
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystalline nanosheets of three-dimensional supramolecular frameworks with uniform thickness and high stability†
Xinxin
Wang
,
Yucheng
Jin
,
Tianyu
Zheng
,
Ning
Li
 ,
Yuesheng
Han
,
Baoqiu
Yu
,
Kang
Wang
*,
Dongdong
Qi
*,
Tianyu
Wang
* and
Jianzhuang
Jiang
*
,
Yuesheng
Han
,
Baoqiu
Yu
,
Kang
Wang
*,
Dongdong
Qi
*,
Tianyu
Wang
* and
Jianzhuang
Jiang
*
Beijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry and Chemical Engineering, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: kangwang@ustb.edu.cn; qdd@ustb.edu.cn; twang@ustb.edu.cn; jianzhuang@ustb.edu.cn
First published on 16th April 2024
Abstract
Fabricating three dimensional (3D) supramolecular frameworks (SMFs) into stable crystalline nanosheets remains a great challenge due to the homogeneous and weak inter-building block interactions along 3D directions. Herein, crystalline nanosheets of a 3D SMF with a uniform thickness of 4.8 ± 0.1 nm immobilized with Pt nanocrystals on the surface (Q[8]/Pt NSs) were fabricated via the solid–liquid reaction between cucurbit[8]uril/H2PtCl6 single crystals and hydrazine hydrate with the help of gas and heat yielded during the reaction process. A series of experiments and theoretical calculations reveal the ultrahigh stability of Q[8]/Pt NSs due to the high density hydrogen bonding interaction among neighboring Q[8] molecules. This in turn endows Q[8]/Pt NSs with excellent photocatalytic and continuous thermocatalytic CO oxidation performance, representing the thus-far reported best Pt nano-material-based catalysts.
Introduction
Organic molecule-based frameworks including supramolecular frameworks (SMFs),1,2 metal–organic frameworks (MOFs),3,4 and covalent organic frameworks (COFs)5–8 have exhibited great application potentials in various areas such as gas storage and separation,9–15 catalysis,16–20 sensing,21–24 enzyme encapsulation,25,26 and proton conduction27,28 due to their periodic porous structure and functional diversity. Crystalline materials with ultrathin nanosheet morphology have aroused significant research interest in the past two decades since the report of graphene in 2004.29 Towards further tailored structures, pores, and functionalities, a wide range of two dimensional (2D) bulk SMFs,30,31 MOFs,4,32,33 and COFs34–39 composed of layered nanostructural components have been easily fabricated into ultrathin crystalline nanosheets depending on the heterogeneous inter-building block interactions, by means of either top-down or bottom-up methods on the basis of the commonly employed strategy through intensifying the intra-layer inter-building block interactions and simultaneously weakening the inter-layer supramolecular interactions. This, however, is not the case for the three dimensional (3D) analogous frameworks due to the homogeneous inter-building block interactions along the 3D directions. Among the 3D organic molecule-based frameworks, 3D MOFs were firstly fabricated into stable crystalline nanosheets by top-down and bottom-up methods in 2016 (ref. 40) and 2018,41 respectively, because of their moderate inter-building block coordination bonding interaction intensity, which is “weak” enough to be broken down but “strong” enough to maintain the fragile continuous networks with few-nanometer thickness.41–43 In 2021, crystalline 3D COF thin films were also successfully fabricated by a bottom-up solid–liquid/liquid–liquid interfacial synthesis approach depending on selectively confining the crystal growth of 3D COFs.44–47 However, pure supramolecular interactions including the hydrogen bond, π–π interaction, and van der Waals force interactions are conventionally considered too weak to support the fabrication and isolation of stable nanosheets of 3D SMFs due to the homogeneous and weak inter-building block interactions. Obviously, realizing the stable crystalline nanosheets for 3D SMFs will significantly expand the range of ultrathin crystalline molecule-based framework materials with enlarged surface and structural features for enhanced functionalities and diverse application potentials.Herein, we describe the fabrication of crystalline nanosheets of a 3D cucurbit[8]uril (Q[8]) SMF via direct chemical synthesis. A solid–liquid reaction between the Q[8]/H2PtCl6 single crystals dispersed in water and liquid hydrazine hydrate affords ultrathin supramolecular nanosheets of Q[8] decorated with platinum particles (Q[8]/Pt NSs) with a uniform thickness of 4.8 ± 0.1 nm and ultrastability, according to the transmission electron microscopy (TEM) and atomic force microscopy (AFM) images. Powder X-ray diffraction (PXRD) analysis discloses the crystalline nature of the Q[8] nanosheets and Pt nanocrystals. Experimental investigations in combination with theoretical calculations reveal the important role of explosive gases (N2 and HCl) and heat yielded during the chemical reaction process in the formation of the nanosheets of 3D SMF and the high density hydrogen bonding interaction in stabilizing the Q[8]/Pt NSs. A series of spectroscopic technologies including Fourier transform infrared (FT-IR), X-ray absorption structure (XAS), and X-ray photoelectron spectroscopy (XPS) unveil the immobilization of Pt nanocrystals onto the surface of Q[8] nanosheets through O–Pt coordinating interactions, endowing the immobilized Pt nanocrystals with significantly enhanced stability. This in turn results in an excellent photocatalytic and, in particular, continuous thermocatalytic CO oxidation performance of Q[8]/Pt NSs with high CO-to-CO2 conversion efficiency and good durability, representing the thus-far reported best Pt nano-material-based catalysts.
