
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDuplex-forming oligocarbamates with tunable nonbonding sites†
R. Kenton
Weigel
 and
Christopher A.
Alabi
*
and
Christopher A.
Alabi
*
Robert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, New York, USA. E-mail: caa238@cornell.edu
First published on 13th May 2024
Abstract
In biopolymers such as proteins and nucleic acids, monomer sequence encodes for highly specific intra- and intermolecular interactions that direct self-assembly into complex architectures with high fidelity. This remarkable structural control translates into precise control over the properties of the biopolymer. Polymer scientists have sought to achieve similarly precise control over the structure and function of synthetic assemblies. A common strategy for achieving this goal has been to exploit existing biopolymers, known to associate with specific geometries and stoichiometries, for the assembly of synthetic building blocks. However, such systems are neither scalable nor amenable to the relatively harsh conditions required by various materials science applications, particularly those involving non-aqueous environments. To overcome these limitations, we have synthesized sequence-defined oligocarbamates (SeDOCs) that assemble into duplexes through complementary hydrogen bonds between thymine (T) and diaminotriazine (D) pendant groups. The SeDOC platform makes it simple to incorporate non-hydrogen-bonding sites into an oligomer's array of recognition motifs, thereby enabling an investigation into this unexplored handle for controlling the hybridization of complementary ligands. We successfully synthesized monovalent, divalent, and trivalent SeDOCs and characterized their self-assembly via diffusion ordered spectroscopy, 1H-NMR titration, and isothermal titration calorimetry. Our findings reveal that the binding strength of monovalent oligomers with complementary pendant groups is entropically driven and independent of monomer sequence. The results further show that the hybridization of multivalent oligomers is cooperative, that their binding enthalpy (ΔH) and entropy (TΔS) depend on monomer sequence, and that sequence-dependent changes in ΔH and TΔS occur in tandem to minimize the overall change in binding free energy.
Introduction
The precise placement of functional groups along a polymer chain plays a key role in encoding specific intra- and intermolecular interactions that direct self-assembly into discrete architectures. Directed folding and self-assembly in biopolymers like proteins and nucleic acids result in a wide array of properties that facilitate various biological functions, including cell scaffolding, templated synthesis, formation of biomolecular condensates, and information storage. Remarkably, these complex tasks are accomplished using polymers composed of a relatively small number of building blocks. Significant research efforts have been dedicated to producing synthetic systems that replicate the exquisite control over structure and function observed in their natural counterparts. An effective approach has been to exploit interactions optimized by nature to direct the assembly of various building blocks. Both protein–protein1,2 and nucleic acid interactions3–5 have been utilized to create precisely engineered nanostructures via programmable self-assembly. DNA in particular has emerged as a powerful tool for constructing highly complex architectures, such as DNA origami,6,7 because its base-pair specificity allows oligonucleotides to assemble with nanometer-scale precision.8 This has been leveraged for directing the assembly of nanoparticles (NPs) into well-organized structures9 with applications as diagnostic10–12 and therapeutic agents.13,14 NP assemblies also hold significant promise for applications beyond the realm of biology, as they provide opportunities to precisely tailor material properties by manipulating NP size, shape, and the local arrangement of interacting ligands.5,9Unfortunately, the widespread adoption of DNA-directed self-assembly for materials science applications is hindered by DNA's insolubility in organic solvents and the prohibitive cost of synthesizing DNA at the scale required for most nonbiological applications.8 One approach to overcoming these limitations involves the synthesis of recognition-encoded oligomers with various chemistries that are soluble in the organic phase.15 Similar to DNA, these ligands possess precisely positioned recognition sites along a sequence-defined backbone, enabling sequence-selective hybridization. Both dynamic-covalent bonds, such as imine,16–19 boronic ester,20–22 and Diels–Alder,23 and noncovalent interactions such as hydrogen bonds,24–27 coordination bonds,28–32 and salt bridges,33–35 have been used to program self-assembly. The key feature of these bonds and interactions is their reversibility, which results in an equilibrium between unbound and hybridized ligands allowing mismatched complexes to reorganize into a preferred, low energy thermodynamic state. This equilibrium is characterized by an association constant, Ka, that quantifies the stability of the assembled structure. Ka is also a valuable metric for assessing selectivity by comparing the binding strengths of competing complexes. It is therefore important to understand the design principles that govern Ka in oligomeric assemblies, as this knowledge forms the foundation for leveraging recognition-encoded oligomers as building blocks for complex architectures.
In this study, we introduce a synthetic platform designed to generate sequence-defined oligomers capable of forming hybridized duplexes through complementary hydrogen bonding pendant groups. In contrast with previous studies that exclusively employ arrays of hydrogen bonding pendant groups to guide assembly,25,36–43 we have incorporated non-hydrogen bonding sites into our oligomer's array of hydrogen bonding moieties to investigate their effect on hybridization. We show that these oligomers readily undergo duplex formation, and intriguingly, we observe that the number and location of nonbonding sites have a significant impact on the thermodynamics of binding.
