
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVibrational spectroscopy and dissociation dynamics of cyclohexyl hydroperoxide†
Tarun Kumar
Roy
 a,
Yujie
Qian
a,
Elizabeth
Karlsson
a,
Rawan
Rabayah
a,
Christopher A.
Sojdak
a,
Marisa C.
Kozlowski
a,
Tolga N. V.
Karsili
b and
Marsha I.
Lester
*a
a,
Yujie
Qian
a,
Elizabeth
Karlsson
a,
Rawan
Rabayah
a,
Christopher A.
Sojdak
a,
Marisa C.
Kozlowski
a,
Tolga N. V.
Karsili
b and
Marsha I.
Lester
*a
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104-6323, USA. E-mail: milester@sas.upenn.edu
bDepartment of Chemistry, University of Louisiana, Lafayette, LA, USA
First published on 20th March 2024
Abstract
Organic hydroperoxides (ROOH) are ubiquitous in the atmospheric oxidation of volatile organic compounds (VOCs) as well as in low-temperature oxidation of hydrocarbon fuels. The present work focuses on a prototypical cyclic hydroperoxide, cyclohexyl hydroperoxide (CHHP). The overtone OH stretch (2νOH) spectrum of jet-cooled CHHP is recorded by IR multiphoton excitation with UV laser-induced fluorescence detection of the resulting OH products. A distinctive IR feature is observed at 7012.5 cm−1. Two conformers of CHHP are predicted to have similar stabilities (within 0.2 kcal mol−1) and overtone OH stretch transitions (2νOH), yet are separated by a significant interconversion barrier. The IR power dependence indicates that absorption of three or more IR photons is required for dissociation of CHHP to cyclohexoxy (RO) and OH radical products. Accompanying high-level single- and multi-reference electronic structure calculations quantitatively support the experimental results. Calculations are extended to a range of organic hydroperoxides to examine trends in bond dissociation energies associated with RO + OH formation and compared with prior theoretical results.
1 Introduction
Organic hydroperoxides (ROOH) are formed in the oxidation of volatile organic compounds (VOCs) in the atmosphere or during combustion.1,2 The primary VOC generally reacts with hydroxyl (OH) radicals to form an alkyl radical that promptly reacts with O2 to generate an alkylperoxy (RO2) radical.3,4 The fate of the nascent RO2 is heavily dependent on its molecular structure as well as the temperature and pressure of the surroundings.5,6In the atmosphere, the dominant removal of nascent RO2 radicals in pristine environments is by reaction with HO2, which forms lower volatility ROOH compounds.7–10 Alternatively, the RO2 radical may undergo hydrogen atom transfer (HAT) to form a carbon-centered hydroperoxyalkyl radical (˙QOOH),5,6,11–13 which generally undergoes unimolecular decay or reacts with O2 to form ˙OOQOOH. Sequential repetition of HAT and O2 addition, termed autoxidation, may eventually form a highly oxidized ROOH compound, which has lower volatility than the precursor VOC.14–19 Such compounds are implicated in the formation of secondary organic aerosols, which affect radiative forcing in the atmosphere and impact public health.20–22 In contrast, reaction with HO2 represents a minor sink for RO2 radicals formed during low temperature combustion (such as in modern-day vehicle engines).23–26 Under these conditions, the higher temperatures and pressures usually promote intramolecular HAT to form ˙QOOH radicals which play a vital role in fuel autoignition.14,23–36
Given their significance in atmospheric and combustion chemistry, a thorough understanding of spectroscopy and dissociation dynamics of ROOH compounds is needed. Prior studies have focused on the vibrationally mediated photodissociation dynamics of hydrogen peroxide and simple organic peroxides.37–41 The fundamental OH stretch and various overtone OH stretch transitions of H2O2 and its partially deuterated analogue HOOD have been characterized with a variety of techniques.42 Excitation at 6νOH provides sufficient energy to promote O–O bond dissociation to form OH X2Π fragments. These studies reveal that ca. 11% of the resulting OH fragments formed are vibrationally excited. This is in stark contrast to near UV photodissociation studies of H2O2 and organic hydroperoxides, which predominantly yield rovibrationally cold OH products.43–48
Studies have been extended to tert-butyl hydroperoxide (TBHP), where a sequence of OH-stretch transitions from the fundamental through the fifth overtone has been characterized.40,41 Recent work revealed a strong feature at ∼7017 cm−1 arising from the first overtone OH stretch (2νOH), which is heavily coupled to COOH torsion.49,50 The νOH = 0 and νOH = 2 levels undergo tunneling splitting due to a double-minimum potential energy well along the COOH torsional coordinate. OH radical products are detected following νOH = 2 excitation of TBHP, arising from the absorption of three or more photons of the same wavelength to promote dissociation, while νOH = 6 excitation results in single photon dissociation. The prevailing mechanism for dissociation involves rapid intramolecular vibrational energy redistribution (IVR) followed by O–O bond fission to form OH + RO radical products. The timescale for dissociation is controlled by unimolecular decay along a barrierless potential. Analogous vibrationally activated dissociation is expected for other ROOH compounds with greater molecular complexity. Bach and Schlegel undertook a systematic computational study of various organic hydroperoxides, revealing bond dissociation energies on the order of 45 kcal mol−1, which are consistent with experimental values for a range of hydroperoxides.51,52
This study characterizes the first overtone OH stretch (2νOH) of cyclohexyl hydroperoxide (cyc–C6H11O–OH, CHHP) and its multiphoton dissociation dynamics with OH products. A new synthetic approach is introduced to safely generate CHHP from cyclohexene. The present work builds on a recently reported high-resolution rotational spectroscopy study of the equatorial and axial conformers of CHHP in a Ne expansion (CHHP 1 and 2, respectively), which determined their structures and mapped out the conformational landscape involving torsional motions of the –OOH group.53 Another recent study focused on the synthesis of a wide range of organic hydroperoxides, including CHHP, and determined their absolute photoionization cross sections and resultant fragmentation (–OOH loss) channels.54 The present experimental study is complemented by high-level single- and multi-reference electronic structure calculations for CHHP and a range of other hydroperoxides.