Results and discussion
Q[8] represents a well-known cucurbit[n]uril compound with a typical 3D SMF structure. Fig. 1 depicts its single crystal structure on the basis of CCDC-1052403. As can be seen, in the Q[8] single crystal, each Q[8] molecule (the central Q[8]) is fixed by four neighbouring Q[8] molecules with 16 bifurcated (C–H)2⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds. The portal carbonyls of the central Q[8] are close to the outer surface of two neighbouring Q[8] molecules, while the outer surface of the central Q[8] is close to the portal carbonyls of the other two neighbouring Q[8] molecules. This reciprocal interaction leads to a quasi-Td symmetry for the unit formed by the five Q[8] molecules, which further extends into a 3D hydrogen-bonded network, Fig. 1. It is worth noting that the multiple hydrogen-bonding interactions existing between neighbouring Q[8] molecules endow this Q[8] SMF with comparable stability to 3D MOFs and 3D COFs, as proved by its unchanged crystal structure after immersing the Q[8] single crystal into various organic solvents (e.g., DMF and DMSO), boiling water, H2O2, strong acid (6.0 M HCl), and strong base (10.0 M NaOH) for 24 h, Fig. S1.† Such strong and homogeneous inter-building block interactions, however, also result in the failure in exfoliating the Q[8] single crystal into nanosheets via conventional strategies including sonication and mechanical delamination, Fig. S2.† Herein, the reaction between the block-shaped Q[8]/H2PtCl6 single crystals with a similar arrangement of Q[8] molecules dispersed in water with liquid N2H4·H2O at 50 °C for 1 hour yields ordered square nanosheets, Q[8]/Pt NSs, as indicated by the SEM images, Fig. 2a, S3 and S4.† In addition, the reaction time and concentration of N2H4·H2O have been varied to optimize the fabrication process of Q[8]/Pt NSs, Fig. S5–S7.† In the case of Q[8]/Pt NSs, the nanosheets appear after reaction for 10 minutes at 50 °C. Along with further increase in the reaction time, the amount of nanosheets increases, accompanied by the gradual disappearance of the block-shaped crystals of Q[8]/H2PtCl6. After reaction for 1 hour, only nanosheets can be observed from TEM and SEM images, revealing the complete conversion of Q[8]/H2PtCl6 to Q[8]/Pt NSs. Moreover, the nanosheets gradually become thinner with the increase in the N2H4·H2O concentration to 12 mmol L−1, Fig. S7.† Further increase in the N2H4·H2O concentration leads to the decrease in the amount of nanosheets accompanied by the gradual increase in the block-shaped crystals of Q[8]/Pt. As a result, Q[8]/Pt NSs prepared from Q[8]/H2PtCl6 with 12 mmol per L N2H4·H2O at 50 °C for 1 hour were therefore used for the detailed investigation in the present work. In particular, the optimized fabrication process of Q[8]/Pt NSs has been carried out for three times, giving almost the same PXRD patterns and almost the same morphology as revealed by the TEM and SEM images, Fig. S8 and S9,† demonstrating the excellent reproducibility of the preparation method. Moreover, the large-scale preparation of Q[8]/Pt NSs can be easily achieved by using the present method, Fig. S10,† fulfilling the requirement for large-scale production. AFM measurements prove the smooth surface of Q[8]/Pt NSs with a uniform thickness of ca. 4.8 ± 0.1 nm, corresponding to the thickness of six Q[8] layers along the b direction in the Q[8] single crystal, Fig. 2b and c. TEM image reveals the transparent square-like morphology of Q[8]/Pt NSs with the edge length of several micrometers, Fig. 3a, confirming their ultrathin nanosheet nature. Further evidence for this point comes from the clear observation of the Tyndall effect when a red laser is passed through the dispersed ethanol solution of Q[8]/Pt NSs, inset in Fig. 3a. The highly ordered periodic structure of Q[8]/Pt NSs was characterized by high-resolution TEM (HR-TEM) and fast Fourier transform (FFT) images, Fig. 3b and c. HR-TEM demonstrates a clear quadrilateral network structure with a pore size of ca. 0.38 nm, consistent with the calculated channel cavity size of ca. 0.37–0.41 nm (ca. 0.22–0.26 nm after deducting the van der Waals radii of the atoms on the channel surface) for Q[8]/Pt NSs along the b axis on the basis of the single crystal structure of Q[8], Fig. S11 and S12.† Moreover, the Q[8] units can be identified as bright spots in the HR-TEM image, while the channel of Q[8]/Pt NSs is identified as slightly darker spots. The rectangle-shaped dark areas adjacent to the bright spots and the dimmer spots correspond to the curved channel in the Q[8]/Pt NSs, Fig. 3d and e, revealing the same arrangement of Q[8] molecules in the nanosheets with that in Q[8] single crystals. In particular, the selected area FFT image shows a highly ordered arrangement with independent diffraction points, confirming the high crystallinity of Q[8]/Pt NSs, inset in Fig. 3c. In addition, HR-TEM images unveil the widely dispersed Pt nanoparticles with a size of 2–10 nm on Q[8]/Pt NSs, revealing the reduction of H2PtCl6 into Pt0, Fig. S13.† It is worth noting that the size of the Pt nanoparticles is significantly larger than the cavity size of the Q[8] molecule, indicating the location of Pt nanoparticles on the surface of Q[8]/Pt NSs. The pore size distribution of Q[8]/Pt NSs is calculated to be centred at ca. 0.3 nm on the basis of small-angle X-ray scattering (SAXS) analysis, consistent with the result according to the single crystal data of Q[8], Fig. S14.† Energy-dispersive X-ray spectroscopy (EDS) elemental mapping displays the homogeneous distribution of C and N atoms within Q[8]/Pt NSs, Fig. 3f. Inductively coupled plasma optical emission spectrometry analysis indicates 4.8% content of Pt in Q[8]/Pt NSs.
C hydrogen bonds. The portal carbonyls of the central Q[8] are close to the outer surface of two neighbouring Q[8] molecules, while the outer surface of the central Q[8] is close to the portal carbonyls of the other two neighbouring Q[8] molecules. This reciprocal interaction leads to a quasi-Td symmetry for the unit formed by the five Q[8] molecules, which further extends into a 3D hydrogen-bonded network, Fig. 1. It is worth noting that the multiple hydrogen-bonding interactions existing between neighbouring Q[8] molecules endow this Q[8] SMF with comparable stability to 3D MOFs and 3D COFs, as proved by its unchanged crystal structure after immersing the Q[8] single crystal into various organic solvents (e.g., DMF and DMSO), boiling water, H2O2, strong acid (6.0 M HCl), and strong base (10.0 M NaOH) for 24 h, Fig. S1.† Such strong and homogeneous inter-building block interactions, however, also result in the failure in exfoliating the Q[8] single crystal into nanosheets via conventional strategies including sonication and mechanical delamination, Fig. S2.† Herein, the reaction between the block-shaped Q[8]/H2PtCl6 single crystals with a similar arrangement of Q[8] molecules dispersed in water with liquid N2H4·H2O at 50 °C for 1 hour yields ordered square nanosheets, Q[8]/Pt NSs, as indicated by the SEM images, Fig. 2a, S3 and S4.† In addition, the reaction time and concentration of N2H4·H2O have been varied to optimize the fabrication process of Q[8]/Pt NSs, Fig. S5–S7.† In the case of Q[8]/Pt NSs, the nanosheets appear after reaction for 10 minutes at 50 °C. Along with further increase in the reaction time, the amount of nanosheets increases, accompanied by the gradual disappearance of the block-shaped crystals of Q[8]/H2PtCl6. After reaction for 1 hour, only nanosheets can be observed from TEM and SEM images, revealing the complete conversion of Q[8]/H2PtCl6 to Q[8]/Pt NSs. Moreover, the nanosheets gradually become thinner with the increase in the N2H4·H2O concentration to 12 mmol L−1, Fig. S7.† Further increase in the N2H4·H2O concentration leads to the decrease in the amount of nanosheets accompanied by the gradual increase in the block-shaped crystals of Q[8]/Pt. As a result, Q[8]/Pt NSs prepared from Q[8]/H2PtCl6 with 12 mmol per L N2H4·H2O at 50 °C for 1 hour were therefore used for the detailed investigation in the present work. In particular, the optimized fabrication process of Q[8]/Pt NSs has been carried out for three times, giving almost the same PXRD patterns and almost the same morphology as revealed by the TEM and SEM images, Fig. S8 and S9,† demonstrating the excellent reproducibility of the preparation method. Moreover, the large-scale preparation of Q[8]/Pt NSs can be easily achieved by using the present method, Fig. S10,† fulfilling the requirement for large-scale production. AFM measurements prove the smooth surface of Q[8]/Pt NSs with a uniform thickness of ca. 4.8 ± 0.1 nm, corresponding to the thickness of six Q[8] layers along the b direction in the Q[8] single crystal, Fig. 2b and c. TEM image reveals the transparent square-like morphology of Q[8]/Pt NSs with the edge length of several micrometers, Fig. 3a, confirming their ultrathin nanosheet nature. Further evidence for this point comes from the clear observation of the Tyndall effect when a red laser is passed through the dispersed ethanol solution of Q[8]/Pt NSs, inset in Fig. 3a. The highly ordered periodic structure of Q[8]/Pt NSs was characterized by high-resolution TEM (HR-TEM) and fast Fourier transform (FFT) images, Fig. 3b and c. HR-TEM demonstrates a clear quadrilateral network structure with a pore size of ca. 0.38 nm, consistent with the calculated channel cavity size of ca. 0.37–0.41 nm (ca. 0.22–0.26 nm after deducting the van der Waals radii of the atoms on the channel surface) for Q[8]/Pt NSs along the b axis on the basis of the single crystal structure of Q[8], Fig. S11 and S12.† Moreover, the Q[8] units can be identified as bright spots in the HR-TEM image, while the channel of Q[8]/Pt NSs is identified as slightly darker spots. The rectangle-shaped dark areas adjacent to the bright spots and the dimmer spots correspond to the curved channel in the Q[8]/Pt NSs, Fig. 3d and e, revealing the same arrangement of Q[8] molecules in the nanosheets with that in Q[8] single crystals. In particular, the selected area FFT image shows a highly ordered arrangement with independent diffraction points, confirming the high crystallinity of Q[8]/Pt NSs, inset in Fig. 3c. In addition, HR-TEM images unveil the widely dispersed Pt nanoparticles with a size of 2–10 nm on Q[8]/Pt NSs, revealing the reduction of H2PtCl6 into Pt0, Fig. S13.† It is worth noting that the size of the Pt nanoparticles is significantly larger than the cavity size of the Q[8] molecule, indicating the location of Pt nanoparticles on the surface of Q[8]/Pt NSs. The pore size distribution of Q[8]/Pt NSs is calculated to be centred at ca. 0.3 nm on the basis of small-angle X-ray scattering (SAXS) analysis, consistent with the result according to the single crystal data of Q[8], Fig. S14.† Energy-dispersive X-ray spectroscopy (EDS) elemental mapping displays the homogeneous distribution of C and N atoms within Q[8]/Pt NSs, Fig. 3f. Inductively coupled plasma optical emission spectrometry analysis indicates 4.8% content of Pt in Q[8]/Pt NSs.
