
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the electronic structure of sub-nanometric Ru clusters via Pt single-atom modification for highly efficient electrocatalytic hydrogen evolution†
Yuzhuang
Song
a,
Yaowen
Zhang
b,
Wenya
Gao
a,
Chengcheng
Yu
a,
Jun
Xing
 a,
Kang
Liu
*a and
Dingxuan
Ma
*a
a,
Kang
Liu
*a and
Dingxuan
Ma
*a
aCollege of Chemistry and Molecular Engineering, Key Laboratory of Optic-Electric Sensing and Analytical Chemistry for Life Science, MOE, Shandong Key Laboratory of Biochemical Analysis, Qingdao University of Science and Technology, Qingdao 266042, Shandong, P. R. China. E-mail: madingxuan640@126.com; liukang82@126.com
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 29th May 2024
Abstract
Developing electrocatalysts with high activity toward the hydrogen evolution reaction (HER) is a prerequisite for hydrogen fuel generation and sustainable development, but current Pt-based catalysts usually suffer from high cost and unsatisfactory performance in non-acidic media. In this work, we report an environmentally friendly and pyrolysis-free synthesis strategy to prepare an efficient catalyst, CNT-NPA-PtRu, with Pt single-atom engineered sub-nanometric Ru clusters anchored at phytic acid-modified carbon nanotubes for electrochemical HER at all pH conditions. The electronic structure of active sub-nanometric Ru clusters was optimized, which further enhanced the HER activity. The synthesized CNT-NPA-PtRu catalyst presents superior performance, reaching the current density of 10 mA cm−2 with only 18.3, 18.7 and 15 mV overpotential in alkaline, acidic and neutral electrolyte, respectively. Experimental results and theoretical calculations reveal that the single Pt atom on the sub-nanometric Ru cluster surface could modulate the electronic structure of Ru and subsequently optimize the adsorption of reaction intermediates, thus promoting HER performance. These findings underscore the importance of engineering the electronic structure of sub-nanometric clusters and offer an effective approach for the generation of high-performance electrocatalysts for HER.
Introduction
Electrochemical water splitting to produce clean hydrogen (H2), powered by renewable solar and wind energy, has been considered as a sustainable and environment-friendly strategy, which may replace the carbon-based fossil fuel energy system.1,2 Currently, highly efficient electrocatalysts to accelerate reaction kinetics for the hydrogen evolution reaction (HER) play a crucial role in promoting this significant energy conversion technology.3–5 The most commonly accepted effective catalysts for the HER are Pt-based catalysts, but material scarcity and unsatisfactory performance in non-acidic media limit their large-scale applications.6–8 Therefore, a lot of efforts to lower the amount of Pt or replace Pt with other earth-abundant materials have been made.9–12 Ru-based catalysts have attracted intensive interest as ideal alternatives to Pt-based catalysts due to their intriguing electrocatalytic performance and lower costs.13,14 Especially, the dispersion of active Ru nanoparticles (NPs) or Ru single atoms (SAs) on supports have been widely developed as promising electrocatalysts for HER, such as Ru@Ni-MOF, Ru1CoP/CDs, and Ru SAs@PN.15–20Although extensive studies have been made, most current Ru-based electrocatalysts still have some significant limitations. For example, many Ru-based electrocatalysts rely on high-temperature pyrolysis, which is very energy-intensive and has high equipment requirements. Meanwhile, some indeterminate structures and unpredictable active sites are formed inevitably under high temperature, which seriously hamper the elucidation of their nature during electrocatalysis.21 Besides, the strong interaction between Ru and H reduces the efficiency of H2 desorption, especially slowing the HER kinetics in acid electrolyte.22 Furthermore, due to the high surface energy of Ru atoms and weak interaction between metal Ru and supports, isolated Ru atoms tend to aggregate spontaneously or fall off from the supports, which not only reduces catalyst efficiency but also deactivates the electrocatalysts.23 Therefore, a rational and pyrolysis-free strategy to generate the Ru-based electrocatalysts with high pH-universal activity and stability is highly desirable but challenging.
Obviously, the composition, structure, synthetic method, dimension of active metal species, as well as the supports are very crucial in the construction of the electrocatalysts.24–26 With these factors controlled properly, the activity and stability of the catalysts in the HER process can be effectively improved. Sub-nanometric metal clusters consist of only several to tens of metal atoms, and they possess a size dimension between metal SAs and NPs.27 Due to the confinement of the electrons of metal atoms in molecular dimensions and discrete energy levels, sub-nanometric metal clusters have demonstrated unique electronic structures and chemical features distinctive from SAs and NPs.28–30 Due to such novel properties, enhanced catalytic activities of the sub-nanometric metal clusters have been found in many important reactions.31–34 For instance, He's group developed the sub-nanometric Cu clusters in the size of ∼1.0 nm supported on a defect-rich carbon with abundant micropores. The obtained sub-nanometric Cu clusters possess high activity and selectivity for the electrochemical reduction of CO2 towards CH4.35 Yu and coworkers developed a ligand-protected direct hydrogen reduction method for encapsulating sub-nanometric bimetallic Pt–Zn clusters inside S-1 zeolite for propane dehydrogenation.36 Recently, the sub-nanometric Ru clusters were also investigated as catalysts for the HER, showing enhanced catalytic activity as compared with Ru NP catalysts.37,38 Although there have been encouraging findings, sub-nanometric cluster catalysts for electrochemical HER are still in their infancy, especially the limited performance at all pH conditions, which requires further investigation.
