
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extended shortwave infrared absorbing antiaromatic fluorenium-indolizine chromophores†
William E.
Meador‡
 a,
Matthew A.
Saucier‡
a,
Max R.
Tucker
a,
Nicholas A.
Kruse
a,
Alexander J.
Mobley
a,
Connor R.
Brower
a,
Sean R.
Parkin
b,
Kensha M.
Clark
a,
Nathan I.
Hammer
a,
Gregory S.
Tschumper
a and
Jared H.
Delcamp§
*a
a,
Matthew A.
Saucier‡
a,
Max R.
Tucker
a,
Nicholas A.
Kruse
a,
Alexander J.
Mobley
a,
Connor R.
Brower
a,
Sean R.
Parkin
b,
Kensha M.
Clark
a,
Nathan I.
Hammer
a,
Gregory S.
Tschumper
a and
Jared H.
Delcamp§
*a
aUniversity of Mississippi, Department of Chemistry and Biochemistry, Coulter Hall, University, MS 38677, USA. E-mail: delcamp@olemiss.edu
bDepartment of Chemistry, University of Kentucky, Lexington, Kentucky 40506, USA
First published on 12th July 2024
Abstract
Shortwave infrared (SWIR, 1000–1700 nm) and extended SWIR (ESWIR, 1700–2700 nm) absorbing materials are valuable for applications including fluorescence based biological imaging, photodetectors, and light emitting diodes. Currently, ESWIR absorbing materials are largely dominated by inorganic semiconductors which are often costly both in raw materials and manufacturing processes used to produce them. The development of ESWIR absorbing organic molecules is thus of interest due to the tunability, solution processability, and low cost of organic materials compared to their inorganic counterparts. Herein, through the combination of heterocyclic indolizine donors and an antiaromatic fluorene core, a series of organic chromophores with absorption maxima ranging from 1470–2088 nm (0.84–0.59 eV) and absorption onsets ranging from 1693–2596 nm (0.73–0.48 eV) are designed and synthesized. The photophysical and electrochemical properties of these chromophores, referred to as FluIndz herein, are described via absorption spectroscopy in 17 solvents, cyclic voltammetry, solution photostability, and transient absorption spectroscopy. Molecular orbital energies, predicted electronic transitions, and antiaromaticity are compared to higher energy absorbing chromophores using density functional theory. The presence of thermally accessible diradical states is demonstrated using density functional theory and EPR spectroscopy, while XRD crystallography confirms structural connectivity and existence as a single molecule. Overall, the FluIndz chromophore scaffold exhibits a rational means to access organic chromophores with extremely narrow optical gaps.
Introduction
Materials that absorb low energy light in the near infrared (NIR, 700–1000 nm),1 shortwave infrared (SWIR, 1000–1700 nm),2–4 and extended SWIR (ESWIR, 1700–2700 nm)5–7 are valuable for a plethora of optoelectronic applications including but not limited to fluorescence based biological imaging, photodetectors, and light emitting diodes. The field of ESWIR absorbing materials is currently dominated by semiconductors like InGaAs, GaInAsSb, GeSn, and HgCdTe8,9 and quantum dots (QDs).10–12 These semiconductors are narrow band gap materials commonly used in ESWIR photodetectors whose properties are tunable via modification of the nanomaterials size with QDs, and doping in bulk semiconductors. Overall, these ESWIR absorbing semiconductors are heavily inhibited by cost related to both the raw materials and the complex manufacturing processes to make them.13 Semiconductor based photodetectors also require cooling to eliminate dark current. While semiconductors dominate commercial applications of SWIR photodetectors, there is active research into the use of organic materials in SWIR photodetectors, especially with regards to organic polymers.14–18 Organic photodetectors exhibit low cost manufacturing through solution processability that promises lower costs compared to inorganic materials. These organic photodetectors also commonly work at non-cryogenic temperatures. However, to date the photocurrent response wavelengths do not compete with inorganic semiconducting materials, limiting their application. In this way, the development of novel chromophores for applications in lower energy organic photodetectors is of importance moving forward (Table S3†).There are several classes of small molecule organic chromophores that absorb light in the NIR and SWIR including cyanines,19–26 BODIPYs,27,28 squaraines,29–31 benzothiadiazoles,32–34 and xanthenes.35–38 While there is an abundance of chromophores from these classes that absorb in the NIR and SWIR, to the best of our knowledge none exhibit the lowest energy absorption maxima (λabs) in the ESWIR. In the past few years, xanthene dyes containing heterocyclic donors in place of traditional alkyl amine donors have emerged as a viable means to access SWIR absorption. Both indolizine heterocycles and styryl based donors have been utilized in this respect and have proven to be successful as SWIR fluorescence contrast agents for in vivo imaging applications. Along with the use of heterocyclic donors to induce bathochromic shifts in λabs of xanthene chromophores, modifications to the central atom of the xanthene core have also been explored in the literature, with derivatives including nitrogen,39 oxygen,40 carbon,41 silicon,41,42 phosphorous,40 sulfur,40 boron,43 and others.44 In general, electron donating atoms like nitrogen and oxygen exhibit shorter wavelength λabs and electron withdrawing groups like phosphorous oxides, sulfur oxides, ketones, and trivalent boron tend to shift λabs towards longer wavelengths. Recently, a unique observation was made upon the synthesis of a xanthene analogue containing a fluorene core. By deleting the central atom altogether, generating a five membered central ring as opposed to the traditional six membered central ring, Grzybowski and co-workers were able to induce a dramatic shift in λabs of the rhodamine derivative to 943 nm in dye 4-H (Fig. 1),36 a nearly 400 nm (0.95 eV) longer wavelength λabs compared to the traditional oxygen containing rhodamine dye (absorption maxima ∼548 nm). This observation was attributed to antiaromaticity which was observed computationally via nucleus-independent chemical shift (NICS) analysis. In the analysis, the central five membered ring of the fluorenium dyes demonstrated a paratropic ring current, resulting in a positive chemical shift indicative of antiaromaticity. Conversely, the six membered central ring of classic oxygen-containing xanthene dyes demonstrated a diatropic ring current, resulting in a negative chemical shift indicative of aromaticity. This was an intriguing use of antiaromaticity which has been shown to induce low energy electronic transitions through a “Jahn–Teller-like” reorganization of molecular orbitals in antiaromatic heterocycles.45–47 With this knowledge in hand, a heterocyclic indolizine donor-based fluorene dye was pursued in an attempt to produce a series of low energy absorbing small molecule organic chromophores, deemed FluIndz (Fig. 1).
