
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable C–H functionalization and dearomatization enabled by an organic photocatalyst†
Bohang
An
a,
Hao
Cui
a,
Chao
Zheng
 *b,
Ji-Lin
Chen
a,
Feng
Lan
a,
Shu-Li
You
*b and
Xiao
Zhang
*a
*b,
Ji-Lin
Chen
a,
Feng
Lan
a,
Shu-Li
You
*b and
Xiao
Zhang
*a
aFujian Key Laboratory of Polymer Materials, Fujian Provincial Key Laboratory of Advanced Materials Oriented Chemical Engineering, College of Chemistry and Materials Science, Fujian Normal University, Fuzhou 350007, China. E-mail: zhangxiao@fjnu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: zhengchao@sioc.ac.cn; slyou@sioc.ac.cn
First published on 6th February 2024
Abstract
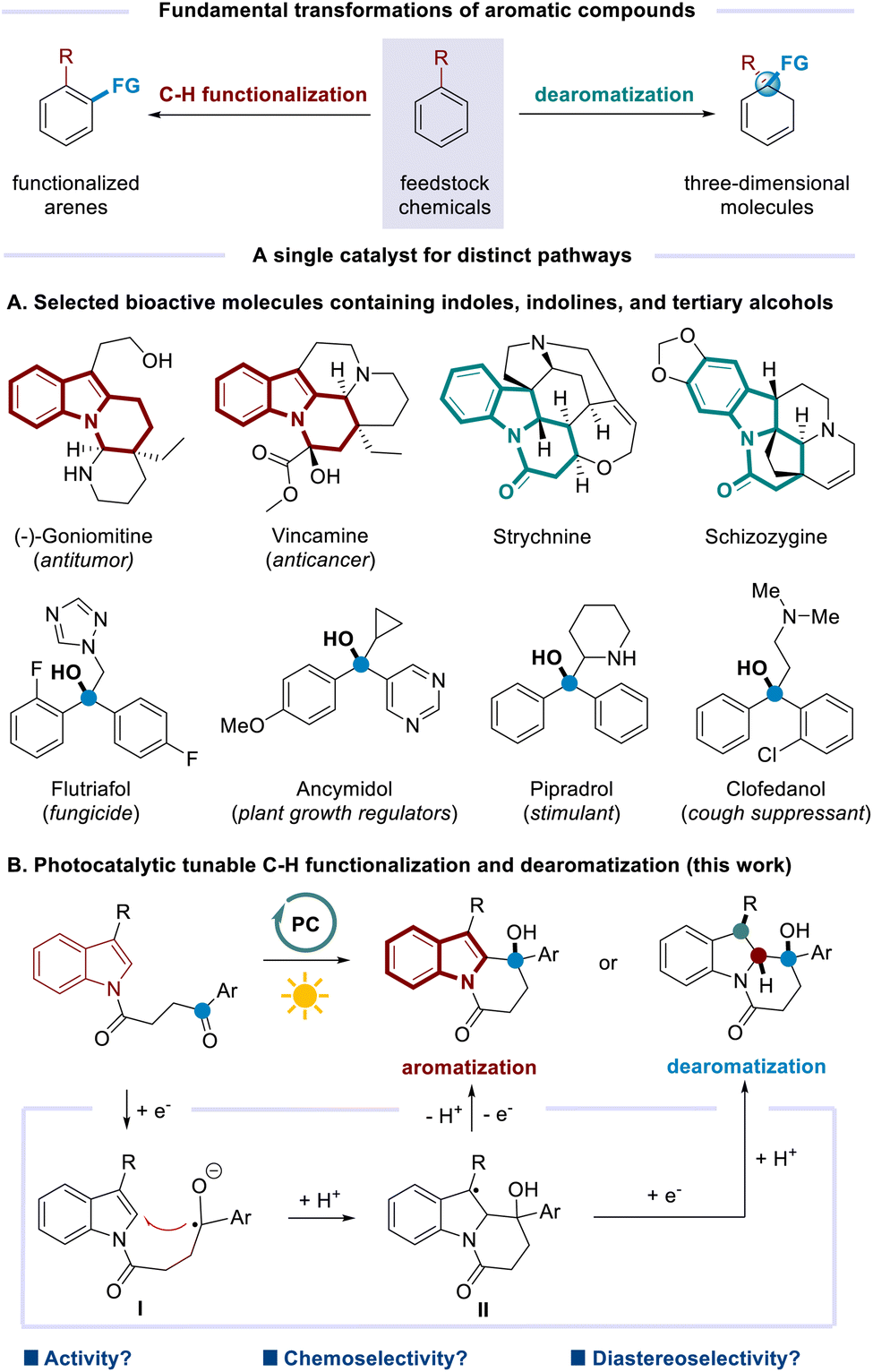
C–H functionalization and dearomatization constitute fundamental transformations of aromatic compounds, which find wide applications in various research areas. However, achieving both transformations from the same substrates with a single catalyst by operating a distinct mechanism remains challenging. Here, we report a photocatalytic strategy to modulate the reaction pathways that can be directed toward either C–H functionalization or dearomatization under redox-neutral or net-reductive conditions, respectively. Two sets of indoles and indolines bearing tertiary alcohols are divergently furnished with good yields and high selectivity. The key to success is the introduction of isoazatruxene ITN-2 as a novel photocatalyst (PC), which outperforms the commonly used PCs. The ready synthesis and high modulability of isoazatruxene type PCs indicate their great application potential.