2 Methods
2.1 Synthesis
A new synthetic approach was chosen for preparing cyclohexyl hydroperoxide (CHHP) to avoid the storage of large quantities of CHHP due to the explosive nature of organic hydroperoxides. It was hypothesized that protecting the peroxide with an appropriate silane group would increase the molecular mass and greatly decrease the risk of unexpected detonation of the material.55 Specifically, as shown in Scheme 1, cobalt-catalyzed silylperoxidation of cyclohexene directly yielded the triethylsilyl protected peroxide, (cyclohexylperoxy) triethylsilane, in excellent yields on a multigram scale.56 From (cyclohexylperoxy) triethylsilane, conventional deprotection with pyridine·HF yielded very pure cyclohexyl hydroperoxide in poor yields. However, the poor yields of this transformation were vastly offset by rapid reaction times and more importantly the avoidance of storing bulk quantities of organic hydroperoxide, thus improving the safety of this approach. Extensive details on the synthetic preparation and analytical characterization of CHHP are presented in the ESI (ESI, see Section S1 and Fig. S1–S4†).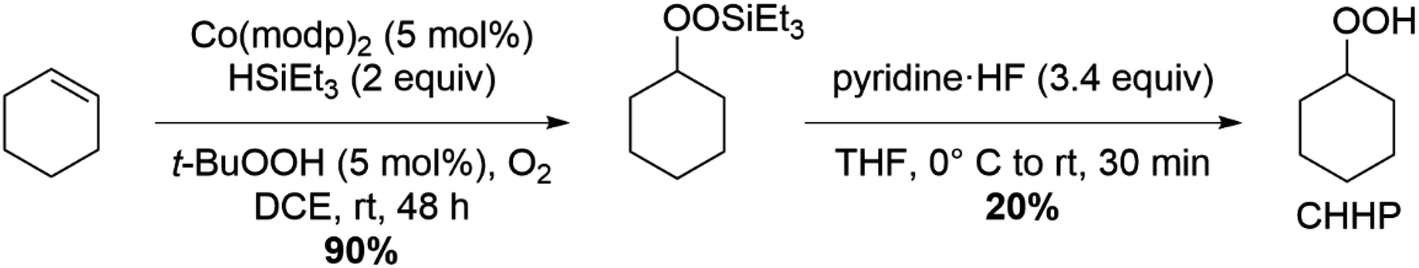 | ||
| Scheme 1 Synthetic route to generate cyclohexyl hydroperoxide (CHHP). | ||
2.2 Spectroscopic methods
The experimental method is similar to that used in a recent IR spectroscopic study of TBHP.49 Freshly synthesized CHHP is seeded in Ar carrier gas at a pressure of 25 psig. The gas mixture is pulsed through a solenoid valve and an affixed quartz capillary tube (∼25 mm length and 1 mm ID) into a vacuum chamber. The resultant supersonic expansion cools CHHP to a rotational temperature of ca. 10 K. Counter propagating IR and UV beams are spatially overlapped and intersect the gas mixture in the collision-free region ca. 1 cm downstream from the exit of the capillary. Multiphoton IR excitation of jet-cooled CHHP in the overtone OH stretch region (2νOH) results in dissociation to OH X2Π3/2 (v = 0) radical products that are detected. The tunable IR beam (with powers up to ca. 28 mJ per pulse) is the signal output of an optical parametric oscillator/amplifier (OPO/OPA, LaserVision; 0.9 cm−1 bandwidth) pumped using an unseeded Nd:YAG laser (1064 nm, Continuum Surelite EX, 5 Hz). The OH radical products are probed via laser-induced fluorescence (LIF) on the A2Σ+–X2Π3/2 (1,0) Q1(3.5) transition at 282 nm utilizing UV radiation (ca. 1 mJ per pulse) generated with the frequency-doubled output of a Nd:YAG (EKSPLA NL300; 532 nm, 10 Hz) pumped dye laser (Continuum ND6000, Rhodamine 590 dye).The fluorescence emitted on the OH A2Σ+–X2Π3/2 (1,1) transition is detected using a gated photomultiplier tube (Electron Tubes 9813QB) and preamplified. Data acquisition involves an active background subtraction scheme (IR on – IR off) to remove a weak OH LIF background signal that arises from UV photodissociation of CHHP at 282 nm.
2.3 Computational methods
The ground state minimum energy geometry for a range of ROOH compounds and their associated anharmonic normal mode wavenumbers were computed at the B2PLYP-D3/cc-pVTZ level of theory. Equivalent calculations were carried out for the OH and RO radicals formed upon O–O bond fission. Single point energies for the resultant optimized geometries were calculated at the CCSD(T)-F12/cc-pVTZ-F12 level of theory. Bond dissociation energies associated with RO + OH formation (D0[RO–OH]) as well as the energy profile along the equatorial to axial isomerization pathway were calculated using the CCSD(T)-F12 energies and harmonic zero-point energies (ZPEs) from the B2PLYP-D3/cc-pVTZ calculations described above. Additional bond dissociation energies for CHHP and other organic hydroperoxides were computed using the W1BD composite method,57 which is known to provide highly accurate thermochemical data within chemical accuracy (ca. 1 kcal mol−1).The ground state relaxed potential energy (PE) profile of CHHP along the O–O stretch coordinate (RO–O) was calculated with single-state complete active space second-order perturbation theory (CASPT2) coupled to Dunning's aug-cc-pVTZ basis set (henceforth CASPT2/AVTZ). This was achieved by fixing RO–O at various values across the range 1.0–5.6 Å and allowing the remainder of the nuclear frameworks to relax to their respective minimum energy configurations. These were based on a 4-state averaged complete active space self-consistent field (SA4-CASSCF) wavefunction employing the same basis set. An active space of 6 electrons in 4 orbitals was utilized to describe all significance short- and long-range interactions of the system while maintaining an appropriate computational expense. The active orbitals are displayed in Fig. S5,† while the orbital occupancies of the ground and excited electronic states of CHHP are shown in Fig. S6 of the ESI.† An imaginary level shift of 0.4 EH was used to aid convergence and to mitigate the involvement of intruder states.