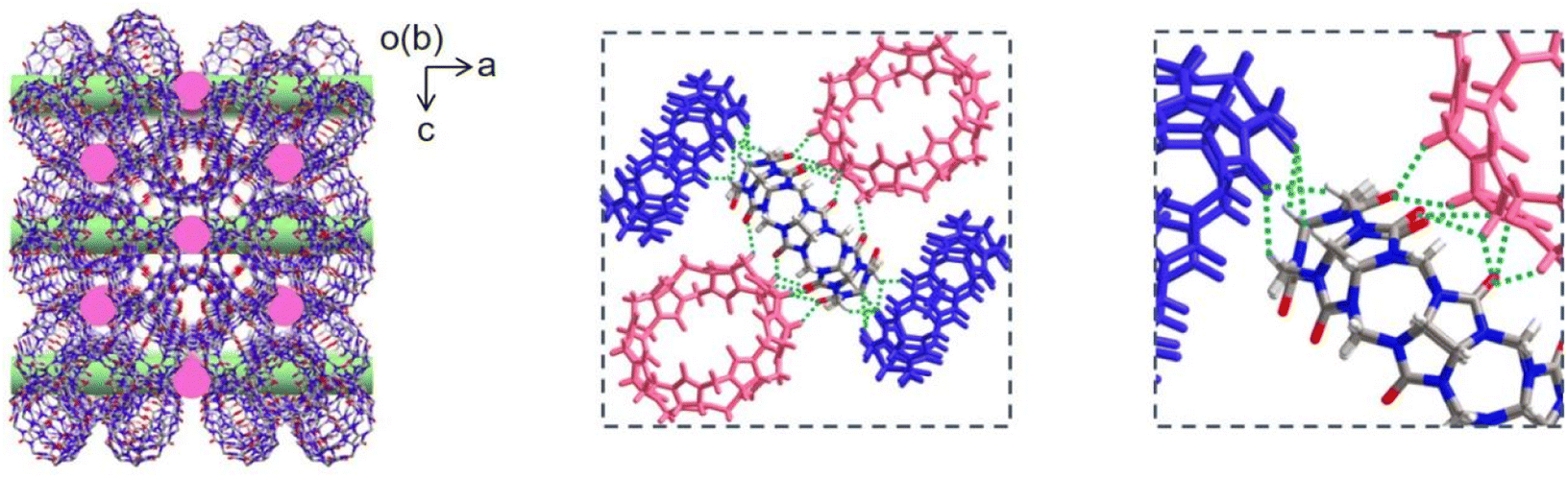 | ||
| Fig. 1 Crystal stacking structure in the Q[8] crystal from the ac plane (left) and detailed interactions between the central Q[8] molecule and four neighboring Q[8] molecules in the Q[8] crystal (middle and right) on the basis of CCDC-1052403. | ||
 | ||
| Fig. 2 Fabricating the crystalline ultrathin Q[8]/Pt NSs from the bulk Q[8]/H2PtCl6 single crystal. (a) Schematic illustration of the formation of Q[8]/Pt NSs via the solid–liquid reaction between Q[8]/H2PtCl6 single crystals and an aqueous solution of N2H4·H2O with the help of a large amount of gas and heat yielded during the chemical reaction process. (b) The AFM image and (c) corresponding height profile of Q[8]/Pt NSs after sonication in ethanol for 5 min. | ||
 | ||
| Fig. 3 Characterization of crystalline ultrathin Q[8]/Pt NSs. (a) TEM image of Q[8]/Pt NSs after sonication in ethanol for 5 min. Inset: the Tyndall effect of Q[8]/Pt NS dispersion in ethanol. (b) HRTEM image of Q[8]/Pt NSs after sonication in ethanol for 5 min. (c) Enlarged image of the area marked in (b) corresponding to the fast Fourier transform image and (d) enlarged HRTEM image of Q[8]/Pt NSs. (e) The curved channel in Q[8]/Pt NSs according to the X-ray crystal structure model of the bc plane. (f) EDS-elemental mapping images of Q[8]/Pt NSs. Experimental PXRD patterns of (g) Q[8] single crystals, (h) Pt nanocrystals, and (i) Q[8]/Pt NSs. | ||
The crystalline structure of Q[8]/Pt NSs was further explored by PXRD. As shown in Fig. 3g and i, the PXRD pattern of Q[8]/Pt NSs displays a series of strong peaks at 8.1, 8.9, 9.2, 10.2, 12.0, 12.5, 13.5, 14.0, 15.3, 16.2, and 18.4°, which match well with the (211), (220), (112), (301), (321), (400), (411), (420), (303), (422), and (224) planes of the pattern of Q[8] single crystals, indicating the same arrangement of Q[8] molecules in the nanosheets as that in Q[8] single crystals, in line with the HR-TEM results. This is further confirmed by the Pawley refinement of the PXRD pattern with small agreement factors of Rp = 4.24% and Rwp = 7.64%, Fig. S15.† Nevertheless, the PXRD pattern of Q[8]/Pt NSs also shows four peaks at 38.4, 44.0, 69.4, and 78.4°, corresponding to the (111), (200), (220), and (311) planes of hcp Pt (PDF card no. 06-0663), disclosing the crystalline nature of the Pt nanoparticles, Fig. 3h and i. In addition, thermogravimetric analysis indicates that Q[8]/Pt NSs can be thermally stable up to 320 °C in air, Fig. S16.† Nevertheless, after being exposed to various organic solvents (e.g., DMF and DMSO), boiling water, H2O2, strong acid (6.0 M HCl), and strong base (10.0 M NaOH) for 24 h, Q[8]/Pt NSs still exhibit unchanged PXRD patterns, Fig. S17,† demonstrating their excellent chemical stability owing to the multiple hydrogen-bonding interactions between neighbouring Q[8] molecules.