 | ||
| Fig. 1 Absorption maxima of indolizine xanthene dyes containing oxygen-based cores (blue), silicon-based cores (purple), and fluorene-based cores (red), and alkyl amine donor-based fluorene dye (green), along with computed orbital energy levels of the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) with the HOMO–LUMO energy gap (EH−Lg). Energy levels obtained from density functional theory (DFT) calculations at the B3LYP/6-311G(d,p) level of theory with dichloromethane (DCM) implicit solvation. | ||
Results and discussion
Synthesis
Synthesis of the FluIndz dyes began with making the indolizine donors. 2Ph,48 1Ph,49 and 7DMA23 indolizine donors (2–4, Scheme 1) were synthesized as previously described, and 1,7DBA indolizine donor (5) was synthesized as shown in Scheme 1. To make 5, a Grignard reaction was run via the formation of the Grignard reagent from bromide 11 and subsequent reaction with aldehyde 10 to yield the alcohol 12 in 58% yield. The alcohol was deoxygenated using acetic acid and hydroiodic acid to yield 13 in 67% yield. 13 was obtained as a mixture after multiple chromatographic separation attempts and was used in the following reaction as a mixture. A Suzuki reaction using 13 and 14 (synthesized as previously described50) yielded 15 in 96% yield, which was subsequently reacted with 2-bromoacetophenone, 16, to yield 1,7DBA indolizine donor, 5, in 74% yield. With the indolizine donors in hand, a palladium catalyzed C–H activation reaction was run utilizing the commercially available dibromofluorenone core (1) and the respective indolizine donor (2–5) to produce the coupled fluorenone products in yields ranging from 18–59% (Scheme 1). From the coupled fluorenones, 6–9, Grignard reactions were run with ortho-xylylmagnesium bromide to yield the intermediate alcohols. Several acidic conditions were evaluated using the 2Ph derivative to remove the alcohol and form the final dye without success. Aqueous acids like HClO4 and HCl commonly used for alcohol removal in xanthene dyes35,36,51 were observed to yield no product via SWIR absorption monitoring. An anhydrous condition using trifluoroacetic acid in DCM solution44,52 was observed to produce the desired product; however, the product was observed to rapidly decompose during isolation. Alternative anhydrous acidic conditions employing triflic acid and hydrofluoroboric acid in DCM solution were also observed to produce the desired product, but at very low yields (<10%) with the predominant product being a green chromophore absorbing near ∼800 nm. Finally, a non-Brønsted acid based condition using POCl3, as used previously for the RhodIndz and VIX series,35,37,53 was observed to work well with a yield of 63% across both the Grignard reaction and alcohol removal step for the 2Ph derivative. The POCl3 conditions yielded the final dyes but with poor solubility and difficult purification, so an anion exchange reaction to give the PF6− salt was performed following alcohol removal to aid in purification and solubility of the FluIndz dyes. The POCl3 conditions were used for the rest of the series and produced the final products in yields ranging from 33–63%. | ||
| Scheme 1 Synthesis of FluIndz dyes (top) and 1,7DBA indolizine donor (bottom). | ||
Photophysical properties
With the chromophores in hand, the FluIndz dyes were studied for their solution absorption profiles in a variety of solvents of varying polarity from dioxane (dielectric constant = 2.21) to dimethyl sulfoxide (DMSO, dielectric constant = 47) to understand how solvent selection affects the photophysical properties of these dyes (Fig. S1–S4†). The absorption spectrum of the FluIndz dyes were observed to be heavily solvent dependent, with some solvents retaining the characteristic “cyanine-like” π → π* electronic features, wherein the lowest energy transition was relatively sharp and intense, while other solvents instead induced broadened features indicative of charge transfer behavior (n → π*) with more intense higher energy transitions. In general, the FluIndz dyes tended to maintain their cyanine-like electronic features in less polar, non-coordinating solvents like toluene, carbon disulfide (CS2), chloroform (CHCl3), chlorobenzene (PhCl), dichloromethane (DCM), and 1,2-dichloroethane (DCE). Exceptions to this include: acetic acid (AcOH), which worked well for 2PhFluIndz and 1PhFluIndz but not 7DMAFluIndz and 1,7DBAFluIndz (likely due to protonation of the aryl amines) and ethanol (EtOH), which worked well for 7DMAFluIndz and 1,7DBAFluIndz, but not 2PhFluIndz and 1PhFluIndz. Ethereal solvents like dioxane and tetrahydrofuran (THF) resulted in broadened features in all dyes except 1PhFluIndz, and despite its low polarity (dielectric constant = 6), ethyl acetate (EtOAc) also resulted in broadened features in all the FluIndz dyes studied. Overall, CS2 was observed to be the best solvent for absorption spectroscopy considering the minimal amount of solvent absorption interference in the SWIR and ESWIR, the sharp features observed, and the lowest energy λabs across all solvents. Therefore it was selected alongside DCM (used for comparison across the literature, Fig. S5†) to collect further photophysical data (Fig. 2). While the absorption bands in CS2 and DCM appear broad for these materials when plotted in nm (especially for the longer wavelength absorbing derivatives 7DMAFluIndz and 1,7DBAFluIndz), when plotted in a scale that is linear with respect to energy, like eV, these compounds exhibit comparable absorption peak widths as similar compounds that absorb at higher energies (Fig. S6†).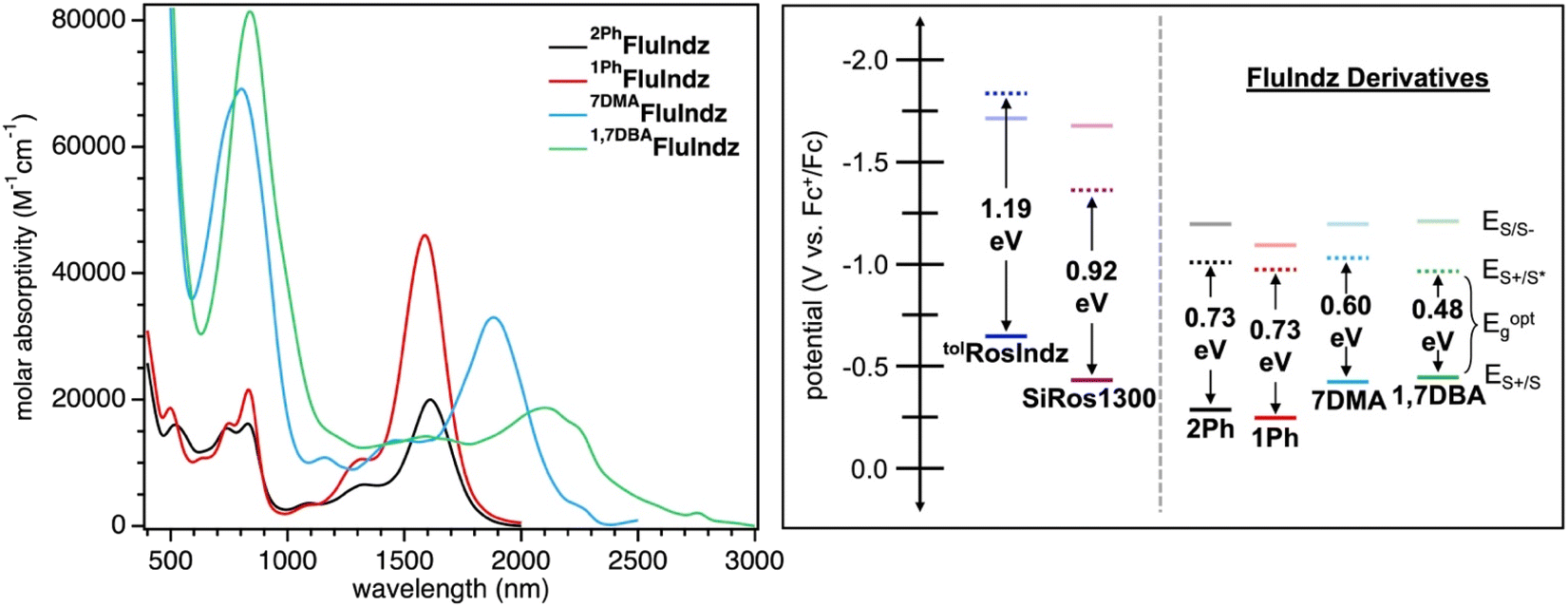 | ||
| Fig. 2 Molar absorptivity of FluIndz dyes in CS2 (left) and electrochemical potentials (ES+/S, ES/S−, and ES+/S*) with optical energy gaps in DCM (right). Electrochemical potentials shown are referenced to Fc+/Fc at 0.00 V in DCM with a 0.1 M Bu4NPF6 supporting electrolyte. | ||
The FluIndz dyes all demonstrated longer wavelength λabs in CS2 compared to DCM, with λabs values of 1590, 1620, 1882, and 2088 nm in CS2 and λabs of 1470, 1500, 1756, and 1956 nm in DCM for 1PhFluIndz, 2PhFluIndz, 7DMAFluIndz, and 1,7DBAFluIndz, respectively (Fig. 2 and S5,†Table 1). The absorption onset values (λonset, determined using the onset program54) were also lower in energy for most of the FluIndz dyes in CS2 compared to DCM. Overall, λabs of the FluIndz dyes are substantially lower in energy than previous indolizine donor-based xanthene derivatives. A shift of λabs to longer wavelengths can be observed in the structurally comparable series (same donor and varied cores) with a 570 nm (0.51 eV) shift from tolRosIndz38 (oxygen-containing core, λabs = 930 nm) to 2PhFluIndz (fluorene core, λabs = 1500 nm) and a 360 nm (0.26 eV) shift from SiRos1300 (ref. 51) (silicon-containing core, λabs = 1140 nm) to 2PhFluIndz (fluorene core, λabs = 1500 nm) (Fig. 1). This demonstrates the value of the antiaromatic cyclopentadienyl cationic core as a means to shift λabs of the xanthene scaffold towards lower energies. The use of indolizine donors in 2PhFluIndz (λabs = 1500 nm) also results in a sizeable shift of 557 nm (0.49 eV) in λabs towards longer wavelengths compared to the alkyl amine donor-based 4-H (ref. 36) (λabs = 943 nm).