The B2PLYP-D3 and W1BD calculations were performed using Gaussian 16 software suite58 while the CCSD(T)-F12 and CASPT2 calculations were performed using the Molpro computation package.59–61
3 Results and discussion
Cyclohexyl hydroperoxide (CHHP) is predicted to have two stable chair conformations with the –OOH group in an equatorial or axial position of the ring. The present calculations indicate that the equatorial conformer is more stable by 0.2 kcal mol−1. A prior investigation of the conformational landscape of CHHP (B3LYP-D3(BJ)/def2-TZVP) also found the equatorial conformer to be lower in energy, although by ca. 1 kcal mol−1.53 As illustrated in Fig. 1, the interconversion between equatorial and axial configurations involves a sequence of geometry changes. The equatorial conformer initially transforms into a twist-boat configuration through a half-chair transition state (TS) barrier of 11.5 kcal mol−1 (Table S1†). Subsequently, interconversion between the two twist-boat configurations occurs via a TS (ca. 1.5 kcal mol−1) with a boat geometry. The second twist-boat configuration converts into the axial conformer through another half-chair TS.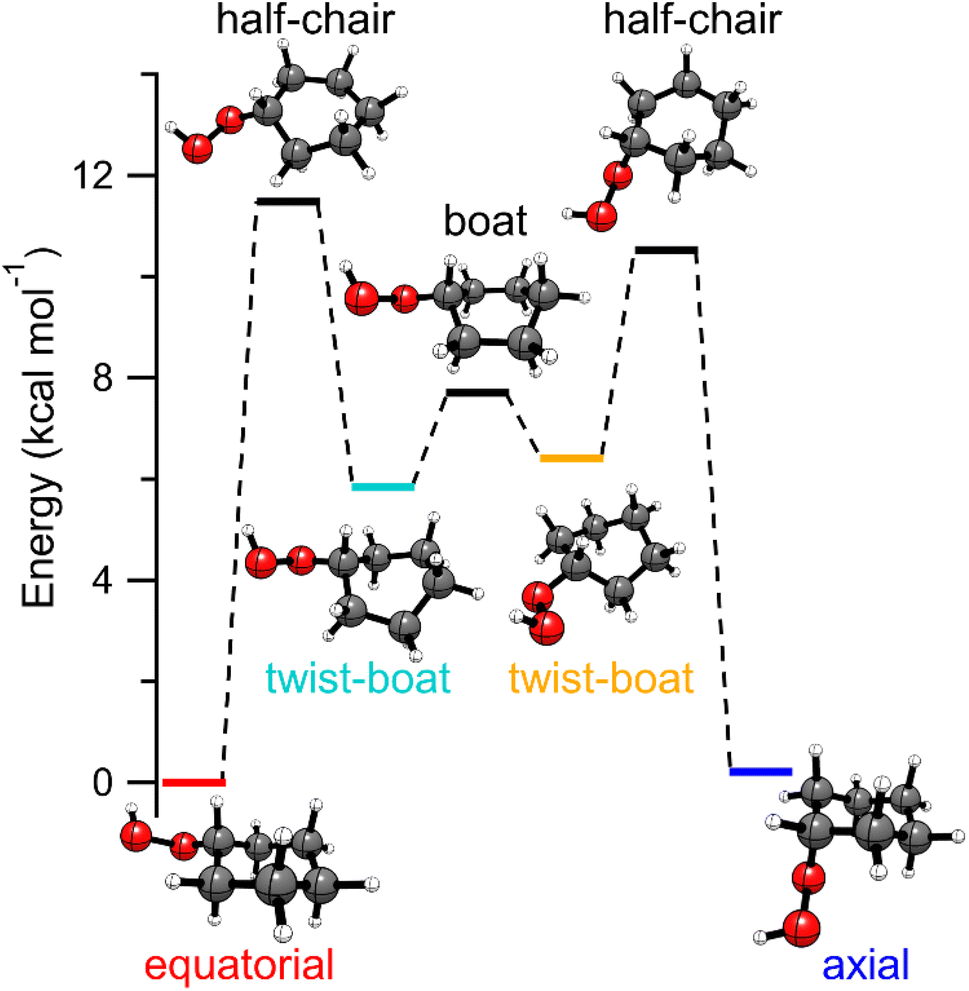 | ||
| Fig. 1 Energy profile connecting the equatorial and axial conformers of CHHP calculated at the CCSD(T)-F12/cc-pVTZ-F12//B2PLYP-D3/cc-pVTZ level of theory. | ||
The relative stability of the equatorial and axial conformers suggests that both conformers will be populated with an estimated 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ratio in favor of the equatorial conformer based on a room temperature Boltzmann population distribution. The significant TS barriers separating the equatorial and axial conformations indicate that both conformers will be populated under jet-cooled conditions, as found in a recent rotational spectroscopy study.53 In addition, the earlier study anticipated a second equatorial conformer (0.67 kcal mol−1 higher in energy) associated with internal rotation about the C–O bond (not shown in Fig. 1) that was not observed, presumably due to relaxation via a low torsional barrier of only 4 kcal mol−1.53
:40 ratio in favor of the equatorial conformer based on a room temperature Boltzmann population distribution. The significant TS barriers separating the equatorial and axial conformations indicate that both conformers will be populated under jet-cooled conditions, as found in a recent rotational spectroscopy study.53 In addition, the earlier study anticipated a second equatorial conformer (0.67 kcal mol−1 higher in energy) associated with internal rotation about the C–O bond (not shown in Fig. 1) that was not observed, presumably due to relaxation via a low torsional barrier of only 4 kcal mol−1.53
In this study, we examine the IR spectrum of CHHP in the first overtone OH stretch region (2νOH), guided by electronic structure calculations performed at the B2PLYP-D3/cc-pVTZ level of theory/basis. The equatorial and axial structures of CHHP are optimized, and the anharmonic vibrational frequencies and IR absorption intensities (Table S2†) are calculated at these geometries using second-order vibrational perturbation theory (VPT2). The first overtone OH stretch for the equatorial and axial conformers of CHHP is predicted at 7006 and 6999 cm−1, respectively, as shown in Fig. 2. IR transitions associated with both equatorial and axial conformers of CHHP are anticipated based on the significant TS barrier separating the two conformers (Fig. 1) and the observation of both conformers in the recent rotational spectroscopy study.53
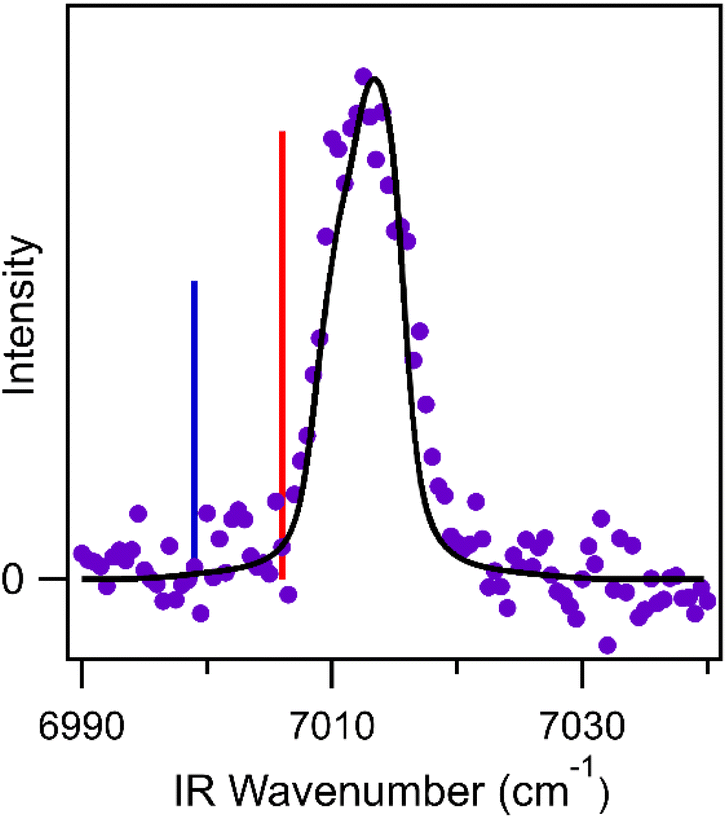 | ||
| Fig. 2 Theoretically predicted 2νOH transitions for the equatorial (red) and axial (blue) conformers of CHHP at 7006 and 6999 cm−1, respectively, shown with relative intensities of 60:40 for a room temperature Boltzmann distribution. The experimental IR action spectrum (purple points) is overlaid with a simulated rotational band contour (black) for a scenario of closely spaced 2νOH transitions (ca. 3 cm−1 apart) for equatorial and axial conformers. A nearly indistinguishable simulation results from an alternate limiting case with IR power broadening of the 2νOH transition for the equatorial conformer that increases the homogeneous linewidth to 6.0 cm−1 (see Fig. S8†). The spectral shift of the observed feature (ca. 10 ± 2 cm−1) is within the accuracy of the theoretical predictions (10–20 cm−1). | ||