In this paper, we developed an extremely facile and pyrolysis-free self-assembly synthesis strategy to prepare an efficient catalyst with atomic Pt-modified sub-nanometric Ru clusters anchored at phytic acid (PA)-modified carbon nanotubes (CNT) for all-pH HER. The synthesized CNT-NPA-PtRu catalyst presents superior HER performance with very small overpotentials of 18.3, 18.7, and 15 mV at 10 mA cm−2 in 1 M KOH, 0.5 M H2SO4, and 1 M PBS, respectively, which are superior to those of commercial Pt/C as well as most Pt-based and Ru-based catalysts. Meanwhile, the mass activity of the CNT-NPA-PtRu catalyst is about 6.6 and 2.6 times better than that of commercial Pt/C catalysts in 1 M KOH and 0.5 M H2SO4 at the overpotential of 100 mV, and the CNT-NPA-PtRu catalyst also exhibits good stability and regenerative property. Based on a thorough electronic structure testing and analysis and theoretical calculations at the atomic level, the remarkably high HER activity of the CNT-NPA-PtRu catalyst can be explained by the introduction of Pt SAs on the sub-nanometric Ru clusters, which could engineer the electronic structure of Ru clusters and further optimize the binding energies of intermediates. Moreover, the pyrolysis-free synthesis strategy ensured the formation of well-defined active sites, and the synergistic effect due to the strong electronic coupling between PA groups and sub-nanometric metal clusters is not only conducive to optimizing the electronic structure of the sub-nanometric metal clusters but also facilitates the stabilization and dispersion of the metal species, which undoubtedly contributes to the compelling electrocatalytic activity and stability.
Results and discussion
Synthesis and characterization
The procedures to synthesize the CNT-NPA-PtRu catalyst are shown in Scheme 1. First, to obtain CNT-NPA, pristine CNT-NH2 was modified by functional PA via chemical reaction. Subsequently, Pt4+ ions and Ru3+ ions are trapped by CNT-NPA because of the strong bonding effect between PA and metal cations.39,40 Finally, atomic Pt-modified sub-nanometric Ru clusters were uniformly assembled on CNT-NPA under mild conditions and denoted as CNT-NPA-PtRu. | ||
| Scheme 1 Schematic presentation of the synthesis process for CNT-NPA-PtRu. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were performed to investigate the morphology of CNT-NPA-PtRu (Fig. S1 and S2†). As shown in Fig. 1a and b, TEM images reveal that the formed sub-nanometric PtRu clusters are uniformly loaded on CNT-NPA, and no obvious NPs are observed. To further elucidate the detailed structure of the sub-nanometric PtRu clusters, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was conducted. As shown in Fig. 1c, numerous sub-nanometric clusters with an average size of about 1.1 nm are found homogeneously dispersed on the CNT support. Furthermore, HAADF-STEM was employed to examine the embedding of Pt SAs in the sub-nanometric Ru clusters. As the brightness levels of Pt and Ru atoms are different, the atomically dispersed Pt atoms on the Ru sub-nanometric clusters can be clearly observed (Fig. 1d, red circles).41,42 A typical HAADF-STEM image of an individual sub-nanometric PtRu cluster is also shown to highlight the embedded Pt atoms (Fig. 1d inside). In addition, energy-dispersive X-ray spectrum (EDS) elemental mapping of the CNT-NPA-PtRu catalyst further presents that N, O, P, Ru and Pt elements are evenly distributed throughout the structure (Fig. 1e). Because of this high dispersion of metals, no crystallized Pt phase or Ru phase can be observed in the XRD spectrum of CNT-NPA-PtRu (Fig. S3†).
