
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fe–Fe bonding in the rhombic Fe4 cores of the Zintl clusters [Fe4E18]4− (E = Sn and Pb)†
Wei-Xing
Chen‡
a,
Zi-Sheng
Li‡
 b,
Harry W. T.
Morgan
b,
Cong-Cong
Shu
a,
Zhong-Ming
Sun
*a and
John E.
McGrady
*b
b,
Harry W. T.
Morgan
b,
Cong-Cong
Shu
a,
Zhong-Ming
Sun
*a and
John E.
McGrady
*b
aState Key Laboratory of Elemento-Organic Chemistry, Tianjin Key Lab for Rare Earth Materials and Applications, School of Materials Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: sunlab@nankai.edu.cn
bDepartment of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QR, UK. E-mail: john.mcgrady@chem.ox.ac.uk
First published on 28th February 2024
Abstract
We report here the synthesis and characterization of two endohedral Zintl-ion clusters, [Fe4Sn18]4− and [Fe4Pb18]4−, which contain rhombic Fe4 cores. The Fe–Fe bond lengths are all below 2.5 Å, distinctly shorter than in the corresponding Cu clusters, indicating the presence of Fe–Fe bonding. Subtle differences in the structure of the Fe4 core between the two clusters suggest that the change in tetrel element causes a change in electronic ground state, with a very short Fe–Fe bond length of 2.328 Å present across the diagonal of the rhombus in the lead case.
1 Introduction
Zintl clusters, and in particular those that contain encapsulated transition metals or f-elements, offer a fascinating insight into chemical bonding on the boundary between molecular systems and nanoparticles/nanoalloys.1–6 Some of the earliest examples, such as icosahedral [M@Pb12]2−,7,8 are relatively simple from an electronic perspective because the endohedral metal has a closed-shell (d10) configuration, and the core-like nature of the d orbitals limits the degree of interaction with the cage. The incorporation of earlier transition metals with fewer d electrons raises the possibility of paramagnetism, as is observed in [Mn@Pb12]3− ((ref. 9) (a triplet)) and [Fe@Ge10]3− ((ref. 10) (a doublet)).11,12 The relative destabilisation of the d orbitals also leads to stronger interactions with the cage (‘back-bonding’) which can, ultimately, drive structural changes, from the deltahedra that are characteristic of the electron deficient regime, to 3-connected ‘fullerene-like’ structures such as that of [Ru@Ge12]3− that are the signature of more electron-rich cages.13–16The extension of these principles to larger clusters containing two or more open-shell transition metal ions opens up a further range of possibilities. If the encapsulated metal ions are dilute, cooperative magnetic phenomena (ferro- and anti-ferromagnetism) may emerge, while a closer approach brings covalent metal–metal bonding into play. In the latter scenario, metal–metal and metal–cage bonding may be complementary: overlap between the metals will destabilise antibonding orbitals, potentially to the extent that electrons may be driven onto the cage. Opportunities to explore this landscape of possibilities have, up until now, been restricted by the relatively small number of well-characterized examples containing two or more endohedral metals. Amongst the very few Zintl-ion clusters with direct covalent metal–metal bonds, the Fe–Fe bond length in [Fe2Ge16]4− (ref. 17) (Fig. 1(a)) is 2.636(3) Å whilst in [Fe2Sn4Bi8]3−,18 it is remarkably short, at 2.396(4) Å. Very recently, we have also reported the tri-iron cluster [Fe3Sn18]4− (Fig. 1(b)) which contains a linear array of Fe centres, again with a very short Fe–Fe bond length of 2.4300(9) Å.19 These Zintl clusters are members of a wider family of iron clusters with direct covalent Fe–Fe bonds, which includes the Fe1Fe1 paddlewheel complex with ultra-short Fe–Fe bonds (2.127 Å), linear Fe3 chains (Fig. 1(b))20,21 and triangular Fe3 (ref. 22 and 23) and octahedral Fe6 units.24–27 Ohki has also reported a series of hydride bridged Fe4 and Fe6 clusters based on a rhombic Fe4 core, with Fe–Fe distances between 2.5 and 2.7 Å (Fig. 1(e)). Neidig and co-workers have also recently characterized the structure of the previously elusive ‘Kochi cluster’, a [Fe8Me12]− (Fig. 1(f)), a distorted cube with a doublet ground state and averaged Fe–Fe bonds lengths of 2.43 Å.28,29 Paramagnetism is ubiquitous in the clusters shown in Fig. 1, and their electronic structure has typically been interpreted in terms of a delocalized manifold of Fe-based orbitals, occupied to maximise the multiplicity (‘Hund's coupling’).30
 | ||
| Fig. 1 Structures of selected Fe–Fe bonded clusters in the literature, including (a) Fe2,17,32–34 (b) linear Fe3,19–21 (c) triangular Fe3,22,23 (d) octahedral Fe6,24–26 (e) rhombic Fe4,35 (f) distorted cubic Fe8.28 | ||
In a previous paper we have reported the structures of the copper-containing clusters, [Cu4Sn18]4− and [Cu4Pb18]4−.31 We now extend that work to the earlier transition metals by reporting the properties of two new Zintl clusters, [Fe4Sn18]4− and [Fe4Pb18]4−, both of which contain rhombic Fe4 cores, isostructural with the Cu analogues. The Fe–Fe separations (2.328 Å to 2.498 Å) are, however, shorter than those in [Cu4E18]4− (2.52–2.55 Å), consistent with the presence of Fe–Fe bonding. The Fe4 cores are superficially very similar, but subtle differences suggest that the nature of the Fe–Fe bonding may be different in the Sn18 and Pb18 clusters. This observation motivates a detailed survey of the electronic structure using density functional theory, where we draw on comparisons both between the Sn and Pb species and also between the Fe and Cu analogues.