The IR action spectrum of CHHP was recorded by scanning the OPO from 6990 to 7040 cm−1 at a scan speed of 0.1 cm−1 s−1 in the first overtone OH stretch (2νOH) region under jet-cooled conditions, as shown in Fig. 2. The IR action spectrum is obtained with LIF detection of OH products on the A-X (1,0) Q1(3.5) transition at an IR-UV delay time of 700 ns (see Section S3 and Fig. S7 of the ESI†). This requires IR multiphoton excitation, which induces dissociation of CHHP and leads to formation of cyclohexoxy (RO) and OH products. The IR action spectrum exhibits a single prominent band at 7012.5 cm−1 with a full width at half maximum (FWHM) of ca. 7.5 cm−1. The breadth of the observed feature is noticeably greater than the ca. 5 cm−1 FWHM band contour expected for an isolated vibrational overtone transition of CHHP under jet-cooled conditions. The anticipated breadth is based on simulations using experimental rotational constants,53 a rotational temperature of 10 K, the OPO bandwidth (0.9 cm−1), and typical homogeneous broadening (1.7 cm−1) associated with rapid (ps) intramolecular vibrational energy redistribution.12,62
The experimental observation of a single broadened feature at 7012.5 cm−1 can result from different population distributions for the equatorial and axial conformers of CHHP. One limiting case is a 60:40 population ratio of equatorial to axial conformers estimated for a Boltzmann distribution at 300 K and assumed to be unchanged upon supersonic expansion. In this case, the 2νOH transitions for the equatorial and axial conformers would need to be more closely spaced, e.g. ca. 3 cm−1 apart, than predicted theoretically such that the two band contours merge into a single feature as shown in Fig. 2. An alternative limiting scenario is that the observed 2νOH feature originates primarily from the lower energy equatorial conformer with IR power broadening increasing the homogeneous linewidth to 6.0 cm−1. The two limiting cases yield nearly indistinguishable spectra as illustrated in Fig. S8.† The observed feature appears at higher energy (ca. 10 ± 2 cm−1) than predicted for the equatorial and axial conformers (7006 and 6999 cm−1, respectively), which is well within the accuracy of the theoretical predictions (10–20 cm−1).
A previous study reported an extensively broadened 2νOH feature (12.2 cm−1 FWHM) for TBHP under jet-cooled conditions, which was attributed to tunneling across the symmetric double-well potential along the OH torsion coordinate.49,50 This resulted in multiple transitions and associated inhomogeneous broadening. Such broadening is not anticipated in CHHP since the double-well potential associated with OH torsion is asymmetric at the global minimum energy geometry of CHHP.
The IR power dependence of the 2νOH transition of CHHP at 7012.5 cm−1 is obtained by varying the output power of the OPO from 13 to 28 mJ per pulse, as shown in Fig. 3 with both axes on a log10 scale. The variation of the OH LIF signal with IR laser power reveals a slope of 2.3 ± 0.3. The nonlinear dependence on IR power implies that the dissociation process requires multiphoton absorption of IR photons in the 2νOH region. The slope indicates that more than two photons are required to promote CHHP above the dissociation limit, yielding the OH radicals that are detected. The energies of the OH fundamental and first overtone transitions are obtained from the VPT2 calculation, while the higher OH overtone levels (3νOH and 4νOH) illustrated in Fig. 3 are estimated from an anharmonic vibrational analysis.
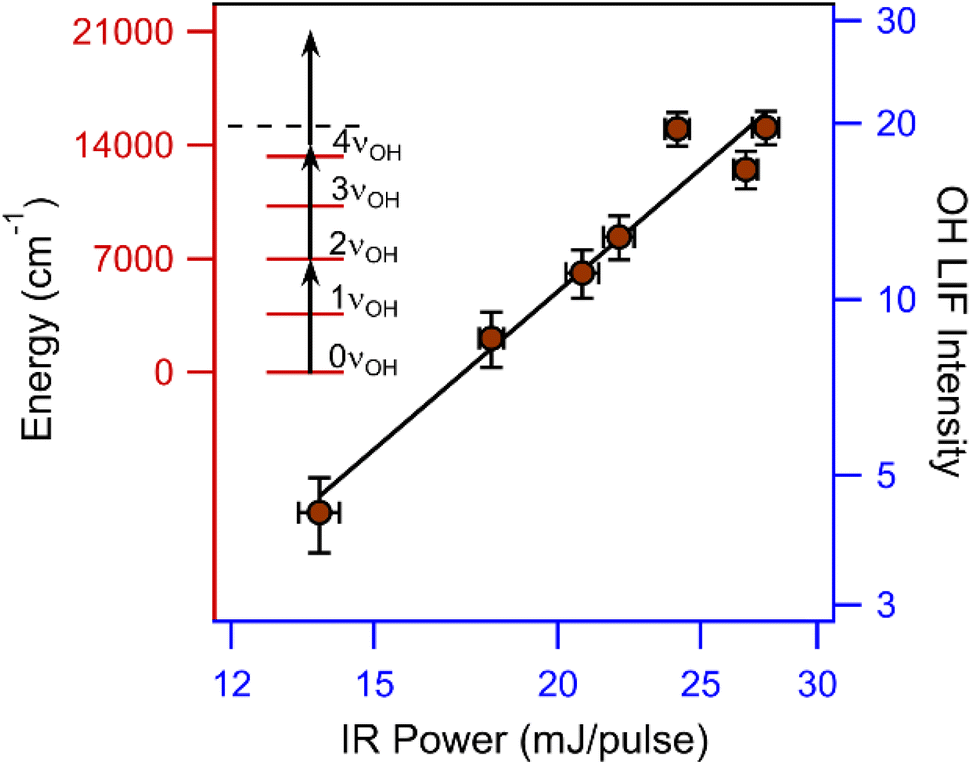 | ||
| Fig. 3 (Left) Energy diagram showing the multiphoton dissociation of CHHP. IR excitation of the 2νOH transition (black arrow) followed by absorption of additional photon(s) provides sufficient energy to exceed the O–O bond dissociation limit of CHHP (dashed line), resulting in cyclohexoxy and OH radicals. The OH radicals are detected using LIF on the A2Σ+–X2Π3/2 (1,0) Q1(3.5) transition at 282 nm. (Right) IR-induced OH LIF signal intensity at 7012.5 cm−1 as a function of IR power (mJ per pulse). Both axes are plotted on a log10 scale, yielding a slope of 2.3 ± 0.3. The nonlinear IR power dependence indicates multiphoton excitation of CHHP. | ||
Fig. 4 presents the CASPT2 relaxed potential energy (PE) profile along the O–O stretch coordinate (ROO) for the equatorial conformer of CHHP. The PE profile shows a weak saddle point at ROO = 2.78 Å followed by a shallow minimum at ROO = 2.8 Å, the latter of which arises from a hydrogen bonding interaction between the O lone pair of the cyclohexoxy radical (RO) and the H-atom of the OH unit. The energy profile along the O–O coordinate beyond ROO > 3.4 Å proceeds smoothly to the RO + OH asymptotic limit, which is consistent with earlier studies of other hydroperoxides.63,64 The corresponding PE profile for the axial conformer of CHHP is shown in Fig. S9.† Using the asymptotic energies, bond dissociation energies (De) of ca. 46 kcal mol−1 are determined for both equatorial and axial conformers. Upon ZPE correction, the resulting D0 values for both conformers are ca. 42 kcal mol−1. The W1BD method predicts a D0 value of 43 kcal mol−1 (Table 1), which agrees well with the ZPE-corrected CASPT2 energies. The W1BD method is known to give thermochemical quantities that are within chemical accuracy (1 kcal mol−1).