Aromatic compounds are among the most fundamental feedstock chemicals, with global production on a million metric ton scale annually. The development of practical methods to increase the valorization of these abundant substances is thus in continuing demand. As classical transformations based on aromatic compounds, C–H functionalization such as Friedel–Crafts alkylation and Minisci-type reaction constitutes a straightforward strategy to incorporate diverse functionalities on the aromatic skeletons.1 In a complementary fashion, dearomatization converts planar aromatics into three-dimensional molecules with increased sp3-hybridized carbons and stereocenters by disturbing the aromaticity (Scheme 1, top). Notable strategies include Birch reduction, oxidative dearomatization of phenols, arene–alkene photocycloadditions, and others.2 Both types of transformations have found broad applications in areas ranging from pharmaceuticals and agrochemicals to materials science.3 Despite the great achievements made to date, the utilization of a single catalyst to promote tunable C–H functionalization and dearomatization processes from the same starting materials is underexplored. Elegant reports mainly involve the substrate control.4 The difficulties might arise from multiple functions required for one catalyst.
 | ||
| Scheme 1 Tunable C–H functionalization and dearomatization. | ||
On the other hand, indoles and indolines are privileged scaffolds in alkaloid natural products and biologically active molecules.5 Meanwhile, tertiary alcohols are important structural units that widely exist in a variety of biologically active natural products, pharmaceuticals, and agrochemicals (Scheme 1A).6 The molecules containing both indole/indolines and tertiary alcohols might have great potential applications. In this regard, divergent syntheses of such compounds are highly desirable. We questioned whether photoredox catalysis7 could engineer C–H functionalization and dearomatization selectively to afford two sets of targets in a catalytic and divergent fashion. It is envisioned that the excited photoredox catalyst (PC) could donate an electron to the ketone moiety. The resultant ketyl radical8 would add to indole nuclei (viaI), which is followed by protonation. Then, intermediate II might undergo single-electron oxidation to achieve aromatization upon loss of a proton. Alternatively, single-electron reduction of II would occur instead to deliver dearomatization products after protonation (Scheme 1B). Although conceptually simple in design, this strategy must contend with problems involving both reactivity and selectivity. Specifically, SET reduction of the ketone moieties is challenging, owing to the significantly negative reduction potentials [e.g. Ered1/2 = −2.11 V vs. SCE for acetophenone].9 A powerful photocatalyst is required to be capable of participating in both SET reduction and SET oxidation events for the C–H functionalization process as well as sequential SET reductions for the dearomatization process. In addition, the reaction might encounter the chemoselective competition from reduction10 or pinacol C–C coupling.11 Furthermore, the diastereoselective issues should be addressed when multiple stereocenters are generated.
Herein, we describe the first photocatalytic method for selective C–H functionalization and dearomatization by operating a distinct mechanism. Starting from the same indole-tethered ketones, intramolecular alkylation and reductive dearomatization have been achieved under redox-neutral and net-reductive conditions, respectively. Two sets of important nitrogen-containing heterocycles are obtained in good yields and excellent selectivity. A new organic photocatalyst, isoazatruxene ITN-2, is discovered to guarantee the success.
Our study was initiated with the investigation of C–H functionalization reaction using indole derivative 1a as the substrate under irradiation with a Kessil lamp (Scheme 2A). Due to the significantly negative reduction potential of a ketone moiety, the highly reducing Ir(ppy)3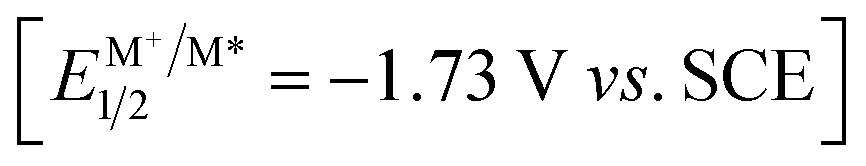 7c was first attempted. However, this photocatalyst failed to give the desired C2-alkylation product 2a, which confirmed that direct SET reduction of a ketone moiety by excited-state
7c was first attempted. However, this photocatalyst failed to give the desired C2-alkylation product 2a, which confirmed that direct SET reduction of a ketone moiety by excited-state 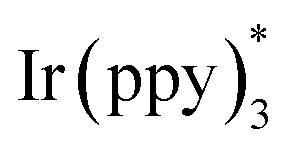 is not feasible. Next, the frequently-used transition-metal-based and organic photocatalysts including Ir(ppy)2(dtbbpy)PF6, Ir[(dFCF3ppy)2(dtbbpy)]PF6, Ru(bpy)3Cl2·6H2O, 4CzIPN [1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene], PTH (N-phenylphenothiazine), rhodamine B, eosin Y, fluorescein, rose bengal, methylene blue, and thioxanthen-9-one were systematically screened, unfortunately all leading to poor results (<15% yield).