Results and discussion
Recognition-encoded oligomers were produced with slight modifications to a scheme previously reported by the Alabi group for the gram-scale synthesis of sequence-defined oligocarbamates (SeDOCs) (Fig. 1).44 In contrast to synthetic pathways where oligomers are synthesized from pre-existing monomers containing the recognition site, the platform employed in this study utilizes iterative reductive amination and carbamation reactions that enable independent tuning of each pendant group with a diverse array of compatible functionalities. The first generation of recognition encoded SeDOCs contain nonbonding methyl units (m) and complementary thymine (T) and diaminotriazine (D) pendant groups. D and T have been shown to form triple hydrogen bonds with a Ka of ∼103 M−1 in chloroform.45,46 The selection of the D–T motif was based on its Ka which allows fine-tuning of the overall binding affinity through incremental adjustment of the D–T valency (i.e., the number of D–T interactions between a pair of oligomers). Furthermore, both D and T exhibit minimal self-dimerization, and their compact steric profiles ensure that they will not impede oligomer binding or negatively impact the synthesis of multivalent chains.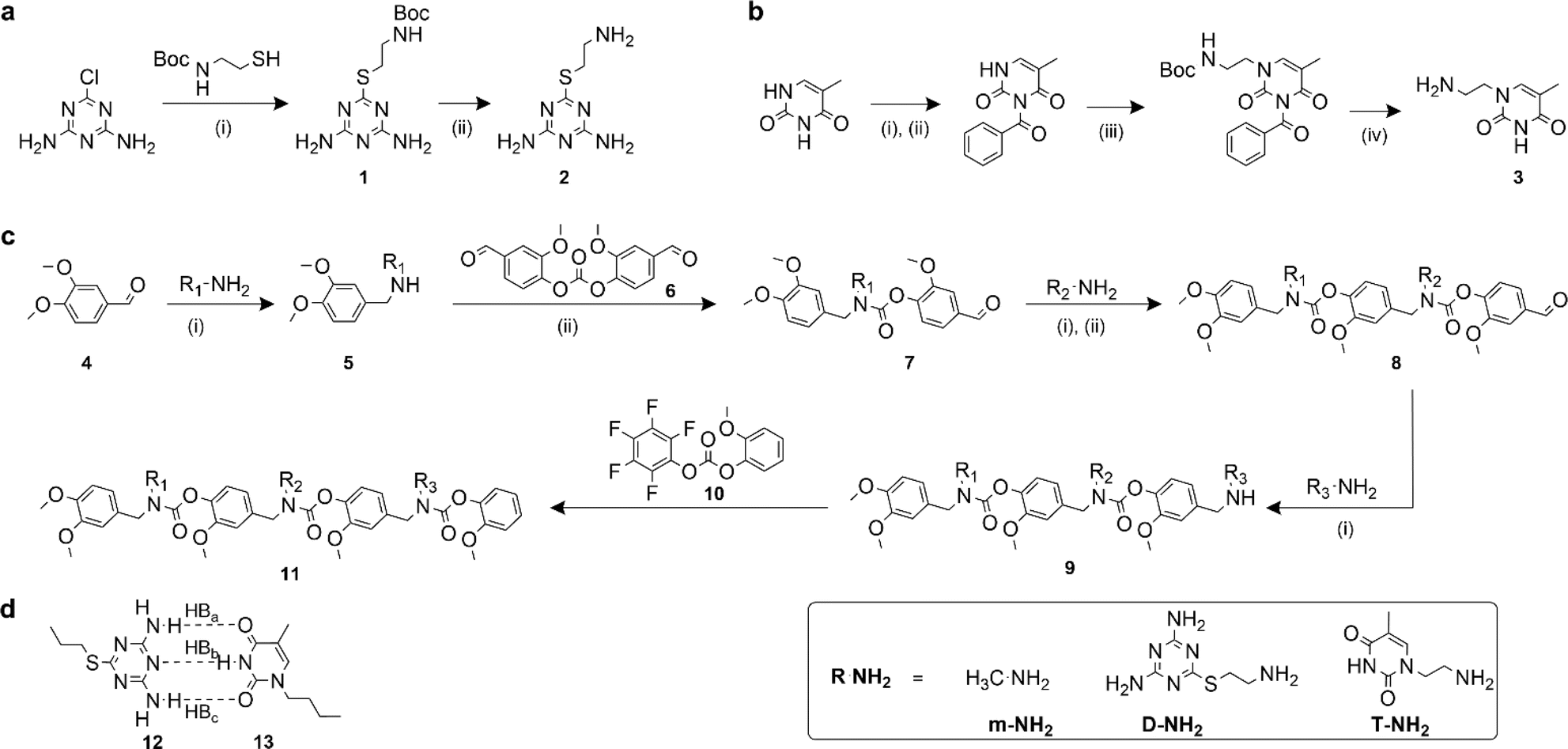 | ||
Fig. 1 Synthesis of SeDOC 3-mers and associated building blocks. (a), Synthesis of an amine-terminated diaminotriazine monomer. (i) Water/ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), KOH; (ii) TFA in CH2Cl2; (b), synthesis of amine-terminated thymine monomer. (i) Acetonitrile, pyridine, benzoyl chloride; (ii) 1,4-dioxane, KOH; (iii) 2-(boc-amino)ethyl bromide, K2CO3; (iv) TFA in CH2Cl2; (c), general SeDOC synthesis scheme (i) MeOH, AcOH, NaBH4; (ii) divanillin carbonate, triethylamine, NH4OH. (d), Watson-Crick-type hydrogen bond paring between a diaminotriazine (compound 12) and thymine (compound 13). :1, v/v), KOH; (ii) TFA in CH2Cl2; (b), synthesis of amine-terminated thymine monomer. (i) Acetonitrile, pyridine, benzoyl chloride; (ii) 1,4-dioxane, KOH; (iii) 2-(boc-amino)ethyl bromide, K2CO3; (iv) TFA in CH2Cl2; (c), general SeDOC synthesis scheme (i) MeOH, AcOH, NaBH4; (ii) divanillin carbonate, triethylamine, NH4OH. (d), Watson-Crick-type hydrogen bond paring between a diaminotriazine (compound 12) and thymine (compound 13). | ||
Synthesis
Primary amine-terminated diaminotriazine 2 and thymine 3 monomers were synthesized according to the reaction scheme in Fig. 1a and b. To obtain compound 2, 2-chloro-4,6-diamino-1,3,5-triazine was refluxed with 2-(boc-amino)ethanethiol and potassium hydroxide. The resulting intermediate 1 was purified via flash chromatography and deprotected with trifluoroacetic acid. Compound 3 was synthesized by benzoyl-protecting the N(3) nitrogen47 on thymine and reacting the N(1) nitrogen with 2-(boc-amino)ethyl bromide. Deprotection with trifluoroacetic acid afforded 3 in good yield. A library of ten SeDOC 3-mers (Fig. 2), each bearing three pendant groups, was generated to facilitate investigations into the effect of monovalent monomer sequence and D–T valency on the binding strength of complementary oligomers. SeDOC 3-mers were synthesized according to the iterative reaction pathway shown in Fig. 1c. 3,4-Dimethoxybenzaldehyde 4 was reacted with a primary amine-terminated m, D, or T (R–NH2, Fig. 1c) and reduced with sodium borohydride. The resulting secondary-amine-terminated molecule 5 was reacted with divanillin carbonate 6 to regenerate the aldehyde end group 7 required for the addition of a second pendant group. Following a second reductive amination with the desired pendant group, the aldehyde was regenerated once again via acylation with 6 to give 8. A final reductive amination was used to install the third desired pendant group and the ensuing secondary amine terminated oligomer 9 was reacted with pentafluorophenyl guaiacol carbonate 10 to generate the desired SeDOC 11.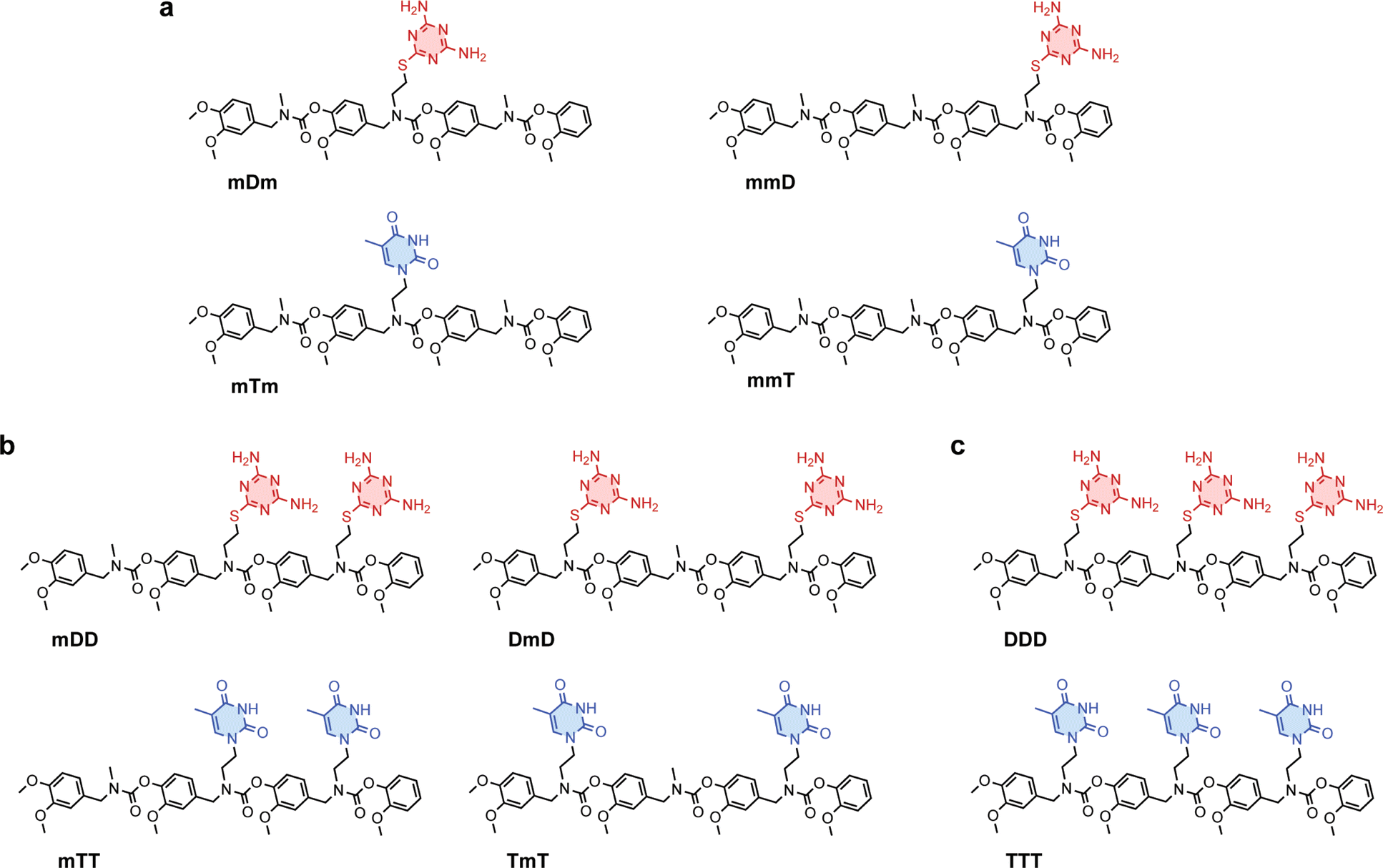 | ||
| Fig. 2 Structures of (a), monovalent (b), divalent and (c), trivalent SeDOC oligomers. | ||
Detection of monovalent SeDOC hybridization via diffusion-ordered spectroscopy (DOSY)