| Dye | Solvent | λ abs (nm) | λ onset (nm) | ε (M−1 cm−1) | E S+/S (V) | E S/S− (V) | E optg (eV) | E S+/S* (V) |
|---|---|---|---|---|---|---|---|---|
| a Reference dye in H2O with ∼5% DMSO cosolvent.36 b Photophysical measurements originally taken in toluene,38 electrochemical measurements taken in DCM herein. | ||||||||
| 2PhFluIndz | DCM | 1500 | 1696 | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
−0.28 | −1.21 | 0.73 | −1.01 |
| CS2 | 1620 | 1794 | 20000 |
— | — | — | — | |
| 1PhFluIndz | DCM | 1470 | 1693 | 39900 |
−0.25 | −1.15 | 0.73 | −0.98 |
| CS2 | 1590 | 1761 | 46000 |
— | — | — | — | |
| 7DMAFluIndz | DCM | 1756 | 2076 | 27500 |
−0.43 | −1.21 | 0.60 | −1.03 |
| CS2 | 1882 | 2166 | 33000 |
— | — | — | — | |
| 1,7DBAFluIndz | DCM | 1956 | 2596 | 23000 |
−0.47 | −1.22 | 0.48 | −0.95 |
| CS2 | 2088 | 2547 | 18700 |
— | — | — | — | |
| 4-H | H2O | 943 | — | 14700 |
— | — | — | — |
| tolRosIndz | Toluene/DCM | 930 | 1042 | 79500 |
−0.65 | −1.70 | 1.19 | −1.84 |
| SiRos1300 (ref. 51) | DCM | 1140 | 1350 | 115000 |
−0.45 | −1.68 | 0.92 | −1.37 |
1,7DBAFluIndz was observed to have the lowest molar absorptivity (ε) for its lowest energy peak in both DCM and CS2, with ε of 23000 and 18700 M−1 cm−1, respectively. This observation is likely due to the steric interaction of the N,N-dimethylaniline (DMA) group at the 1-position of the indolizine donor and the phenyl group at the 2-position of the indolizine donor resulting in a lowering of the ε as observed previously.23,512PhFluIndz had the next lowest ε values of 23400 and 20000 M−1 cm−1 in DCM and CS2, respectively. 1PhFluIndz and 7DMAFluIndz both had higher ε values in CS2 compared to DCM, where 1PhFluIndz had a ε of 39900 and 46000 M−1 cm−1 in DCM and CS2, respectively, and 7DMAFluIndz had a ε of 27500 and 33000 M−1 cm−1 in DCM and CS2, respectively. The removal of the phenyl group at the 2-position is thus observed to result in a sizeable increase in ε of 1PhFluIndz, with a ε in CS2 more than double that of 2PhFluIndz. Removal of the phenyl group at the 2-position likely allows for increased planarization across the chromophore π-system, resulting in better orbital overlap of the frontier molecular orbitals leading to a higher ε. Overall, the FluIndz chromophores are observed to have lower ε than both tolRosIndz (ε = 79500 M−1 cm−1) and SiRos1300 (ε = 115000 M−1 cm−1) which is expected given the typically low ε values antiaromatic chromophores display in the literature.36,43 The trend observed across the series (antiaromatic < oxygen < silicon), with respect to ε, is consistent with previous reports.43,44 Higher energy absorption peaks and associated ε values are given in Tables S1 and S2.† Emission spectroscopy was not collected herein due to the predicted emission range of the chromophores (1640–2320 nm based on a stokes shift of 0.10 eV) requiring highly optimized spectrometer setups as described previously.51 However, transient absorption spectroscopy (TAS) was obtained for 1PhFluIndz in DCM. The dye shows excited state absorption peaks at 443 and 1050 nm within 5 ps after excitation with 1500 nm light (Fig. S7†). Excited state lifetime values were not obtained due to poor fitting to exponential decay equations.
Electrochemical properties
The FluIndz derivatives were also studied via electrochemistry in DCM solution to better understand the redox properties of these materials (Tables 1 and S4,†Fig. 2 and S8–S12†). The ground state oxidation potentials (ES+/S) of the FluIndz derivatives were observed to exhibit a trend based on indolizine donor strength. Added peripheral amine donors shifted potentials more negative versus ferrocene, where 1PhFluIndz was the most positively shifted and 1,7DBAFluIndz was the most negatively shifted. ES+/S was thus observed to be −0.25, −0.28, −0.43, and −0.47 V vs. Fc+/Fc55–57 for 1PhFluIndz, 2PhFluIndz, 7DMAFluIndz, and 1,7DBAFluIndz, respectively. In comparing 2PhFluIndz to tolRosIndz and SiRos1300, the ES+/S was observed to be most negative for tolRosIndz at −0.65 V (Fig. S12†), second most negative for SiRos1300 at −0.45 V,51 and least negative for 2PhFluIndz at −0.28 V. In this way, switching the core from the donating oxygen to the neutral silicon to the antiaromatic fluorene results in a positive shift in ES+/S. For the ground state reduction potentials (ES/S−), 2PhFluIndz, 7DMAFluIndz, and 1,7DBAFluIndz were all grouped within 10 mV of one another (−1.21 to −1.22 V), with 1PhFluIndz being shifted towards a more positive potential at −1.15 V. 2PhFluIndz values are considerably more positive than ES/S− of both tolRosIndz and SiRos1300, which have ES/S− values of −1.70 and −1.68 V, respectively. The optical energy gap of the materials (Eoptg) was determined from λonset in DCM solution via the equation 1240/λonset = Eoptg. Eoptg was thus determined to be 0.73, 0.73, 0.60, and 0.48 eV for 2PhFluIndz, 1PhFluIndz, 7DMAFluIndz, and 1,7DBAFluIndz, respectively. The excited state oxidation potential (ES+/S*) could then be calculated for the FluIndz dyes via the equation ES+/S* = ES+/S − Eoptg. ES+/S* does not follow a trend based on the indolizine donor and is observed to be −0.95, −0.98, −1.01, and −1.03 for 1,7DBAFluIndz, 1PhFluIndz2PhFluIndz, and 7DMAFluIndz, respectively. The large shift in ES/S− and ES+/S* towards more positive potentials observed in the antiaromatic FluIndz dyes compared to previous indolizine xanthenes is consistent with previous computational predictions, which indicate that the primary source of the lower energy absorption comes from a decrease in the lowest unoccupied molecular orbital (LUMO) energy.36,44Computational analysis
Consistent with previous studies of NIR and SWIR dyes,23,38,48,51,58 all computational calculations were conducted at the B3LYP/6-311G(d,p)59–61 level of theory using a DCM polarizable continuum model62–65 as implicit solvent with Gaussian16 (ref. 66) software. Visual analysis of the frontier molecular orbitals (Fig. 3), derived from density functional theory (DFT), demonstrates the HOMO is primarily located on the indolizine donors, with some distribution on the core, whereas the LUMO is heavily distributed across the core of the chromophore as well as the indolizine heterocycle. The DMA groups are observed to have significant contribution to the HOMO, but little contribution to the LUMO. This coincides with the electrochemical measurements wherein the addition of the DMA groups shifts ES+/S towards more negative potentials but has little effect on ES/S− or ES+/S*. Time dependent density functional theory (TD-DFT) corroborates the similar maximum λabs of 2PhFluIndz and 1PhFluIndz at 1500 and 1470 nm (0.83 and 0.84 eV), respectively, with vertical transitions (VT) observed at 1131 and 1151 nm (1.10 and 1.08 eV), respectively (Table S5†). 7DMAFluIndz is accurately predicted to be the second lowest energy absorbing chromophore, with a VT observed at 1405 nm (0.88 eV), and 1,7DBAFluIndz is accurately predicted to be the lowest energy absorbing chromophore, with a VT observed at 1635 nm (0.76 eV). TD-DFT predicts the lowest energy absorbing dye 1,7DBAFluIndz to have the highest oscillator strength in the FluIndz series (0.68), followed by 1PhFluIndz (0.63), then 7DMAFluIndz (0.55), and finally 2PhFluIndz (0.52) having the lowest. TD-DFT also predicts higher energy transitions with significant oscillator strengths (>0.2) that correspond to higher energy features from 700–1000 nm in the solution absorption spectra (Table S5†).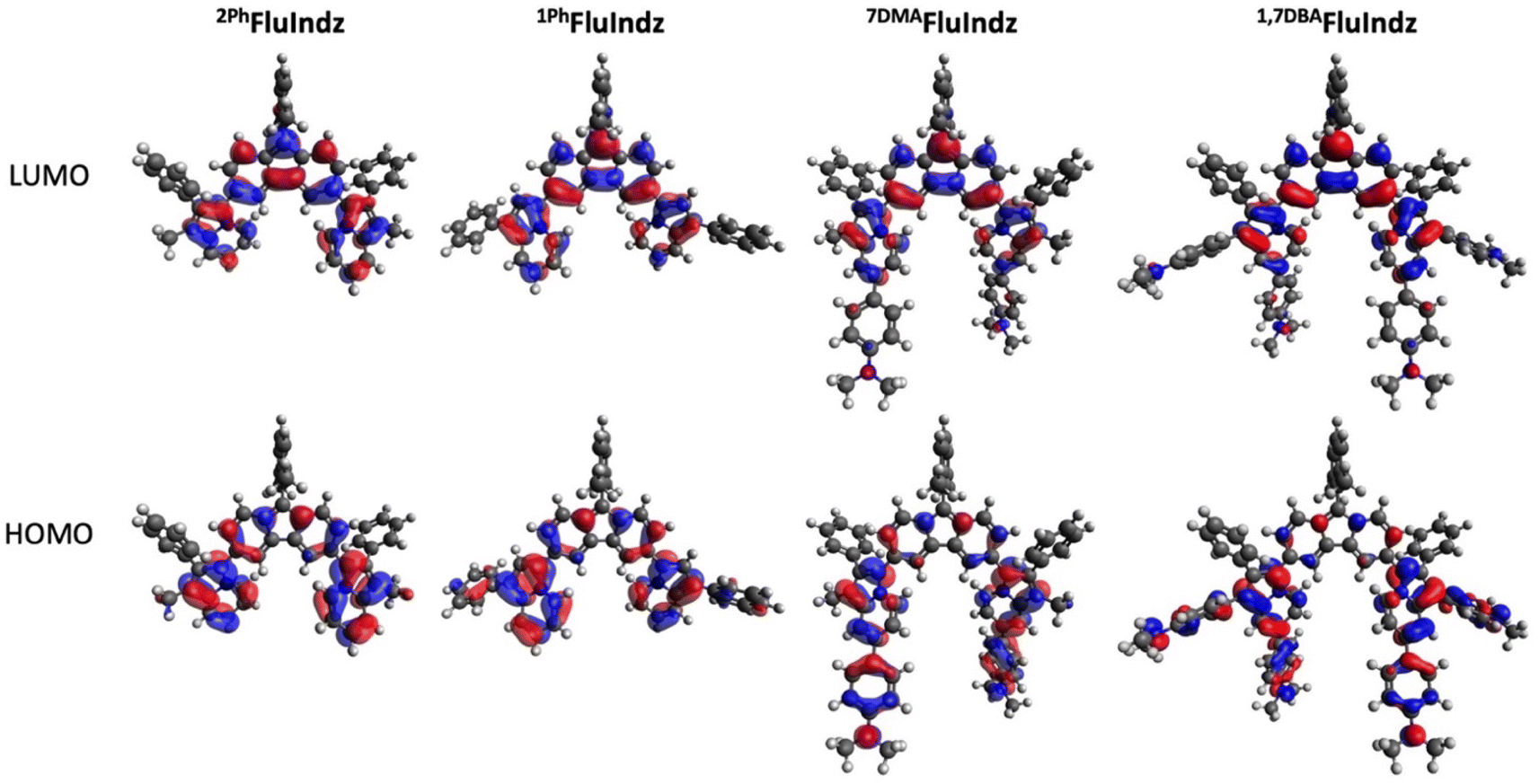 | ||