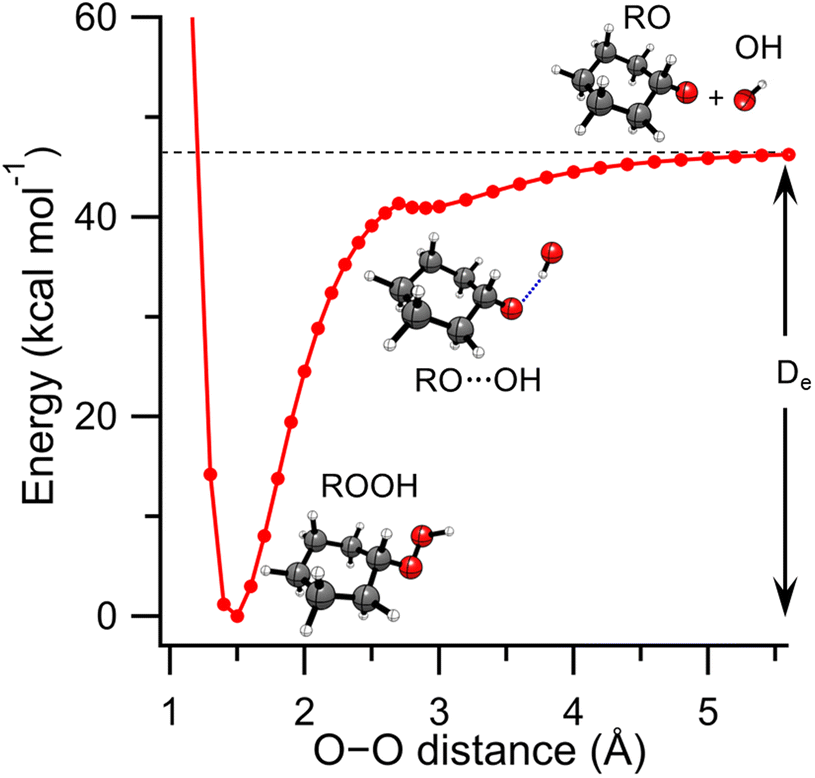 | ||
| Fig. 4 Energy diagram for the equatorial conformer of CHHP (ROOH) along the O–O stretch coordinate (ROO) to form cyclohexoxy (RO) + OH products. The potential is calculated at the CASPT2/aug-cc-pVTZ level of theory. A bond dissociation energy (De) of 46 kcal mol−1 is obtained for the equatorial conformer of CHHP. | ||
| This work | Bach et al.a | Expt. | |||
|---|---|---|---|---|---|
| W1BD | CCSD(T)-F12/cc-pVTZ-F12b | CBS-APNO | G4 | ||
| a Ref. 52. b ZPE corrected with vibrational wavenumbers derived at the B2PLYP-D3/cc-pVTZ level. c Ref. 65. d Ref. 66. | |||||
| HO–OH | 48.4 | 48.1 | 50.2 | 49.0 | 50.30c |
| CH3O–OH | 41.9 | 42.4 | 44.6 | 43.2 | 44.67c |
| (CH3)3CO–-OH | 44.0 | 43.5 | 45.8 | 43.9 | 44.1d |
| (CH3)2CHO–OH | 43.9 | 44.1 | — | — | — |
| cyc–C6H11O–OH | 43.4 | — | — | — | — |
Given that the IR excitation is tuned to 7012.5 cm−1 (ca. 20 kcal mol−1), which corresponds to the 2νOH transition of CHHP, multiphoton dissociation is required to initiate O–O bond fission. With a W1BD derived D0 value of 43 kcal mol−1, at least three photons of the OH first overtone transition would be required to promote O–O bond fission. The third photon would impart an excess energy of ca. 17 kcal mol−1 into CHHP, which upon intramolecular vibrational energy redistribution would provide sufficient energy in O–O stretch to promote O–O bond fission.
