
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of waste poly(vinyl chloride) to branched polyethylene mediated by silylium ions†
Zachary A.
Wood
 a,
Eunice C.
Castro
a,
Angelyn N.
Nguyen
a and
Megan E.
Fieser
*ab
a,
Eunice C.
Castro
a,
Angelyn N.
Nguyen
a and
Megan E.
Fieser
*ab
aDepartment of Chemistry, University of Southern California, Los Angeles, CA 91706, USA
bWrigley Institute for Environment and Sustainability University of Southern California, Los Angeles, CA 91706, USA. E-mail: fieser@usc.edu
First published on 14th May 2024
Abstract
Full dechlorination of poly(vinyl chloride) (PVC) in a controlled manner to yield useful polymeric and chlorinated products is of great interest for the processing of PVC waste. Forming polyethylene (PE) without corrosive by-products would allow for a pre-treatment of PE wastes that are often contaminated with PVC. Herein, full dechlorination of PVC has been achieved via generation of silylium ions in situ, to furnish PE products. Complete dechlorination of PVC can be achieved in 2 hours, yielding organic polymer that has similar spectroscopic and thermal signatures of branched PE, with no observable chlorine. The degree of branching can be tuned between 31 and 57 branches per 1000 carbons, with melting temperatures ranging from 51 to 93 °C. This method is applicable to not only pure PVC, but also commercial PVC products. Depending on if the PVC products are separated from plasticizers, different melting points of the resulting PE are observed. PVC dechlorination in the presence of PE waste is also shown. This is the first report of being able to cleanly convert PVC waste to PE in high yields and tune the thermal properties of the PE product, highlighting the remarkable control that silylium ion mediated transformations enables compared to past chemical methods.
Introduction
As the third-most produced plastic worldwide, the end-of-life of poly(vinyl chloride) (PVC) has garnered particular interest in addressing the plastic waste crisis due to its toxicity and the barriers it presents to recycling of mixed plastic streams.1 For example, mechanical recycling of PVC contaminated polyethylene (PE) waste results in release of HCl and Cl2, corroding recycling plants.2 Moreover, waxes and fuels from pyrolyzed polymers containing just 10 ppm Cl are unusable.2 Methods for recycling or upcycling PVC, as well as its compatibilization with other plastic waste recycling, are therefore vital.Because polyolefin waste commonly has PVC (and other plastic) contamination, it is imperative to find multiple ways to process this plastic waste that can yield a variety of useful products. While processing of polyolefin waste to a distributions of small molecules and oligomers (e.g., waxes and fuels) can add more value to contaminated waste,3,4 researching ways to convert such waste to practical materials that can be used for other applications would also be of importance. Converting PVC to PE, without the generation of corrosive byproducts, would be valuable as the product would promote more selective pyrolysis with longer-lived pyrolysis catalysts. An added benefit of producing PE compared to waxes and fuels is that the products can also be mechanically recycled.
There are many strategies that have been reported to dechlorinate PVC waste, yet two problems persist: full dechlorination is rarely achieved, and even if high dechlorination is achieved, the C–C backbone left behind is often not clean or selective, severely limiting its applications. A few emerging strategies include productive addition of the chlorine to other organic molecules,5 tandem dehydrochlorination3,6 and π-bond metathesis to achieve smaller molecules and oligomers,7 or tandem substitution of the Cl for H and arenes to form upcycled copolymers.8 While important in developing more ways to process PVC waste, these methods can't be used to convert PE/PVC waste into PE that can be mechanically recycled or pyrolyzed. Methods to process PVC contaminated PE waste can be separated into three categories. One method is the dechlorination of PVC-PE waste streams over solid material, such as Mg3AlO4.5, before pyrolysis is performed with a catalyst, often with Ru-based catalysts.4,9–17 However, in a recent example, the product selectivity of the Ru catalyst changes in the presence of chlorinated contamination.4 Additionally, the solid pyrolysis product still has Cl content (∼10 wt%) while the liquid fraction has <0.1 wt% (Fig. 1A). A second method is to perform sequential dehydrochlorination/hydrogenation to yield PE-like polymers. Only partial dechlorination (<90% dechlorination) is achieved in one case,6 and another case reports full dechlorination by Fourier-transform infrared spectroscopy (FT-IR) but product selectivity and yield are poor compared to other methods (Fig. 1B).3 The third method is to dechlorinate PVC selectively through reduction or hydrodechlorination to form PE-like materials using catalytic methods or stoichiometric reagents such as Bu3SnH.18–20 However, these reports either don't achieve full dechlorination, or if full dechlorination is achieved the polymer product does not have the necessary purity and thermal properties to enter the PE mechanical recycling stream (Fig. 1C).
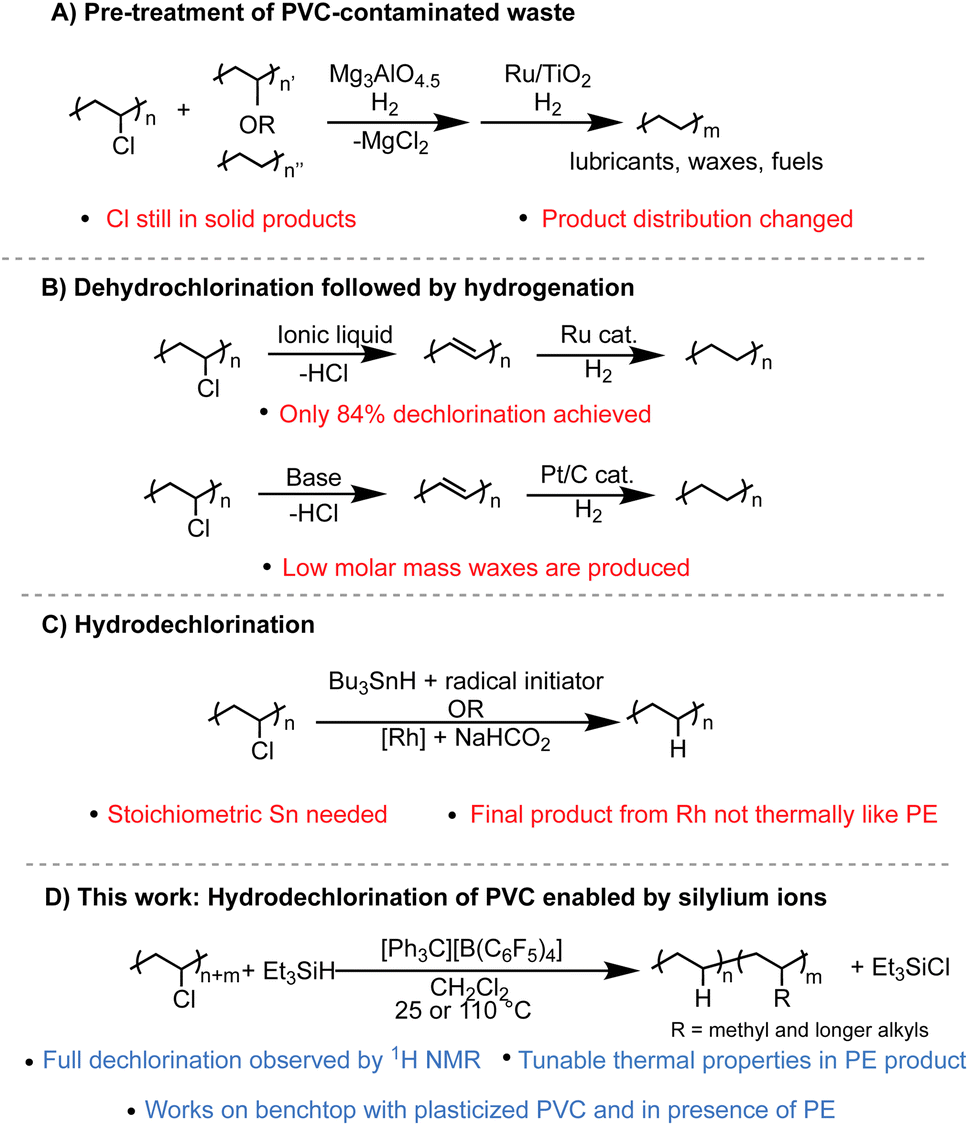 | ||
| Fig. 1 Summary of different methods (A–D) to dechlorinate PVC to PE-like materials. | ||
We therefore sought to develop a system in which PVC can be dechlorinated fully to PE-like products, while also being purified of any reactants and matching the thermal properties of PE. Our recent report on using silylium ions generated in situ to dechlorinate PVC rapidly (5 minute reaction time) was a new strategy to convert PVC waste to polymers (in this case poly(ethylene-co-styrene) copolymers) with the potential for higher value applications.8 To form these copolymers, tandem hydrodechlorination/Friedel–Crafts reactivity is hypothesized. While full dechlorination is observed by 1H nuclear magnetic resonance spectroscopy (NMR), the resulting polymers cannot be recycled with PE due to the poly(styrene) (PS) moiety. Considering silylium ions were able to rapidly dechlorinate PVC in arene solvents, we questioned if the reaction conditions could be modified to fully dechlorinate PVC to PE as a proof-of-concept in mixed waste treatment before recycling. Not only would this furnish fully dechlorinated products, but Et3SiCl is also generated, which is much more benign than HCl and is a valuable chemical and a convenient starting point for many useful silicon containing compounds.21 However, the hydrodechlorination/Friedel–Crafts strategy was hypothesized to work so well due to the polymer products being soluble in the aromatic solvent, allowing for all the chlorine to be reached.8 Molecular analogues often use neat substrate, which would not be an option for PVC. With a very distinct solubility difference between PVC and PE, it was unclear if a method could be optimized to easily reach all chlorine in the polymer.