| Fig. 3 Frontier molecular orbitals including HOMO (bottom) and LUMO (top) of FluIndz dyes 2PhFluIndz (left), 1PhFluIndz (center left), 7DMAFluIndz (center right), and 1,7DBAFluIndz (right). | ||
Calculations were also performed on the previous oxygen (tolRosIndz) and silicon (SiRos1300) derivatives to compare with 2PhFluIndz (Fig. 1 and Table S5†). The trend in VT energy correlates to the decrease in maximum λabs energy according to the trend tolRosIndz > SiRos1300 > 2PhFluIndz. The oscillator strength values follow the trend SiRos1300 > tolRosIndz > 2PhFluIndz. Frontier molecular orbital energies of these three dyes show the HOMOs being close in energy within 64 mV (Fig. 1 and Table S5†). The primary contribution to the decreasing HOMO–LUMO energy gap (EH−Lg) is the decreasing LUMO energy according to the trend: tolRosIndz > SiRos1300 > 2PhFluIndz (455 mV difference, Fig. 1 and Table S5†).
The adiabatic singlet–triplet energy gaps (ΔEadST) were determined as the difference between the total energies of the optimized singlet ground state (S0) and first triplet state (T1) for each dye (Table S6†). Interestingly, the FluIndz series displayed relatively small ΔEadST gaps (3.6–9.7 kcal mol−1) compared to tolRosIndz (22.6 kcal mol−1) and SiRos1300 (16.2 kcal mol−1), implying possible thermally accessible triplet states for the FluIndz dyes. Materials with thermally accessible diradical triplet states in the literature commonly exhibit an experimentally determined ΔEST of ∼4–5 kcal mol−1, while a ΔEST > 15 kcal mol−1 is commonly observed to exist exclusively as a paired singlet.67 Although ΔEST is likely overestimated herein since the B3LYP functional often overestimates this energy gap,68,69 the data suggest that the FluIndz chromophores do not exist exclusively as a paired singlet while tolRosIndz and SiRos1300 have lower diradical triplet populations.
NICS calculations were performed using the optimized geometries for the singlet ground state (S0) and first triplet state (T1) for tolRosIndz, SiRos1300, and 2PhFluIndz (Fig. 4). Negative NICSZZ values at 1 Å above/below a ring correspond to aromaticity in that ring while positive values correspond to antiaromaticity and values near zero signify that the ring is nonaromatic. NICSZZ(avg) values are reported as the average of the values at 1 Å above (NICSZZ(+1)) and below (NICSZZ(−1)) each ring for the 7 rings of interest. Similar to previous reports,58,70 the T1 state is used as an approximation of the excited state since the magnetic shielding tensors are not available via TD-DFT for the S1 state with our computational software. The central ring (Ring D, see Fig. S13† for ring naming scheme, which proceeds alphabetically starting from the bottom left ring and moving clockwise) is the primary site of aromaticity or antiaromaticity induced by the core. In the ground state, tolRosIndz is aromatic in all 7 rings. In the excited state, ring D flips from being aromatic (−4.8 ppm) to being antiaromatic (+11.0 ppm), while the adjacent rings (rings C and E) remain aromatic, albeit less so (S0 = −16.1 and −16.0 ppm, T1 = −12.0 and −11.8 ppm). Interestingly, ring D in SiRos1300 is antiaromatic (+9.1 ppm) with all the other rings being aromatic in the ground state. In the excited state, ring D remains antiaromatic (+7.5 ppm) and the adjacent rings (rings C and E) become slightly more aromatic than in the ground state (S0 = −12.4 and −12.4 ppm, T1 = −14.8 and 14.6 ppm). Lastly, 2PhFluIndz is antiaromatic in ring D (+25.6 ppm) with nonaromatic adjacent rings (rings C and E). In the excited state, ring D becomes nonaromatic (−0.5 ppm) and rings C and E both become more aromatic than in the ground state (S0 = +0.1 ppm each, T1 = −11.6 ppm each). The indolizine rings (rings A, B, F, and G) in all three dyes behave nearly the same: aromatic in the ground state and less aromatic in the excited state. The trend observed in aromaticity of ring D between the ground and excited states is similar to the trend expected from Baird's rule71 for closely related systems where a 4n π-electron antiaromatic ground state becomes more aromatic in the excited state, and vice versa. Here, the aromatic ground state of tolRosIndz flips to being antiaromatic in the excited state, SiRos1300 remains antiaromatic in both states, and the antiaromatic ground state of 2PhFluIndz becomes nonaromatic in the excited state. NICSZZ(avg) values and profiles can be found in the ESI (Tables S7–S9, Fig. S14–S20).†
 | ||
| Fig. 4 NICSZZ(avg) (in ppm) for both the singlet ground state (S0) and first triplet state (T1) as the excited state. Ar = o-tolyl (tolRosIndz) or o-xylyl (SiRos1300 and 2PhFluIndz). | ||
Paramagnetic properties
Upon investigation into other fluorene-based materials in the literature, it was observed that some fluorene materials exhibit broad 1H-NMR spectra with no discernible peaks at room temperature72 or elevated temperatures73 due to thermally accessible diradical triplet states. Unlike previous indolizine–xanthene35,38,51 and fluorenium36 dyes with well resolved 1H-NMR spectra, the FluIndz 1H-NMR spectra are similarly broad and featureless in the aromatic region. Both this NMR observation and the computational evidence of possible thermally accessible triplet states led to the investigation of possible paramagnetic properties in these dyes.First, the 1H-NMR spectra of the FluIndz dyes were taken in DCM at both room temperature (25 °C) and at −30 °C (the lower limit of the available instrumentation) to see if the resolution of the spectrum could be improved as seen in the literature. However, little to no change was observed in the 1H-NMR spectra, possibly due to not reaching low enough temperatures (Fig. S63, S65, S67, and S69†). Next, electron paramagnetic resonance (EPR) spectroscopic measurements were conducted for the FluIndz dyes in DCM (Fig. S25–S28†). All four FluIndz dyes demonstrated an EPR signal, exhibiting unpaired electrons indicative of diradical behavior. EPR measurements were also conducted for SiRos1300 and tolRosIndz to see if these materials exhibited diradical nature (Fig. S29 and S30†). Interestingly, SiRos1300 was observed to be EPR active, indicating the presence of unpaired electrons, while tolRosIndz was not.