2 Results and discussion
2.1 Synthesis and properties of [Fe4E18]4−, E = Sn, Pb
[K(2.2.2-crypt)]4[Fe4Sn18]·4Py (1) and [K(2.2.2-crypt)]4[Fe4Pb18]·4Py (2), were obtained from the reactions of ethylenediamine solutions of K4E9 with ferrocene in the presence of 2.2.2-crypt (further details are given in the experimental section). The reaction protocol differs from that used previously19 to synthesise the [Fe3Sn18]4− cluster only in the source of Fe: ferrocene here but [K(thf)Fe(OtBu)3]2 (thf = tetrahydrofuran). The two clusters, [Fe3Sn18]4− and [Fe4Sn18]4−, appear not to interconvert, even in the presence of excess Fe. 1 crystallizes in the monoclinic space group P21/n and contains a single anionic stannaspherene unit [Fe4Sn18]4− along with four [K(2.2.2-crypt)]+ cations and four pyridine molecules in the asymmetric unit (Fig. 2(a)). The [Fe4Pb18]4− unit 2 is isostructural with 1 (Fig. 2(b)), both having approximate D2h symmetry, as do their copper analogues, [Cu4Sn18]4− and [Cu4Pb18]4−.31 Two of the Fe atoms (Fe1 and Fe1′) are encapsulated in 8-vertex E8 polyhedra while Fe2 and Fe2′ sit in approximately trigonal prismatic sites either side of the waist of the cluster. Averaged bond lengths for all four clusters are collected together in Table 1 for comparison. The gross features of the E18 cage are largely unaffected by the change from Cu to Fe, with E–E bond lengths deviating by less than 0.05 Å in all cases. There are, however, subtle changes in M–M distances that reflect the emergence of metal–metal bonding in the iron clusters. As we have noted above, the d10 configuration of the Cu+ ions limits direct Cu–Cu bonding to the cuprophilic type, and so the short Cu1–Cu2 distances of ∼2.53 Å must reflect, at least to some extent, the constraints imposed by the E18 framework. Irrespective of whether the main-group element is Sn or Pb, the Fe1–Fe2 bond lengths are shorter than the Cu1–Cu2 analogues by approximately 0.1 Å. Differences in the M2–M2′ distances are, in contrast, rather more variable: in the Sn clusters, the Fe2–Fe2′ is shorter than Cu2–Cu2′ by 0.135 Å, but this difference increases to 0.233 Å for the Pb pair. Indeed the Fe2–Fe2′ distance of 2.328 Å in [Fe4Pb18]4− is comparable to the shortest Fe–Fe bonds shown in Fig. 1. The correlation of bond length and bond strength is not always straightforward in clusters with bridging ligands, but we note that for a given oxidation state the radius of Fe is intrinsically larger than that of Cu, so the shorter Fe–Fe distances within a constant E18 framework are a strong a priori indication of direct Fe–Fe bonding. Moreover, the wider variation in the M2–M2′ bond lengths is probably a reflection of the fact that these atoms are not fully encapsulated by the cluster, and are therefore more free to move in response to subtle changes in Fe–Fe bonding. We explore the origins of these structural differences as part of a detailed analysis of electronic structure presented in the following sections.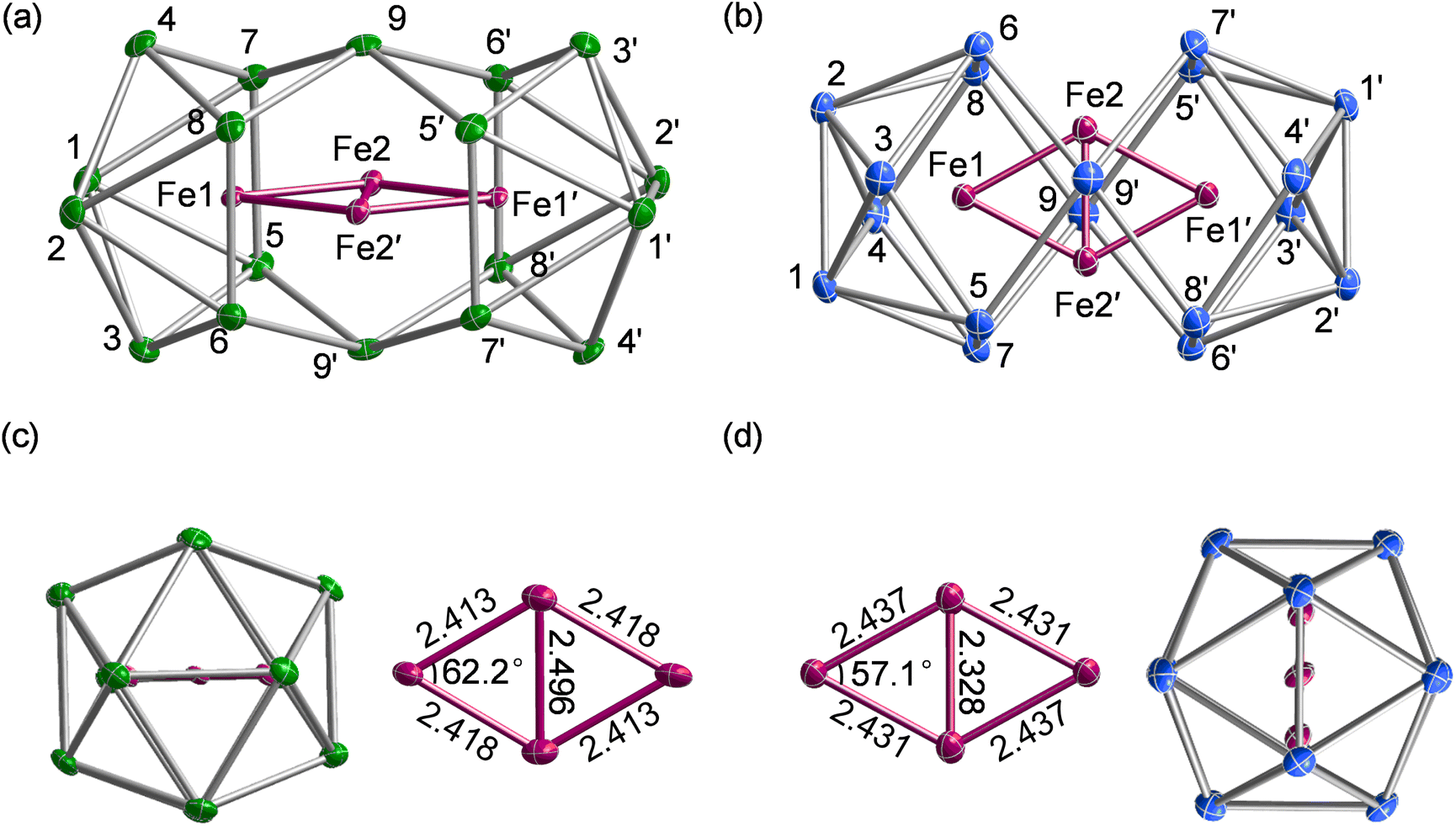 | ||
| Fig. 2 Molecular structures of the anionic components, [Fe4Sn18]4− and [Fe4Pb18]4−. The two anions are isostructural: [Fe4Sn18]4− is viewed along the x axis in (a) while [Fe4Pb18]4− is viewed along z in (b). Fe–Fe bond lengths in the Fe4 rhombus are also shown in (c) and (d). | ||
| E rel/eV | M1–M2 | M2–M2′ | M1–E3 | M2–E9 | E2–E6 | E5–E9′ | E7–E8′ | Ref | ||
|---|---|---|---|---|---|---|---|---|---|---|
| [Fe4Sn18]4− | X-ray (100 K) | 2.413 | 2.496 | 2.828 | 2.739 | 3.142 | 3.225 | 4.898 | This work | |