Monovalent SeDOCs (i.e., bearing a single diaminotriazine or thymine pendant group) were synthesized to study the effect of monovalent monomer sequence on Ka. We first confirmed via diffusion-ordered spectroscopy (DOSY) that SeDOCs with complementary pendant groups could hybridize.48 According to the Stokes–Einstein equation, the diffusion coefficient scales with size. As such, unhybridized SeDOCs are expected to exhibit faster diffusion compared to the larger hybrid. The formation of a slow-diffusing species when ligands with complementary pendant groups are mixed is therefore indicative of hybridization. However, since rapid exchange between the unbound and bound ligands outpaces the NMR timescale, the calculated diffusion coefficient from a specific peak in the NMR spectrum represents an ensemble average, incorporating contributions from both the hybrid and unbound ligand depending on their respective populations. To overcome this limitation, pairwise mixtures of oligomers with complementary pendant groups were prepared with an excess of one ligand (∼3:1 mole ratio) to bias the limiting component towards the hybridized state. The diffusion coefficient was then calculated using resonances specific to the limiting component. Results of the DOSY experiments are shown in Fig. 3. The Stejskal–Tanner equation describes the relationship between the NMR signal strength (I), parameters related to the NMR experiment (b), and the diffusion coefficient (D0):| I = I0e0−bD | (1) |
| b = γ2G2δ2(Δ − δ/3) | (2) |
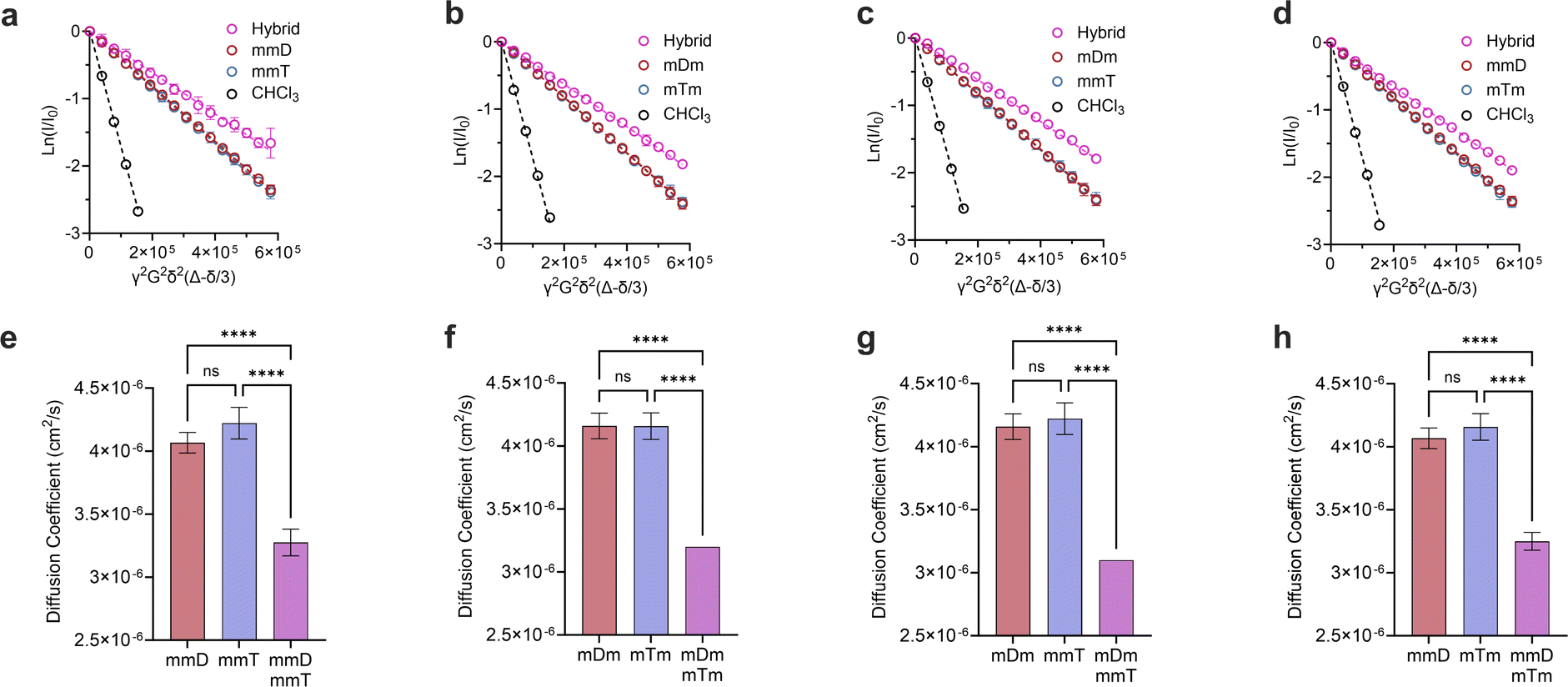 | ||
| Fig. 3 Stesjkal–Tanner plots of (a), mmD·mmT; (b), mDm mTm; (c), mDm mmT; (d), mmD·mTm and their corresponding diffusion coefficients in (e–h). All DOSY experiments were performed in duplicates. Data are displayed as mean ± SD by one-way ANOVA. *p < 0.033, **p < 0.0021, ***p < 0.0002, ****p < 0.0001. | ||
γ is the gyromagnetic ratio, G is the gradient strength, δ is the gradient pulse length, and Δ is the mixing time. According to eqn (1), a plot of ln(I/I0) versus b should give a line with a negative slope that can be used to obtain the diffusion coefficient (Fig. 3a–d). The DOSY results in Fig. 3a–h show that the diffusion coefficients of the pure ligands were similar, as anticipated given their similar size (mDm or mmD = 872 g mol−1, mTm or mmT = 855 g mol−1). The data further reveals that within each mixture, the resonance specific to the limiting component exhibited a smaller diffusion coefficient than the corresponding pure ligand (Fig. 3e–h). This suggests that hybridization occurred between each complementary SeDOC pair. The diffusion coefficient of chloroform was the same across all samples, indicating that the observed reduction in SeDOC diffusion coefficient is not due to an elevated sample viscosity (Fig. 3a–d).
Hydrogen bonding between D and T confirmed via1H-NMR titration experiments
Having confirmed that monovalent SeDOCs with complementary pendant groups were assembling into larger hybrids, 1H-NMR titration experiments were performed in CDCl3 to determine whether hybridization was due to Watson-Crick hydrogen bonding between D and T. In these experiments, a D ligand (the titrant, mDm or mmD) was titrated into a T ligand (the titrand, mTm or mmT), and changes in the chemical shift of protons at the titrand's binding site were monitored as a function of mole ratio. A downfield shift upon titration is a classic indicator of hydrogen bonding between the titrand and titrant.49 The T ligand was used as the titrand given the convenient location of thymine's NH peak, which appeared in a region of the spectrum with minimal spectral overlap. The NH peak in mmT alone gave rise to double signals which we attribute either to the presence of rotamers resulting from hindered rotation along the oligocarbamate backbone, or keto/enol thymine tautomers in relatively equal populations. Titrations were performed for each pairwise combination of monovalent oligomers with complementary pendant groups. In every experiment, the thymine NH proton shifted downfield from approximately 8.4 ppm in pure T ligand to approximately 12 ppm in a 3.5:1 