In the Pt 4f XPS spectrum, Q[8]/Pt NSs display two major peaks at 70.3 and 73.7 eV attributed to Pt0 together with two weak peaks at 71.6 and 74.9 eV due to Pt2+,48–50 Fig. S18.† The Pt0/Pt2+ ratio amounts to 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 in Q[8]/Pt NSs, indicating the highly abundant zero-valence Pt in the nanocrystals. In the O 1s XPS spectrum of Q[8]/Pt NSs, in addition to a peak at 531.3 eV owing to CO in Q[8],50 a sign ascribed to C–O–Pt is also observed at 533.0 eV,51 revealing the strong interaction between Pt nanocrystals and Q[8] in Q[8]/Pt NSs, Fig. S19.† Furthermore, a typical absorption peak due to the carbonyl band of Q[8] was observed at 1734 cm−1 in the FT-IR spectrum of pure Q[8], which shifts to 1724 cm−1 for Q[8]/Pt NSs, Fig. S20,† proving the effective bonding of Pt nanocrystals with the portal carbonyl groups of Q[8] in Q[8]/Pt NSs.52 In addition, in the Pt L3-edge X-ray absorption near-edge structure (XANES) spectrum, Fig. S21,† the energy position and spectral features of Q[8]/Pt NSs are similar to those for Pt foil, confirming the mainly zero valence metal state of Pt in Q[8]/Pt NSs. Moreover, the Pt L3 extended X-ray absorption fine structure (EXAFS) spectrum in the R space of Q[8]/Pt NSs shows the peaks at 2.4 and 1.8 Å, Fig. S22,† which are indexed as the Pt–Pt interaction and Pt–O coordination, respectively.53–55 These results further demonstrate the domination of metallic Pt species and the existence of coordination interaction between Pt nanocrystals and Q[8] molecules in Q[8]/Pt NSs.
:19 in Q[8]/Pt NSs, indicating the highly abundant zero-valence Pt in the nanocrystals. In the O 1s XPS spectrum of Q[8]/Pt NSs, in addition to a peak at 531.3 eV owing to CO in Q[8],50 a sign ascribed to C–O–Pt is also observed at 533.0 eV,51 revealing the strong interaction between Pt nanocrystals and Q[8] in Q[8]/Pt NSs, Fig. S19.† Furthermore, a typical absorption peak due to the carbonyl band of Q[8] was observed at 1734 cm−1 in the FT-IR spectrum of pure Q[8], which shifts to 1724 cm−1 for Q[8]/Pt NSs, Fig. S20,† proving the effective bonding of Pt nanocrystals with the portal carbonyl groups of Q[8] in Q[8]/Pt NSs.52 In addition, in the Pt L3-edge X-ray absorption near-edge structure (XANES) spectrum, Fig. S21,† the energy position and spectral features of Q[8]/Pt NSs are similar to those for Pt foil, confirming the mainly zero valence metal state of Pt in Q[8]/Pt NSs. Moreover, the Pt L3 extended X-ray absorption fine structure (EXAFS) spectrum in the R space of Q[8]/Pt NSs shows the peaks at 2.4 and 1.8 Å, Fig. S22,† which are indexed as the Pt–Pt interaction and Pt–O coordination, respectively.53–55 These results further demonstrate the domination of metallic Pt species and the existence of coordination interaction between Pt nanocrystals and Q[8] molecules in Q[8]/Pt NSs.
To understand the formation mechanism of the Q[8]/Pt NSs, both experimental and theoretical investigations have been carried out. According to eqn (1), drastic reduction of H2PtCl6 revealing the strong penetration not only produces the Q[8] crystalline layer in the same depth, but simultaneously yields a large amount of N2 and HCl gases and releases a large amount of heat.
| H2PtCl6 + N2H4·H2O → Pt + N2↑ + H2O + 6HCl↑, ΔrHm = −127 kJ mol−1 | (1) |
Combination of the gas-releasing explosive effect with the simultaneously yielding heat in turn leads to the successful exfoliation of the Q[8] crystalline nanosheets. This process proceeds in a repeated manner until the complete reduction of Q[8]/H2PtCl6 single crystals. Additional support for this point comes from the failed fabrication of the crystalline nanosheets starting from the same Q[8]/H2[PtCl6] single crystals using weak reducing agents like methanol and glycol without yielding gas during the reduction reaction process, which however induces the isolation of Pt nanocrystal-immobilized Q[8] single crystals adopting the unchanged bulk morphology to the starting Q[8]/H2PtCl6 single crystals, Fig. S23–S26.† Actually, N2 gas and heat release measurement result gives further support for the above-mentioned gas-releasing explosive effect. As shown in Fig. S27,† N2 gas was detected as the sole gas product by GC spectroscopy and bright-field microscopy images since HCl yielded would dissolve in the aqueous reaction system once it leaves from the single crystals. In addition, as can be found, at the very beginning step of the reaction, the amount of N2 gas increases in a gradual manner. However, after the reaction proceeds for 10 min, the amount of N2 gas suddenly increases, suggesting the yielding process of the gas in a microscopic explosive manner at this time scale with a large amount of nanosheets formed, Fig. S28.† The temperature change in the reaction system was revealed by the thermal imager. As shown in Fig. S29,† the temperature increases from 50.1 to 52.6 °C after the reaction between 10.0 mg Q[8]/H2PtCl6 single crystals and 0.02 mL N2H4·H2O for 10 min. On the basis of the above analysis, a two-step fabrication process for the nanosheets, Q[8]/Pt NSs, was proposed, Fig. S30 and S31:† (1) the reduction process from PtIV to Pt0 together with the formation of Pt nanocrystals and (2) the formation of the crystalline ultrathin nanosheets of 3D SMF together with the formation of more Pt nanocrystals.
CO oxidation was selected as the probe reaction to investigate the catalytic performance of the Q[8]/Pt NSs due to the critical role of this reaction in cleaning air and fuel-cell applications.55 Before the catalysis investigation, the free-standing flexible films of the Q[8]/Pt NSs, named Q[8]/Pt NS film-10/20/30/40, were fabricated through filtering 10/20/30/40 mg of Q[8]/Pt NS suspension in ethanol on the filter membrane, respectively, Fig. 4a and b. The cross-sectional SEM images of each membrane sample show a uniform thickness of 10, 20, 30, and 40 μm, respectively, Fig. 4c and S32.† For comparison, the film of 20 mg Q[8]/Pt material isolated from reducing Q[8]/H2PtCl6 using CH3OH was also fabricated in the same manner, affording the CH3OH-reduced Q[8]/Pt film-20.