In regard to this goal, a recent report was published by Carta et al. that utilizes Lewis-acidic cations (Si+ and Zr+) to hydrodechlorination PVC up to 91%.22 By using a bromobenzene as the solvent, Friedel–Crafts reactivity is supressed and hydrodechlorination is the favored reaction. However, the polymer product is ill-defined, as many unwanted side reactions such as dehydrochlorination, chain scission, and cross linking are proposed to occur. Additionally, this report highlights how difficult it is to achieve >99% hydrodechlorination of PVC without the presence of deleterious side reactions.
Herein, we present the formation of branched PE products generated from the complete or near complete dechlorination of PVC in as little as two hours and as mild as room temperature (Fig. 1D). Adjusting reaction conditions was found to alter the production of PE with tunable degrees of branching, and therefore thermal properties, suggesting this process could work in the presence of different PE waste streams. Branched PE polymers have been of great interest, especially for use as thermoplastic elastomers, and are traditionally synthesized through metal catalyzed polymerization of ethylene and/or other olefins. This work represents a new pathway to achieve branched PE products from waste PVC material. Additionally, this method with silylium ions is remarkably robust to common plasticizers such as phthalate esters. Finally, the dechlorination of PVC in the presence of other plastics is also shown as a proof-of-concept of repurposing of PVC-contaminated waste streams.
Results and discussion
Solvents were first screened that would enable hydrodechlorination without the presence of Friedel–Crafts reactivity (Scheme 1). Initial experiments were performed with 0.5 mol% of an initiator ([Ph3C][B(C6F5)4]) and 1.1 equivalence of Et3SiH at 110 °C for 18 h. Tetrahydrofuran (THF), dimethylacetamide (DMA), and cyclopentyl methyl ether (CpOMe) were all chosen first due to precedence for their ability to solubilize PVC. THF did not show any conversion of PVC to PE, which is unsurprising due to [Ph3C][B(C6F5)4] being able to ring-open polymerize THF.23 DMA and CpOMe also did not show any dechlorination, which is hypothesized to be due to the heteroatoms present forming strong interactions with silylium ions, rendering the catalyst in an unreactive dormant state.24,25 Attempts in non-polar, weakly coordinating solvents, such as hexanes and methylcyclohexane, showed little-to-no conversion observed by FT-IR spectroscopy even after 18 h at 110 °C. This can likely be due to the observation that no PVC dissolved in the solvent during the reaction time.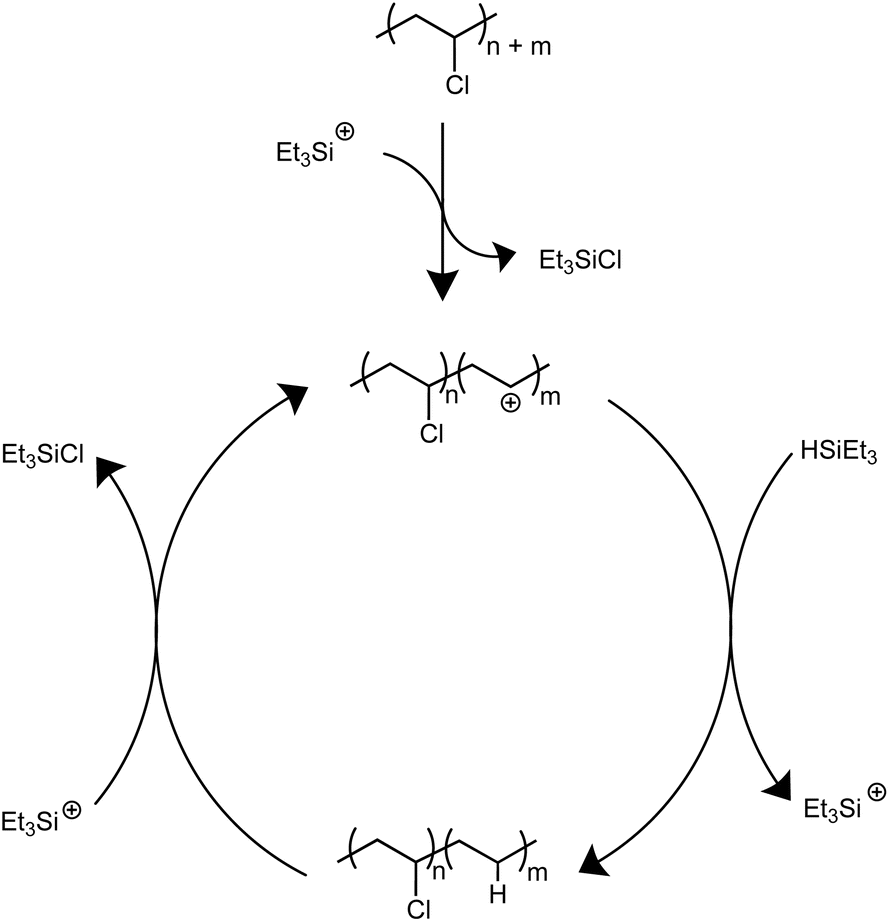 | ||
| Scheme 1 Proposed mechanism for dechlorination of PVC mediated by silylium ions. | ||
Dechlorination in the presence of dichloromethane (CH2Cl2),26 at 110 °C, showed >99% dechlorination after 18 h (by 1H NMR spectroscopy), see ESI† for details.27–33Scheme 1 represents the proposed mechanism for the hydrodechlorination of PVC mediated by silylium ions, which is based on the work of Ozerov and coworkers for the dechlorination of small molecule substrates.34 Gratifyingly, this reaction was performed on the benchtop with no drying needed of the PVC, Et3SiH, or CH2Cl2 used for the reaction. The [Ph3C][B(C6F5)4] can initiate the reaction even when stored in a desiccator on the benchtop for days, and the Et3SiH is still an active hydride source even when stored exposed to normal atmosphere. This is surprising given the highly Lewis-acidic nature of the presumed silylium ions being generated, and past researchers noting the necessity to rigorously dry all solvents and reagents in order to achieve high degrees of dechlorination.24,25,34 Even more surprising is the stability of the reaction in CH2Cl2, as past methods have mentioned activation of the C–Cl bond when generating silylium ions. CH2Cl2 activation in prior literature occurs with more activated (e.g., more Lewis acidic) silylium ions that are sterically hindered, while our system likely never forms large concentrations of bare silylium ions due to excess Si–H present that forms [Si–H–Si]+ dimers, or silylium ions coordinated to CH2Cl2, both of which are less Lewis acidic. To test if the CH2Cl2 is dechlorinated to form CH3Cl or CH4, control reactions were performed in a Parr reactor, in which pressure build-up could be monitored. These reactions confirmed no evidence for CH2Cl2 activation (see ESI for more details, Table S7†).
Further investigation of the 1H NMR of the polymer product shows the C–H resonance of the C–Cl bond in PVC is insignificant and integrating it (relative to the CH2 repeat unit of the PE repeat unit) gives a >99% dechlorination value. Elemental analysis was also performed on the resulting PE product, in addition to pure high-density polyethylene (HDPE) from Sigma Aldrich, but even experimental data from pure PE was not in agreement with theoretical (Table S6†). Therefore, 1H NMR spectroscopy was primarily used to determine percent dechlorination in the resulting PE products from PVC. For this study, full dechorination is defined as >99%, which is when no C–H associated with the C–Cl of PVC can be observed in the 1H NMR and FT-IR spectra.18 Combination of 1H NMR spectroscopy, FT-IR spectroscopy, and thermogravimetric analysis (TGA) can all be used to distinguish chlorine still remaining in the polymer product (Fig. 2).