D:T mixture (Fig. 4a, S42–S52†). The downfield shift of the thymine NH resonance was evidence that HBb (Fig. 1d) was forming. This indicated that the slower diffusing species attributed to the hybridized oligomers in the DOSY experiments was due, at least in part, to hydrogen bonding between the T pendant group and D ligand. However, these titrations did not reveal the identity of thymine's hydrogen bonding partner, which could be the expected D pendant group, or the SeDOC backbone. To confirm that association was not caused by nonspecific hydrogen bonds with backbone acceptor sites, an additional experiment was performed in which mmm was titrated into mmT. The mmm SeDOC lacks hydrogen bonding pendant groups, so a downfield shift in T's donor proton upon titration would indicate the presence of hydrogen bonds with the oligocarbamate backbone. Fig. S53† shows that the addition of mmm did not cause a change in the chemical shift of thymine's NH, indicating that the downfield shift observed in titrations with complementary pendant groups (Fig. 4a, S42–S52†) could only be due to hydrogen bonds between D and T. While T does not hydrogen bond nonspecifically, 1H-NMR titration (with mmm) and dilution experiments showed that D forms weak hydrogen bonds with the SeDOC backbone. However, these interactions have minimal impact on SeDOC hybridization because they are much weaker than the hydrogen bonds between D and T (Fig. S55–S58 and S65†).
Effect of monovalent monomer sequence on Ka assessed via isothermal titration calorimetry (ITC)
After establishing that monovalent SeDOCs undergo hybridization primarily driven by hydrogen bonding interactions between complementary D and T pendant groups, our next objective was to quantify the strength of hybridization and investigate how it varies with monomer sequence. As the exchange between the labile protons at the D–T binding site (including thymine's NH) could introduce errors in estimating binding strength via NMR titration (Fig. S44, S47, S50 and S52†), we opted instead for isothermal titration calorimetry (ITC), which quantifies the heat absorbed or released during a binding event.50 ITC experiments in this work were performed in chloroform and the resulting data were fit to an independent binding model to calculate Ka, ΔH, and the binding stoichiometry (n). This model describes the association of ligands due to interactions at a single recognition site or multiple equivalent sites. ΔG and ΔS were then calculated from eqn (3).| −RTlnKa = ΔG = ΔH − TΔS | (3) |
ITC revealed that monovalent SeDOCs with complementary pendant groups formed 1:1 duplexes (Fig. S63†). The binding strength of these duplexes (logKa ≈ 3.2 M−1) was independent of monomer sequence (Fig. 4b). In other words, a monovalent SeDOC, such as mDm, did not exhibit a strong preference for independent equilibrium binding to one sequence isomer, like mTm, over another, such as mmT. This suggests that there were no significant conformational differences between each pair of monovalent sequence isomers. ITC also showed that hybridization was both enthalpically (ΔH < 0) and entropically (ΔS > 0) favorable (Fig. 4c). While we expected that ΔH would be negative given the formation of hydrogen bonds between D and T pendant groups, the positive ΔS was unexpected. Hybridization typically leads to a decrease in entropy because it reduces the translational and conformational entropy of the binding partners. However, in the case of monovalent SeDOCs, hybridization was not only entropically favorable but also entropically driven. The value of |TΔS| was approximately three to four times greater than |ΔH|. While it is unusual to observe an increase in entropy upon hybridization, there are examples of systems that display this phenomenon.51,52 A common feature of these systems is that they have small binding enthalpies (ΔH > −15 kJ mol−1), which is theorized to limit the loss of translational and conformational entropy experienced by the binding partners upon hybridization.51,53 If this decrease is sufficiently small, it can be overcome by favorable sources of entropy so that the overall ΔS is positive.