Variable temperature (VT) EPR was conducted for 2PhFluIndz from −95 to 127 °C. As the temperature decreased to −95 °C (which is 65° lower than the VT 1H-NMR experiment), the integrated absorption intensity decreased to 65% of the signal at room temperature (25 °C, Fig. S35†). When 2PhFluIndz was heated to 127 °C in PhCl solution, the integrated absorption intensity significantly increased (500%) compared to the signal at room temperature (Fig. 5 and S36†). This type of behavior is common for molecules with an open-shell singlet ground state and a thermally accessible triplet state.74,75 A radical control sample of TEMPO was observed to have a nearly identical integrated absorption intensity for both the elevated and room temperature spectra indicating no temperature dependence (Fig. S37†).
 | ||
| Fig. 5 Variable temperature EPR experiment: spectrum of the first derivative absorption of 2PhFluIndz in PhCl solution from 25 °C (g = 2.0034) up to 127 °C (g = 2.0035). | ||
Evans method NMR measurements were also performed on all four dyes to determine their respective spin counts (Fig. S31–S34†). The effective magnetic moments (μeff) at room temperature were calculated in the same way as previous literature76–79 and are 0.624 μB for 2PhFluIndz, 0.726 μB for 1PhFluIndz, 0.405 μB for 7DMAFluIndz, and 0.792 μB for 1,7DBAFluIndz. These small μeff values indicate that the dyes do not exist as purely diradical single molecules (μeff = ≥2.0 μB), which is a phenomenon referred to as the “biradical paradox” and is commonly observed in conjugated organic biradical systems.67,73,78,80 The reason for this observation is most likely either aggregation leading to antiferromagnetic intermolecular spin pairing80,81 or thermal population of the diradical triplet state.67,72,73 Characterization of the FluIndz dyes including XRD crystallography, mass spectroscopy (MS), and elemental analysis (EA) suggest that the dyes exist as single molecules and rules out radical polymerization leading to the lower than expected μeff values (as seen for Tschitschibabin's hydrocarbon which forms oligomers in solution).67
Importantly, the trend observed in computational ΔEST is consistent with the paramagnetic data. As the optical gap decreases, ΔEST decreases in magnitude, and the population of thermally accessed diradical states becomes more prevalent. We have previously observed lower energy fluorophores exhibiting longer excited state lifetimes.51 This inverse phenomenon was suggested to be due to mixing of singlet and triplet states, which these EPR measurements strongly support.
X-ray crystallography
X-ray diffraction (XRD) crystallography was collected for 1PhFluIndz to better understand the geometry of the antiaromatic chromophore (Fig. 6), especially due to the lack of discernible NMR signals observed. Herein, XRD crystallography was only successfully gathered for 1PhFluIndz due to 2PhFluIndz growing in very small, thin needles that were not suitable for adequate diffraction and 7DMAFluIndz and 1,7DBAFluIndz not preferentially forming crystals due their long alkyl chains. Crystals of 1PhFluIndz could be grown via vapor diffusion of anhydrous diethyl ether into 1.0 mL of anhydrous acetonitrile containing ∼10 mg of the dye (with the vials being backfilled with N2 prior to sealing as a precaution). The chromophore formed dark colored, shiny crystals. For 1PhFluIndz, the dye molecules were observed to crystallize in an orientation where the indolizine donors were face to face on top of one another orienting in a head-to-tail direction (Fig. S21–S24†). The result of this packing was a non-symmetric orientation of the indolizine donors with the core (39.1° dihedral angle for C6, C5, C22, and C23 and a 13.8° dihedral angle for C9, C10, C36, and C37) resulting in differing bond lengths between the two donors (0.01–0.05 Å difference between the same bonds of each donor). Prior crystals of indolizine donor-based fluorophores have exhibited greater symmetry in their molecular orientation, with the indolizine donors having nearly identical dihedral angles and bond lengths.35,48,82 The average bond length of the indolizine donors in the 1PhFluIndz was still observed to be nearly identical to previously reported values for indolizine donor-based fluorophores. The bond lengths within the core were in good agreeance with previously observed fluorenium cation crystals,36,72,83 and exhibited the same variation in bond length on the two sides of the fluorenium core. Atomic coordinates and bond lengths and angles can be found in Tables S10–S12 in the ESI.† | ||
| Fig. 6 XRD crystal structure of 1PhFluIndz dye (the counteranion and solvent molecules have been omitted to enhance clarity). | ||
Photostability
The FluIndz dyes were studied for their relative photostability in DCM solution under ambient atmosphere and 1 sun irradiation using a Pico Solar Simulator from G2V Optics Inc. The photostability was determined by taking the absorption of the samples (done in triplicate) at regular time intervals over a 24 hour period (Fig. 7). 1PhFluIndz was observed to have the greatest photostability of all the derivatives tested herein, maintaining ∼95% of its initial absorbance over the 24 hour period. Once the 24 hour period was over, the samples were left on the benchtop under ambient conditions for an additional week and observed to maintain >90% of their initial absorbance. The other FluIndz derivatives were observed to have no detectable absorbance signal after 24 hours, and their half-lives are estimated from exponential fits of the data. 2PhFluIndz was observed to have the second greatest photostability with a half-life of ∼11.5 hours, followed by 7DMAFluIndz with a half-life of ∼7.5 hours, and lastly 1,7DBAFluIndz with a half-life of ∼6 hours. Interestingly, the trend in photo stability aligns with the trends in both λabs and ES+/S, wherein the lower energy absorbing and more negatively shifted ES+/S have the shortest half-lives. While this relative stability study gives a comparison of the chromophore stability based on structural changes, there is no practical threshold described herein which would support or prevent these dyes' use in a practical application. Stability is highly environmentally dependent, and many chromophores are used in high performing devices for photodetectors and encapsulated in nanoemulsions for biological imaging that are not stable in ambient environments for extended periods. The improved relative stability of 1PhFluIndz does suggests this molecular design will impart greater stability in ambient environments, but this does not imply the other chromophores are unsuitable for specialized environments such as inert environments where most IR photodetectors are prepared for example. In an inert environment, these dyes are likely indefinitely stable since they have been stored without degradation for over a year.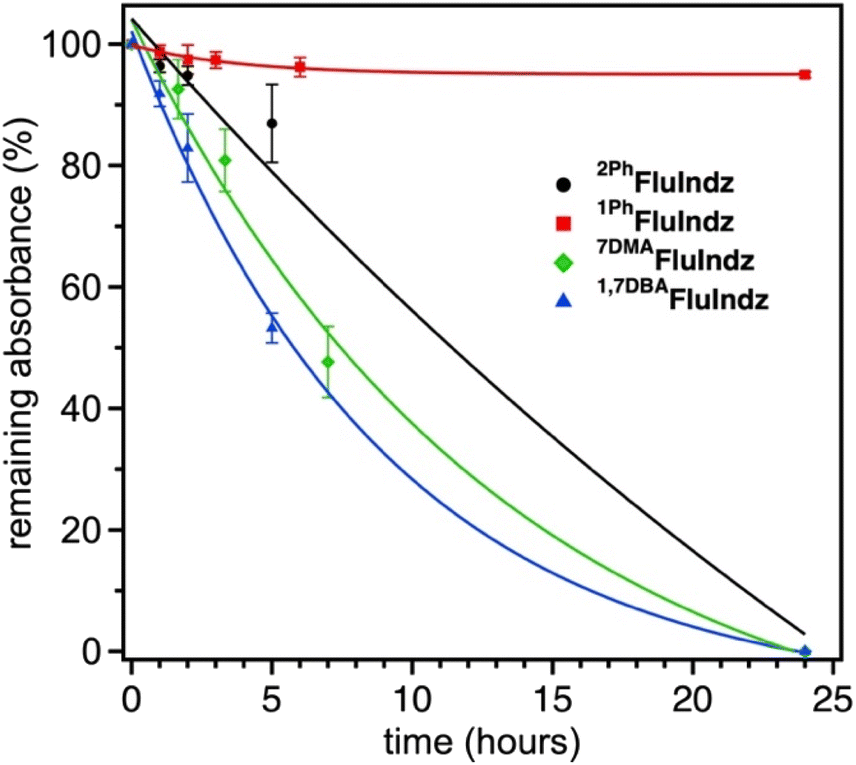 | ||
| Fig. 7 Photostability of FluIndz dyes under 1 sun irradiation in DCM solution. Trials were done in triplicate (error bars given as the standard deviation in % absorbance remaining at each time point) and each data set is fit with an exponential function. | ||
Experimental
General information