The O–O bond dissociation energy (D0) for CHHP (cyc–C6H11O–OH) is compared with those computed for four simpler hydroperoxides (ROOH) using the same W1BD method in Table 1. The D0 values of the representative hydroperoxides are also evaluated with the CCSD(T)-F12/cc-pVTZ-F12 method (Table 1). The calculated dissociation energies using the W1BD and CCSD(T)-F12 methods compare favorably with those previously reported with the CBS-APNO and G4 methods.52 Prior theoretical studies of O–O bond dissociation energies for ROOH with various substituents ranged from ca. 35 to 50 kcal mol−1 with a typical O–O bond dissociation energy of ca. 45 kcal mol−1.51,52
The present calculations, along with those conducted previously,51,52 show that the organic hydroperoxides have D0(O–O) values that are ca. 4–7 kcal mol−1 lower than that of hydrogen peroxide. This can be qualitatively understood from the relative stabilities of the OH vs. RO radical products. Alkyl groups are well-known σ-electron donors and the H-atoms of the CH3 moiety participate in hyperconjugation. Both effects are expected to stabilize the oxygen-centered odd-electron (the electron deficient p-orbital) of the RO radical. Equivalent stabilizations are not available for OH, resulting in more stable OR radicals. Interestingly, the organic hydroperoxide with a primary carbon-center (CH3O–OH) has a D0 value that is ca. 1–2 kcal mol−1 lower than that of secondary [(CH3)2CHO–OH and cyc–C6H11O–OH] and tertiary ((CH3)3CO–OH) carbon-centered hydroperoxides. The secondary and tertiary carbon-centers are more electron-rich than primary carbon-centers due to the +I inductive effects imparted by the alkyl-substituents; this is expected to increase the nucleophilicity of the carbon-center. Additionally, hyperconjugation is not available in the secondary and tertiary carbon centers. Both effects are expected to destabilize secondary and tertiary carbon-centered organic hydroperoxides, consistent with their higher bond dissociation energies.
4 Conclusions
A new synthetic approach for generating CHHP is utilized in the present study of its IR spectroscopy and multiphoton dissociation dynamics. The IR spectrum of jet-cooled CHHP has been recorded in the first overtone OH stretch (2νOH) region. Quantum chemical studies reveal that the equatorial conformer of CHHP is slightly more stable than the axial conformer (ca. 0.2 kcal mol−1) and separated by a substantial TS barrier for isomerization (ca. 11.5 kcal mol−1). As a result, both conformers may be populated under jet-cooled conditions, in accord with a recent rotational spectroscopy investigation.53 IR multiphoton excitation promotes CHHP above the O–O dissociation limit, resulting in OH X2Π (v = 0) radical products that are detected via UV-LIF. IR action spectroscopy yields a distinctive, yet broadened, vibrational overtone feature for CHHP at 7012.5 cm−1. This can be ascribed to various scenarios ranging from a single homogeneously broadened feature dominated by the equatorial conformer to overlapping band contours for the equatorial and axial conformers that merge into a single feature. The nonlinear IR power dependence of this spectral feature confirms that a minimum of three photons with energies corresponding to 2νOH are required to promote O–O bond fission. This observation is confirmed by computation of relaxed CASPT2 potential energy profiles along the O–O bond stretch coordinate for the equatorial and axial conformers, and determination of the O–O bond dissociation energy using the W1BD method.The present study adds to the small but growing body of work on larger organic hydroperoxides. In the future, we anticipate the emergence of synthetic procedures for laboratory investigation of the spectroscopy and dissociation dynamics of more complex and atmospherically relevant organic hydroperoxides.
Data availability
All supporting data and methods are available in the ESI.†Author contributions
M. I. L. and T. N. V. K. designed the experimental and theoretical aspects of the research project, respectively. C. S. and M. C. K. synthesized cyclohexyl hydroperoxide (CHHP). T. K. R and Y. Q. conducted the experiments and analyzed the experimental data. L. K., R. R., and T. N. V. K. carried out the theoretical calculations. All authors discussed the results and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research in the Lester laboratory was supported by the National Science Foundation under grants CHE-1955068 and CHE-2301298. M. C. K. is grateful to the NSF (CHE-2102626) for financial support of this research. Partial instrumentation support (NMR spectrometer) was provided by NSF CHE-1827457 and the Vagelos Institute for Energy Science and Technology. T. K. R. was primarily supported by a Walter-Benjamin Scholarship funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 508074809. T. N. V. K. is grateful to the NSF (OIA-2229695) for financial support. This work used the Advanced Cyberinfrastructure Coordination Ecosystem: Service & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296, through the allocation TG-CHE190088.References
- S. Enami, Fates of Organic Hydroperoxides in Atmospheric Condensed Phases, J. Phys. Chem. A, 2021, 125, 4513–4523 CrossRef CAS PubMed.
- F. Bianchi, O. Garmash, X. He, C. Yan, S. Iyer, I. Rosendahl, Z. Xu, M. P. Rissanen, M. Riva, R. Taipale, N. Sarnela, T. Petäjä, D. R. Worsnop, M. Kulmala, M. Ehn and H. Junninen, The role of highly oxygenated molecules (HOMs) in determining the composition of ambient ions in the boreal forest, Atmos. Chem. Phys., 2017, 17, 13819–13831 CrossRef CAS.
- L. Vereecken and B. Nozière, H-migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS.
- J. Lelieveld, T. M. Butler, J. N. Crowley, T. J. Dillon, H. Fischer, L. Ganzeveld, H. Harder, M. G. Lawrence, M. Martinez, D. Taraborrelli and J. Williams, Atmospheric oxidation capacity sustained by a tropical forest, Nature, 2008, 452, 737–740 CrossRef CAS PubMed.
- D. L. Osborn, Reaction mechanisms on multiwell potential energy surfaces in combustion (and atmospheric) chemistry, Annu. Rev. Phys. Chem., 2017, 68, 233–260 CrossRef CAS PubMed.
- J. D. Savee, E. Papajak, B. Rotavera, H. Huang, A. J. Eskola, O. Welz, L. Sheps, C. A. Taatjes, J. Zádor and D. L. Osborn, Direct observation and kinetics of a hydroperoxyalkyl radical (QOOH), Science, 2015, 347, 643–646 CrossRef CAS PubMed.
- L. Vereecken and J. S. Francisco, Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- G. Meloni, P. Zou, S. J. Klippenstein, M. Ahmed, S. R. Leone, C. A. Taatjes and D. L. Osborn, Energy-Resolved Photoionization of Alkylperoxy Radicals and the Stability of Their Cations, J. Am. Chem. Soc., 2006, 128, 13559–13567 CrossRef CAS PubMed.
- W. P. L. Carter, A detailed mechanism for the gas-phase atmospheric reactions of organic compounds, Atmos. Environ. Part A. General Topics, 1990, 24, 481–518 CrossRef.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson Jr, J. A. Kerr, M. J. Rossi and J. Troe, Evaluated Kinetic, Photochemical and Heterogeneous Data for Atmospheric Chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 1997, 26, 521–1011 CrossRef CAS.
- A. S. Hansen, T. Bhagde, K. B. Moore, D. R. Moberg, A. W. Jasper, Y. Georgievskii, M. F. Vansco, S. J. Klippenstein and M. I. Lester, Watching a hydroperoxyalkyl radical (•QOOH) dissociate, Science, 2021, 373, 679–682 CrossRef CAS PubMed.
- A. S. Hansen, T. Bhagde, Y. Qian, A. Cavazos, R. M. Huchmala, M. A. Boyer, C. F. Gavin-Hanner, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Infrared spectroscopic signature of a hydroperoxyalkyl radical (•QOOH), J. Chem. Phys., 2022, 156, 014301 CrossRef CAS PubMed.