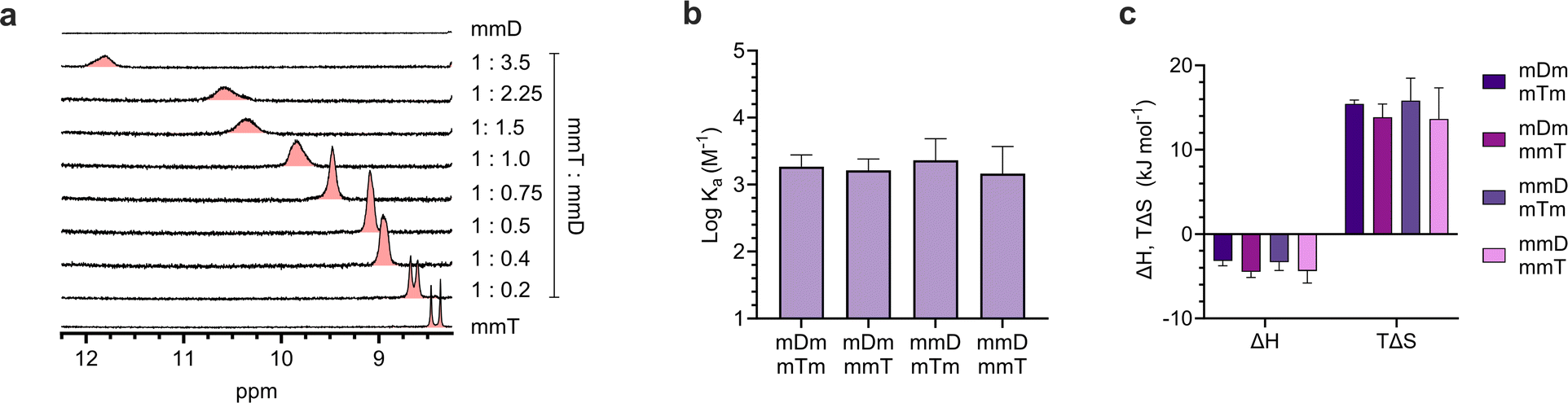 | ||
| Fig. 4 (a). 1H-NMR titration experiment between mmT and mmD in CDCl3 showed a downfield shift in mmT's NH proton. (b). Binding strength, i.e. association constants (Ka), of monovalent hybrids measured by ITC. (c). Thermodynamic binding parameters of monovalent SeDOCs with complementary pendant groups measured via ITC in chloroform. | ||
Given that ITC measures the change in entropy of the entire system (i.e., oligomers and solvent), the observed increase in entropy could arise from an increase in the solvent's entropy, the oligomer(s)’ entropy, or a combination of both. An increase in solvent entropy could occur upon hybridization if solvent ordering was present around the SeDOCs in the unbound state.
Solvent release upon hybridization would then contribute to an increase in both translational and configurational entropy. To investigate further, an ITC experiment was performed in acetonitrile to determine whether hybridization remained entropically favorable (Fig. S66†). Interestingly, the ITC results showed that binding of mDm to mTm in acetonitrile still led to an increase in entropy. Acetonitrile, like chloroform, can hydrogen bond54 with the oligomers and so this experiment does not rule out solvent effects as the source of ΔS > 0. However, it demonstrated that the phenomenon is not unique to chloroform. Unfortunately, limited SeDOC solubility in inert solvents prevented hybridization experiments from being investigated more broadly.
To investigate the SeDOC backbone's role in hybridization, free-standing thymine (compound 12, T) and diaminotriazine (compound 13, D) monomers were synthesized (Fig. 1D). Hybridization of these monomers was analyzed by ITC experiments in chloroform. The results showed that hybridization was still entropically favorable (Fig. S67†) indicating that the positive change in entropy cannot be solely attributed to backbone effects. Additional computational studies are underway to shed light on the factors that contribute to the entropically driven hybridization of monovalent SeDOCs.
Assessing Ka, ΔH and ΔS of multivalent ligands
Divalent and trivalent SeDOCs (Fig. 2) were synthesized to investigate the impact of valency and sequence on Ka. In cases where interactions between a pair of ligands are cooperative, increasing the valency can lead to a substantial increase in binding strength of an order of magnitude or more. Hybridization of mDD with mTT, DmD with TmT, and DDD with TTT was confirmed via DOSY in CDCl3 for mDD·mTT and DmD·TmT, and in CDCl3/MeOD (9:1, v/v) for DDD·TTT. Stejskal–Tanner plots revealed that the hybrids diffused slower than their pure, unbound constituents, as evidenced by the hybrids' shallower slope (Fig. 5a–c) and the resulting quantified diffusion coefficients (Fig. 5e–g). ITC experiments in chloroform were used to measure the thermodynamic parameters of each binding event. Fitting the ITC data to an independent binding model revealed that mDD and mTT formed 1:1 duplexes with a logKa of 3.8 M−1 (Fig. 5d). ΔH decreased from approximately −4 kJ mol−1 in the monovalent hybrids to −20.8 kJ mol−1 in mDD·mTT (Fig. 5h). This significant decrease in binding enthalpy points to the formation of an additional set of hydrogen bonds between mDD and mTT relative to their monovalent counterparts. However, the strengthening of the enthalpic term did not lead to as large increase in Ka as expected because mDD·mTT had a diminished entropic driving force. TΔS was significantly reduced, from roughly 14 kJ mol−1 in the monovalent hybrids to 0.9 kJ mol−1 in mDD·mTT.