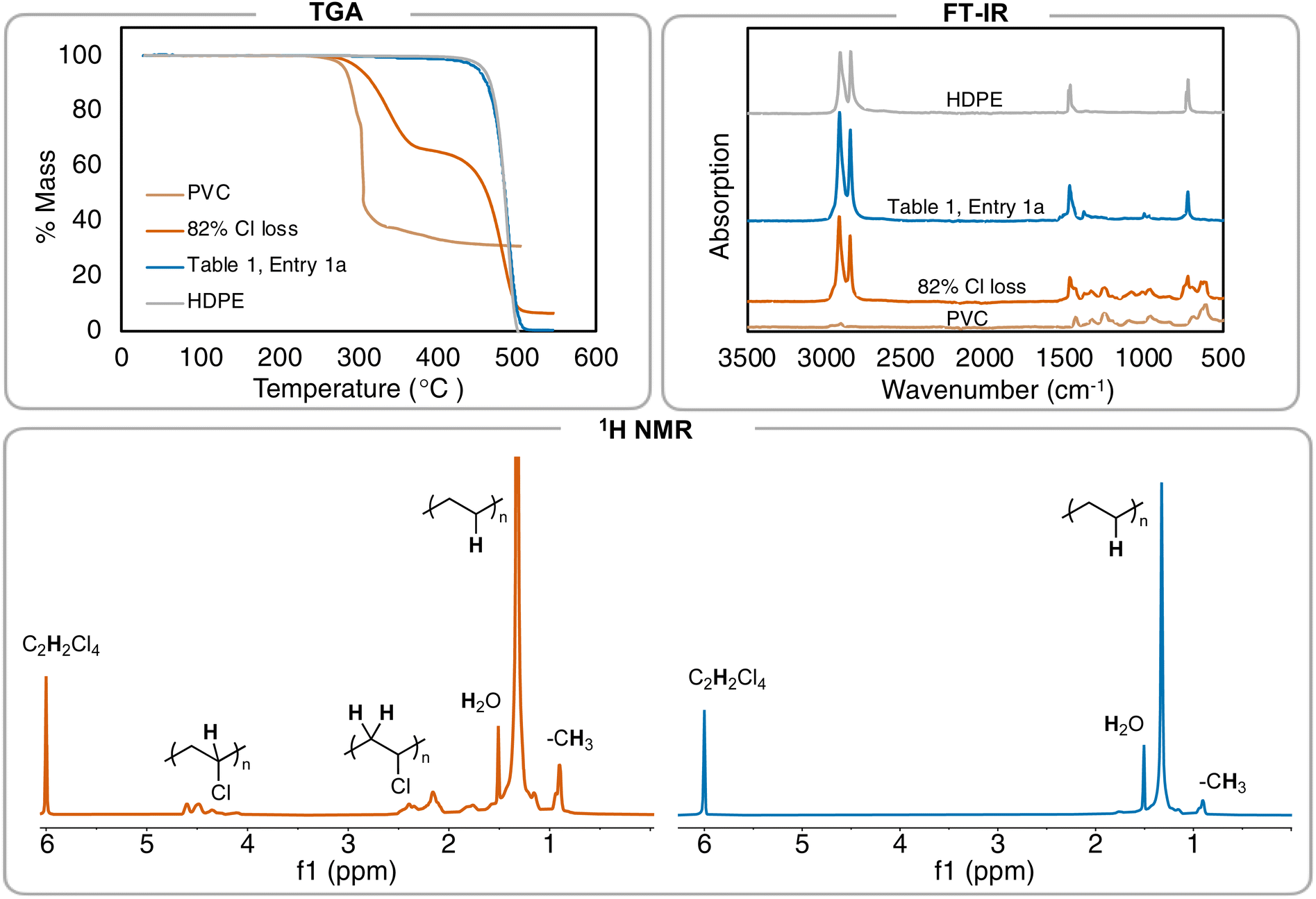 | ||
| Fig. 2 Summary of different methods to analyze branched PE formed from PVC. TGA of the resulting products (performed with a scan rate of 10 °C min−1). FT-IR spectra of varying amounts of PVC, PE, and the resulting 82% and >99% dechlorinated products. 1H NMR spectra of 82% dechlorinated PVC (left) vs.Table 1, entry 1 (right). Peak at 6.0 ppm is tetrachloroethane-d2. | ||
To ensure full dechlorination, the reaction was performed for 2 hours in slight excess of Et3SiH (1.2 equivalence to PVC) and a higher initiator loading (0.8 mol% vs. 0.5 mol%) to achieve no observable 1H NMR resonance ∼4.00 ppm (in d2-tetrachloroethane) (Table 1, entries 1a–b). FT-IR spectroscopy confirms near full dechlorination, as the vibration ∼600 cm−1 disappears.35,3629Si and 19F NMR spectroscopy were performed to confirm no contamination of the initiator and any by-products of the reaction (Fig. S61b and c†). A near quantitative isolated yield is achieved for this conversion of PVC to PE, which is a significant increase from other reports only able to achieve up to 69% yield of dechlorinated oils and waxes that are rid of Cl.4
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Time (h) | Init.b (mol%) | Et3SiHc (equiv.) | % Cl loss (1H NMR)d | B/1000 Ce | M n (kDa) | Đ | T m (°C) | T d,5% (°C) | Isolated yield (%) |
| a Entries 1a–2b and performed in 3 mL of CH2Cl2. Entries 2a–6b were performed in 9 mL of CH2Cl2. b Initiator mol% relative to moles of PVC. c Et3SiH equivalence relative to PVC. d NMR performed in tetrachloroethane-d2 at 80 °C. e Calculated by 1H NMR at 80 °C in tetrachlorethane-d2. f Performed at 140 °C in trichlorobenzene. Mark–Houwink constants of HDPE (k = 3.23 × 102 mL g−1, a = 0.735) were applied as a correction. Instrument calibrated with polystyrene standards using RI detector.42 g DSC performed at a scan rate of 10 °C min−1. Tm measured in second heating cycle. h TGA performed and Td,5% calculated from a heating rate of 10 °C min−1. | ||||||||||
| 1a | 2 | 0.8 | 1.2 | >99 | 49 | 13 | 2.4 | 56 | 444 | >99 |
| 1b | 57 | 12 | 3.1 | 50 | 450 | 81 | ||||
| 2a | 2 | 0.8 | 2.4 | >99 | 47 | 21.8 | 3.1 | 67 | 443 | 69 |
| 2b | 40 | 18.7 | 2.2 | 70 | 441 | 96 | ||||
| 3a | 2 | 0.8 | 3.6 | >99 | 47 | 18 | 2.9 | 62 | 450 | 87 |
| 3b | 46 | 27.1 | 2.6 | 68 | 445 | 84 | ||||
| 4a | 2 | 0.8 | 6 | >99 | 41 | 24.3 | 2.7 | 81 | 453 | 89 |
| 4b | 37 | 36.1 | 2.4 | 85 | 442 | 58 | ||||
| 5a | 2 | 0.8 | 12 | >99 | 31 | 51.1 | 2.3 | 93 | 450 | 94 |
| 5b | 36 | 40.1 | 2.7 | 90 | 444 | 34 | ||||
| 6a | 2 | 0.8 | 6 | >99 | 48 | 13.3 | 2.3 | 67 | 449 | >99 |
| 6b | 39 | 21 | 2.5 | 82 | 446 | 69 | ||||
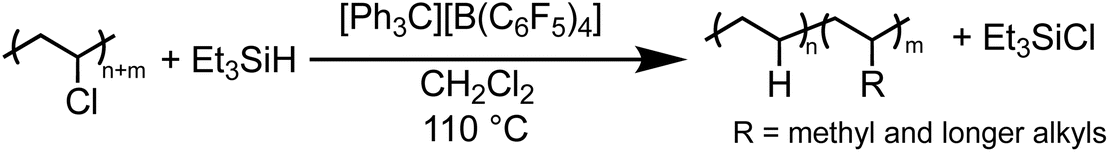
1H NMR analysis indicates 49 branches per 1000 carbons (49/1000 C),37,38 which is much higher than what PVC is suggested to have (∼2–4/1000 C) based on prior reports.18,39 This additional branching can be due to carbocation rearrangement of the PE chain, which would produce methyl (and other alkyl) branches along the polymer backbone (Scheme 2A).40,4113C NMR analysis of a resulting PE product (dechlorination = 94%) reveals 81% of branches are methyls, 14% are ethyls, and the remaining 5% are propyl and longer (Fig. S169†). Vibrations of branching in PE are also consistent with those reported in past literature.43 Hydrodechlorination using silylium ions on small molecules have exhibited carbocation rearrangement, supporting this can happen on larger molecules, such as organic polymers.441H NMR spectroscopy also indicates a very minor presence of aromatic and alkene environments <1% compared to the CH2 repeat unit of PE. Alkene resonances can appear through chain scission events caused by carbocation rearrangements (Scheme 2B), and aromatic resonances can appear through further degradation involving the double bonds.6,32,45 Weak C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C frequencies ∼1500 cm−1 are also present in the FT-IR spectrum, supporting the chemical shifts seen in 1H NMR spectrum.46 Chain scission is further supported by the final polymer molar mass (Mn) of 13.0 kDa, acquired through high temperature gel permeation chromatography (GPC), which is lower than the theoretical molar mass of 20.4 kDa if dechlorination without chain scission of PVC occurred. Chain scission is also consistent with the increase in dispersity (Đ) (2.4 from the original, 1.7 from the PVC starting material). Importantly, the molar mass numbers were calculated in the GPC software with Mark–Houwink correction factors that have been used in studies for the synthesis of branched PE. Scrambling of the carbocation between polymer chains is also possible (Scheme 2C).47 To test if chain scission was consistent between runs, a duplicate reaction was performed (Table 1, entry 1a vs. 1b). Only slight variation in the molar mass and dispersity of the resulting product can be seen. These results suggest that while carbonation rearrangements occur quickly, reasonable repeatability can be obtained.