| DFT (9Ag) | 0.59 | 2.41 | 2.47 | 2.90 | 2.75 | 3.17 | 3.18 | 4.89 | ||
| DFT (11B1g) | 0.0 | 2.42 | 2.31 | 2.90 | 2.79 | 3.20 | 3.19 | 4.88 | ||
| DFT (11B3g) | 0.05 | 2.41 | 2.52 | 2.88 | 2.75 | 3.17 | 3.24 | 4.86 | ||
| DFT (13B2g) | 0.25 | 2.43 | 2.63 | 2.94 | 2.75 | 3.14 | 3.27 | 5.06 | ||
| [Fe4Pb18]4− | X-ray (100 K) | 2.437 | 2.328 | 2.974 | 2.892 | 3.247 | 3.287 | 4.941 | This work | |
| DFT (9B1u) | 0.79 | 2.41 | 2.46 | 2.99 | 2.90 | 3.32 | 3.37 | 4.99 | ||
| DFT (11B1g) | 0.0 | 2.44 | 2.28 | 3.03 | 2.90 | 3.35 | 3.32 | 4.99 | ||
| DFT (11B3g) | 0.18 | 2.43 | 2.48 | 3.00 | 2.86 | 3.32 | 3.36 | 4.98 | ||
| DFT (13B3g) | 0.18 | 2.46 | 2.35 | 3.01 | 2.86 | 3.34 | 3.37 | 5.05 | ||
| [Cu4Sn18]4− | X-ray (100 K) | 2.525 | 2.631 | 2.842 | 2.760 | 3.108 | 3.219 | 4.915 | 31 | |
| DFT (1Ag) | 2.52 | 2.58 | 2.86 | 2.76 | 3.14 | 3.24 | 4.91 | |||
| [Cu4Pb18]4− | X-ray (100 K) | 2.547 | 2.561 | 2.935 | 2.825 | 3.224 | 3.299 | 5.012 | 31 | |
| DFT (1Ag) | 2.55 | 2.52 | 2.99 | 2.88 | 3.30 | 3.35 | 5.02 | |||
Electrospray ionization mass spectra (ESI-MS) of freshly prepared samples of 1 and 2, measured in negative-ion mode, are shown in Fig. S9–15.† For 1, the spectrum shows an intense feature due to the dianion [Fe4Sn18]2−, as well as smaller features due to the monoanion, [Fe4Sn18]− and the dianion paired with a single [K(2.2.2-crypt)]+ cation, [K(2.2.2-crypt)Fe4Sn18]−. The corresponding spectrum for 2, in contrast, shows a single dominant peak due to the decomposition product [FePb12]−, most likely an endohedrally encapsulated icosahedron, similar in structure to the known [Mn@Pb12]3− cluster. The only possible trace of the intact cluster is a very small peak, just above the baseline, centreed at m/z = 1977: it is clear that the parent cluster does not survive in any significant quantities under ESI-MS conditions. The magnetic susceptibility of 1 was measured between 2 and 300 K using a superconducting quantum interference device (SQUID), Fig. 3. The measured magnetic moments (μeff) decreases with decreasing temperature from a value of 9.37 μB (χMT = 10.97 cm3 K mol−1) at 300 K to ∼9 μB at 100 K, before dropping to 8.6 μB at 10 K, and, finally, to much lower values below this temperature. The drop off below 10 K may be the result of zero-field splitting and/or weak antiferromagnetic coupling between cluster units. Limiting spin-only (g = 2) values of μeff for S = 4, S = 5 and S = 6 are 8.94 μB, 10.95 μB and 12.96 μB, respectively, and the absence of a plateau at 300 K suggests that the ground spin state is not isolated. Despite multiple attempts on freshly prepared samples, we were not able to make reproducible magnetic measurements on the lead analogue, 2: this probably reflect the difficulties in separating crystals of 2 from other side products of the reaction while preparing the sample for magnetic measurements and/or rapid decomposition of the sample, as is apparent in the ESI-MS experiment.
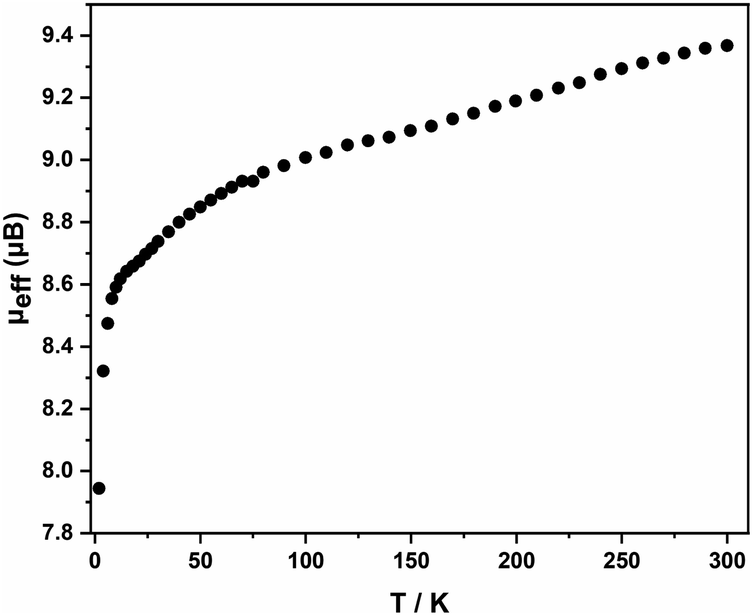 | ||
| Fig. 3 Effective magnetic moment, μeff, as a function of temperature for 1. | ||
2.2 Electronic structure