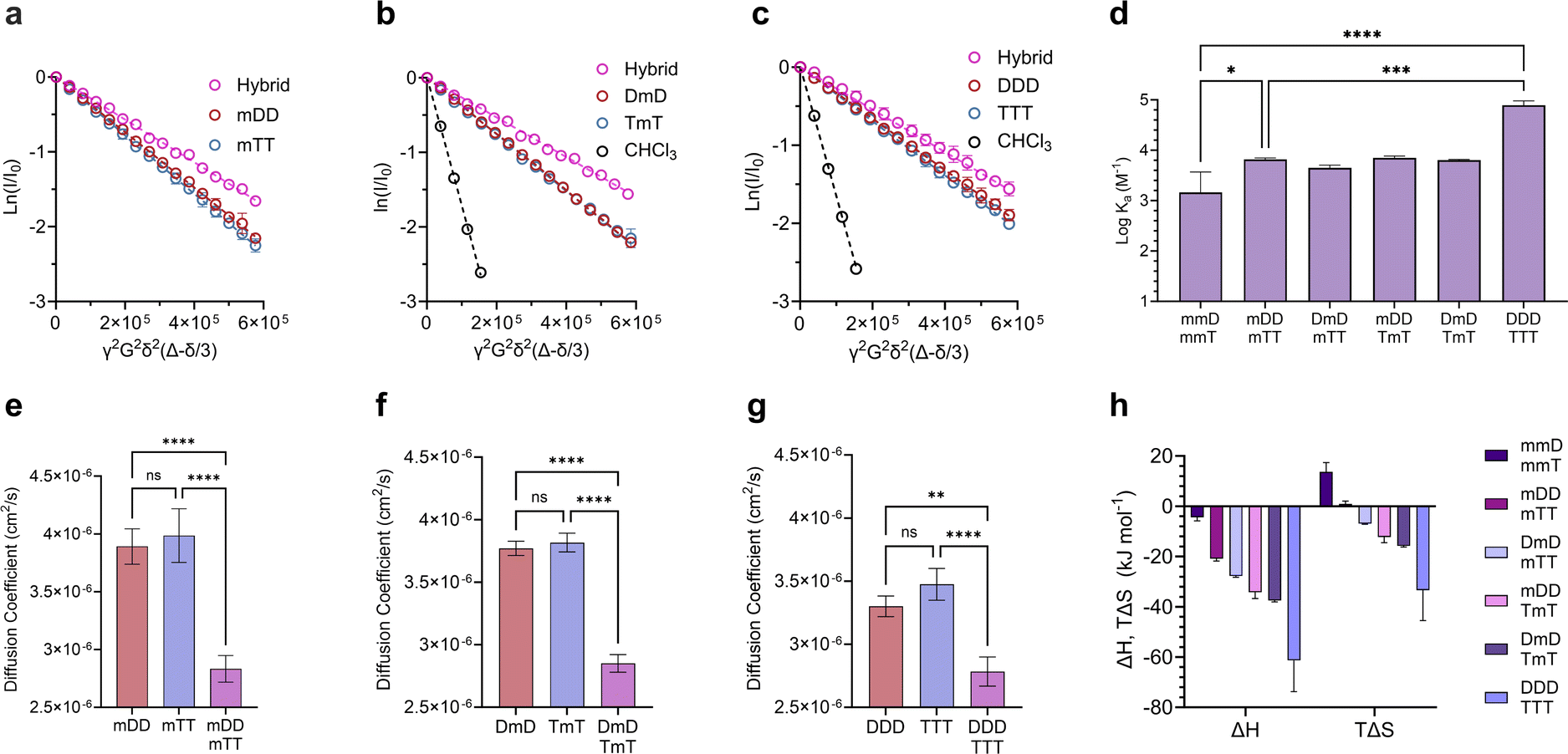 | ||
| Fig. 5 Characterization of di- and trivalent SeDOC hybrids. Stejskal–Tanner plots comparing diffusion coefficients of (a), mDD, mTT, and mDD·mTT. (b), DmD, TmT, and DmD·TmT. (c), DDD, TTT, and DDD·TTT. (d), logKa values of di- and trivalent SeDOC hybrids measured by ITC. (e–g), Diffusion coefficients of pure mono, di- and trivalent SeDOCs and the corresponding hybrids. Diffusion coefficients obtained from slopes in plots in (a–c). (h), Thermodynamic binding parameters of multivalent SeDOCs with complementary pendant groups measured via ITC. Data are displayed as mean ± SD by one-way ANOVA. *p < 0.033, **p < 0.0021, ***p < 0.0002, ****p < 0.0001. | ||
LogKa values of DmD·TmT and mDD·TmT were similar to that of mDD·mTT (logKa = 3.8 M−1). DmD·mTT (logKa = 3.6 M−1) was slightly less stable than the other divalent duplexes, however this difference was not statistically significant (Fig. 5f). Though logKa did not vary with sequence, there were sequence-dependent differences in the entropic and enthalpic contributions to their binding free energies. The complexes were equally stable despite these differences because stronger enthalpic interactions were offset by increased entropic penalties. For example, DmD·TmT had both the strongest enthalpic interactions (ΔH = −37.2 kJ mol−1) of the three and the greatest adverse entropy (TΔS = −15.4 kJ mol−1). Similarly, mDD·TmT (ΔH = −34.2 kJ mol−1) had both a weaker enthalpic driving force than DmD·TmT and a smaller entropic cost (TΔS = −12.2 kJ mol−1) (Fig. 5f). These differences in ΔH and ΔS were observed despite each complex having the same maximum number of D–T interactions, and could be attributed to electronic or conformational effects that vary with sequence.
Adding a third D–T interaction between the 3-mers, as represented in DDD·TTT, increased the logKa to 4.9 M−1. This large increase in binding strength was indicative of a cooperative binding mechanism as Ka was over an order of magnitude greater than that of mDD·mTT. The increase in Ka was due to a substantial decrease in ΔH, from −20.8 kJ mol−1 in mDD·mTT and −37.2 kJ mol−1 in DmD·TmT to −61.3 kJ mol−1 in DDD·TTT. The large change in ΔH suggests that DDD and TTT formed three sets of hydrogen bonds to give a fully zipped-up duplex. The formation of this duplex came with a significant entropic penalty, with TΔS = −33.4 kJ mol−1 (Fig. 5h).
We hypothesize that TΔS was diminished in the divalent duplexes compared to the monovalent hybrids because the second D–T interaction created an additional anchor point between the ligands which limited the conformations accessible to the duplex. The enhanced enthalpic interactions also rigidified the structure by limiting vibrations between the hybridized ligands.55 For similar reasons, ΔS decreased even further in the trivalent case. The reduction in conformational flexibility of the di- and trivalent duplexes compared to their unbound states, along with the presence of strong hydrogen bonds, significantly outweighed other effects likely responsible for the positive entropy observed in the monovalent ligands.