 | ||
| Fig. 1 (a and b) TEM images of CNT-NPA-PtRu; (c) HAADF-STEM image of CNT-NPA-PtRu, and the inset is the corresponding size distribution of sub-nanometric PtRu clusters; (d) atomic-resolution HAADF-STEM image; the inset image derives from an individual sub-nanometric PtRu cluster, and Pt single atoms are marked by red arrows; (e) EDS maps of different elements in CNT-NPA-PtRu. | ||
X-ray photoelectron spectroscopy (XPS) was conducted to analyze the detailed elemental composition and electronic structure. The XPS survey spectrum further confirmed the presence of C, N, O, P, Ru and Pt elements (Fig. S4†). The high-resolution spectrum of Ru 3p shows peaks at 464.2 and 486.5 eV corresponding to 3p3/2 and 3p1/2 of Ru0 (Fig. 2a). Two peaks, located at 466.0 and 488.3 eV, are characteristic of 3p3/2 and 3p1/2 of Rux+. Compared to the contrast sample, CNT-NPA-Ru, the CNT-NPA-PtRu catalyst shows that the proportion of Rux+ decreases with the addition of Pt atoms. The Pt 4f XPS spectrum of CNT-NPA-PtRu in Fig. 2b shows two peaks at 76.01 eV and 72.72 eV, which are assigned to Pt 4f5/2 and 4f7/2, respectively. In contrast, there are no obvious peaks of CNT-NPA-Ru in Pt 4f regions. CNT-NPA-Pt was also prepared as contrast sample. As shown in Fig. S6b,† Pt in CNT-NPA-Pt and CNT-NPA-PtRu show the combined chemical valences of Pt0 and Pt2+; CNT-NPA-PtRu has a higher atomic ratio of Pt2+/Pt0 than CNT-NPA-Pt. These results illustrate that there is a strong electronic interaction between Pt and Ru atoms.
 | ||
| Fig. 2 XPS spectra of as-prepared CNT-NPA-PtRu and CNT-NPA-Ru for (a) Ru 3p and (b) Pt 4f; (c) Pt L3-edge XANES spectra of Pt foil, PtO2, and CNT-NPA-PtRu; (d) EXAFS spectra of Pt foil, PtO2, and CNT-NPA-PtRu; (e) WT-EXAFS contour plots of Pt foil, PtO2, and CNT-NPA-PtRu. | ||
To further investigate the electronic structure and coordination state of Pt SAs on the sub-nanometric Ru clusters, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analysis were performed.43 The XANES spectra of Pt L3-edge for CNT-NPA-PtRu and references are presented in Fig. 2c. The valence states of Pt for different samples are compared based on the intensity of white line (WL) peaks. The valence state of Pt for CNT-NPA-PtRu is between Pt foil and PtO2, implying the hybridization and slight charge transfer between Pt and Ru.44 Besides, the isolated Pt atom structure across the sub-nanometric Ru clusters is confirmed by the absence of a Pt–Pt peak in EXAFS spectra (Fig. 2d). The lower main peak position indicates the formation of a Pt–Ru bond.45 For further corroborant results, EXAFS wavelet transform (WT) was also carried out. As shown in Fig. 2e, compared with the Pt foil and PtO2 references, the WT contour plot of Pt for CNT-NPA-PtRu is distinctly different, which corresponds to the Pt–Ru bond.46 The above results, combined with HAADF-STEM images, demonstrate the successful introduction of atomically dispersed Pt on the sub-nanometric Ru clusters and further confirm the interaction between Pt and Ru atoms. These ultrafine sub-nanometric PtRu clusters may accelerate the HER electrocatalytic process due to the unique electronic structures and extraordinary physical and chemical features distinctive from common SA or NP catalysts.