The electronic structure of the two Cu clusters, [Cu4Sn18]4− and [Cu4Pb18]4−, was the subject of a previous study31 where we concluded that the ground states were singlets, with Cu+ ions in a d10 configuration and no direct Cu–Cu interactions. In contrast, the open-shell Fe centres generate several closely-spaced electronic states that differ both in multiplicity and spatial symmetry, and the energies of the lowest-lying states with 2S + 1 = 9, 11 and 13, calculated with the M06-L functional, are summarized in Table 1. Amongst these, the two lowest share a common spin, S = 5, but have different spatial symmetries, 11B1g and 11B3g: the lowest-lying state with S = 4 lies more than 0.5 eV higher in energy. The measured magnetic moment of 9.37 μB for [Fe4Sn18]4− at 300 K lies considerably below the spin-only value for S = 5 (10.95 μB), and in fact is more consistent with a ground-state with S = 4. The apparent discrepancy between computed and experimental values may be a consequence of the well-known sensitivity of ground-state multiplicity to choice of density functional or, alternatively, may indicate the presence of diamagnetic impurities in the sample that serve to depress the measured moment. Of the two states with 2S + 1 = 11, 11B1g is the most stable for both [Fe4Pb18]4− and [Fe4Sn18]4−, although the energetic difference is too small to allow a confident assignment of the ground state, particularly in the Sn cluster where the two differ by only 0.05 eV. However, a comparison of optimized and measured bond lengths offers important clues to the identity of the electronic ground states. The optimised Fe2–Fe2′ bond is short in the 11B1g state (2.31 Å and 2.28 Å for Sn and Pb, respectively), while the Fe1–Fe2 bond is relatively long (2.42 Å and 2.44 Å for Sn and Pb, respectively). In the 11B3g state, the opposite pattern is found: the Fe2–Fe2′ bond is long (2.52 Å for Sn and 2.48 Å for Pb) while the Fe1–Fe2 bond is somewhat shorter (2.41 Å for Sn and 2.43 Å for Pb). When compared to experiment, it is clear that the 11B3g state offers a much better match to the X-ray structure of [Fe4Sn18]4− (Fe1–Fe2 = 2.413 Å, Fe2–Fe2′ = 2.496 Å) while the 11B1g state offers a better match to the X-ray structure of [Fe4Pb18]4− (Fe1–Fe2 = 2.437 Å, Fe2–Fe2′ = 2.328 Å). On that basis, and despite the fact that the 11B3g state is 0.05 eV less stable than 11B1g, we believe that former is the best candidate for the electronic ground state of the Sn cluster.A schematic representation of the Kohn–Sham orbitals for [Fe4Sn18]4− and [Fe4Pb18]4− in their respective 11B3g and 11B1g states is shown in Fig. 4, and a full analysis of the orbitals, projected densities of states (PDOS), and overlap-projected densities of states (OPDOS) is presented in the ESI.† The interpretation of the electronic structure is a complex challenge due to the open-shell character, and a consideration from a symmetry-based perspective is a useful starting point. The rhombic Fe44+ unit has a total of 4 × 5 = 20 linear combinations of Fe 3d orbitals, which can be separated into in-plane (ag, b1g, b2u, b3u symmetries) and out-of-plane (au, b1u, b2g and b3g) subsets. The in-plane set support Fe–Fe interactions of local σ and πip symmetry, while the out-of-plane set have local Fe–Fe πop and δ symmetry.§ The formal oxidation state, and therefore the number of electrons occupying the Fe 3d manifold, is not straightfoward due to the multiple redox states available to Fe. However, the structures of the E18 units in 1 and 2 and also the Sn/Pb densities of states (ESI, Fig. S22†) are striking similar to those in the [Cu4E18]4− cases, where the charge state of the E18 unit was assigned as 8-.31 The relationship between structure and charge in the family of 18-vertex tetrel clusters has been discussed by us and others in the recent literature,19,36–40 and we provide further justification for the assignment of an 8- charge state in the ESI.