C frequencies ∼1500 cm−1 are also present in the FT-IR spectrum, supporting the chemical shifts seen in 1H NMR spectrum.46 Chain scission is further supported by the final polymer molar mass (Mn) of 13.0 kDa, acquired through high temperature gel permeation chromatography (GPC), which is lower than the theoretical molar mass of 20.4 kDa if dechlorination without chain scission of PVC occurred. Chain scission is also consistent with the increase in dispersity (Đ) (2.4 from the original, 1.7 from the PVC starting material). Importantly, the molar mass numbers were calculated in the GPC software with Mark–Houwink correction factors that have been used in studies for the synthesis of branched PE. Scrambling of the carbocation between polymer chains is also possible (Scheme 2C).47 To test if chain scission was consistent between runs, a duplicate reaction was performed (Table 1, entry 1a vs. 1b). Only slight variation in the molar mass and dispersity of the resulting product can be seen. These results suggest that while carbonation rearrangements occur quickly, reasonable repeatability can be obtained.
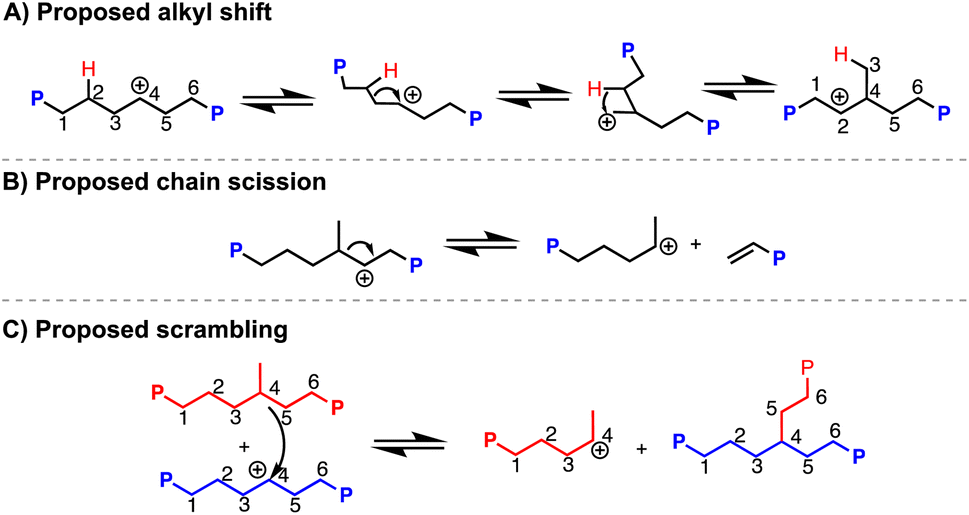 | ||
| Scheme 2 Proposed mechanisms of carbocation rearrangement (alkyl shift (A), chain scission (B), scrambling (C)) along polymer backbone in dechlorination of PVC mediated by silylium ions. | ||
To confirm the branching content, differential scanning calorimetry (DSC) analysis was performed to measure the melting point of the PE product (Table 1 and Fig. 3). The resulting PE product has a melting point (Tm) = 56 °C, which agrees with a relatively high branching content calculated by 1H NMR spectroscopy. For comparison, HDPE (Sigma Aldrich) has a Tm = 128 °C (Fig. S75†). The melting point depression is due to the short chain branches (which are likely placed randomly in this case) causing the PE to become less crystalline and more amorphous.37,38,48 When compared to reports with primarily methyl branching on the PE backbone, the Tm is lower than expected, further supporting the presence of longer alkyl branches (see ESI† for details). Regarding a duplicate run, the similar number of branches is achieved with the same conditions (57 B/1000 C) and a similar Tm as well (50 °C vs. 56 °C).
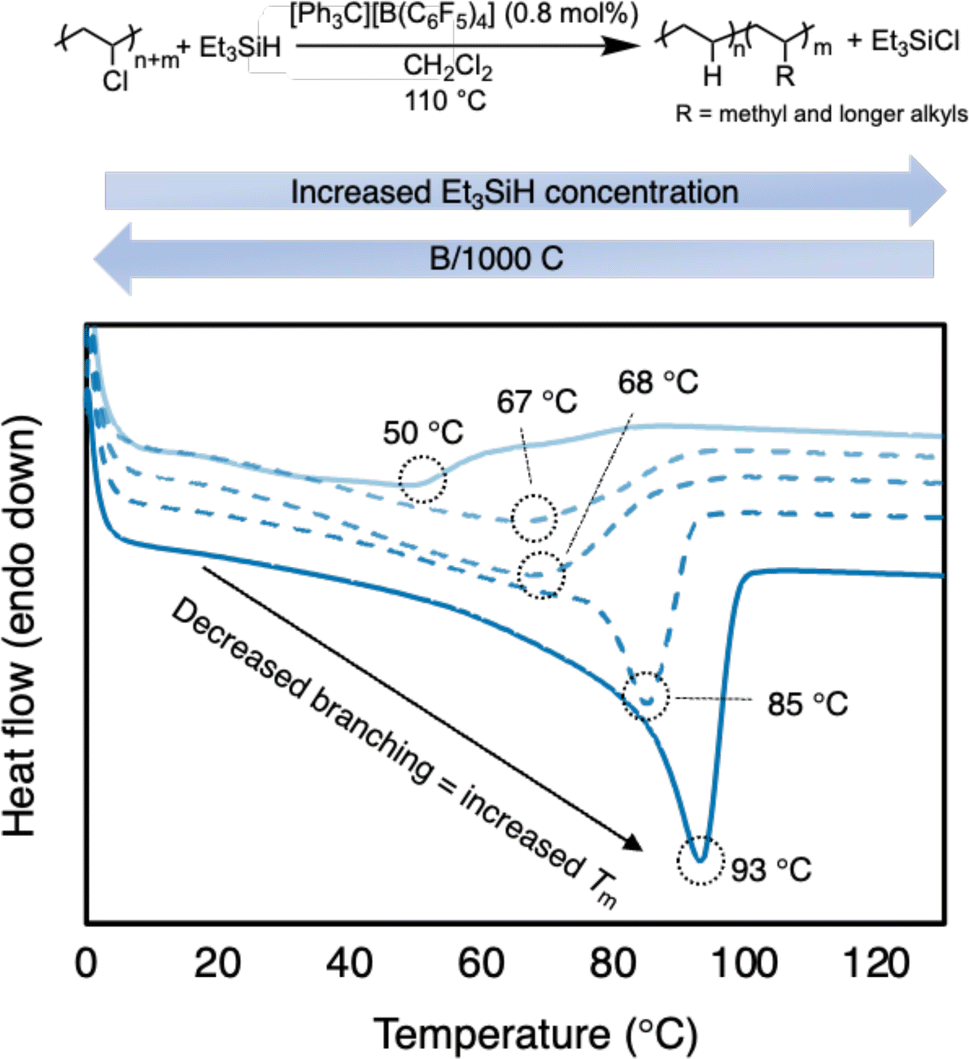 | ||
| Fig. 3 Plot of second heating curve of resulting branched PE products from Table 1 (entries 1b, 2a, 3b, 4b, and 5b). DSC traces offset on y-axis for clarity. | ||
Additionally, the resulting PE product displays a Td,5% = 444 °C, which is near virgin HDPE from Sigma Aldrich (Td,5% = 457 °C) and matches that from other literature reports for PE polymers (Fig. 2A). A sample with 82% dechlorination by 1H NMR gives a Td,5% = 302 °C, which is noticeably lower than the sample with >99% dechlorination. These data indicate that the Td,5% of PE from PVC is a useful secondary measure measurement that can be used to see if Cl content is still in the polymer. TGA is secondary to NMR in quantifying the amount of Cl left, as a duplicate reaction (Table 1, entry 1b) that reached full dechlorination by 1H NMR has a Td,5% of 450 °C, suggesting there is variability in the TGA of individual samples.