Electrochemical characterization
The HER catalytic performance of as-prepared CNT-NPA-PtRu was first investigated by a typical three-electrode configuration in 1 M KOH electrolyte. For comparison, CNT-NPA-Ru, CNT-NPA-Pt, and commercial Pt/C (20 wt%) were also measured under same conditions. Linear sweep voltammetric (LSV) curves of the above electrocatalysts are shown in Fig. 3a; CNT-NPA-PtRu exhibits a HER overpotential of 18.3 mV to deliver a current density of 10 mA cm−2, which is lower than Pt/C (33 mV). For single-metal contrast catalysts, CNT-NPA-Ru and CNT-NPA-Pt, the overpotentials at 10 mA cm−2 current density are obviously larger than that of CNT-NPA-PtRu, indicating that the introduction of atomically dispersed Pt can modulate the electronic structure of sub-nanometric Ru clusters and enhance the HER activity. To study the effect of PA on the catalyst, O 1s XPS and comparative experiments were conducted. The O 1s XPS spectra of CNT-NPA and CNT-NPA-PtRu are displayed in Fig. S7.† In the presence of the alloy, the binding energy of O 1s moves toward lower binding energy, which reflects that the metal species have strong interactions with O atoms in PA.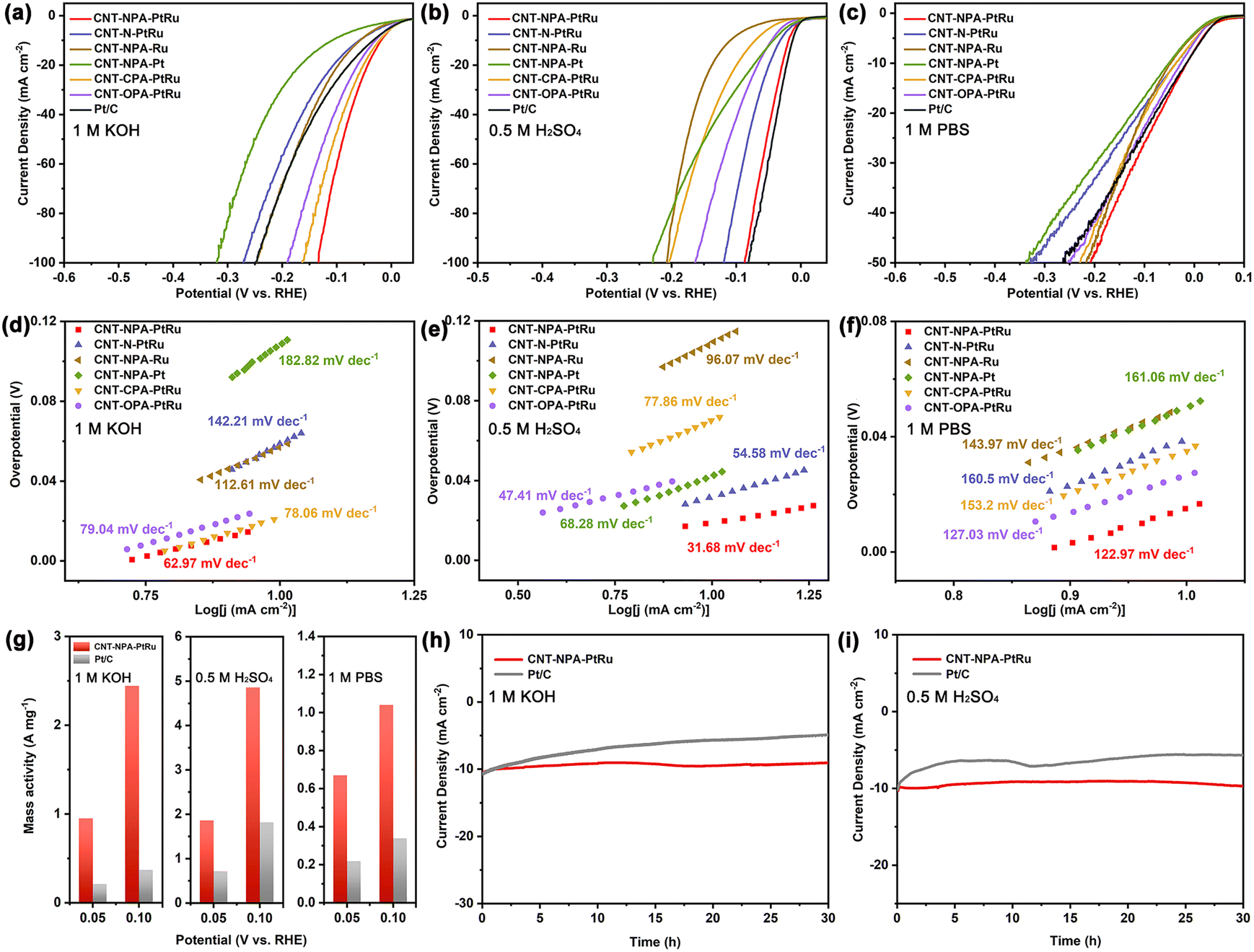 | ||
| Fig. 3 HER LSV curves for CNT-NPA-PtRu and other contrast catalysts in (a) 1 M KOH, (b) 0.5 M H2SO4 and (c) 1 M PBS; Tafel plots for CNT-NPA-PtRu and the other catalysts in (d) 1 M KOH, (e) 0.5 M H2SO4 and (f) 1 M PBS; (g) mass activity of CNT-NPA-PtRu and Pt/C in 1 M KOH, 0.5 M H2SO4 and 1 M PBS; chronopotentiometry curves of CNT-NPA-PtRu and Pt/C in (h) 1 M KOH and (i) 0.5 M H2SO4. | ||
In addition, CNT-N-PtRu was prepared with CNT-NH2 as substrate material directly. Relatively large metal NPs (2–3 nm) are observed in TEM images (Fig. S8†), which is probably due to the higher affinity of PA to metal cations, promoting the formation of more distributed sub-nanometric alloy clusters. The lower HER catalytic activity of CNT-N-PtRu (η10 = 59 mV) indicates that PA modification and sub-nanometric structure have a promoting effect on catalyst performance. Meanwhile, the influence of CNT substrate was also taken into consideration. In addition to CNT-NH2, CNT-COOH and CNT-OH were used as substrate material. As shown in Fig. 3a, the η10 of CNT-CPA-PtRu (21 mV) and CNT-OPA-PtRu (28 mV) were close to that of CNT-NPA-PtRu, and both are better than Pt/C, demonstrating the excellent capability for HER catalysis. However, with the increase of current density, the overpotential gap between CNT-NPA-PtRu and the other two electrocatalysts, CNT-CPA-PtRu and CNT-OPA-PtRu, increases significantly. This may be attributed to the much stronger bonding interaction between amine groups and PA, which contribute to increase the stability of the electrocatalyst, especially at a large current density. The corresponding Tafel slopes in Fig. 3d further indicate the same HER activity sequence.