† Assuming the same 8- charge for the E18 unit in [Fe4E18]4−, we then have a net 4+ charge for the Fe4 unit and hence 28 valence electrons in the Fe 3d manifold. Distributing these 28 valence electrons across the 20 orbitals generates a maximum possible spin of S = 6 (the 13B2g state identified in Table 1). In the ground states of the two clusters with 2S + 1 = 11, (11B1g and 11B3g) the highest of these 20 linear combinations, 7b1g, is vacant in both spin-α and spin-β manifolds, leaving 9 doubly-occupied and 10 singly-occupied orbitals. The doubly-occupied levels are largely Fe1–Fe2 and Fe2–Fe2′ bonding while the singly-occupied levels are largely antibonding, giving rise to net Fe–Fe bonding, in contrast to the Cu analogues where all bonding and antibonding combinations are filled (see the discussion of the PDOS and OPDOS in the ESI† for more details). We can identify three distinct bonding/antibonding orbital pairs that make substantial contributions to the Fe2–Fe2′ bonding in both the 11B1g and 11B3g states: the 8ag28b2u1 configuration (Fe2–Fe2′σ), the 4b1u25b3g1 configuration (πop) and the 7b3u27b1g0 configuration (πip). The key difference between the 11B1g and 11B3g states lies in the reversal of the 6b1g and 4b3g orbitals (highlighted in red and blue, respectively, in Fig. 4) the first of which has significant Fe2–Fe2′ πip* character while the second is localised largely on the Fe1 centres with little amplitude on the central Fe2–Fe2′ unit. The in-plane component of the Fe2–Fe2′ bond is therefore stronger in the 11B1g state, leading to compressed bond in [Fe4Pb18]4−. The differences between Cu and Fe, and between [Fe4Sn18]4− and [Fe4Pb18]4−, are captured in the Mayer bond orders (MBO),41 delocalization indices (DI)42 and bond critical point ellipticities (ε) shown in the ESI, Table S6.† The Mayer bond orders of 0.51 and 0.91 for the Fe2–Fe2′ bonds of [Fe4Sn18]4− and [Fe4Pb18]4− in their respective ground states are a clear indication of the stronger bond in the latter while the Fe1–Fe2 bonds show the opposite trend (0.85 and 0.69). The corresponding values are, on average, less than 0.2 for the Cu clusters, where formal Cu–Cu bond orders are zero. The ellipticity at the critical point, ε, provides a measure of the balance between σ, πip and πop character: values close to 0 are indicative of a cylindrically symmetric environment while larger values indicate an anisotropic π component to the bond. The values of ε ∼0.2 for the Cu clusters again establish a useful reference point. The ellipticity of 0.46 the Fe2–Fe2′ bond of [Fe4Pb18]4− in its ground state is much lower than the value of 2.11 for the corresponding bond in the 11B3g ground state of [Fe4Sn18]4−, the high value in the latter case reflecting the substantial contribution of the πop bonding mediated by the 4b1u/5b3g pair. In the 11B1g ground state of [Fe4Pb18]4−, this out-of-plane π component is supplemented by an additional πip component resulting from the removal of one electron from the Fe–Fe π antibonding 4b3g orbital. In summary, our analysis of the electronic structure reveals that (a) There is significant Fe–Fe covalent bonding in both 1 and 2, in contrast to the situation in the Cu analogues where bonding is absent and (b) [Fe4Sn18]4− and [Fe4Pb18]4− have different electronic configurations, leading to a marked change in the structure of the Fe4 rhombus with a significantly contracted Fe2–Fe2′ bond in the latter.