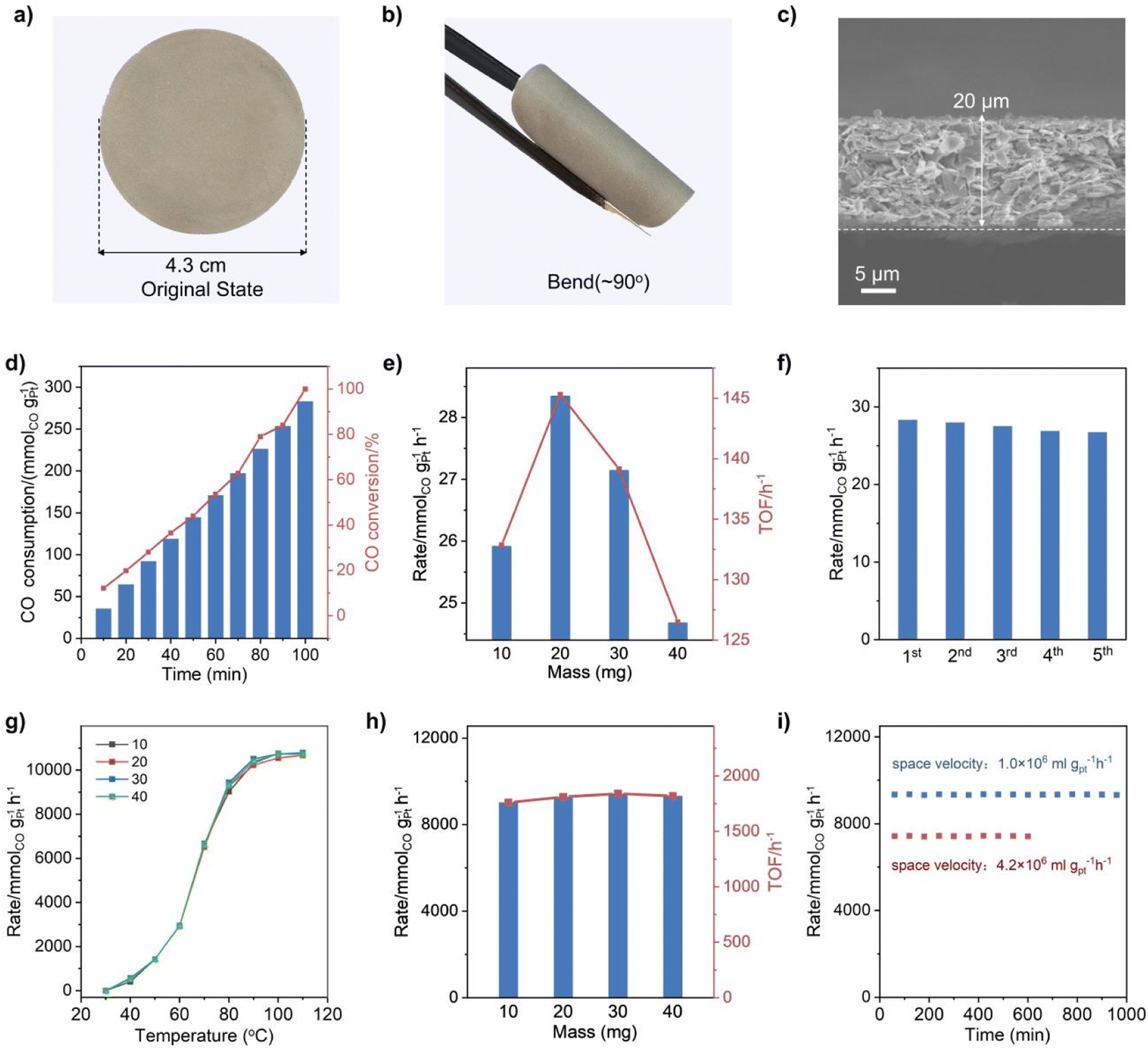 | ||
| Fig. 4 Photocatalytic and continuous thermocatalytic CO oxidation studies. (a and b) Optical images of the Q[8]/Pt NS film with a thickness of 20 μm as a typical representative. (c) Cross-sectional SEM images of Q[8]/Pt NS film-20. (d) Photocatalytic CO oxidation activities of Q[8]/Pt NS film-20 at 25 °C in an idealized gas mixture (0.8% CO, 20% O2, He balance). (e) Mass dependence of the reaction rates and TOFs for photocatalytic CO oxidation by Q[8]/Pt NS films. (f) Durability tests of Q[8]/Pt NS film-20 for photocatalysis. (g) Thermocatalytic CO oxidation activities of Q[8]/Pt NS film-10/20/30/40 at 30–80 °C in an idealized gas mixture (0.8% CO, 20% O2, and He balance). (h) Mass dependence of the reaction rates and TOFs for thermocatalytic CO oxidation by Q[8]/Pt NS films at 80 °C. (i) Durability tests of Q[8]/Pt NS film-40 at 80 °C for thermocatalysis. | ||
The visible-light-driven photocatalytic CO oxidation performance of these films was investigated using an idealized gas mixture (0.8% CO, 20% O2, and He balance) at 25 °C. GC spectroscopy was used to quantify the gaseous product. As shown in Fig. 4d and S33–S35,† CO could be oxidized to CO2 under the photocatalysis of the Q[8]/Pt NS films with a CO2 selectivity of 100%. The CO-to-CO2 conversion rates of the Q[8]/Pt NS films with different thicknesses amount to 25.9, 28.3, 27.1, and 24.6 mmolCO gPt−1 h−1, respectively, Fig. 4e, corresponding to a turnover frequency (TOF) of 132, 145, 139, and 126 h−1. In contrast, the CH3OH-reduced Q[8]/Pt film-20 shows a CO conversion rate of 0.13 mmolCO gPt−1 h−1 with a TOF of 6.5 h−1, Fig. S36,† much inferior to the Q[8]/Pt NS film-20. In particular, the result of the Q[8]/Pt NS films is superior to that of the commercial P25 TiO2, and actually represents one of the best visible-light-driven photocatalytic CO oxidation performances in terms of catalytic efficiency, Table S1,† revealing the significant advantage of the Q[8]/Pt NSs in the heterogeneous photocatalysis. It is worth noting that no CO2 was detected when N2 instead of CO was used as the starting material, Fig. S37,† unveiling the origin of CO2 from the CO molecules in the photosynthesis system of the Q[8]/Pt NS films. Besides, the Q[8] support itself was essentially inactive for the photocatalytic CO oxidation reaction under the same reaction conditions, disclosing the essential role of the Pt nanocrystals immobilized at the surface of Q[8] nanosheets in the high activity of the Q[8]/Pt NS films. In addition, the performance of the Q[8]/Pt NS film-20 shows only a negligible drop after five consecutive runs with each run lasting for 40 min under light irradiation, indicating the excellent photostability of the Q[8]/Pt NS films, Fig. 4f and Table S1.† This is further supported by the unchanged morphology, XPS spectrum, and XRD pattern of Q[8]/Pt NS film-20 after the stability tests, Fig. S38–S41.†
Towards further extending the application potential of the present Q[8]/Pt NSs, the continuous thermocatalytic CO oxidation performance of the Q[8]/Pt NS films was also evaluated by a dynamic test with the feed gas (0.8% CO, 20% O2, and He balance) passing through the Q[8]/Pt NS films in a fixed-bed reactor at a flow rate of 25 mL min−1. As displayed in Fig. 4g, the Q[8]/Pt NS film-10/20/30/40 can achieve the CO oxidation at 40 °C with a specific reaction rate of 396, 540, 576, and 504 mmolCO gPt−1 h−1, respectively, and a CO2 selectivity of 100%. Along with the increase in temperature, the CO conversion rate increases. At the typical working temperature for polymer electrolyte membrane fuel cells of 80 °C, the CO-to-CO2 conversion rate of the Q[8]/Pt NS film-10/20/30/40 reaches 9026, 9284, 9437, and 9330 mmolCO gPt−1 h−1, respectively, corresponding to a TOF of 1760, 1810, 1840, and 1819 h−1, Fig. 4h and S42.† In good contrast, no CO2 was detected for the CH3OH-reduced Q[8]/Pt film-40 under the same conditions, Fig. S43.† In particular, the catalytic efficiency of the Q[8]/Pt NS films for CO oxidation is 1–2 orders of magnitude higher than that of the thus-far reported best supported Pt catalysts and is also much superior to the standard gold nanocatalyst Au/Fe2O3, Table S2,† indicating the outstanding performance of the Q[8]/Pt NS films for continuous CO oxidation. Moreover, the CO conversion and the CO2 selectivity over the Q[8]/Pt NS film-40 remain at constant values over a 1000 minute run under a space velocity of 1.0 × 106 mL gPt−1 h−1 at 80 °C, Fig. 4i. Further studies reveal that Q[8]/Pt NS film-40 remains stable for 300 minutes even at a 5 times higher space velocity of 4.2 × 107 mL gPt−1 h−1. Nevertheless, no obvious change could be observed from the TEM, SEM, XRD, and XPS data of the Q[8]/Pt NS films after the stability tests, Fig. S44–S47,† demonstrating the excellent durability of the Q[8]/Pt NS films, compared to the reported state-of-the-art Pt nano-material-based catalysts, Table S2.†