An ideal electrocatalyst should be able to work in a pH-universal electrolyte to meet the needs of application in different environments. Hence, the HER performance of CNT-NPA-PtRu and contrast catalysts was examined in 0.5 M H2SO4 and 1 M phosphate-buffered saline (PBS) solutions, respectively. As demonstrated in Fig. 3b and c, CNT-NPA-PtRu exhibits outstanding HER activity in 0.5 M H2SO4 (η10 = 18.7 mV) and 1 M PBS (η10 = 15 mV), comparable with Pt/C and superior to the values of the other catalysts. Moreover, CNT-NPA-PtRu displays Tafel slopes of 31.68 and 122.97 mV dec−1 in acidic and neutral media (Fig. 3e and f), which are lower than the slope values of the other control samples, consistent with the results demonstrated on LSV curves.
To illustrate the influence of particle size on the electrocatalytic performance, we synthesized CNT-NPA-PtRu-n1 and CNT-NPA-PtRu-n2 with larger PtRu NPs loaded on CNT-NPA. Relatively large metal NPs can be observed in the TEM images (Fig. S9 and S10†). As displayed in Fig. S11 and S12,† we investigated the water dissociation ability of CNT-NPA-PtRu-n1 and CNT-NPA-PtRu-n2; with increasing particle size of PtRu alloys from sub-nanometric scale to nanoscale, the electrocatalytic performance significantly decreases in alkaline and acidic media. This can be attributed to the distinctive chemical properties and more surface-exposed atoms of the sub-nanometric clusters. In addition, the sub-nanometric clusters with lower average metal–metal coordination number often display a d band center closest to the Fermi level, which results in a stronger adsorption strength towards H2O molecules and H* intermediates, markedly accelerating the HER performance.37,38 We also evaluated the electrochemical impedance spectroscopy (EIS) and electroactive surface area (ECSA) of CNT-NPA-PtRu and different contrast electrocatalysts in all pH solutions (Fig. S13–S16†). Apparently, CNT-NPA-PtRu shows a small charge transfer resistance (Rct) and the largest double-layer capacitance (Cdl) values among all the samples, which suggests the rapid electron transfer and highly exposed ultrafine sub-nanometric PtRu clusters, further confirming its intrinsic electrocatalytic activity and enhanced kinetics for HER. Encouragingly, even when compared with some recently reported Ru-based and Pt-based catalysts, the small overpotential of CNT-NPA-PtRu at 10 mA cm−2 also manifests itself as a remarkable electrocatalyst towards HER over the whole pH range (Table S2†).47–55
To further reveal the intrinsic activity of the catalyst, the mass activity and turnover frequency (TOF) of CNT-NPA-PtRu were calculated. As shown in Fig. 3g, the mass activities of CNT-NPA-PtRu are 0.95 and 2.44 A mg−1 at 50 mV and 100 mV, respectively, about 4.5 and 6.6 times higher than that of Pt/C (0.21 and 0.37 A mg−1) in 1.0 M KOH electrolyte. Meanwhile, we further investigated the mass activity of CNT-NPA-PtRu in acidic and neutral media. CNT-NPA-PtRu still shows a larger mass activity value than Pt/C at the overpotential of 50 mV and 100 mV in 0.5 M H2SO4 and 1 M PBS solutions, indicating the high utilization efficiency of the noble metal. Furthermore, we compared the HER activity of CNT-NPA-PtRu and Pt/C based on TOF values. CNT-NPA-PtRu delivers larger TOF values (1.53, 3.03, and 0.65 s−1) than Pt/C (0.38, 1.84, and 0.34 s−1) at the overpotential of 100 mV in 1 M KOH, 0.5 M H2SO4 and 1 M PBS solutions. These results suggest that CNT-NPA-PtRu has superior catalytic activity in all pH solutions.