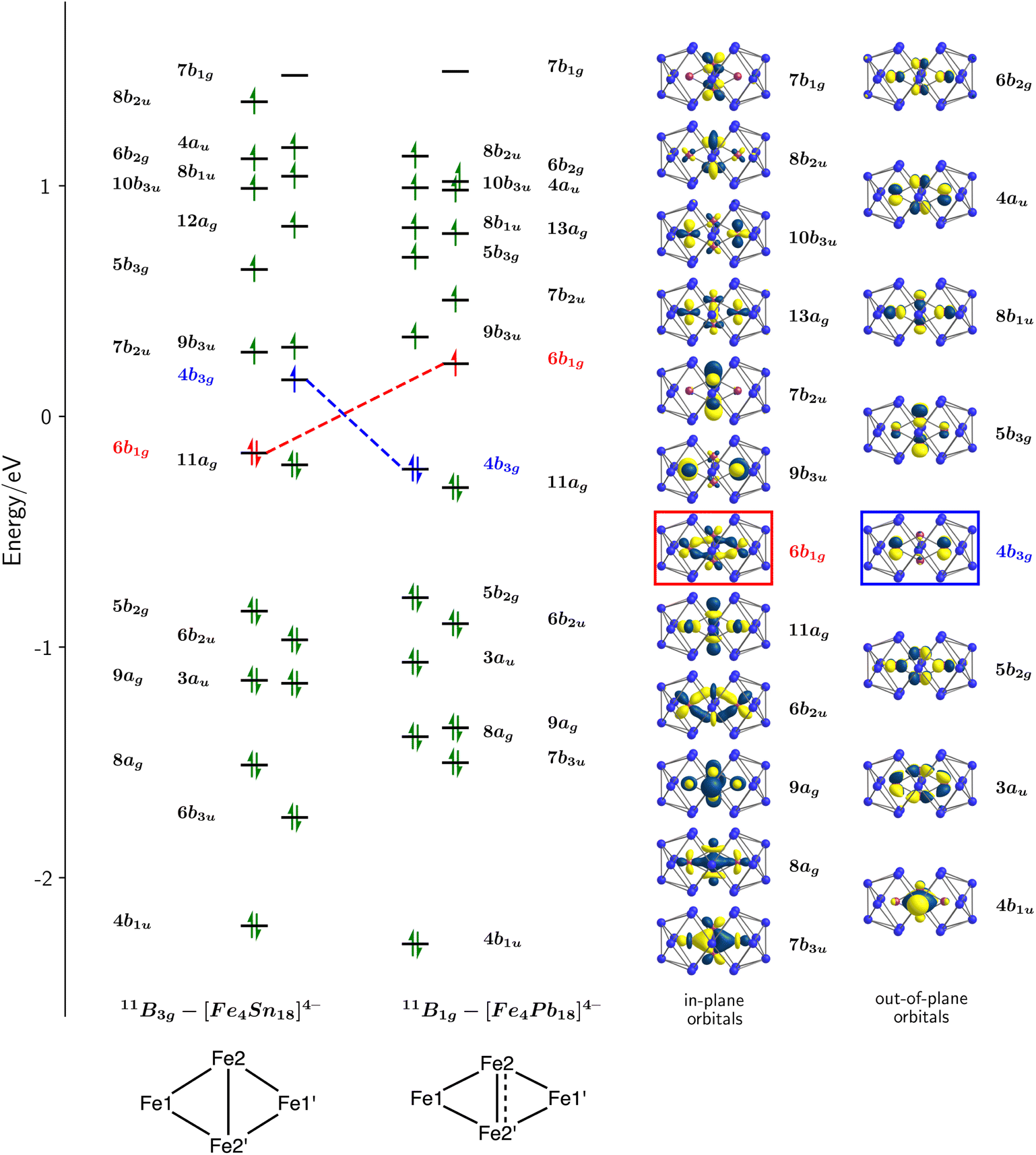 | ||
| Fig. 4 Kohn–Sham molecular orbitals for the 11B3g of [Fe4Sn18]4− and the 11B1g state of [Fe4Pb18]4−. The eigenvalues correspond to the spin-β manifold in both cases, and they are shifted such that E = 0 is defined as the mid-point between the eigenvalues of HOMO and LUMO. The orbitals highlighted in red and blue are those whose occupations differ in the 11B3g and 11B1g states. The isosurfaces shown correspond to the spin-β set of [Fe4Pb18]4−. | ||
3 Summary and conclusions
In this paper we have reported the synthesis and characterization of two new Fe-based Zintl clusters, [Fe4Sn18]4−, [Fe4Pb18]4−, both of which have rhombic Fe4 cores. The clusters are isostructutal with the previously-reported Cu analogues, but the Fe–Fe bonds, all of which are below 2.5 Å, are substantially shorter than the corresponding Cu–Cu distances, indicating the presence of Fe–Fe bonding. There are also significant differences between the Fe4 cores in the Sn and Pb clusters, with the Fe2–Fe2′ bond across the diagonal of the rhombus being notably shorter in the latter (2.328 Å vs. 2.498 Å). Magnetic susceptibility measurements for [Fe4Sn18]4− indicate a high-spin ground state, with limiting values of μeff consistent with either S = 5 or S = 6. A survey of the potential energy surface using density functional theory identifies two almost degenerate states with S = 5 as the most stable (M06-L functional), with very different Fe2–Fe2′ bond lengths that map onto the differences seen in the crystallography. On that basis we believe that the two clusters adopt ground states with the same multiplicity but different spatial symmetry, with Fe–Fe π bonding more developed in the [Fe4Pb18]4− case.4 Materials and methods
4.1 Materials and reagents
All manipulations and reactions were performed under a nitrogen atmosphere using standard Schlenk or glovebox techniques. Ethylenediamine (en) (Aldrich, 99%) and DMF (Aldrich, 99.8%) were freshly distilled by CaH2 prior to use, and stored in N2 prior to use. Tol (Aldrich, 99.8%) was distilled from sodium/benzophenone under nitrogen and stored under nitrogen. 2.2.2-crypt (4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo (8.8.8) hexacosane, purchased from Sigma-Aldirich, 98%) was dried in vacuum for one day prior to use. FeCp2 was purchased from Energy Chemical (China). K4E9 was synthesized by heating a stoichiometric mixture of the elements (K: +99%, Sn: 99.99% and Pb: 99.99% all from Aladdin) at 850 °C for 36 h in a niobium tube.4.2 Synthesis of 1 and 2
4.3 Experimental analysis
4.4 Computational details