To tune the degree of branching in the resulting PE, it was hypothesized that performing the reaction in excess Et3SiH would favor hydride transfer from Et3SiH rather than carbocation rearrangement (Scheme 2). Indeed, as the Et3SiH loading was increased (Table 1, entries 2–5), the degree of branching was decreased. Satisfyingly, the branch content can be lowered to 31/1000 C (Table 1, entry 5a). The resulting PE displays a Tm of 93 °C. The increase in Tm with subtle decreases in branching content can likely be attributed to the presence of more methyl branches and fewer longer chain branches, which are known to impact the melt temperature more (Fig. 3).49 However, even at 12 equivalence of Et3SiH to PVC, carbocation rearrangement is still difficult to inhibit. This is likely due to carbocation rearrangement existing in a rapid equilibrium. Any reagents that can trap carbocations (e.g., nucleophiles) will also likely trap the highly Lewis-acidic silylium ions, rendering the reaction dormant.24,25 It should be noted that the PVC materials were less soluble in conditions with higher Et3SiH content, requiring additional solvent (Table 1, entries 3–5).
Lowering the temperature of the reaction to 25 °C was also tried under more dilute conditions of Table 1, entries 1a–b (e.g., 9 mL of CH2Cl2 was used instead of 3 mL). However, complete dechlorination was difficult to achieve, with only up to 87% being achieved even at longer reaction times of 72 hours (ESI Table S6,† entries 2a–b). Using the concentrated conditions, directly analogous to Table 1, entries 1a–b, and run for 18 hours at 25 °C, only one reaction reach full dechlorination. While full dechlorination was achieved in one run, only 92% dechlorination was observed in the second and third runs (Table S4,† entries 1a–c). This inconsistent conversion is hypothesized to be due to the insolubility of PVC and PE in CH2Cl2 at lower temperatures, and also being below the melting point of the resulting polymer products, making it difficult for silylium ions to reach all Cl atoms in the polymer backbone.
The initiator loading was also increased from 0.8 mol% to 1.6 mol%, and the branching content was subsequently increased from 31 to 40 B/1000 C (Table 1, entry 6). This increase in branching could be due to more cations being generated in solution, leading to more rearrangement.
Across duplicate reactions, the branching content, Tm, and Td,5% all stayed quite close to each other. The molar mass of the polymer did have variability between runs, which could indicate that chain scission reactions are not entirely controllable.
Dilution of the reaction from Table 1, entries 4a–b, did not change the branching in the product and therefore the Tm was the same (Table S3,† entries 5a–b). However, The 1H NMR spectrum of the dilute conditions indicates no arene or alkene environments, suggesting little chain scission happens with excess Et3SiH and dilute conditions.
Since Cl accounts for 57 wt% of PVC, it is important to determine its fate in these reactions. When the reaction from Table 1, entries 2a–b is characterized by 1H NMR spectroscopy before quenching, it can be seen that there is a resulting 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of Et3SiCl:Et3SiH in the reaction, with a minor presence of hydrolyzed products (Fig. S171†).50 This is exciting given that near quantitative conversion of Et3SiH to Et3SiCl is seen, which means that the Cl atoms are going to a valuable source in addition to the PVC backbone forming a valued product.
:50 mixture of Et3SiCl:Et3SiH in the reaction, with a minor presence of hydrolyzed products (Fig. S171†).50 This is exciting given that near quantitative conversion of Et3SiH to Et3SiCl is seen, which means that the Cl atoms are going to a valuable source in addition to the PVC backbone forming a valued product.
In most cases, additives are considered a concern for the lifetime or stability of a catalyst. PVC is rarely used in its pure form commercially, as plasticizers (sometimes >50 wt% of the product) are added to give PVC properties for different applications.51–54 In particular, many additives have polar functional groups that could react with the silylium cations, triethyl silane or the trityl initial catalyst. Therefore, reactions were performed on four commercial items that varied in appearance and flexibility (including a colorful toy lizard, rigid PVC plumbing pipe, flexible and colorless PVC tubing, and a vinyl record, Fig. 4). Reactions were performed with the four items used without purification, other than to break the items into smaller pieces in a coffee grinder or with scissors. Using the same optimized conditions from Table 1, entries 4a–b (assuming the items were 100% PVC), near complete hydrodechlorination was realized for all four items (Table 2 and Fig. 4).
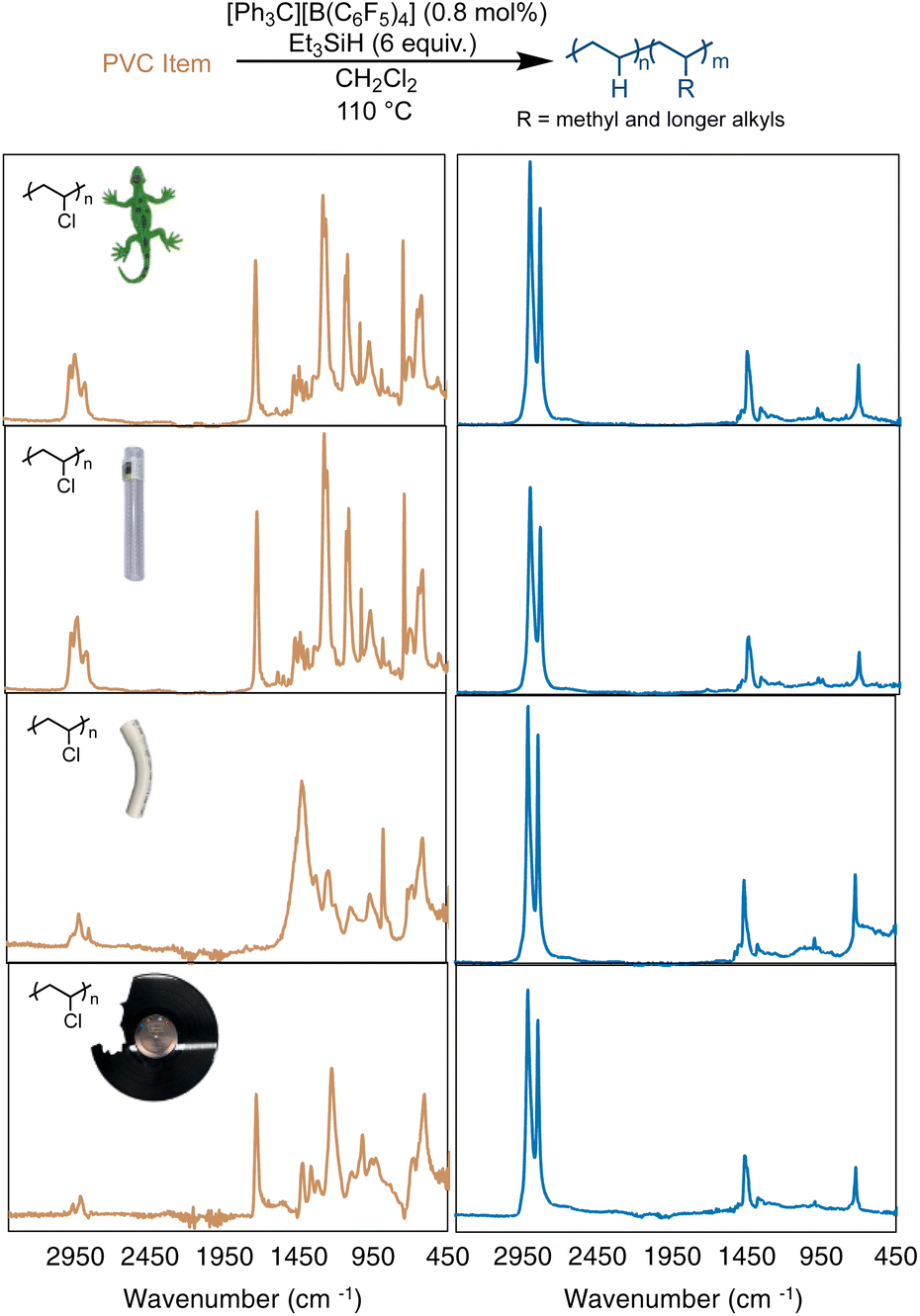 | ||
| Fig. 4 Dechlorination of a toy lizard, flexible PVC pipe, rigid PVC pipe, and used vinyl record (top-to-bottom), as measured by FT-IR spectroscopy. | ||
|
|
|||||
|---|---|---|---|---|---|
| PVC item | % Cl lossa (1H NMR) | B/1000 Cb | M n (Đ) | T m (°C) | T d,5% (°C) |
| a NMR performed in tetrachloroethane-d2 at 80 °C. b Calculated by 1H NMR spectroscopy at 80 °C in tetrachlorethane-d2. c Performed at 140 °C in trichlorobenzene. Mark–Houwink constants of HDPE (k = 3.23 × 102 mL g−1, a = 0.735) were applied as a correction. Instrument calibrated with polystyrene standards using RI detector. d DSC performed at a scan rate of 10 °C min−1. Tm measured in second heating cycle. e TGA performed and Td,5% calculated from a heating rate of 10 °C min−1. f Pure PVC from Table 1, entry 4a. | |||||
| Toy lizard | >99 | 53 | 9.5 (2.1) | 65 | 431 |
| Extracted toy lizard | >99 | 34 | 26.7 (4.1) | 83102 |
451 |
| Rigid PVC pipe | 98 | 42 | 26.9 (2.4) | 82 | 434 |
| Soft PVC pipe | >99 | 58 | 4.6 (3.2) | 42 | 386 |
| Vinyl record | >99 | 55 | 4.6 (2.1) | 69 | 437 |
| Extracted vinyl record | >99 | 55 | 4.1 (2.3) | 76 | 441 |
| Pure PVCf | >99 | 41 | 24.3 (2.7) | 81 | 453 |