Reagents and solvents used in this study were purchased from Ambeed, TCI, Sigma Aldrich, Acros Organics, VWR, and Thermo Fischer Scientific and were used as received without further purification. Thin-layer chromatography (TLC) was conducted with Sorbtech Silica XHL TLC Plates (Support: Glass backed and Thickness: 250 μm) and visualized with a UV lamp. Flash column chromatography was performed using a Teledyne CombiFlash Rf + system. The silica gel cartridges were purchased from Luknova SuperSep (FC003012, 50 μm). 1H and 13C NMR spectra were recorded on a Bruker Ascend-300 (300 MHz) and a Bruker Ascend-400 (400 MHz) spectrometer using deuterated solvents. J values are expressed in Hz and chemical shifts are in ppm using residual solvent as a reference (CDCl3 at 7.26 ppm, DMSO-d6 at 2.50 ppm, CD3CN at 1.94 ppm, and C6D6 at 7.16 ppm). Singlet (s), doublet (d), doublet of doublets (dd), triplet (t), multiplet (m), multiple signals (ms), broad (br), and apparent (ap) are designated as 1H-NMR multiplicity patterns. EPR measurements were collected using X-band Magnettech ESR5000 spectrometer. The hyperfine coupling was not resolved and g-values are calculated according to the apparent isotropic signals observed. For electrospray ionization (ESI) high-resolution mass spectrometry (HRMS), quadruple-TOF was used to obtain the data, both in positive and negative modes, with a Waters Synapt HDMS or Orbitrap Exploris 240 to obtain the data in positive mode with a spray voltage of 3600 V, a resolution of 240000, the ion transfer tube temperature set at 300 °C, and the mass analyzer set to the 200–2000 Da range. Infrared spectra were recorded with an Agilent Cary 660 attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectrometer. All the absorption profiles were recorded on a Cary 5000 UV-Vis-NIR spectrophotometer set to double beam mode. All spectra were taken with the dyes at a concentration of 1.8 × 10−5 M in a 1.0 cm pathlength cuvette and are smoothed with a LOESS functional to bring clarity to some of the noise arising from solvent absorption in the SWIR and ESWIR regions. The same experimental setup used for transient absorption spectroscopy has been described previously.84 Briefly, the 800 nm, 100 fs fundamental output from a femtosecond amplifier (Coherent Astrella, Santa Clara, California) was split with an 85–15 beamsplitter to generate pump and probe beams. To generate the pump excitation wavelength for these samples (λexc = 1500 nm), the reflected portion of the fundamental output was routed through an optical parametric amplifier (Light Conversion Topas OPerA Solo, Vilnius, Lithuania). The excitation wavelength of 1500 nm was selected based on the ground state absorption of 1PhFluIndz. Both the output of the OPerA Solo and the residual fundamental output were directed into commercial transient absorption spectrometers (Ultrafast Systems Helios, Sarasota, Florida), where the pump pulse was routed through an 850 nm long pass filter (Thorlabs, Newton, NJ) and focused with a 350 mm focal length lens to the sample position. To generate the probe pulse, the residual fundamental output was routed through mechanical delay lines and OPAs (Ultrafast Systems, Sarasota, Florida) that created continua with ranges of 300 to 600 nm and 800 to 1300 nm, respectively. Samples were held in 2 mm or 1 mm quartz cuvettes (FireflySci, Inc., Staten Island, New York), and ultrafast data were corrected with a polynomial to account for temporal chirp. Cyclic voltammetry curves were measured with a C–H Instruments Electrochemical Analyzer (Model CHI602E). All measurements were conducted under an argon atmosphere and taken using a platinum counter electrode, silver pseudo reference electrode, and glassy carbon working electrode. The electrolyte solution used was 0.1 M Bu4NPF6 in DCM. Ferrocene was used as the reference, taken as 0.00 V and oxidation and reduction potentials of the materials are reported versus Fc+/Fc in DCM. For all computations, all molecules were first drawn in ChemDraw (20.0.0.41) with all alkyl chains truncated to methyl groups to minimize computational expense, then saved as MDL Molfile. The geometries of those molecules were then optimized with the MMFF94 force field using Avogadro (1.2.0). All single bonds between rings were confirmed to have a dihedral angle between 0 and 90° to avoid parallel or perpendicular relative orientations of adjacent rings (thereby avoiding local minima conformations). Then, sequential geometry optimizations were performed by DFT using Gaussian 16 (ref. 66) employing the default convergence criteria, numerical integrations grids, and implicit solvation parameters with the B3LYP60,61 functional and the following basis sets: first 3-21G, then 6-31G(d,p),85,86 and finally 6-311G(d,p)59 with a dichloromethane polarizable continuum model.62–65 Harmonic vibrational frequency computations confirmed that all optimized structures correspond to minima. After generating the optimized geometries, TD-DFT was performed with the B3LYP functional and 6-311G(d,p) basis set to compute the vertical transition energies and oscillator strengths. The aromatic character associated with each optimized structure was investigated using Nucleus-Independent Chemical Shifts (NICS87). The gauge-independent atomic orbital (GIAO88–91) method was used to compute the B3LYP/6-311G(d,p) magnetic shielding tensors at points 1.0 Å above and below the geometric center of each ring A–G (NICSZZ(±1)). See ESI† for further NICS & crystallographic information. The Crystallographic Information File (CIF) for the crystal reported in this manuscript has been deposited with the Cambridge Crystallographic Data Centre (CCDC) with deposition number 2329750. Stability studies were performed using a G2V Optics Inc. Pico Solar Simulator calibrated to 1 sun intensity as the light source. Dye concentration was adjusted so the absorbance value of the lowest energy feature was measured to be 1.0 in DCM solution in a 1 cm path length cuvette. The samples were monitored at regular time intervals over 24 hours and each study was performed in triplicate. Error bars are given as standard deviation of the remaining absorbance at each time point over the three trials.
Conclusions
A series of four FluIndz dyes varying in the indolizine donor used were designed, synthesized, and their molecular properties studied. The FluIndz dyes have λabs located in the SWIR and ESWIR and behave most “cyanine-like” in CS2 with λabs ranging from 1590–2088 nm. 1PhFluIndz has the greatest ε of the series, more than doubling that of 2PhFluIndz (ε = 46000 versus 20000 M−1 cm−1 in CS2), while 1,7DBAFluIndz has the lowest ε of the series at 18700 M−1 cm−1 in CS2. The large increase in ε observed for 1PhFluIndz is important since antiaromatic dyes have characteristically low ε values. Cyclic voltammetry measurements and DFT calculations reveal that donor modifications within the FluIndz series primarily impact ES+/S and HOMO energy levels. As electron density increases in the indolizine donor from 1Ph ≈ 2Ph < 7DMA < 1,7DBA, ES+/S shifts to more negative potentials and the HOMO energy level increases. Core modifications primarily impact ES+/S* and ES/S− and LUMO energy levels, where both ES+/S* and ES/S− shift to more positive potentials and LUMO energy levels decrease from tolRosIndz > SiRos1300 > 2PhFluIndz. These trends in electrochemical and molecular orbital energy levels lead to smaller Eoptg and EH−Lg energy gaps producing the longer wavelength absorptions observed. TD-DFT computational analysis of vertical transitions corroborates the trends observed in maximum λabs where the lowest energy feature shifts to lower energy with increased electron density in the indolizine donor across the FluIndz series 1Ph ≈ 2Ph > 7DMA > 1,7DBA. Core modifications also shift VT and maximum λabs to lower energies from tolRosIndz > SiRos1300 > 2PhFluIndz. NICS analysis demonstrates how core modifications affect aromaticity in the central ring of the chromophore core. In the ground state, 2PhFluIndz is antiaromatic while tolRosIndz is aromatic. In the excited state, Baird's rule is observed where 2PhFluIndz becomes more aromatic while tolRosIndz becomes more antiaromatic. XRD crystallography of 1PhFluIndz reveals unequivocal determination of the chromophore structure and existence as a single molecule. Photostability studies of the FluIndz dyes in DCM solution reveal that 1PhFluIndz is remarkably more photostable than the other analogues which contain a phenyl group at the 2-position of the indolizine, with ∼95% absorption remaining after 24 hours of 1 sun exposure and >90% absorption over an additional week of ambient exposure. EPR spectroscopy reveals that the FluIndz dyes exist to some extent as diradicals and demonstrates how the diradical triplet population in 2PhFluIndz increases with increased temperature. Furthermore, these dyes exhibit lower-than-expected spin counts that are attributed to the “biradical paradox.” Overall, these materials are exceptionally low energy absorbing small molecule organic chromophores and exhibit unique properties that could be exploited in infrared optoelectronic sensing applications. This study provides a rational design framework to influence the future development of ESWIR absorbing organic molecules.