The stability of electrocatalysts is very important for their practical application. The long-time durability of CNT-NPA-PtRu was demonstrated by chronopotentiometry testing in alkaline and acidic environments. As shown in Fig. 3h and i, CNT-NPA-PtRu has stable current density during the 30 hours of continuous I–t testing in 1 M KOH and 0.5 M H2SO4 solution, while significantly decreased activity for Pt/C is observed. In addition, the durability of CNT-NPA-PtRu was examined with long-term cyclic voltammetry (CV) measurement in alkaline and acidic media. Fig. S17† shows the LSV of CNT-NPA-PtRu measured before and after 5000 CV cycles, and polarization curves exhibit a very similar activity as the initial test, which indicates the stability of CNT-NPA-PtRu. After the durability test, the sub-nanometric PtRu clusters were still uniformly dispersed on the CNT substrate and retained their original elements and oxidation state (Fig. S18 and S19†).
Mechanism research
To gain theoretical insights and the underlying mechanism into the excellent HER performance of CNT-NPA-PtRu, first-principles density functional theory (DFT) calculations were performed. As shown in Fig. 4a, we established simplified models, CNPA-PtRu and CNPA-Ru, for DFT calculation. It is commonly accepted that catalysts with hydrogen adsorption free energy (ΔGH*) values close to 0 eV have good HER performance.56 Fig. S20† shows H* adsorption theoretical models on different Ru sites of CNPA-Ru and CNPA-PtRu, and the ΔGH* value of these Ru sites were calculated (Table S3†). As shown in Fig. 4b, the ΔGH* value was changed from −0.577 eV to −0.482 eV after the introduction of Pt SA to the sub-nanometric Ru cluster, demonstrating that Pt SA modification can optimize the HER activity of the sub-nanometric Ru cluster. Meanwhile, the ΔGH* value of the Pt site in CNPA-PtRu is closest to 0 eV among these sites, indicating a moderate binding with H*, which is also of great benefit to the promotion of HER. In addition, the projected density of states (PDOS) and d-band center (εd) of Ru sites in two different models were calculated (Fig. 4d). With the assistance of Pt SA, the εd of Ru1 atom in the CNPA-PtRu model reveals higher occupation than CNPA-Ru near the Fermi level, which indicates the strong adsorption capability of reaction intermediates during HER catalysis.57,58 To understand how the electronic structure change of Pt SA affects the sub-nanometric Ru cluster, differential charge density analysis was carried out. The obvious charge density redistribution at the junction of Pt and Ru atoms is observed in Fig. 4c. A clear charge transfer from Pt to Ru, as evidenced by the electron depletion (cyan areas) of Pt and electron accumulation (yellow areas) of Ru, is consistent with XPS and XANES analysis results, indicating the strong interaction between Pt and Ru. All these results proved that the introduction of Pt SA to the sub-nanometric Ru cluster could engineer the electronic structure of the Ru cluster and subsequently optimize the adsorption of reaction intermediates, thus leading to excellent performance for all-pH HER. | ||
| Fig. 4 (a) Chemisorption models of CNPA-PtRu and CNPA-Ru; (b) hydrogen adsorption energy of different models; (c) charge density difference of CNPA-PtRu; (d) calculated PDOS of Ru sites at CNPA-PtRu and CNPA-Ru. | ||
Conclusions
In summary, this work proposed sub-nanometric Ru cluster catalysts with atomic Pt modification through a pyrolysis-free route for electrocatalytic water splitting at all pH conditions. The obtained CNT-NPA-PtRu catalyst with adequate surface-exposed active sites can effectively drive HER throughout the entire pH range, better than the commercial Pt/C catalyst and many other reported Pt-based and Ru-based catalysts. Experimental results and DFT calculations confirmed that the engineering of the electronic structure of sub-nanometric Ru cluster by Pt SA is the key contributor to the superior HER performance. This project is of great significance for developing highly efficient sub-nanometric cluster electrocatalysts for pH-universal water splitting and highlights the ability of engineering the electronic structure at the atomic scale to provide further impetus for sub-nanometric cluster catalysts design.Data availability
All experimental and computational data are available in the ESI.†Author contributions
D. Ma, K. Liu and Y. Song designed the study. Y. Song, C. Yu, W. Gao and Y. Zhang performed the experiments and analysed the data. Y. Song, J. Xing, K. Liu and D. Ma prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province, China (No. ZR2022MB022).Notes and references
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC
.