To get an insight into the catalysis mechanism, the adsorption behavior of CO on the catalyst surface was investigated by in situ DRIFTS spectroscopy. Fig. 5a and b show the visible light-driven and thermo-driven in situ DRIFTS spectra, respectively, of Q[8]/Pt NS films under an atmosphere of 0.8% CO, 20% O2, and remaining He as a function of time. At the beginning of the visible light-driven reaction, the band due to the Pt–CO bond appeared at 2064 cm−1, Fig. 5a, indicating the strong CO adsorption of the Pt nanocrystals.56,57 In addition, the band due to the C–O stretching vibration of CO2 at 2349 cm−1 gradually increases, revealing the CO-to-CO2 conversion under the catalysis of Q[8]/Pt NSs.58,59 This is also true for the thermo-driven reaction, Fig. 5b and S48a,† in line with the DFT calculation results. To further clarify the CO → CO2 reaction mechanism, DFT calculation was carried out using the optimized Q[8]/Pt NS model, in which Pt nanoparticles are located in the shallow pits at the surface of Q[8] NSs and constructed by three Q[8] molecules, Fig. S48a.† According to the calculation results from this model, when one O2 molecule is absorbed onto two adjacent Pt atoms, the O–O bond is activated along with the formation of a transition state [*O⋯O⋯*CO] with the potential barrier of 1.17 eV, Fig. S48b,† which could be easily driven by the visible light irradiation or thermal vibration at ca. 80 °C. This transition state then gets split into O* and CO2 with a significant Gibbs free energy decrease as high as 2.49 eV. Along with the leaving of this newly generated CO2, another CO molecule is attracted onto the same Pt catalytic site and oxidized to CO2 by the O* with an energy decrease of 1.02 eV, finally completing one catalytic cycle.
 | ||
| Fig. 5 In situ DRIFTS spectroscopic investigation for the CO oxidation performance of the Q[8]/Pt NS films. (a) The light-driven in situ DRIFTS spectra of CO adsorption for Q[8]/Pt NS film under visible light irradiation (λ > 400 nm). (b) The thermo-driven in situ DRIFTS spectra of CO adsorption for Q[8]/Pt NS film. All DRIFTS measurements were obtained after CO adsorption under 0.8% CO, 20% O2, and He balance with a flow rate of 100 mL min−1. | ||
Conclusions
In summary, crystalline ultrathin nanosheets of the 3D Q[8] SMF immobilized with Pt nanocrystals on the surface have been fabricated from a solid–liquid chemical reaction under the help of a large amount of gas and heat yielded. This composite with ultrahigh chemical stability exhibits excellent photo-/thermo-catalytic CO oxidation performance with high CO-to-CO2 conversion efficiency and good durability due to the synergistic effect between Pt nanocrystals and Q[8] molecules in the nanosheets. Crystalline ultrathin nanosheets of 3D SMFs represent a new family of organic molecule-based materials with exceptional structural features and properties for diverse application potentials. This work will hopefully inspire further efforts towards designing and preparing crystalline ultrathin nanosheets of 3D molecule-based frameworks for enhanced functionalities.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
J. Jiang, K. Wang, D. Qi, T. Wang and X. Wang conceived and supervised the project, X. Wang performed the experiments, D. Qi and Y. Jin performed the DFT calculation, T. Zheng performed the X-ray absorption structural data analysis, D. Qi and Ning Li worked on part of the diagrams and models and participated in the discussion of the experimental results. Y. Han performed the SEM experimental characterization, B. Yu performed the single crystal test and analysis, J. Jiang, K. Wang, D. Qi, T. Wang and X. Wang linked the experiments, analysis and calculations, and all the authors discussed and wrote the manuscript.Conflicts of interest
There is no conflict of interest to report.Acknowledgements
Financial support from the Natural Science Foundation of China (No. 22235001 and 22175020) and the University of Science and Technology Beijing is gratefully acknowledged. The authors wish to thank the facility support of the 1W2A beamline of the Beijing Synchrotron Radiation Facility (BSRF).References
- J. Dong, L. Liu, C. Tan, Q. Xu, J. Zhang, Z. Qiao, D. Chu, Y. Liu, Q. Zhang, J. Jiang, Y. Han, A. P. Davis and Y. Cui, Free-standing homochiral 2D monolayers by exfoliation of molecular crystals, Nature, 2022, 602, 606–611 CrossRef CAS PubMed.
- M. Pfeffermann, R. Dong, R. Graf, W. Zajaczkowski, T. Gorelik, W. Pisula, A. Narita, K. Müllen and X. Feng, Free-Standing Monolayer Two-Dimensional Supramolecular Organic Framework with Good Internal Order, J. Am. Chem. Soc., 2015, 137, 14525–14532 CrossRef CAS PubMed.
- B. E. R. Snyder, A. B. Turkiewicz, H. Furukawa, M. V. Paley, E. O. Velasquez, M. N. Dods and J. R. Long, A ligand insertion mechanism for cooperative NH3 capture in metal-organic frameworks, Nature, 2023, 613, 287–291 CrossRef CAS PubMed.
- L.-H. Xu, S.-H. Li, H. Mao, Y. Li, A.-S. Zhang, S. Wang, W.-M. Liu, J. Lv, T. Wang, W.-W. Cai, L. Sang, W.-W. Xie, C. Pei, Z.-Z. Li, Y.-N. Feng and Z.-P. Zhao, Highly flexible and superhydrophobic MOF nanosheet membrane for ultrafast alcohol-water separation, Science, 2022, 378, 308–313 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Single-crystal x-ray diffraction structures of covalent organic frameworks, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- C. Kang, Z. Zhang, S. Kusaka, K. Negita, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, X. Zou, Z. Huang, R. Matsuda and D. Zhao, Covalent organic framework atropisomers with multiple gas-triggered structural flexibilities, Nat. Mater., 2023, 22, 636–643 CrossRef CAS.
- B. Yu, R.-B. Lin, G. Xu, Z.-H. Fu, H. Wu, W. Zhou, S. Lu, Q.-W. Li, Y. Jin, J.-H. Li, Z. Zhang, H. Wang, Z. Yan, X. Liu, K. Wang, B. Chen and J. Jiang, Linkage conversions in single-crystalline covalent organic frameworks, Nat. Chem., 2023, 16, 114–124 CrossRef PubMed.