Data availability
The data underlying this study are available in the published article and its ESI.† Crystallographic data reported herein was deposited with the Cambridge Crystallographic Data Centre (CCDC) with deposition number 2329750.Author contributions
Conceptualization: WEM, MAS, JHD; formal analysis: WEM, MAS, MRT, AJM, NAK, CRB, KMC, SRP, GST, and JHD; funding acquisition: JHD, NIH, and GST; investigation: WEM, MAS, MRT, AJM, NAK, CRB, KMC, and SRP; methodology: WEM, MAS, MRT, NAK, KMC, and SRP; project administration: JHD, NIH, and GST; writing – original draft: WEM; writing – review & editing: WEM, MAS, and JHD. All authors have read and agreed to the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to thank the National Science Foundation (NSF) for awards OIA-1757220 and CHE-2154403 for financial support. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program awarded to WEM. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under award number P20GM130460.Notes and references
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- H. Li, X. Wang, X. Li, S. Zeng and G. Chen, Chem. Mater., 2020, 32, 3365–3375 CrossRef CAS.
- J. B. Barton, J. C. Demro, R. Amber, G. Gasparian and M. Lange, Defense Public Release: Technical Report, ADA399438, 1998 Search PubMed.
- S. Chinnathambi and N. Shirahata, Sci. Technol. Adv. Mater., 2019, 20, 337–355 CrossRef CAS PubMed.
- H. Wen and E. Bellotti, J. Appl. Phys., 2016, 119, 205702 CrossRef.
- L. Vittadello, J. Klenen, K. Koempe, L. Kocsor, Z. Szaller and M. Imlau, Nanomaterials, 2021, 11, 3193 CrossRef CAS PubMed.
- D. C. Sordillo, L. A. Sordillo, P. P. Sordillo, L. Shi and R. R. Alfano, J. Biomed. Opt., 2017, 22, 045002 CrossRef.
- B. Chen, Y. Chen and Z. Deng, Photonics, 2021, 8, 14 CrossRef CAS.
- A. Attiaoui, É. Bouthillier, G. Daligou, A. Kumar, S. Assali and O. Moutanabbir, Phys. Rev. Appl., 2021, 15, 014034 CrossRef CAS.
- C. Gregory, A. Hilton, K. Violette and E. J. D. Klem, in SID Symposium Digest of Technical Papers, 2021, pp. 982–986 Search PubMed.
- V. Pejovic, E. Georgitzikis, J. Lee, I. Lieberman, D. Cheyns, P. Heremans and P. E. Malinowski, IEEE Trans. Electron Devices, 2022, 69, 2840–2850 CAS.
- K. Ba and J. Wang, Mater. Today, 2022, 58, 119–134 CrossRef CAS.
- Y. Yang, Y. H. Zhang, W. Z. Shen and H. C. Liu, Prog. Quantum Electron., 2011, 35, 77–108 CrossRef.
- Z. Wu, Y. Zhai, H. Kim, J. D. Azoulay and T. N. Ng, Acc. Chem. Res., 2018, 51, 3144–3153 CrossRef CAS PubMed.
- N. Li, Z. Lan, Y. S. Lau, J. Xie, D. Zhao and F. Zhu, Adv. Sci., 2020, 7, 2000444 CrossRef CAS.
- N. Li, I. Park, J. H. Vella, S. J. Oh, J. D. Azoulay, D.-S. Leem and T. N. Ng, ACS Appl. Mater. Interfaces, 2022, 14, 53111–53119 CrossRef CAS PubMed.
- A. E. London, H. Chen, M. A. Sabuj, J. Tropp, M. Saghayezhian, N. Eedugurala, B. A. Zhang, Y. Liu, X. Gu, B. M. Wong, N. Rai, M. K. Bowman and J. D. Azoulay, Sci. Adv., 2019, 5, eaav2336 CrossRef CAS PubMed.
- K. S. Mayer, D. J. Adams, N. Eedugurala, M. M. Lockart, P. Mahalingavelar, L. Huang, L. A. Galuska, E. R. King, X. Gu, M. K. Bowman and J. D. Azoulay, Cell Rep. Phys. Sci., 2021, 2, 100467 CrossRef CAS.
- S. Wang, Y. Fan, D. Li, C. Sun, Z. Lei, L. Lu, T. Wang and F. Zhang, Nat. Commun., 2019, 10, 1058 CrossRef PubMed.
- E. D. Cosco, B. A. Arus, A. L. Spearman, T. L. Atallah, I. Lim, O. S. Leland, J. R. Caram, T. S. Bischof, O. T. Bruns and E. M. Sletten, J. Am. Chem. Soc., 2021, 143, 6836–6846 CrossRef CAS.
- Y. Yang, C. Sun, S. Wang, K. Yan, M. Zhao, B. Wu and F. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202117436 CrossRef CAS PubMed.
- B. Ding, Y. Xiao, H. Zhou, X. Zhang, C. Qu, F. Xu, Z. Deng, Z. Cheng and X. Hong, J. Med. Chem., 2019, 62, 2049–2059 CrossRef CAS PubMed.
- D. Ndaleh, C. Smith, M. Loku Yaddehige, A. K. Shaik, D. L. Watkins, N. I. Hammer and J. H. Delcamp, J. Org. Chem., 2021, 86, 15376–15386 CrossRef CAS.
- E. D. Cosco, A. L. Spearman, S. Ramakrishnan, J. G. P. Lingg, M. Saccomano, M. Pengshung, B. A. Arús, K. C. Y. Wong, S. Glasl, V. Ntziachristos, M. Warmer, R. R. McLaughlin, O. T. Bruns and E. M. Sletten, Nat. Chem., 2020, 12, 1123–1130 CrossRef CAS PubMed.
- B. Li, L. Lu, M. Zhao, Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7483–7487 CrossRef CAS.
- M.-H. Liu, Z. Zhang, Y.-C. Yang and Y.-H. Chan, Angew. Chem., Int. Ed., 2021, 60, 983–989 CrossRef CAS PubMed.
- K. Dou, W. Feng, C. Fan, Y. Cao, Y. Xiang and Z. Liu, Anal. Chem., 2021, 93, 4006–4014 CrossRef CAS.
- L. Bai, P. Sun, Y. Liu, H. Zhang, W. Hu, W. Zhang, Z. Liu, Q. Fan, L. Li and W. Huang, Chem. Commun., 2019, 55, 10920–10923 RSC.
- Y. Wang, M. Wang, G. Xia, Y. Yang, L. Si, H. Wang and H. Wang, Chem. Commun., 2023, 59, 3598–3601 RSC.
- D. Yao, Y. Wang, R. Zou, K. Bian, P. Liu, S. Shen, W. Yang, B. Zhang and D. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 4276–4284 CrossRef CAS PubMed.
- K. Ilina, W. M. MacCuaig, M. Laramie, J. N. Jeouty, L. R. McNally and M. Henary, Bioconjugate Chem., 2020, 31, 194–213 CrossRef CAS PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS.
- Y. Fang, J. Shang, D. Liu, W. Shi, X. Li and H. Ma, J. Am. Chem. Soc., 2020, 142, 15271–15275 CrossRef CAS PubMed.