- S. Sanati, A. Morsali and H. García, Energy Environ. Sci., 2022, 15, 3119–3151 RSC
- C. Walter, P. W. Menezes and M. Driess, Chem. Sci., 2021, 12, 8603–8631 RSC
- Y. Zhu, Q. Lin, Y. Zhong, H. A. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 13, 3361–3392 RSC
- J. Kundu, H. J. Kim, M. Li, H. Huang and S. Choi, Mater. Chem. Front., 2023, 7, 6366–6388 RSC
- J. Li, J. Zhang, J. Zhang, K. Pan, H. Xu, H. Chen, G. Liu, N. Wu, C. Yuan and X. Liu, J. Mater. Chem. A, 2023, 11, 19812–19844 RSC
- S. Khamgaonkar, M. Okasha and V. Maheshwari, Inorg. Chem. Front., 2023, 10, 6812–6848 RSC
- W. Ma, X. Zhang, W. Li, M. Jiao, L. Zhang, R. Ma and Z. Zhou, Nanoscale, 2023, 15, 11759–11776 RSC
- Y. Yao, X. K. Gu, D. He, Z. Li, W. Liu, Q. Xu, T. Yao, Y. Lin, H. J. Wang, C. Zhao, X. Wang, P. Yin, H. Li, X. Hong, S. Wei, W. X. Li, Y. Li and Y. Wu, J. Am. Chem. Soc., 2019, 141, 19964–19968 CrossRef CAS PubMed
- Y. Wang, W. Wu, R. Chen, C. Lin, S. Mu and N. Cheng, Nano Res., 2022, 15, 4958–4964 CrossRef CAS
- J. Chen, G. Qian, H. Zhang, S. Feng, Y. Mo, L. Luo and S. Yin, Adv. Funct. Mater., 2021, 2107597 Search PubMed
- X. Zhang, H. Zhao, C. Li, S. Li, K. Liu and L. Wang, Appl. Catal., B, 2021, 299, 120641 CrossRef CAS
- S. Bae, J. Mahmood, I. Jeon and J. Baek, Nanoscale Horiz., 2020, 5, 43–56 RSC
- J. Zhu, L. Cai, Y. Tu, L. Zhang and W. Zhang, J. Mater. Chem. A, 2022, 10, 15370–15389 RSC
- L. Deng, F. Hu, M. Ma, S. Huang, Y. Xiong, H. Chen, L. Li and S. Peng, Angew. Chem., Int. Ed., 2021, 60, 22276–22282 CrossRef CAS PubMed
- H. Zhang, W. Zhou, X. F. Lu, T. Chen and X. W. Lou, Adv. Energy Mater., 2020, 2000882 CrossRef CAS
- J. Yang, B. Chen, X. Liu, W. Liu, Z. Li, J. Dong, W. Chen, W. Yan, T. Yao, X. Duan, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 9495–9500 CrossRef CAS
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed
- S. Wang, M. Wang, Z. Liu, S. Liu, Y. Chen, M. Li, H. Zhang, Q. Wu, J. Guo, X. Feng, Z. Chen and Y. Pan, ACS Appl. Mater. Interfaces, 2022, 14, 15250–15258 CrossRef CAS PubMed
- H. Zhao, X. Zhang, Y. Zhang, Y. Song, C. Li, K. Liu and D. Ma, J. Mater. Chem. A, 2022, 10, 25272 RSC
- B. Huang, L. Li, X. Tang, W. Zhai, Y. Hong, T. Hu, K. Yuan and Y. Chen, Energy Environ. Sci., 2021, 14, 2789–2808 RSC
- C. Cai, K. Liu, Y. Zhu, P. Li, Q. Wang, B. Liu, S. Chen, H. Li, L. Zhu, H. Li, J. Fu, Y. Chen, E. Pensa, J. Hu, Y. Lu, T. Chan, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202113664 CrossRef CAS
- Z. Li, D. Wang, Y. Wu and Y. Li, Natl. Sci. Rev., 2018, 5, 673–689 CrossRef CAS
- X. Li, C. Deng, Y. Kong, Q. Huo, L. Mi, J. Sun, J. Cao, J. Shao, X. Chen, W. Zhou, M. Lv, X. Chai, H. Yang, Q. Hu and C. He, Angew. Chem., Int. Ed., 2023, e202309732 CAS
- Q. Hu, S. Qi, Q. Huo, Y. Zhao, J. Sun, X. Chen, M. Lv, W. Zhou, C. Feng, X. Chai, H. Yang and C. He, J. Am. Chem. Soc., 2024, 146, 2967–2976 CrossRef CAS PubMed
- C. Li, M. Yuan, Y. Liu, H. Lan, Y. Chen, Z. Li, K. Liu and L. Wang, Chem. Eng. J., 2023, 477, 146988 CrossRef CAS
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC
- Z. Ou, Y. Li, W. Wu, Y. Bi, E. Xing, T. Yu and Q. Chen, Chem. Eng. J., 2022, 430, 132925 CrossRef CAS
- Q. Liu and X. Wang, Chem Catal., 2022, 2, 1257–1266 CrossRef CAS
- Y. Deng, Y. Guo, Z. Jia, J. Liu, J. Guo, X. Cai, C. Dong, M. Wang, C. Li, J. Diao, Z. Jiang, J. Xie, N. Wang, H. Xiao, B. Xu, H. Zhang, H. Liu, J. Li and D. Ma, J. Am. Chem. Soc., 2022, 144, 3535–3542 CrossRef CAS