All calculations are performed using density functional theory as implemented in the ADF 2021.104 package.43 The local functional of Minnesota 06 family (M06-L) functionals44,45 was used and triple-zeta basis sets included with two polarization functions are used for all atoms.46 All electrons are treated as valence in the calculations. Relativistic effect was considered with The Zeroth-Order Regular Approximation (ZORA).47 The numerical quality was set to ‘verygood’.48 A Conductor-like Screening Model (COSMO) with dielectric constant of 78.39 was used to simulate the confining environment of the ionic lattice.49 Optimized structures were confirmed to be minima through the absence of imaginary frequencies.50 The sensitivity of the results to choice of functional was also explored by repeating the calculations using the PBE and PBE0 functionals.51,52Data availability
Yes, the original data that supports the finding in the article are available in the ESI† and CCDC databases.Author contributions
W.-X. C. and C.-C. S. performed the synthesis and characterisation, Z. L. and H. W. T. M. performed the computational analysis. Z.-M. S. and J. E. M. conceived the project and supervised the experimental and computational aspects of the research, respectively. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 92161102 and 22371140), the Natural Science Foundation of Tianjin City (no. 21JCZXJC00140) and the PhD Candidate Research Innovation Fund of NKU School of Materials Science and Engineering, and 111 project (B18030) from China (MOE).Notes and references
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- B. Weinert and S. Dehnen, in Binary and Ternary Intermetalloid Clusters, ed. S. Dehnen, Springer International Publishing, Cham, 2017, pp. 99–134 Search PubMed.
- B. Oelkers, M. V. Butovskii and R. Kempe, Chem.–An Euro. J., 2012, 18, 13566–13579 CrossRef CAS PubMed.
- N. Lichtenberger, R. J. Wilson, A. R. Eulenstein, W. Massa, R. Clérac, F. Weigend and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 9033–9036 CrossRef CAS PubMed.
- X. Min, I. A. Popov, F.-X. Pan, L.-J. Li, E. Matito, Z.-M. Sun, L.-S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2016, 55, 5531–5535 CrossRef CAS PubMed.
- L.-F. Cui, X. Huang, L.-M. Wang, J. Li and L.-S. Wang, Angew. Chem., Int. Ed., 2007, 46, 742–745 CrossRef CAS PubMed.
- E. N. Esenturk, J. Fettinger and B. Eichhorn, J. Am. Chem. Soc., 2006, 128, 9178–9186 CrossRef CAS PubMed.
- L.-F. Cui and L.-S. Wang, Int. Rev. Phys. Chem., 2008, 27, 139–166 Search PubMed.
- B. Zhou, T. Krämer, A. L. Thompson, J. E. McGrady and J. M. Goicoechea, Inorg. Chem., 2011, 50, 8028–8037 CrossRef CAS PubMed.
- B. Zhou, M. S. Denning, D. L. Kays and J. M. Goicoechea, J. Am. Chem. Soc., 2009, 131, 2802–2803 CrossRef CAS PubMed.
- X. Jin and J. E. McGrady, Adv. Inorg. Chem., 2019, 73, 265–304 CrossRef CAS.
- K. Mayer, J. Weßing, T. F. Fässler and R. A. Fischer, Angew. Chem., Int. Ed. Engl., 2018, 57, 14372–14393 CrossRef CAS PubMed.
- G. Espinoza-Quintero, J. C. A. Duckworth, W. K. Myers, J. E. McGrady and J. M. Goicoechea, J. Am. Chem. Soc., 2014, 136, 1210–1213 CrossRef CAS PubMed.
- J. E. McGrady, F. Weigend and S. Dehnen, Chem. Soc. Rev., 2022, 51, 628–649 RSC.