- X. Yang, L. Gong, X. Liu, P. Zhang, B. Li, D. Qi, K. Wang, F. He and J. Jiang, Mesoporous Polyimide-Linked Covalent Organic Framework with Multiple Redox-Active Sites for High-Performance Cathodic Li Storage, Angew. Chem., Int. Ed., 2022, 61, e202207043 CrossRef CAS PubMed.
- D. Gupta, G. Chanteux, G. Kumar, K. Robeyns and A. Vlad, Modular metallotecton for engineering permanently porous frameworks: supernumerary role of ancillary ion, Chem. Sci., 2023, 14, 9780–9786 RSC.
- Y. Zhang, L. Yang, L. Wang, X. Cui and H. Xing, Pillar iodination in functional boron cage hybrid supramolecular frameworks for high performance separation of light hydrocarbons, J. Mater. Chem. A, 2019, 7, 27560–27566 RSC.
- G. Zhang, B. Li, Y. Zhou, X. Chen, B. Li, Z.-Y. Lu and L. Wu, Processing supramolecular framework for free interconvertible liquid separation, Nat. Commun., 2020, 11, 425 CrossRef CAS PubMed.
- G. Zhang, X. Li, G. Chen, Y. Zhang, M. Wei, X. Chen, B. Li, Y. Wu and L. Wu, Supramolecular framework membrane for precise sieving of small molecules, nanoparticles and proteins, Nat. Commun., 2023, 14, 975 CrossRef CAS PubMed.
- S. Laha, R. Haldar, N. Dwarkanath, S. Bonakala, A. Sharma, A. Hazra, S. Balasubramanian and T. K. Maji, A Dynamic Chemical Clip in Supramolecular Framework for Sorting Alkylaromatic Isomers using Thermodynamic and Kinetic Preferences, Angew. Chem., Int. Ed., 2021, 60, 19921–19927 CrossRef CAS PubMed.
- Y. Zhang, G. Zhang, B. Li and L. Wu, Non-Stop Switching Separation of Superfine Solid/Liquid Dispersed Phases in Oil and Water Systems Using Polymer-Assisted Framework Fiber Membranes, Small Methods, 2023, 7, 2201455 CrossRef CAS PubMed.
- H. Li, C. Chen, Q. Li, X. J. Kong, Y. Liu, Z. Ji, S. Zou, M. Hong and M. Wu, An Ultra-stable Supramolecular Framework Based on Consecutive Side-by-side Hydrogen Bonds for One-step C2H4/C2H6 Separation, Angew. Chem., Int. Ed., 2024, e202401754 CAS.
- A. Ozden, J. Li, S. Kandambeth, X.-Y. Li, S. Liu, O. Shekhah, P. Ou, Y. Zou Finfrock, Y.-K. Wang, T. Alkayyali, F. Pelayo García de Arquer, V. S. Kale, P. M. Bhatt, A. H. Ip, M. Eddaoudi, E. H. Sargent and D. Sinton, Energy- and carbon-efficient CO2/CO electrolysis to multicarbon products via asymmetric ion migration–adsorption, Nat. Energy, 2023, 8, 179–190 CrossRef CAS.
- X. Chen, H. Wu, X. Shi and L. Wu, Polyoxometalate-based frameworks for photocatalysis and photothermal catalysis, Nanoscale, 2023, 15, 9242–9255 RSC.
- X. Liu, J. Zhang, Y. Lan, Q. Zheng and W. Xuan, Infinite building blocks for directed self-assembly of a supramolecular polyoxometalate–cyclodextrin framework for multifunctional oxidative catalysis, Inorg. Chem. Front., 2022, 9, 6534–6543 RSC.
- X. Lu, Y. Tang, G. Yang and Y.-Y. Wang, Multifunctional lanthanide MOFs with active sites as new platforms for smart sensing of methylmalonic acid and anti-counterfeiting applications, J. Mater. Chem. C, 2023, 11, 2328–2335 RSC.
- S. Mondal, N. Alam, S. Sahoo and D. Sarma, A “heat set” Zr-Diimide based Fibrous Metallogel: Multiresponsive Sensor, Column-based Dye Separation, and Iodine Sequestration, J. Colloid Interface Sci., 2023, 633, 441–452 CrossRef CAS PubMed.
- Q. Liu, J. Lu, H. Lin, X. Wang, Z. Chang, Y. Chen and Y. Zhang, Polyoxometalate-based metal–organic complexes constructed from a new bis-pyrimidine-amide ligand with high capacitance performance and selectivity for the detection of Cr(VI), Chin. Chem. Lett., 2022, 33, 4389–4394 CrossRef CAS.
- B. Li, Q.-Q. Yan and G.-P. Yong, A new porous coordination polymer reveals selective sensing of Fe3+, Cr2O72−, CrO42−, MnO4− and nitrobenzene, and stimuli-responsive luminescence color conversions, J. Mater. Chem. C, 2020, 8, 11786–11795 RSC.
- W. Jia, S. Ren, H. Xia, C. Zhang and J. Zhang, An ultra-stable Cd coordination polymer based on double-chelated ligand for efficient dual-response of TNP and MnO4, Sens. Actuators, B, 2020, 317, 128230 CrossRef CAS.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: from biomimetics to artificial enzymes, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- S. Li, B. Wang, G. Liu, X. Li, C. Sun, Z. Zhang and X. Wang, Achieving ultra-trace analysis and multi-light driven photodegradation toward phenolic derivatives via a bifunctional catalyst derived from a Cu(i)-complex-modified polyoxometalate, Inorg. Chem. Front., 2024, 11, 1561–1572 RSC.
- Z. A. Siddiqi, M. Shahid, M. Khalid and S. Kumar, Antimicrobial and SOD activities of novel transition metal ternary complexes of iminodiacetic acid containing α-diimine as auxiliary ligand, Eur. J. Med. Chem., 2009, 44, 2517–2522 CrossRef CAS PubMed.
- X. Yu, S. U. Khan, Z. Jin, Q. Wu, H. Pang, H. Ma, X. Wang, L. Tan and G. Yang, Water cluster encapsulated polyoxometalate-based hydrogen-bonded supramolecular frameworks (PHSFs) as a new family of high-capacity electrode materials, J. Energy Storage, 2022, 53, 105192 CrossRef.
- Z.-Q. Shi, N.-N. Ji, K.-M. Guo and G. Li, Crystalline hydrogen-bonded supramolecular frameworks (HSFs) as new class of proton conductive materials, Appl. Surf. Sci., 2020, 504, 144484 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric Field Effect in Atomically Thin Carbon Films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- L. P. Skala, C. L. Stern, L. Bancroft, C. M. Moisanu, C. Pelkowski, X. Aguilar-Enriquez, J. L. Swartz, M. R. Wasielewski and W. R. Dichtel, A modular platform for the precise assembly of molecular frameworks composed of ion pairs, Chem, 2023, 9, 1208–1220 CAS.
- B. Baris, V. Luzet, E. Duverger, P. Sonnet, F. Palmino and F. Cherioux, Robust and Open Tailored Supramolecular Networks Controlled by the Template Effect of a Silicon Surface, Angew. Chem., Int. Ed., 2011, 50, 4094–4098 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Two-dimensional metal-organic framework nanosheets: synthesis and applications, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Metal-organic framework nanosheets as building blocks for molecular sieving membranes, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed.