Formation of mixed-mode hybrids
Mixed-mode ITC experiments were performed to assess how Ka, ΔH, and TΔS varied in hybrids of ligands with differing valency (Fig. 6). As expected, increasing the total number of D–T units in a hybridized duplex led to an increase in Ka (Fig. 6a). However, the relative size of Ka was dependent on the magnitudes of ΔH and TΔS. By comparing the ΔH values of “matched” complexes (e.g., mmD·mmT, mDD·mTT) with those of “mismatched” complexes (e.g., mmD·mTT, mDD·TTT), we determined that matched hybrids were indeed engaging in the maximum number of available hydrogen bonds. Hybridization of mmD with divalent mTT or trivalent TTT was expected to have a slightly stronger enthalpic term than the mmD·mmT hybrid (ΔH = −4.4 kJ mol−1) due to the presence of a neighboring T binding site. We also anticipated that mmD·mTT (ΔH = −11.8 kJ mol−1) would have a weaker ΔH than mDD·mTT (ΔH = −20.8 kJ mol−1) and mDD·TmT (ΔH = −34.2 kJ mol−1) because mmD can only form a single set of hydrogen bonds. While it appeared that these trends were observed, the error associated with the measurements prevented the binding enthalpy of mmD·mTT from being statistically different from those of mmD·mmT or mDD·mTT (Fig. 6b). Similarly, the ΔH of mDD·mTT and mDD·TmT was weaker than mDD·TTT (ΔH = −33.6 kJ mol−1) due to the latter's additional T site. Finally, a statistically significant difference was observed between the enthalpic terms of mDD·TTT and DDD·TTT, thus confirming that DDD·TTT had formed a three-rung molecular ladder. We also examined the entropic favorability of both matched and mismatched hybrids. We observed minimal changes in TΔS in duplexes formed between mmD and thymine-containing SeDOCs, i.e., mmT, mTT and TTT (Fig. 6c). In contrast, a significant decrease in entropy was observed in duplexes formed between two multivalent SeDOCs. Since strengthening the enthalpic interactions reduces the amount of residual motion retained by the complex, we expected the binding entropy of mDD·mTT to be more positive than mDD·TTT, and this was observed. Similarly, DDD·TTT was predicted to be the least conformationally flexible oligomer, i.e., most negative TΔS, because it has the greatest number of interactions between binding partners.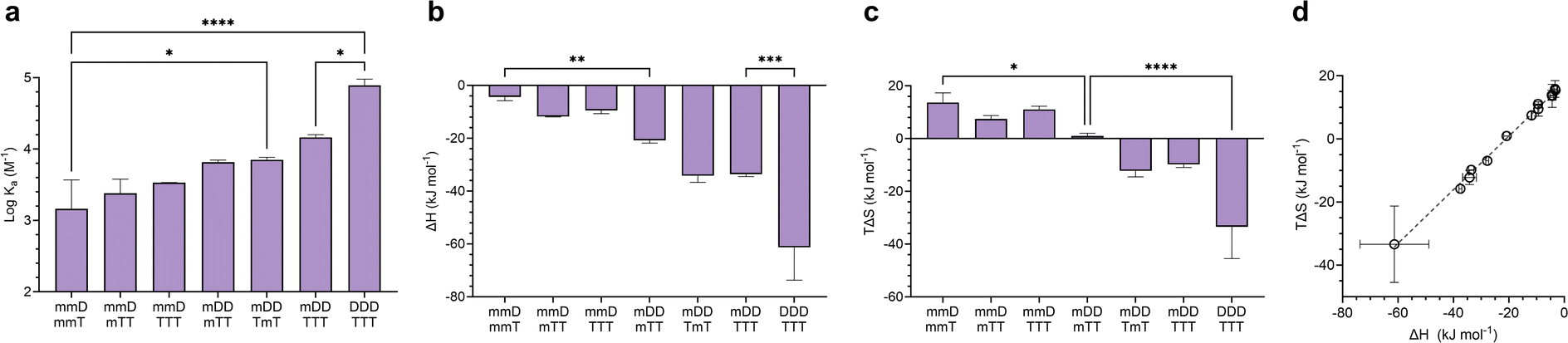 | ||
| Fig. 6 (a–c), Binding thermodynamics of mixed-mode hybrids compared to complementary mono-, di-, and trivalent complexes. (d), Enthalpy versus entropy plot of D–T and SeDOC complexes in chloroform. Data are displayed as mean ± SD by one-way ANOVA. *p < 0.033, **p < 0.0021, ***p < 0.0002, ****p < 0.0001. | ||
A plot of all ΔH values against TΔS is shown in Fig. 6d. This data shows that SeDOCs exhibit enthalpy-entropy compensation, a phenomenon characterized by a linear relationship between ΔH and TΔS.56,57 Enthalpy–entropy compensation is observed within a family of similar compounds, as seen here with our SeDOCs, when an increase in the exothermicity of a noncovalent association leads to a decrease in entropy (or vice versa) that offsets the resulting change in binding free energy.58 This compensation effect has been observed in a number of other systems, including in the binding of neutral molecules to macrocycles in dichloromethane,51,59 the binding of protein-ligand complexes,60 and the gas-phase complexation of iodine with organic donor molecules,61 just to name a few. The molecular basis of this phenomenon is still under debate, with possible explanations invoking solvent structure, a consequence of finite specific heat capacities, multiple weak interactions, or even an evolutionary driven condition for thermodynamic homeostasis. Molecular dynamic simulations are key to providing the connection between these macroscopic extra-thermodynamic correlations and their molecular structure and dynamic determinants.
Conclusions
This body of work demonstrates that SeDOCs encoded with D and T moieties have the capacity to assemble into macromolecular duplexes through the formation of hydrogen bonds between complementary pendant groups. Hybridization of mono-, di-, and trivalent SeDOC 3-mers with complementary pendant groups was first identified using DOSY, as evidenced by a reduction in the diffusion coefficient of the duplex. 1H-NMR titration experiments provided further evidence that these hybrids resulted from hydrogen bonding interactions between D and T units, rather than non-specific interactions with the SeDOC backbone. Thermodynamic binding parameters measured via ITC revealed that Ka, ΔH, and ΔS did not vary with monovalent monomer sequence. Surprisingly, hybridization of monovalent oligomers was entropically driven (TΔS ≫ |ΔH|). Divalent oligomers formed stronger hybrids that were facilitated by the second set of hydrogen bonds that formed between the ligands. There were sequence-dependent differences in the enthalpic and entropic contributions to the binding free energies of divalent hybrids though their logKa remained similar. The Ka of the trivalent SeDOC hybrids, DDD·TTT (logKa = 4.9 M−1), was over an order of magnitude greater than that of the divalent duplexes, indicative of a cooperative binding mechanism. Mixed-mode ITC experiments provided evidence that the duplexes were forming the maximum possible number of available hydrogen bonds. Increasing the number of hydrogen bonding motifs in a duplex eventually led to a decrease in entropy upon hybridization. We attribute this negative ΔS to the increased rigidity of the duplex and the strong collective hydrogen bonds that limit residual motion. Finally, we observe a linear relationship between ΔH and TΔS, commonly characterized as enthalpy–entropy compensation, which minimizes large changes to the resulting free energy term. In conclusion, this study offers a comprehensive exploration of the thermodynamic principles governing SeDOC association through D–T interactions and establishes a solid groundwork for harnessing recognition-encoded SeDOCs as fundamental building blocks for the assembly of higher-order architectures.
Data availability
The data supporting the findings, including NMR and LCMS characterization of SeDOC building blocks and 3-mers, 1H-NMR titration data, and ITC data are available in the ESI.†Author contributions
RKW performed the experiments, analyzed the data, and wrote the manuscript. CAA conceptualized the research, validated the data, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ivan Keresztes and Anthony Condo Jr. at the Cornell NMR facility for assistance with setting up and interpreting the DOSY experiments. This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF under Award Number CHE-1531632. This work was supported financially by the NSF Division of Chemistry under award number CHE-2105834.Notes and references
- K.-H. Kim, D.-K. Ko, Y.-T. Kim, N. H. Kim, J. Paul, S.-Q. Zhang, C. B. Murray, R. Acharya, W. F. DeGrado, Y. H. Kim and G. Grigoryan, Nat. Commun., 2016, 7, 11429 CrossRef CAS.