- J. M. Goicoechea and J. E. McGrady, Dalton Trans., 2015, 44, 6755–6766 RSC.
- X. Jin, V. Arcisauskaite and J. E. McGrady, Dalton Trans., 2017, 46, 11636–11644 RSC.
- H. W. T. Morgan, K.-S. Csizi, Y.-S. Huang, Z.-M. Sun and J. E. McGrady, J. Phys. Chem. A, 2021, 125, 4578–4588 CrossRef CAS PubMed.
- Y.-N. Yang, Z.-S. Li, S. Mondal, L. Qiao, C.-C. Wang, W.-J. Tian, Z.-M. Sun and J. E. McGrady, Chin. Chem. Lett., 2023, 109048 CrossRef.
- Z.-S. Li, W.-X. Chen, H. W. T. Morgan, C.-C. Shu, J. E. McGrady and Z.-M. Sun, Chem. Sci., 2024, 15, 1018–1026 RSC.
- A. Srinivasan, R. A. Musgrave, M. Rouzières, R. Clérac, J. E. McGrady and E. A. Hillard, Chem. Commun., 2021, 57, 13357–13360 RSC.
- G. L. Guillet, K. Y. Arpin, A. M. Boltin, J. B. Gordon, J. A. Rave and P. C. Hillesheim, Inorg. Chem., 2020, 59, 11238–11243 CrossRef CAS PubMed.
- E. V. Eames, T. D. Harris and T. A. Betley, Chem. Sci., 2012, 3, 407–415 RSC.
- R. Hernández Sánchez, A. K. Bartholomew, T. M. Powers, G. Ménard and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 2235–2243 CrossRef PubMed.
- Q. Zhao, T. D. Harris and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 8293–8306 CrossRef CAS PubMed.
- R. Hernández Sánchez and T. A. Betley, J. Am. Chem. Soc., 2015, 137, 13949–13956 CrossRef PubMed.
- R. H. Sánchez and T. A. Betley, J. Am. Chem. Soc., 2018, 140, 16792–16806 CrossRef PubMed.
- J. Nehrkorn, S. M. Greer, B. J. Malbrecht, K. J. Anderton, A. Aliabadi, J. Krzystek, A. Schnegg, K. Holldack, C. Herrmann, T. A. Betley, S. Stoll and S. Hill, Inorg. Chem., 2021, 60, 4610–4622 CrossRef CAS PubMed.
- S. B. I. Muñoz, S. L. Daifuku, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2016, 138, 7492–7495 CrossRef PubMed.
- C. L. Kwan and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 4903–4912 CrossRef CAS.
- J. A. Chipman and J. F. Berry, Chem. Rev., 2020, 120, 2409–2447 CrossRef CAS PubMed.
- L. Qiao, C. Zhang, C.-C. Shu, H. W. T. Morgan, J. E. McGrady and Z.-M. Sun, J. Am. Chem. Soc., 2020, 142, 13288–13293 CrossRef CAS PubMed.
- C. Hess, T. Weyhermüller, E. Bill and K. Wieghardt, Angew. Chem., Int. Ed. Engl., 2009, 48, 3703–3706 CrossRef CAS PubMed.
- T. Nguyen, W. Merrill, C. Ni, H. Lei, J. Fettinger, B. Ellis, G. Long, M. Brynda and P. Power, Angew. Chem., Int. Ed. Engl., 2008, 47, 9115–9117 CrossRef CAS PubMed.
- L. Fohlmeister, S. Liu, C. Schulten, B. Moubaraki, A. Stasch, J. D. Cashion, K. S. Murray, L. Gagliardi and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8294–8298 CrossRef CAS PubMed.
- R. Araake, K. Sakadani, M. Tada, Y. Sakai and Y. Ohki, J. Am. Chem. Soc., 2017, 139, 5596–5606 CrossRef CAS PubMed.
- F. K. Sheong, W.-J. Chen, J.-X. Zhang, Y. Li and Z. Lin, Dalton Trans., 2017, 46, 2214–2219 RSC.
- J.-X. Zhang, F. K. Sheong and Z. Lin, Inorg. Chem., 2019, 58, 3473–3478 CrossRef CAS PubMed.
- J.-X. Zhang, F. K. Sheong and Z. Lin, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 10, e1469 CrossRef CAS.
- F. S. Geitner, W. Klein and T. F. Fässler, Dalton Trans., 2017, 46, 5796–5800 RSC.
- J.-Q. Wang, B. Wahl and T. Fässler, Angew. Chem., Int. Ed., 2010, 49, 6592–6595 CrossRef CAS PubMed.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- J. Poater, M. Solà, M. Duran and X. Fradera, Theor. Chem. Acc., 2002, 107, 362–371 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- E. Van Lenthe and E.-J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- M. Franchini, P. H. T. Philipsen and L. Visscher, J. Comput. Chem., 2013, 34, 1819–1827 CrossRef CAS PubMed.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- L. Versluis and T. Ziegler, J. Chem. Phys., 1988, 88, 322–328 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental and computational details, along with the optimized coordinates. CCDC 2206397, 2170117, 2101583. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00165f |
| ‡ These authors contributed equally to this work. |
| § ip and op denote in-plane and out-of-plane, respectively. |
| This journal is © The Royal Society of Chemistry 2024 |