- D. Rodríguez-San-Miguel, C. Montoro and F. Zamora, Covalent organic framework nanosheets: preparation, properties and applications, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Oriented 2D Covalent Organic Framework Thin Films on Single-Layer Graphene, Science, 2011, 332, 228–231 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, A Scalable General Synthetic Approach toward Ultrathin Imine-Linked Two-Dimensional Covalent Organic Framework Nanosheets for Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Bulk COFs and COF nanosheets for electrochemical energy storage and conversion, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- X. Li, H.-S. Xu, K. Leng, S. W. Chee, X. Zhao, N. Jain, H. Xu, J. Qiao, Q. Gao, I.-H. Park, S. Y. Quek, U. Mirsaidov and K. P. Loh, Partitioning the interlayer space of covalent organic frameworks by embedding pseudorotaxanes in their backbones, Nat. Chem., 2020, 12, 1115–1122 CrossRef CAS PubMed.
- J. Yang, B. Tu, G. Zhang, P. Liu, K. Hu, J. Wang, Z. Yan, Z. Huang, M. Fang, J. Hou, Q. Fang, X. Qiu, L. Li and Z. Tang, Advancing osmotic power generation by covalent organic framework monolayer, Nat. Nanotechnology, 2022, 17, 622–628 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Ultrathin metal-organic framework nanosheets for electrocatalytic oxygen evolution, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- A. Pustovarenko, M. G. Goesten, S. Sachdeva, M. Shan, Z. Amghouz, Y. Belmabkhout, A. Dikhtiarenko, T. Rodenas, D. Keskin, I. K. Voets, B. M. Weckhuysen, M. Eddaoudi, L. C. P. M. de Smet, E. J. R. Sudhölter, F. Kapteijn, B. Seoane and J. Gascon, Nanosheets of Nonlayered Aluminum Metal-Organic Frameworks through a Surfactant-Assisted Method, Adv. Mater., 2018, 30, 1707234 CrossRef.
- S. J. Datta, A. Mayoral, N. Murthy Srivatsa Bettahalli, P. M. Bhatt, M. Karunakaran, I. D. Carja, D. Fan, P. G. M. Mileo, R. Semino, G. Maurin, O. Terasaki and M. Eddaoudi, Rational design of mixed-matrix metal-organic framework membranes for molecular separations, Science, 2022, 376, 1080–1087 CrossRef CAS.
- P. Falcaro, A. J. Hill, K. M. Nairn, J. Jasieniak, J. I. Mardel, T. J. Bastow, S. C. Mayo, M. Gimona, D. Gomez, H. J. Whitfield, R. Riccò, A. Patelli, B. Marmiroli, H. Amenitsch, T. Colson, L. Villanova and D. Buso, A new method to position and functionalize metal-organic framework crystals, Nat. Commun., 2011, 2, 237 CrossRef PubMed.
- S. Chen, C. Zhu, W. Xian, X. Liu, X. Liu, Q. Zhang, S. Ma and Q. Sun, Imparting Ion Selectivity to Covalent Organic Framework Membranes Using de Novo Assembly for Blue Energy Harvesting, J. Am. Chem. Soc., 2021, 143, 9415–9422 CrossRef CAS PubMed.
- Y. Yang, M. Ratsch, A. M. Evans and K. Börjesson, Layered 3D Covalent Organic Framework Films Based on Carbon-Carbon Bonds, J. Am. Chem. Soc., 2023, 145, 18668–18675 CrossRef CAS PubMed.
- T. Zhu, Y. Kong, B. Lyu, L. Cao, B. Shi, X. Wang, X. Pang, C. Fan, C. Yang, H. Wu and Z. Jiang, 3D covalent organic framework membrane with fast and selective ion transport, Nat. Commun., 2023, 14, 5926 CrossRef CAS PubMed.
- X. Shi, Z. Zhang, C. Yin, X. Zhang, J. Long, Z. Zhang and Y. Wang, Design of Three-Dimensional Covalent Organic Framework Membranes for Fast and Robust Organic Solvent Nanofiltration, Angew. Chem., Int. Ed., 2022, 61, e202207559 CrossRef CAS PubMed.
- G. Yun, Z. Hassan, J. Lee, J. Kim, N.-S. Lee, N. H. Kim, K. Baek, I. Hwang, C. G. Park and K. Kim, Highly Stable, Water-Dispersible Metal-Nanoparticle-Decorated Polymer Nanocapsules and Their Catalytic Applications, Angew. Chem., Int. Ed., 2014, 53, 6414–6418 CrossRef CAS PubMed.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- C. Guo, Y. Jiao, Y. Zheng, J. Luo, K. Davey and S.-Z. Qiao, Intermediate Modulation on Noble Metal Hybridized to 2D Metal-Organic Framework for Accelerated Water Electrocatalysis, Chem, 2019, 5, 2429–2441 CAS.
- M. Wang, Y. Xu, C.-K. Peng, S.-Y. Chen, Y.-G. Lin, Z. Hu, L. Sun, S. Ding, C.-W. Pao, Q. Shao and X. Huang, Site-Specified Two-Dimensional Heterojunction of Pt Nanoparticles/Metal-Organic Frameworks for Enhanced Hydrogen Evolution, J. Am. Chem. Soc., 2021, 143, 16512–16518 CrossRef CAS PubMed.
- H. You, D. Wu, Z.-n. Chen, F. Sun, H. Zhang, Z. Chen, M. Cao, W. Zhuang and R. Cao, Highly Active and Stable Water Splitting in Acidic Media Using a Bifunctional Iridium/Cucurbit[6]uril Catalyst, ACS Energy Lett., 2019, 4, 1301–1307 CrossRef CAS.
- T. Ma, H. Cao, S. Li, S. Cao, Z. Zhao, Z. Wu, R. Yan, C. Yang, Y. Wang, P. A. van Aken, L. Qiu, Y.-G. Wang and C. Cheng, Crystalline Lattice-Confined Atomic Pt in Metal Carbides to Match Electronic Structures and Hydrogen Evolution Behaviors of Platinum, Adv. Mater., 2022, 34, 2206368 CrossRef CAS.
- X. Shi, C. Dai, X. Wang, J. Hu, J. Zhang, L. Zheng, L. Mao, H. Zheng and M. Zhu, Protruding Pt single-sites on hexagonal ZnIn2S4 to accelerate photocatalytic hydrogen evolution, Nat. Commun., 2022, 13, 1287 CrossRef CAS PubMed.
- J. Wei, K. Xiao, Y. Chen, X.-P. Guo, B. Huang and Z.-Q. Liu, In situ precise anchoring of Pt single atoms in spinel Mn3O4 for a highly efficient hydrogen evolution reaction, Energy Environ. Sci., 2022, 15, 4592–4600 RSC.
- X. Li, X. I. Pereira-Hernández, Y. Chen, J. Xu, J. Zhao, C.-W. Pao, C.-Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Functional CeOx nanoglues for robust atomically dispersed catalysts, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, S. Impeng, J. Bansmann, J. Rabeah, S. Chen, T. Häring, S. Namuangrak, K. Faungnawakij, A. Brückner and R. J. Behm, Unveiling the CO Oxidation Mechanism over a Molecularly Defined Copper Single-Atom Catalyst Supported on a Metal–Organic Framework, Angew. Chem., Int. Ed., 2023, e202301920 CAS.
- V. Muravev, A. Parastaev, Y. van den Bosch, B. Ligt, N. Claes, S. Bals, N. Kosinov and E. J. M. Hensen, Size of cerium dioxide support nanocrystals dictates reactivity of highly dispersed palladium catalysts, Science, 2023, 380, 1174–1179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00656a |
| This journal is © The Royal Society of Chemistry 2024 |