, J. Samsonowicz-Górski, K. Kramkowski, A. Wypych, M. Szymczak and R. Ostaszewski, Materials, 2022, 15, 3846 CrossRef CAS PubMed;
(b) M. G. Nowak, A. S. Skwarecki and M. J. Milewska, ChemMedChem, 2021, 16, 3513 CrossRef CAS.
- K. U. M. Rao, S. Swapna, D. M. Manidhar, K. M. K. Reddy and C. S. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 232 CrossRef CAS.
- J. J. Uparkar, P. P. Dhavan, B. L. Jadhav and S. D. Pawar, J. Iran. Chem. Soc., 2022, 19, 3103 CrossRef CAS.
- S. Hkiri, M. Mekni-Toujani, E. Üstün, K. Hosni, A. Ghram, S. Touil, A. Samarat and D. Sémeril, Pharmaceutics, 2023, 15, 114 CrossRef CAS.
-
(a) Q. Wang, L. Yang, H. Ding, X. Chen, H. Wang and X. Tang, Bioorg. Chem., 2016, 69, 132 CrossRef CAS;
(b) F. Bahrami, F. Panahi, F. Daneshgar, R. Yousefi, M. B. Shahsavanib and A. Khalafi-Nezhad, RSC Adv., 2016, 6, 5915 RSC.
- Reviews:
(a) P. Kafarski and B. Lejczak, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 63, 193 CrossRef CAS;
(b) A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955 CrossRef CAS;
(c) M. Talma, M. Maślanka and A. Mucha, Bioorg. Med. Chem. Lett., 2019, 29, 1031 CrossRef CAS PubMed;
(d) For a review of the synthesis of enantioenriched derivatives see: P. R. Varga and G. Keglevich, Molecules, 2023, 28, 6150 CrossRef CAS.
-
(a) A. Aubry, J.-P. Mangeot, J. Vidal, A. Collet, S. Zerkout, M. Marraud and J. Pept, Protein Res., 1994, 43, 305 CAS;
(b) T. Katoh and H. Suga, J. Am. Chem. Soc., 2021, 143, 18844 CrossRef CAS.
- E. F. Ewies, M. El-Hussieny, N. F. El-Sayed and M. A. Fouad, Eur. J. Med. Chem., 2019, 180, 310 CrossRef CAS.
- S. V. Tiwari, N. S. Sharif, R. I. Gajare, J. A. Seijas Vazquez, J. N. Sangshetti, M. D. Damale and A. P. G. Nikalje, Molecules, 2018, 23, 1981 CrossRef PubMed.
- L.-X. Xiao, K. Li, D.-Q. Shi and J. Heterocyclic, Chem, 2009, 46, 555 CAS.
- Pyrazolo[4,3-e][1,2,4]-triazolo[1,5-d]pyrimidines: L.-X. Xiao, K. Li and D.-Q. Shi, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 3156 CrossRef CAS.
- L. Bernardi, W. Zhuang and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 5772 CrossRef CAS PubMed.
-
(a) N. Huang, X. Tong, S. Zhou, Q. Guo and Y. Peng, Adv. Synth. Catal., 2019, 361, 4805 CrossRef CAS;
(b) W. Wu, X. Yan, X. Li, Y. Ning, L. Hu, L. Zhu, Q. Ouyang and Y. Peng, Org. Lett., 2022, 24, 3766 CrossRef CAS PubMed.
- L.-P. Kong, N.-K. Li, S.-Y. Zhang, X. Chen, M. Zhao, Y.-F. Zhang and X.-W. Wang, Org. Biomol. Chem., 2014, 12, 8656 RSC.
- N. S. Goulioukina, I. A. Shergold, V. B. Rybakov and I. P. Beletskaya, Adv. Synth. Catal., 2017, 359, 153 CrossRef CAS.
-
(a) M. Velázquez, S. Alberca, J. Iglesias-Sigüenza, R. Fernández, J. M. Lassaletta and D. Monge, Chem. Commun., 2020, 56, 5823 RSC;
(b) S. Alberca, M. Velázquez, J. Trujillo-Sierra, J. Iglesias-Sigüenza, R. Fernández, J. M. Lassaletta and D. Monge, Adv. Synth. Catal., 2022, 364, 2373 CrossRef CAS.
- For a review of pyridine-oxazolines as chiral ligands for asymmetric catalysis, see:
(a) G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783 RSC ; Selected examples on asymmetric addition of organoboronic acids to C=N bonds:;
(b) H. Dai, M. Yang and X. Lua, Adv. Synth. Catal., 2008, 350, 249 CrossRef CAS;
(c) J. Chen, X. Lu, W. Lou, Y. Ye, H. Jiang and W. Zeng, J. Org. Chem., 2012, 77, 8541 CrossRef CAS PubMed;
(d) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540 CrossRef CAS;
(e) M. Quan, G. Yang, F. Xie, I. D. Gridnev and W. Zhang, Org. Chem. Front., 2015, 2, 398 RSC;
(f) M. Quan, X. Wang, L. Wu, I. D. Gridnev, G. Yang and W. Zhang, Nat. Commun., 2018, 9, 2258 CrossRef;
(g) C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 53, 9936 CrossRef CAS PubMed.
-
(a) Y. Álvarez-Casao, D. Monge, E. Álvarez, R. Fernández and J. M. Lassaletta, Org. Lett., 2015, 17, 5104 CrossRef PubMed;
(b) M. Retamosa, Y. Álvarez-Casao, E. Matador, A. Gómez, D. Monge, R. Fernández and J. M. Lassaletta, Adv. Synth. Catal., 2019, 361, 176 CrossRef . and reference 19.
- J. I. Seeman, Chem. Rev., 1983, 83, 83–134 CrossRef CAS.
- P. Kaushik, B. Lai, C. D. Raghuveeran and R. Vaidyanathaswamy, J. Org. Chem., 1982, 47, 3503 CrossRef.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
-
(a) I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953 RSC;
(b) L. P. Wolters and F. M. Bickelhaupt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 324 CAS;
(c) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS PubMed . See also;;
(d)
I. Fernández, in Discovering the Future of Molecular Sciences ed. B. Pignataro, Wiley-VCH, Weinheim, 2014, pp. 165–187 Search PubMed.
- For reviews on the EDA method, see:
(a)
F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 1–86 Search PubMed;
(b) M. von Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43 CAS;
(c)
I. Fernández, in Applied Theoretical Organic Chemistry, ed. D. J. Tantillo, World Scientific, New Jersey, 2018, pp. 191–226 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional optimization results, full experimental procedures, characterization data, NMR spectra for new compounds, HPLC traces, detailed DFT calculations. See DOI: https://doi.org/10.1039/d4sc00822g |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Javier
Romero-Parra
b,
Israel
Fernández
a,
Javier
Romero-Parra
b,
Israel
Fernández