As seen in Fig. 4, all 4 PVC items were converted to PE-like products, based on FT-IR spectroscopy, with all items reaching >98% dechlorination by 1H NMR. This result was particularly exciting, as many of the PVC items showed a vibration at 1715 cm−1, which is indicative of a carbonyl stretching mode of an ester. For example, DEHP (di(2-ethylhexyl)phthalate) exhibits a carbonyl absorption also at 1725 cm−1.551H NMR analysis (of the soluble fraction of the flexible PVC pipe) reveals one aromatic environment, indicative of a plasticizer more like bis(2-ethylhexyl) terephthalate (DEHT). As mentioned before, highly Lewis-acidic silylium ions are likely being formed in this reaction, so the observation that the reaction is not halted by the interaction of carbonyl and ester moieties of any plasticizers is unexpected. This is even more surprising given the fact that all the PVC items used in Fig. 4 were not purified or dried before the dechlorination, and likely retained some moisture. Surprisingly, the vinyl record showed no remaining Cl in NMR or FT-IR studies. Although rigid PVC pipe showed the least dechlorination (98.3%), the ability to achieve that high of dechlorination was unexpected, due to the large amount of additives and low solubility of this material. These results show that silylium ions are surprisingly robust to plasticizers and additives in commercial PVC items.
While near full dechlorination of almost all PVC items in Fig. 4 was achieved, the resulting polyethylene products differed quite greatly. Depending on the item selected, there were different degrees of calculated branching, with all items leading to more branching in the PE product than that of the pure PVC. Both the toy lizard and vinyl record produced PE materials with >50 branches per 1000 C, resulting in Tm's of 65 and 69, respectively. Rigid PVC pipe produced a PE product with 42 branches per 1000 C, leading to a higher Tm of 82 °C. A hypothesis for this observation is that there are a lot of additives by weight in rigid PVC pipe, making the Et3SiH loading higher than 6 equivalence, which was shown previously to decrease the branches in Fig. 3.
For comparison, PVC was extracted from the toy lizard and vinyl record, with additives separated, to identify if the additives influence the reaction. Upon using the same reaction conditions as Table 1, entries 4a–b (still assuming the materials are 100% PVC), near full dechlorination was observed for both purified PVC items. Notably, the vinyl record showed a slightly raised Tm measured by DSC (76 °C vs. 69 °C), yet the PVC extracted from the lizard showed a signficantly higher Tm (83 °C vs. 65 °C), when compared to the product from the item directly (Fig. S64 and 65†). The product from the reaction with the extracted PVC from the lizard also showed a second thermal transition at 102 °C, which could be due to insolubility of the PVC until a few hours into the reaction. These data suggest that depending on the PVC item being dechlorinated, the final properties of the polymer can be greatly impacted by additives/plasticizers present.
The dechlorination of PVC to PE in the presence of other plastics was also pursued in efforts to simulate what would happen in a plastic waste stream (Fig. 5). Dechlorination of PVC was attempted in the presence of poly(ethylene terephthalate) (PET) or PS to first test the compatibility of the reaction conditions with other plastics. The reaction in the presence of PET did not yield any dechlorination by FT-IR spectroscopy, which is hypothesized to be due to the high concentration of ester bonds in PET. This is surprising, as the method seemed to be robust to the ester-containing plasticizers in the commercial items. Dechlorination of PVC in the presence of PS yielded a very insoluble polymer product. FT-IR and 1H NMR analysis indicate presence of arenes in the polymer product, which supports cross linking between the C–C backbone of the PVC chain and the phenyl rings on PS, yielding cross-linked PS. This can be possible due to Friedel–Crafts alkylation of the carbocations generated on the PVC backbone, which is supported by past silylium ion chemistry on small molecules and PVC.8 Nonetheless, FT-IR and 1H NMR spectroscopy identify a significant loss of Cl in the presence of PS.
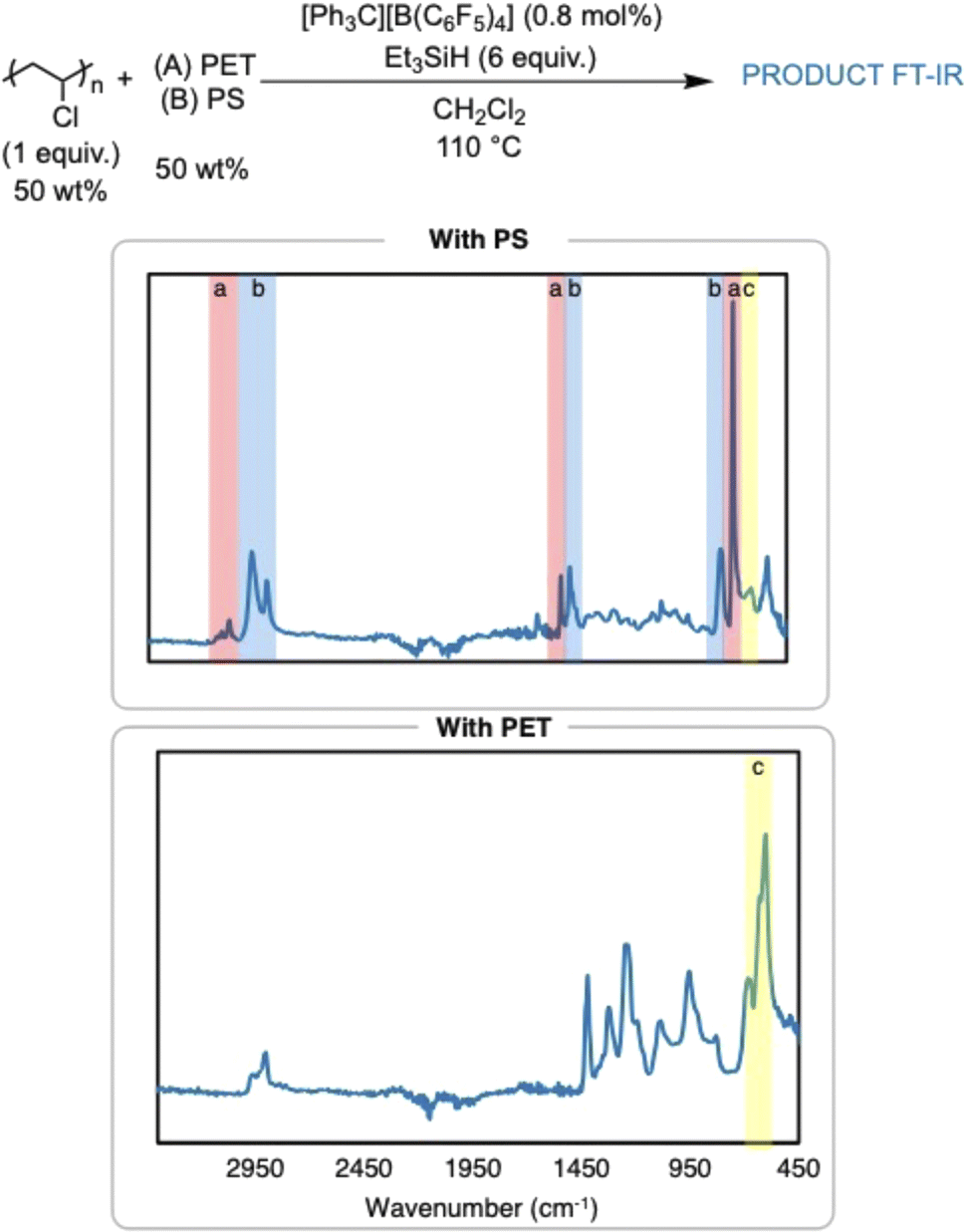 | ||
| Fig. 5 FT-IR spectra of PVC dechlorination in the presence of 50 wt% PET, and 50% PS (top-to-bottom): highlights are used to label (a) (red highlight) PS stretches, (b) (blue high-light) PE stretches, and (c) (yellow highlight) C–Cl stretch. | ||
When dechlorination of PVC was attempted in the presence of PE (4 wt% of PVC, 96 wt% PE) to mimic the common impurity of PVC in PE waste, the reaction yielded near fully dechlorinated PVC (>99% Cl loss by 1H NMR) (Fig. 6). The reaction was performed in CH2Cl2, and the CH2Cl2 fraction was separated from the insoluble PE beads. The resulting CH2Cl2 fraction was purified (see ESI† for details), and the PE product was isolated in a 108% yield. In the reaction with pure PVC, a branching degree of 31 B/1000 C was observed and a Tm = 84 °C (Table 1, entry 4a). The PE achieved from the dechlorination in the presence of excess PE had a branching content of 32 B/1000 C, but a Tm = 91 °C. A very minor thermal transition at 123 °C is also observed in this sample. When the reaction conditions from Fig. 6 were replicated without the presence of HDPE beads, a similar molar mass and dispersity was achieved compared to Table 1, entries 4a–b. To probe whether the PE product and HDPE phase separates, PE from reaction Table 1, entry 4a was analyzed by DSC in the presence of HDPE (83 wt% vs. 17 wt%, respectively). The DSC trace shows two distinct melting points, with a Tm = 84 °C and Tm = 126 °C (Fig. S79†). These data show that the products obtained from mixed PVC/PE waste are separable, leading to pure PE products with different thermal properties. More importantly, the silylium ions generated in situ are able to dechlorinate PVC in reaction conditions that are dilute, which are like what recycling streams would see with small PVC impurities.