- T. Bhagde, A. S. Hansen, S. Chen, P. J. Walsh, S. J. Klippenstein and M. I. Lester, Energy-resolved and time-dependent unimolecular dissociation of hydroperoxyalkyl radicals (•QOOH), Faraday Discuss., 2022, 238, 575–588 RSC.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- T. Jokinen, M. Sipilä, S. Richters, V.-M. Kerminen, P. Paasonen, F. Stratmann, D. Worsnop, M. Kulmala, M. Ehn, H. Herrmann and T. Berndt, Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem., Int. Ed., 2014, 53, 14596–14600 CrossRef CAS PubMed.
- T. Berndt, S. Richters, T. Jokinen, N. Hyttinen, T. Kurtén, R. V. Otkjær, H. G. Kjaergaard, F. Stratmann, H. Herrmann, M. Sipilä, M. Kulmala and M. Ehn, Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 2016, 7, 13677 CrossRef CAS PubMed.
- E. Praske, R. V. Otkjær, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard and P. O. Wennberg, Atmospheric autoxidation is increasingly important in urban and suburban North America, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 64–69 CrossRef CAS PubMed.
- D. J. Medeiros, M. A. Blitz, P. W. Seakins and L. K. Whalley, Direct Measurements of Isoprene Autoxidation: Pinpointing Atmospheric Oxidation in Tropical Forests, J. Am. Chem. Soc., 2022, 2, 809–818 Search PubMed.
- M. Ge, S. Tong, W. Wang, W. Zhang, M. Chen, C. Peng, J. Li, L. Zhou, Y. Chen and M. Liu, Important Oxidants and Their Impact on the Environmental Effects of Aerosols, J. Phys. Chem. A, 2021, 125, 3813–3825 CrossRef CAS PubMed.
- C. Li and R. Signorell, Understanding vapor nucleation on the molecular level: A review, J. Aerosol Sci., 2021, 153, 105676 CrossRef CAS.
- V. F. McNeill, Virtual Special Issue: New Advances in Organic Aerosol Chemistry, ACS Earth Space Chem., 2020, 4, 491–494 CrossRef CAS.
- E. W. Kaiser, Temperature and Pressure Dependence of the C2H4 Yield from the Reaction C2H5 + O2, J. Phys. Chem., 1995, 99, 707–711 CrossRef CAS.
- E. W. Kaiser, Formation of C3H6 from the Reaction C3H7 + O2 between 450 and 550 K, J. Phys. Chem. A, 1998, 102, 5903–5906 CrossRef CAS.
- E. W. Kaiser and T. J. Wallington, Formation of C3H6 from the Reaction C3H7 + O2 and C2H3Cl from C2H4Cl + O2 at 297 K, J. Phys. Chem., 1996, 100, 18770–18774 CrossRef CAS.
- I. C. Plumb and K. R. Ryan, Kinetic studies of the reaction of C2H5 with O2 at 295 K, Int. J. Chem. Kinet., 1981, 13, 1011–1028 CrossRef CAS.
- E. P. Clifford, J. T. Farrell, J. D. DeSain and C. A. Taatjes, Infrared Frequency-Modulation Probing of Product Formation in Alkyl + O2 Reactions: I. The Reaction of C2H5 with O2 between 295 and 698 K, J. Phys. Chem. A, 2000, 104, 11549–11560 CrossRef CAS.
- J. D. DeSain, E. P. Clifford and C. A. Taatjes, Infrared Frequency-Modulation Probing of Product Formation in Alkyl + O2 Reactions: II. The Reaction of C3H7 with O2 between 296 and 683 K, J. Phys. Chem. A, 2001, 105, 3205–3213 CrossRef CAS.
- J. D. DeSain, C. A. Taatjes, J. A. Miller, S. J. Klippenstein and D. K. Hahn, Infrared frequency-modulation probing of product formation in alkyl + O2 reactions. Part IV. Reactions of propyl and butyl radicals with O2, Faraday Discuss., 2002, 119, 101–120 RSC.
- E. W. Kaiser, I. M. Lorkovic and T. J. Wallington, Pressure dependence of the ethene yield from the reaction ethyl radical + oxygen, J. Phys. Chem., 1990, 94, 3352–3354 CrossRef CAS.
- E. G. Estupiñán, J. D. Smith, A. Tezaki, S. J. Klippenstein and C. A. Taatjes, Measurements and Modeling of DO2 Formation in the Reactions of C2D5 and C3D7 Radicals with O2, J. Phys. Chem. A, 2007, 111, 4015–4030 CrossRef PubMed.
- J. Zádor, C. A. Taatjes and R. X. Fernandes, Kinetics of elementary reactions in low-temperature autoignition chemistry, Prog. Energy Combust. Sci., 2011, 37, 371–421 CrossRef.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- R. W. Walker and C. Morley, Chapter 1 Basic chemistry of combustion, In Comprehensive Chemical Kinetics, ed. M. J. Pilling, Elsevier, 1997, vol. 35, pp. 1–124 Search PubMed.
- S. Sharma, S. Raman and W. H. Green, Intramolecular Hydrogen Migration in Alkylperoxy and Hydroperoxyalkylperoxy Radicals: Accurate Treatment of Hindered Rotors, J. Phys. Chem. A, 2010, 114, 5689–5701 CrossRef CAS PubMed.
- J. D. DeSain and C. A. Taatjes, Infrared Frequency-Modulation Probing of Product Formation in Alkyl + O2 Reactions: III. The Reaction of Cyclopentyl Radical (c-C5H9) with O2 between 296 and 723 K, J. Phys. Chem. A, 2001, 105, 6646–6654 CrossRef CAS.
- H. R. Dübal and F. F. Crim, Vibrational overtone predissociation spectroscopy of hydrogen peroxide, J. Chem. Phys., 1985, 83, 3863–3872 CrossRef.
- T. M. Ticich, M. D. Likar, H. R. Dübal, L. J. Butler and F. F. Crim, Vibrationally mediated photodissociation of hydrogen peroxide, J. Chem. Phys., 1987, 87, 5820–5829 CrossRef CAS.
- D. T. Colbert and E. L. Sibert III, Theory of vibrationally mediated photodissociation of HOOH: Delocalized tails in a localized wave function, J. Chem. Phys., 1991, 94, 6519–6545 CrossRef CAS.
- M. D. Likar, J. E. Baggott and F. F. Crim, Vibrationally mediated photodissociation of t-butyl hydroperoxide: Vibrational overtone spectroscopy and photodissociation dynamics, J. Chem. Phys., 1989, 90, 6266–6274 CrossRef CAS.