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Nat. Catal., 2020, 3, 628–638 CrossRef CAS
- X. Wang, L. Zhao, X. Li, Y. Liu, Y. Wang, Q. Yao, J. Xie, Q. Xue, Z. Yan, X. Yuan and W. Xing, Nat. Commun., 2022, 13, 1596 CrossRef CAS PubMed
- S. Rong and X. Wang, Chem. Commun., 2022, 58, 11475–11487 RSC
- Q. Hu, Z. Han, X. Wang, G. Li, Z. Wang, X. Huang, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Angew. Chem., Int. Ed., 2020, 59, 19054–19059 CrossRef CAS PubMed
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS
- Y. Pi, Z. Qiu, Y. Sun, H. Ishii, Y. Liao, X. Zhang, H. Chen and H. Pang, Adv. Sci., 2023, 10, 2206096 CrossRef CAS PubMed
- Q. Liang, Q. Li, L. Xie, H. Zeng, S. Zhou, Y. Huang, M. Yan, X. Zhang, T. Liu, J. Zeng, K. Liang, O. Terasaki, D. Zhao, L. Jiang and B. Kong, ACS Nano, 2022, 16, 7993–8004 CrossRef CAS PubMed
- Z. Pu, I. S. Amiinu, Z. Kou, W. Li and S. Mu, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed
- Y. Chen, J. Li, N. Wang, Y. Zhou, J. Zheng and W. Chu, Chem. Eng. J., 2022, 448, 137611 CrossRef CAS
- A. R. Poerwoprajitno1, L. Gloag, J. Watt, S. Cheong, X. Tan, H. Lei, H. A. Tahini, A. Henson, B. Subhash, N. M. Bedford, B. K. Miller, P. B. O'Mara1, T. M. Benedetti, D. L. Huber, W. Zhang, S. C. Smith, J. J. Gooding, W. Schuhmann and R. D. Tilley, Nat. Catal., 2022, 5, 231–237 CrossRef
- B. Ravel and M. Newxille, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- B. Pang, X. Liu, T. Liu, T. Chen, X. Shen, W. Zhang, S. Wang, T. Liu, D. Liu, T. Ding, Z. Liao, Y. Li, C. Liang and T. Yao, Energy Environ.
Sci., 2022, 15, 102–108 RSC
- Y. Li, W. Pei, J. He, K. Liu, W. Qi, X. Gao, S. Zhou, H. Xie, K. Yin, Y. Gao, J. He, J. Zhao, J. Hu, T. Chan, Z. Li, G. Zhang and M. Liu, ACS Catal., 2019, 9, 10870–10875 CrossRef CAS
- B. Yuan, Z. Yao, C. Qiu, H. Zheng, Y. Yan, Q. Zhang, X. Sun, Y. Gu, X. Zhong and J. Wang, J. Energy Chem., 2020, 51, 312–322 CrossRef
- Y. Wu, X. Li, Y. Wei, Z. Fu, W. Wei, X. Wu, Q. Zhu and Q. Xu, Adv. Mater., 2021, 33, 2006965 CrossRef CAS PubMed
- C. Hong, X. Li, W. Wei, X. Wu and Q. Zhu, Appl. Catal., B, 2021, 294, 120230 CrossRef CAS
- R. Tang, Y. Yang, Y. Zhou and X. Yu, Adv. Funct. Mater., 2023, 2301925 Search PubMed
- Y. Liu, X. Li, Q. Zhang, W. Li, Y. Xie, H. Liu, L. Shang, Z. Liu, Z. Chen, L. Gu, Z. Tang, T. Zhang and S. Lu, Angew. Chem., Int. Ed., 2020, 59, 1718–1726 CrossRef CAS PubMed
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed
- Y. Dang, T. Wu, H. Tan, J. Wang, C. Cui, P. Kerns, W. Zhao, L. Posada, L. Wen and S. L. Suib, Energy Environ. Sci., 2021, 14, 5433–5443 RSC
- X. Ma, H. Xiao, Y. Gao, M. Zhao, L. Zhang, J. Zhang, J. Jia and H. Wu, J. Mater. Chem. A, 2023, 11, 3524–3534 RSC
- R. Samanta, R. Mishra and S. Barman, ACS Sustainable Chem. Eng., 2022, 10, 3704–3715 CrossRef CAS
- Y. Wang, H. Lv, L. Sun, F. Jia and B. Liu, Adv. Energy Mater., 2022, 2201478 CrossRef CAS
- Y. Zhao, X. Wang, G. Cheng and W. Luo, ACS Catal., 2020, 10, 11751–11757 CrossRef CAS
- Y. Xie, C. Chang, F. Luo and Z. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 20081–20088 CrossRef CAS PubMed
- J. Zhang, X. Zhang, C. Shi, G. Xia, H. Li, P. Wang and L. Di, Nanoscale, 2022, 14, 15942–15949 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00182f |
| This journal is © The Royal Society of Chemistry 2024 |