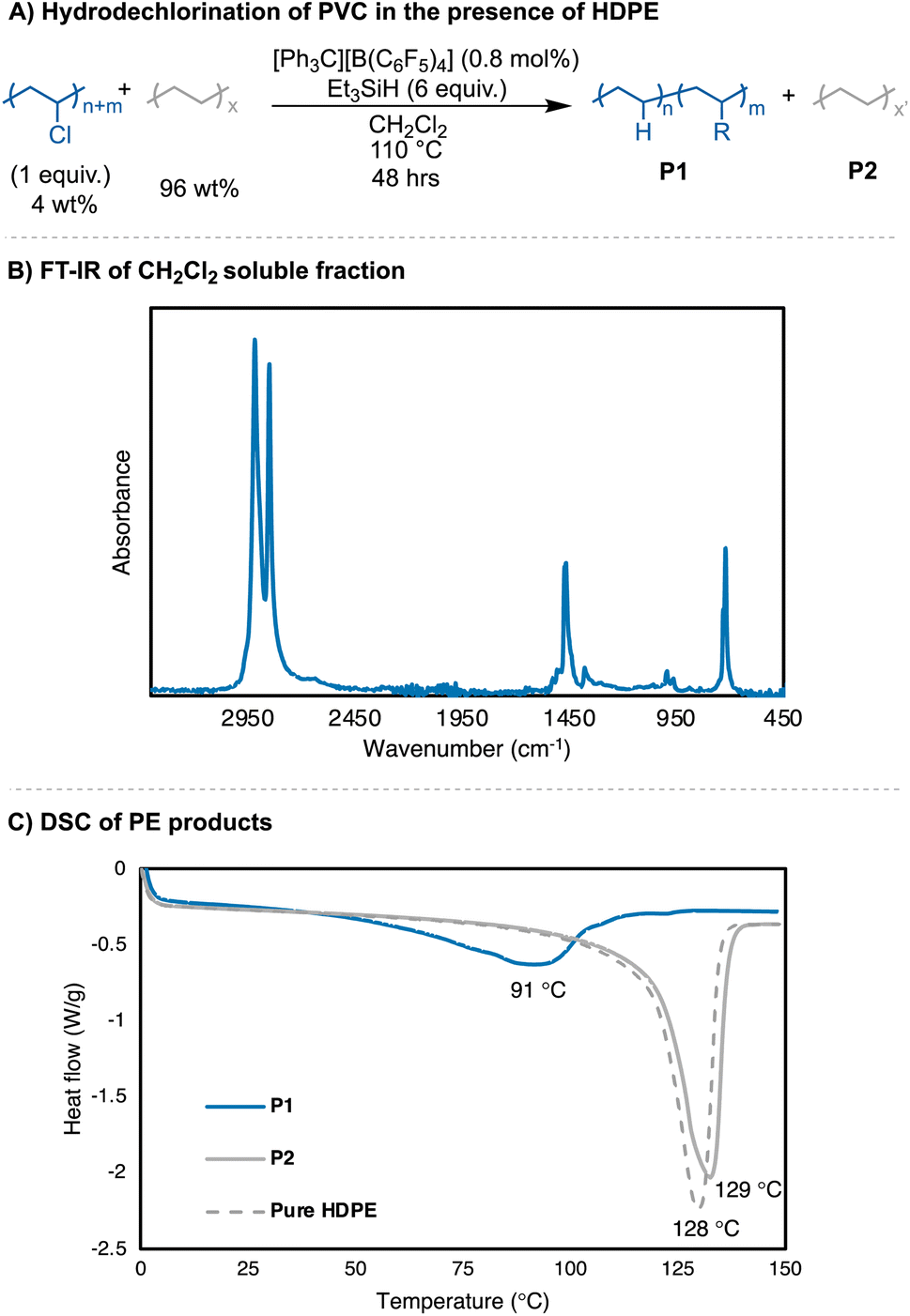 | ||
| Fig. 6 (A) Reaction conditions for PVC hydrodechlorination in the presence of HDPE; (B) FT-IR spectra of PE product; (C) DSC traces of PE products from mixed PVC/PE waste. CH2Cl2 soluble fraction has a Tm = 91 °C, with very little phase separation. | ||
Conclusions
We have demonstrated the ability to fully dechlorinate PVC to PE within two hours products by generating silylium ions in situ. By varying Et3SiH concentration, the degree of branching, and therefore thermal properties, can be tuned of the resulting PE product. No corrosive products are formed, with useful Et3SiCl being the chlorine-containing product, which is sold at a higher price than the silane precursor. Although not considered a great solvent for PVC or PE, dichloromethane was found to be the only solvent that could support dechlorination above 99%. Branched polyethylene products can be characterized fully, with thermal properties providing key insight into the degree and selectivity of hydrodechlorination. Importantly, this reaction is robust to common plasticizers and additives found in commercial PVC items, and full dechlorination can even occur in the presence of PE waste. This dechlorination strategy offers a controlled way of converting PVC contaminated PE waste to useful products that can be used for upcycled applications or also passed on to pyrolysis without the worry of corroding reactors, decomposing catalysts or altering the product distribution.Data availability
All relevant datasets supporting this article including general considerations for chemicals, solvents, reaction conditions and results, characterization data (including 1H, 13C, 19F, and 29Si NMR spectra, FT-IR spectra, DSC curves, and TGA curves) are available in the ESI.†Author contributions
Z. A. W., E. C. C., and A. N. N. contributed to experimental work, devising experiments, and writing the manuscript. M. E. F. directed the project, and helped write the manuscript. All authors discussed results.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported as part of the Institute for Cooperative Upcycling of Plastics (iCOUP), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under under Contract DE-AC-02-07CH11358 (Ames National Laboratory). Instrumentation in the USC Chemistry Instrument Facility was acquired with support from the USC Research and Innovation Instrumentation Award Program. Additionally, funds were provided by the National Science Foundation (DBI-0821671, CHE-0840366) and National Institute of Health (S10 RR25432) supported the acquisition of the NMR spectrometers used in our work, and polymer analysis instrumentation was supported by the National Institute of Standards and Technology (NIST Award 60NANB22D134). A. N. N. thanks the Arnold and Mabel Beckman Foundation for its research support and funding through the Beckman Scholars Program and USC for Provost Fellowships.Notes and references
- S. Xu, Z. Han, K. Yuan, P. Qin, W. Zhao, T. Lin, T. Zhou and F. Huang, Nat. Rev. Methods Primers, 2023, 3, 44 CrossRef CAS.