- M. D. Likar, J. E. Baggott, A. Sinha, T. M. Ticich, R. L. V. Wal and F. F. Crim, Vibrationally mediated photodissociation, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 1483–1497 RSC.
- M. Brouard, M. T. Martinez, J. O'Mahony and J. P. Simons, Energy and angular momentum disposals in the vibrationally mediated photodissociation of HOOH and HOOD, Mol. Phys., 1990, 69, 65–84 CrossRef CAS.
- J. August, M. Brouard, M. P. Docker, C. J. Milne, J. P. Simons, R. Lavi, S. Rosenwaks and D. Schwartz-Lavi, Photodissociation dynamics of tert-butyl nitrite (S2) and tert-butyl hydroperoxide at 248-250 nm, J. Phys. Chem., 1988, 92, 5485–5491 CrossRef CAS.
- Y. Inagaki, Y. Matsumi and M. Kawasaki, Formation of a Hydrogen Atom from the Photodissociation of Hydrogen Peroxide at 193 nm, Bull. Chem. Soc. Jpn., 1993, 66, 3166–3170 CrossRef CAS.
- S. K. Shin, S. O. Park, Y. S. Choi, H. L. Kim and C. R. Park, Photodissociation Dynamics of Cumene Hydroperoxide at 248 and 193 nm, J. Phys. Chem. A, 2001, 105, 10018–10024 CrossRef CAS.
- B.-G. Ryu, C. R. Park, Y. Lee, S. K. Shin and H. L. Kim, Photodissociation dynamics of tert-butyl hydroperoxide at 266 nm, J. Photochem. Photobiol., 2002, 149, 15–21 CrossRef CAS.
- S. Hsieh, R. Vushe, Y. T. Tun and J. L. Vallejo, Trends in organic hydroperoxide photodissociation and absorption cross sections between 266 and 377nm, Chem. Phys. Lett., 2014, 591, 99–102 CrossRef CAS.
- M. D. Likar, J. E. Baggott and F. F. Crim, Vibrationally mediated photodissociation of t-butyl hydroperoxide: Vibrational overtone spectroscopy and photodissociation dynamics, J. Chem. Phys., 1989, 90, 6266–6274 CrossRef CAS.
- A. S. Hansen, R. M. Huchmala, E. Vogt, M. A. Boyer, T. Bhagde, M. F. Vansco, C. V. Jensen, A. Kjærsgaard, H. G. Kjaergaard, A. B. McCoy and M. I. Lester, Coupling of torsion and OH-stretching in tert-butyl hydroperoxide. I. The cold and warm first OH-stretching overtone spectrum, J. Chem. Phys., 2021, 154, 164306 CrossRef CAS PubMed.
- E. Vogt, R. M. Huchmala, C. V. Jensen, M. A. Boyer, J. Wallberg, A. S. Hansen, A. Kjærsgaard, M. I. Lester, A. B. McCoy and H. G. Kjaergaard, Coupling of torsion and OH-stretching in tert-butyl hydroperoxide. II. The OH-stretching fundamental and overtone spectra, J. Chem. Phys., 2021, 154, 164307 CrossRef CAS PubMed.
- R. D. Bach, P. Y. Ayala and H. B. Schlegel, A Reassessment of the Bond Dissociation Energies of Peroxides. An ab Initio Study, J. Am. Chem. Soc., 1996, 118, 12758–12765 CrossRef CAS.
- R. D. Bach and H. B. Schlegel, Bond Dissociation Energy of Peroxides Revisited, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRef CAS PubMed.
- P. Pinacho, W. Sun, D. A. Obenchain and M. Schnell, Conformational analysis of cyclohexyl hydroperoxide by rotational spectroscopy, J. Mol. Spectrosc., 2023, 392, 111758 CrossRef CAS.
- Z. Hu, Q. Di, B. Liu, Y. Li, Y. He, Q. Zhu, Q. Xu, P. Dagaut, N. Hansen, S. M. Sarathy, L. Xing, D. G. Truhlar and Z. Wang, Elucidating the photodissociation fingerprint and quantifying the determination of organic hydroperoxides in gas-phase autoxidation, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2220131120 CrossRef CAS PubMed.
- J. B. Sperry, M. Azuma and S. Stone, Explosive Hazard Identification in Pharmaceutical Process Development: A Novel Screening Method and Workflow for Shipping Potentially Explosive Materials, Org. Process Res. Dev., 2021, 25, 212–224 CrossRef CAS.
- T. Tokuyasu, S. Kunikawa, A. Masuyama and M. Nojima, Co(III)−Alkyl Complex- and Co(III)−Alkylperoxo Complex-Catalyzed Triethylsilylperoxidation of Alkenes with Molecular Oxygen and Triethylsilane, Org. Lett., 2002, 4, 3595–3598 CrossRef CAS PubMed.
- E. C. Barnes, G. A. Petersson, J. A. Montgomery Jr, M. J. Frisch and J. M. L. Martin, Unrestricted Coupled Cluster and Brueckner Doubles Variations of W1 Theory, J. Chem. Theory Comput., 2009, 5, 2687–2693 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji and et al., Gaussian 16, Rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, et.al., MOLPRO 2020.1, a package of ab initio programs, 2020, see https://www.molpro.net Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: a general-purpose quantum chemistry program package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro quantum chemistry package, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- A. S. Hansen, Y. Qian, C. A. Sojdak, M. C. Kozlowski, V. J. Esposito, J. S. Francisco, S. J. Klippenstein and M. I. Lester, Rapid Allylic 1,6 H-Atom Transfer in an Unsaturated Criegee Intermediate, J. Am. Chem. Soc., 2022, 144, 5945–5955 CrossRef CAS PubMed.
- N. M. Kidwell, H. Li, X. Wang, J. M. Bowman and M. I. Lester, Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products, Nat. Chem., 2016, 8, 509–514 CrossRef CAS PubMed.
- B. Marchetti, V. J. Esposito, R. E. Bush and T. N. V. Karsili, The states that hide in the shadows: the potential role of conical intersections in the ground state unimolecular decay of a Criegee intermediate, Phys. Chem. Chem. Phys., 2022, 24, 532–540 RSC.
- Active Thermochemical Tables, https://atct.anl.gov/Thermochemical/Data/version-1.130/index.php, (accessed February 2024) Search PubMed.
- W. Reints, D. A. Pratt, H.-G. Korth and P. Mulder, O−O Bond Dissociation Enthalpy in Di(trifluoromethyl) Peroxide (CF3OOCF3) as Determined by Very Low Pressure Pyrolysis. Density Functional Theory Computations on O−O and O−H Bonds in (Fluorinated) Derivatives, J. Phys. Chem. A, 2000, 104, 10713–10720 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00151f |
| This journal is © The Royal Society of Chemistry 2024 |