- A. L. Antaris, H. Chen, S. Diao, Z. Ma, Z. Zhang, S. Zhu, J. Wang, A. X. Lozano, Q. Fan, L. Chew, M. Zhu, K. Cheng, X. Hong, H. Dai and Z. Cheng, Nat. Commun., 2017, 8, 15269 CAS.
- S. Chatterjee, A. K. Shaik, K. H. Wijesinghe, D. Ndaleh, A. Dass, N. I. Hammer and J. H. Delcamp, J. Org. Chem., 2022, 87, 11319–11328 CrossRef CAS PubMed.
- M. Grzybowski, O. Morawski, K. Nowak and P. Garbacz, Chem. Commun., 2022, 58, 5455–5458 RSC.
- D. Liu, Z. He, Y. Zhao, Y. Yang, W. Shi, X. Li and H. Ma, J. Am. Chem. Soc., 2021, 143, 17136–17143 CrossRef CAS PubMed.
- S. Chatterjee, W. E. Meador, C. Smith, I. Chandrasiri, M. F. Zia, J. Nguyen, A. Dorris, A. Flynt, D. L. Watkins, N. I. Hammer and J. H. Delcamp, RSC Adv., 2021, 11, 27832–27836 RSC.
- B. L. Van Duuren, B. M. Goldschmidt and H. H. Seltzman, J. Chem. Soc. B, 1967, 814–819 RSC.
- L. Wang, W. Du, Z. Hu, K. Uvdal, L. Li and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14026–14043 CrossRef CAS PubMed.
- Q. Zheng, A. X. Ayala, I. Chung, A. V. Weigel, A. Ranjan, N. Falco, J. B. Grimm, A. N. Tkachuk, C. Wu, J. Lippincott-Schwartz, R. H. Singer and L. D. Lavis, ACS Cent. Sci., 2019, 5, 1602–1613 CrossRef CAS.
- J. B. Grimm, T. A. Brown, A. N. Tkachuk and L. D. Lavis, ACS Cent. Sci., 2017, 3, 975–985 CrossRef CAS.
- N. Ando, H. Soutome and S. Yamaguchi, Chem. Sci., 2019, 10, 7816–7821 RSC.
- H. C. Daly, S. S. Matikonda, H. C. Steffens, B. Ruehle, U. Resch-Genger, J. Ivanic and M. J. Schnermann, Photochem. Photobiol., 2022, 98, 325–333 CrossRef CAS.
- S. Cho, Z. S. Yoon, K. S. Kim, M. Yoon, D. Cho, J. L. Sessler and D. Kim, J. Phys. Chem. Lett., 2010, 1, 895–900 CrossRef CAS.
- H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC.
- F. Dietz, N. Tyutyulkova and M. Rabinovitzb, J. Chem. Soc., Perkin Trans. 2, 1993, 157–164 RSC.
- L. E. McNamara, T. A. Rill, A. J. Huckaba, V. Ganeshraj, J. Gayton, R. A. Nelson, E. A. Sharpe, A. Dass, N. I. Hammer and J. H. Delcamp, Chem.–Eur. J., 2017, 23, 12494–12501 CrossRef CAS PubMed.
- E. Pohjala, Tetrahedron Lett., 1972, 13, 2585–2588 CrossRef.
- L. Han and A. Islam, European patent, EP2752935A1, 2014 Search PubMed.
- W. E. Meador, E. Y. Lin, I. Lim, H. C. Friedman, D. Ndaleh, A. K. Shaik, N. I. Hammer, B. Yang, J. R. Caram, E. M. Sletten and J. H. Delcamp, Nat. Chem., 2024, 1–9 Search PubMed.
- I. Rajapaksha, H. Chang, Y. Xiong, S. Marder, S. R. Gwaltney and C. N. Scott, J. Org. Chem., 2020, 85, 12108–12116 CrossRef CAS.
- C. S. L. Rathnamalala, J. N. Gayton, A. L. Dorris, S. A. Autry, W. Meador, N. I. Hammer, J. H. Delcamp and C. N. Scott, J. Org. Chem., 2019, 84, 13186–13193 CrossRef CAS PubMed.
- A. M. Wallace, C. Curiac, J. H. Delcamp and R. C. Fortenberry, J. Quant. Spectrosc. Radiat. Transfer, 2021, 265, 107544 CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- R. D. Foltz, Handbook of Physics and Chemistry, CRC Press, Boca Raton, FL, 63rd edn, 1982 Search PubMed.
- N. Ree, C. L. Andersen, M. D. Kilde, O. Hammerich, M. B. Nielsen and K. V. Mikkelsen, Phys. Chem. Chem. Phys., 2018, 20, 7438–7446 RSC.
- A. L. Dorris, J. Watson, J. J. Mosely, E. C. Lambert, G. S. Tschumper, J. H. Delcamp and N. I. Hammer, J. Phys. Chem. C, 2023, 127, 649–659 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef.
- J. L. Pascual-ahuir, E. Silla and I. Tuñon, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. Ravat and M. Baumgarten, Phys. Chem. Chem. Phys., 2015, 17, 983–991 RSC.
- J. M. Anglada, J. Poater, I. de P. R. Moreira and J. M. Bofill, J. Org. Chem., 2023, 88, 8553–8562 CrossRef CAS PubMed.
- I. Badía-Domínguez, S. Canola, V. Hernández Jolín, J. T. López Navarrete, J. C. Sancho-García, F. Negri and M. C. Ruiz Delgado, J. Phys. Chem. Lett., 2022, 13, 6003–6010 CrossRef.
- P. Brogdon, F. Giordano, G. A. Puneky, A. Dass, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel, G. S. Tschumper and J. H. Delcamp, Chem.–Eur. J., 2016, 22, 694–703 CrossRef CAS PubMed.
- J. Yan, T. Slanina, J. Bergman and H. Ottosson, Chem.–Eur. J., 2023, 29, e202203748 CrossRef CAS.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., 2013, 52, 6076–6079 CrossRef CAS.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gomez-Garcia, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS.
- D. J. Adams, K. S. Mayer, M. Steelman and J. D. Azoulay, J. Phys. Chem. C, 2022, 126, 5701–5710 CrossRef CAS.
- Y. Han, J. Zhu, S. Dong, Y. Eng, T. Tao, T. Y. Gopalakrishna and C. Chi, Org. Lett., 2023, 25, 3380–3385 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- M. Imran, C. M. Wehrmann and M. S. Chen, J. Am. Chem. Soc., 2020, 142, 38–43 CrossRef CAS PubMed.
- C. M. Wehrmann, R. T. Charlton and M. S. Chen, J. Am. Chem. Soc., 2019, 141, 3240–3248 CrossRef CAS PubMed.
- R. Rausch, D. Schmidt, D. Bialas, I. Krummenacher, H. Braunschweig and F. Würthner, Chem.–Eur. J., 2018, 24, 3420–3424 CrossRef CAS PubMed.
- Y. Kanzaki, D. Shiomi, K. Sato and T. Takui, J. Phys. Chem. B, 2012, 116, 1053–1059 CAS.
- H. Han, D. Zhang, Z. Zhu, R. Wei, X. Xiao, X. Wang, Y. Liu, Y. Ma and D. Zhao, J. Am. Chem. Soc., 2021, 143, 17690–17700 CrossRef CAS.
- J. Gayton, S. A. Autry, W. Meador, S. R. Parkin, G. A. Jr. Hill, N. I. Hammer and J. H. Delcamp, J. Org. Chem., 2019, 84, 687–697 CrossRef CAS.
- D. Duvinage, S. Mebs and J. Beckmann, Chem.–Eur. J., 2021, 27, 8105–8109 CrossRef CAS PubMed.
- N. E. Sparks, S. M. Vijayan, J. K. Roy, A. Dorris, E. Lambert, D. Karunathilaka, N. I. Hammer, J. Leszczynski and D. L. Watkins, ACS Omega, 2023, 8, 24513–24523 CrossRef CAS PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CAS.
- T. Helgaker, M. Jaszuński and K. Ruud, Chem. Rev., 1999, 99, 293–352 CrossRef CAS.
- F. London, J. Phys. Radium, 1937, 8, 397–409 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures, experimental information, characterization, and crystal structure files (CIF). CCDC 2329750. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00733f |
| ‡ These authors contributed equally to this work. |
| § Present address: Air Force Research Laboratory, Materials and Manufacturing Directorate (RXNC), 2230 Tenth Street B655, Wright-Patterson AFB, OH 45433, USA. |
| This journal is © The Royal Society of Chemistry 2024 |