- M. Roosen, N. Mys, M. Kusenberg, P. Billen, A. Dumoulin, J. Dewulf, K. M. Van Geem, K. Ragaert and S. De Meester, Environ. Sci. Technol., 2020, 54, 13282–13293 CrossRef CAS PubMed.
- S. Svadlenak, S. Wojcik, O. Ogunlalu, M. Vu, M. Dor, B. W. Boudouris, D. Wildenschild and K. A. Goulas, Appl. Catal., B, 2023, 338, 123065 CrossRef CAS.
- P. A. Kots, B. C. Vance, C. M. Quinn, C. Wang and D. G. Vlachos, Nat. Sustain., 2023, 6, 1258–1267 CrossRef.
- D. E. Fagnani, D. Kim, S. I. Camarero, J. F. Alfaro and A. J. McNeil, Nat. Chem., 2023, 15, 222–229 CrossRef CAS PubMed.
- G. O’Rourke, T. Hennebel, M. Stalpaert, A. Skorynina, A. Bugaev, K. Janssens, L. Van Emelen, V. Lemmens, R. De Oliveira Silva, C. Colemonts, P. Gabriels, D. Sakellariou and D. De Vos, Chem. Sci., 2023, 4401–4412 RSC.
- D. Glas, J. Hulsbosch, P. Dubois, K. Binnemans and D. E. De Vos, ChemSusChem, 2014, 7, 610–617 CrossRef CAS PubMed.
- M. K. Assefa and M. E. Fieser, J. Mater. Chem. A, 2023, 11, 2128–2132 RSC.
- Q. Zhou, C. Tang, Y.-Z. Wang and L. Zheng, Fuel, 2004, 83, 1727–1732 CrossRef CAS.
- T. Bhaskar, R. Negoro, A. Muto and Y. Sakata, Green Chem., 2006, 8, 697–700 RSC.
- K.-B. Park, S.-J. Oh, G. Begum and J.-S. Kim, Energy, 2018, 157, 402–411 CrossRef CAS.
- T. Okada, S. Sutoh, K. Sejima, H. Tomohara and S. Mishima, Polym. Degrad. Stab., 2020, 171, 109040 CrossRef CAS.
- A. Marino, A. Aloise, H. Hernando, J. Fermoso, D. Cozza, E. Giglio, M. Migliori, P. Pizarro, G. Giordano and D. P. Serrano, Catal. Today, 2022, 390–391, 210–220 CrossRef CAS.
- L. Guo, G. Shi and Y. Liang, Polymer, 2001, 42, 5581–5587 CrossRef CAS.
- Y. Chen, S. Zhang, X. Han, X. Zhang, M. Yi, S. Yang, D. Yu and W. Liu, Energy Fuels, 2018, 32, 2407–2413 CrossRef CAS.
- P. Zhao, J. Clean. Product., 2017, 15, 38–46 CrossRef.
- P. Zhao, T. Li, W. Yan and L. Yuan, Environ. Technol., 2018, 39, 977–985 CrossRef PubMed.
- T. Hjertberg and A. Wendel, Polymer, 1982, 23, 1641–1645 CrossRef CAS.
- N. G. Bush, M. K. Assefa, S. Bac, S. Mallikarjun Sharada and M. E. Fieser, Mater. Horiz., 2023, 10, 2047–2052 RSC.
- A. E. Kynman, S. Christodoulou, E. T. Ouellette, A. Peterson, S. N. Kelly, L. Maron and P. Arnold, Chem. Comm., 2023, 59, 10924–10927 RSC.
- A. D. Beck, S. Haufe and S. R. Waldvogel, ChemElectroChem, 2023, 10, e202201149 CrossRef CAS.
- A. T. Normand, Y. Wu, T. Régnier, P. Fleurat-Lessard, Y. Rousselin, B. Théron, P. Le Gendre and M. Carta, Chem.–Eur. J, 2024, e202304005 CrossRef CAS PubMed.
- M. P. Dreyfuss, J. C. Westfahl and P. Dreyfuss, Macromolecules, 1968, 1, 437–441 CrossRef CAS.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- Z. Xie, D. J. Liston, T. Jelínek, V. Mitro, R. Bau and C. A. Reed, J. Chem. Soc., Chem. Commun., 1993, 384–386 RSC.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- A. Misono, Y. Uchida and K. Yamada, Bull. Chem. Soc. Jpn., 1966, 39, 2458–2463 CrossRef CAS.
- D. Braun, W. Mao, B. Böhringer and R. W. Garbella, Angew. Makromol. Chem., 1986, 141, 113–129 CrossRef CAS.
- N. Pourahmady, P. I. Bak and R. A. Kinsey, J. Macromol. Sci., Part A: Pure Appl.Chem., 1992, 29, 959–974 CrossRef.
- P. V. C. Rao and V. K. Kaushik*, Polym. Test., 1999, 18, 429–438 CrossRef CAS.
- A. Misono, Y. Uchida and K. Yamada, Bull. Chem. Soc. Jpn., 1970, 43, 3597–3601 CrossRef CAS.
- M.-F. Llauro-Darricades, N. Bensemra, A. Guyot and R. Petiaud, Makromol. Chem., Macromol. Symp., 1989, 29, 171–184 CrossRef CAS.
- F. C. Schilling, A. E. Tonelli and M. Valenciano, Macromolecules, 1985, 18, 356–360 CrossRef CAS.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- S. Krimm, C. Y. Liang and G. B. B. M. Sutherland, J. Chem. Phys., 1956, 25, 549–562 CrossRef CAS.
- J. M. Contreras, G. Martínez and J. Millán, Polymer, 2001, 42, 09867–09876 CrossRef CAS.
- F. A. Hicks, J. C. Jenkins and M. Brookhart, Organometallics, 2003, 22, 3533–3545 CrossRef CAS.
- O. Padilla-Vélez, K. S. O’Connor, A. M. LaPointe, S. N. MacMillan and G. W. Coates, Chem. Commun., 2019, 55, 7607–7610 RSC.
- P. d’Antuono, E. Botek, B. Champagne, J. Wieme, M.-F. Reyniers, G. B. Marin, P. J. Adriaensens and J. M. Gelan, J. Phys. Chem. B, 2008, 112, 14804–14818 CrossRef PubMed.
- D. J. S. Sandbeck, D. J. Markewich and A. L. L. East, J. Org. Chem., 2016, 81, 1410–1415 CrossRef PubMed.
- G. A. Olah, G. K. S. Prakash, A. Molnár and J. Sommer, in Superacid Chemistry, Wiley, Hoboken, NJ, 2nd ed., 2009, pp. 83–310 Search PubMed.
- L. Wild, R. Ranganath and T. Ryle, J. Polym. Sci. A-2 Polym. Phys., 1971, 9, 2137–2150 CrossRef CAS.
- F. A. Bovey, K. B. Abbås, F. C. Schilling and W. H. Starnes, Macromolecules, 1975, 8, 437–439 CrossRef CAS.
- C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed.
- C. A. Bennett and M. T. Klein, Energy Fuels, 2012, 26, 41–51 CrossRef CAS.
- C. Y. Liang and S. Krimm, J. Polym. Sci., 1958, 27, 241–254 CrossRef CAS.
- G. A. Olah and A. M. White, J. Am. Chem. Soc., 1969, 91, 5801–5810 CrossRef CAS.
- R. G. Alamo and L. Mandelkern, Macromolecules, 1989, 22, 1273–1277 CrossRef CAS.
- D. J. Lohse, S. T. Milner, L. J. Fetters, M. Xenidou, N. Hadjichristidis, R. A. Mendelson, C. A. García-Franco and M. K. Lyon, Macromolecules, 2002, 35, 3066–3075 CrossRef CAS.
- N. Almenara, S. Azpeitia, M. A. Garralda and M. A. Huertos, Dalton Trans., 2018, 47, 16225–16231 RSC.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- K. Pivnenko, M. K. Eriksen, J. A. Martín-Fernández, E. Eriksson and T. F. Astrup, Waste Manage., 2016, 54, 44–52 CrossRef CAS PubMed.
- M. Rahman and C. Brazel, Prog. Polym. Sci., 2004, 29, 1223–1248 CrossRef CAS.
- D. Lithner, Å. Larsson and G. Dave, Sci. Total Environ., 2011, 409, 3309–3324 CrossRef CAS PubMed.
- X. Zhang and Z. Chen, Langmuir, 2014, 30, 4933–4944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00130c |
| This journal is © The Royal Society of Chemistry 2024 |