- B. E. Barnes, T. A. Jenkins, L. M. Stein, R. T. Mathers, M. Wicaksana, M. A. Pasquinelli and D. A. Savin, Biomacromolecules, 2020, 21, 2463–2472 CrossRef CAS PubMed.
- Z. J. Gartner and D. R. Liu, J. Am. Chem. Soc., 2001, 123, 6961–6963 CrossRef CAS PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- D. Han, X. Qi, C. Myhrvold, B. Wang, M. Dai, S. Jiang, M. Bates, Y. Liu, B. An, F. Zhang, H. Yan and P. Yin, Science, 2017, 358, 6369 CrossRef.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS PubMed.
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- N. W. S. Kam, M. O'Connell, J. A. Wisdom and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600–11605 CrossRef CAS PubMed.
- G. von Maltzahn, J.-H. Park, K. Y. Lin, N. Singh, C. Schwöppe, R. Mesters, W. E. Berdel, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Nat. Mater., 2011, 10, 545–552 CrossRef CAS PubMed.
- R. K. Weigel, A. Rangamani and C. A. Alabi, Nat. Rev. Chem, 2023, 7, 875–888 CrossRef CAS.
- C. S. Hartley, E. L. Elliott and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 4512–4513 CrossRef CAS PubMed.
- E. L. Elliott, C. S. Hartley and J. S. Moore, Chem. Commun., 2011, 47, 5028 RSC.
- M. M. Cencer, A. J. Greenlee and J. S. Moore, J. Am. Chem. Soc., 2020, 142, 162–168 CrossRef CAS PubMed.
- T. Wei, J. C. Furgal, J. H. Jung and T. F. Scott, Polym. Chem., 2017, 8, 520–527 RSC.
- M. Hebel, A. Riegger, M. M. Zegota, G. Kizilsavas, J. Gačanin, M. Pieszka, T. Lückerath, J. A. S. Coelho, M. Wagner, P. M. P. Gois, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2019, 141, 14026–14031 CrossRef CAS PubMed.
- M. F. Dunn, T. Wei, R. N. Zuckermann and T. F. Scott, Polym. Chem., 2019, 10, 2337–2343 RSC.
- M. M. Zegota, M. A. Müller, B. Lantzberg, G. Kizilsavas, J. A. S. Coelho, P. Moscariello, M. Martínez-Negro, S. Morsbach, P. M. P. Gois, M. Wagner, D. Y. W. Ng, S. L. Kuan and T. Weil, J. Am. Chem. Soc., 2021, 143, 17047–17058 CrossRef CAS.
- S. C. Leguizamon, A. F. Alqubati and T. F. Scott, Polym. Chem., 2020, 11, 7714–7720 RSC.
- E. A. Archer and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 5074–5083 CrossRef CAS PubMed.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 94–101 RSC.
- G. Iadevaia and C. A. Hunter, Acc. Chem. Res., 2023, 56, 712–727 CrossRef CAS PubMed.
- H. Zeng, X. Yang, A. L. Brown, S. Martinovic, R. D. Smith and B. Gong, Chem. Commun., 2003, 1556 RSC.
- A. Marquis, V. Smith, J. Harrowfield, J. Lehn, H. Herschbach, R. Sanvito, E. Leize-Wagner and A. Van Dorsselaer, Chem.–Eur. J., 2006, 12, 5632–5641 CrossRef CAS PubMed.
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS PubMed.
- R. Kramer, J. M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 CrossRef CAS PubMed.
- V. C. M. Smith and J.-M. Lehn, Chem. Commun., 1996, 2733 RSC.
- B. Hasenknopf and J.-M. Lehn, Helv. Chim. Acta, 1996, 79, 1643–1650 CrossRef CAS.
- H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008–14015 CrossRef CAS PubMed.
- J. Sánchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277–278 CrossRef.
- Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., 2005, 117, 3935–3938 CrossRef.
- G. Iadevaia, A. E. Stross, A. Neumann and C. A. Hunter, Chem. Sci., 2016, 7, 1760–1767 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 5686–5691 RSC.
- D. Núñez-Villanueva, G. Iadevaia, A. E. Stross, M. A. Jinks, J. A. Swain and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6654–6662 CrossRef.
- A. E. Stross, G. Iadevaia, D. Núñez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 12655–12663 CrossRef CAS PubMed.
- G. Iadevaia, D. Núñez-Villanueva, A. E. Stross and C. A. Hunter, Org. Biomol. Chem., 2018, 16, 4183–4190 RSC.
- F. T. Szczypiński and C. A. Hunter, Chem. Sci., 2019, 10, 2444–2451 RSC.
- F. T. Szczypiński, L. Gabrielli and C. A. Hunter, Chem. Sci., 2019, 10, 5397–5404 RSC.
- P. Troselj, P. Bolgar, P. Ballester and C. A. Hunter, J. Am. Chem. Soc., 2021, 143, 8669–8678 CrossRef PubMed.
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371–6380 CrossRef CAS PubMed.
- T. K. Park, J. Schroeder and J. Rebek, J. Am. Chem. Soc., 1991, 113, 5125–5127 CrossRef CAS.
- J. Zhou and P. B. Shevlin, Synth. Commun., 1997, 27, 3591–3597 CrossRef CAS.
- P. Groves, Polym. Chem., 2017, 8, 6700–6708 RSC.
- M. N. C. Zarycz and C. Fonseca Guerra, J. Phys. Chem. Lett., 2018, 9, 3720–3724 CrossRef CAS PubMed.
- W. R. Archer and M. D. Schulz, Soft Matter, 2020, 16, 8760–8774 RSC.
- D. H. Williams and M. S. Westwell, Chem. Soc. Rev., 1998, 27, 57 RSC.
- A. Camara-Campos, C. A. Hunter and S. Tomas, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3034–3038 CrossRef CAS PubMed.
- J. D. Dunitz, Chem. Biol., 1995, 2, 709–712 CrossRef CAS PubMed.
- R. Gopi, N. Ramanathan and K. Sundararajan, J. Mol. Struct., 2015, 1094, 118–129 CrossRef CAS.
- D. H. Williams, N. L. Davies and J. J. Koivisto, J. Am. Chem. Soc., 2004, 126, 14267–14272 CrossRef CAS.
- U. Ryde, Med. Chem. Commun., 2014, 5, 1324–1336 RSC.
- D. M. Ford, J. Am. Chem. Soc., 2005, 127, 16167–16170 CrossRef CAS PubMed.
- F. Peccati and G. Jiménez-Osés, ACS Omega, 2021, 6, 11122–11130 CrossRef CAS PubMed.
- R. M. Izatt, J. S. Bradshaw, K. Pawlak, R. L. Bruening and B. J. Tarbet, Chem. Rev., 1992, 92, 1261–1354 CrossRef CAS.
- C. H. Reynolds and M. K. Holloway, ACS Med. Chem. Lett., 2011, 2, 433–437 CrossRef CAS PubMed.
- M. S. Searle, M. S. Westwell and D. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1995, 141–151 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00242c |
| This journal is © The Royal Society of Chemistry 2024 |