is not feasible. Next, the frequently-used transition-metal-based and organic photocatalysts including Ir(ppy)2(dtbbpy)PF6, Ir[(dFCF3ppy)2(dtbbpy)]PF6, Ru(bpy)3Cl2·6H2O, 4CzIPN [1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene], PTH (N-phenylphenothiazine), rhodamine B, eosin Y, fluorescein, rose bengal, methylene blue, and thioxanthen-9-one were systematically screened, unfortunately all leading to poor results (<15% yield).
 | ||
| Scheme 2 Performance of various photocatalysts in C–H functionalization and characterization of ITNs. | ||
Given our interest in organic electronic materials,12 we turned attention to isoazatruxenes (ITNs), which can be readily synthesized via cyclotrimerization of indoles.13 Despite the easy access, the application of ITNs in photocatalysis remained elusive. Thus, the photophysical and electrochemical properties of isoazatruxenes (ITNs) possessing varied substituents on the nitrogen atoms (ITN-1: R = H; ITN-2: R = nBu; ITN-3: R = Ph) were measured and are summarized in Scheme 2B (Fig. S1–S18†).14 In addition, DFT calculations (B3LYP-D3(BJ)/def2-SVP) were performed to obtain the HOMO/LUMO energies of ITNs (Fig. S20†).14 While ITN-1 shows weak absorption in the visible region, the introduction of alkyl- or aryl groups results in a bathochromic shift with respect to the unsubstituted counterpart based on the ultraviolet-visible (UV-vis) spectroscopy, possibly due to the inductive effect of the alkyl groups and the conjugative effect of the aryl groups (Scheme 2C). Electron-excitation analyses on the basis of TD-DFT calculations suggested that the S0-to-S1 excitation of ITNs was mainly contributed by the electron transition from the corresponding HOMOs to LUMOs. The calculated vertical excitation energies of ITNs qualitatively reproduced the bathochromic shift order of the highest absorption peaks (ITN-1 < ITN-3 < ITN-2) (Fig. S21†).14 Notably, ITNs exhibit highly negative excited state reductive potentials (spanning from −2.17 to −2.54 V vs. SCE) as well as comparable excited-state lifetimes (4.15–5.84 ns), which allowed them to be promising candidates for promoting the above transformation (Scheme 2E).
To our delight, the C2 alkylation proceeded when ITNs were used as the photocatalysts. Particularly, ITN-2 bearing nbutyl substituents on the nitrogen atoms outperformed all the other photocatalysts, providing the corresponding polycyclic indole 2a in 86% yield with toluene as the solvent. The superior catalytic efficiency of ITN-2 in comparison with other ITNs might correspond to its enhanced visible-light absorption, which resulted in better visible photon harvesting. Furthermore, control experiments demonstrated that the reaction requires a photocatalyst and visible light.14
Having determined the optimal reaction conditions, we next examined the scope of C–H functionalization. As shown in Scheme 3, the tethered aromatic ketones bearing either electron-withdrawing or electron-donating substituents (–Cl, –F, –Ph, –Me, –Et, –nPr, –iPr, –tBu, –OMe) on the para-position of arenes were well tolerated, delivering the corresponding polycyclic indoles (2b–2j) in good to excellent yields (71–92%). The structure of 2a was determined unambiguously by X-ray crystallographic analysis.14 Interestingly, the more challenging substrates with multi-electron-donating groups [2,4-(Me)2, 2,5-(Me)2, 3,4-(Me)2, 3,4-(OMe)2] that possess lower reduction potentials could be accommodated, highlighting the power of ITN-2 (2k–2n, 52–83% yields). Moreover, both naphthyl and thienyl-derived ketones were converted to their corresponding polycyclic indoles bearing tertiary alcohols (2o and 2p) with high efficiency under the catalysis of ITN-2. This method is also compatible with the substrates bearing ethyl ester or acetyl groups at the C-3 position of indole skeletons (2q–2r, 82–87% yields). Finally, the reactions of substrates bearing cyano, fluorine, and methyl substituents at the indole cores also proceeded smoothly, providing 2s–2u in 76–90% yields.
 | ||
| Scheme 3 Substrate scope for C–H functionalization.a,b aReaction conditions: a solution of ITN-2 (5.2 mg, 5.0 mol%) and 1 (0.2 mmol, 1.0 equiv.) in toluene (2.0 mL, 0.1 M) was irradiated by a 427 nm Kessil lamp (40 W) at room temperature under a nitrogen atmosphere for 4–36 h. bIsolated yield. | ||
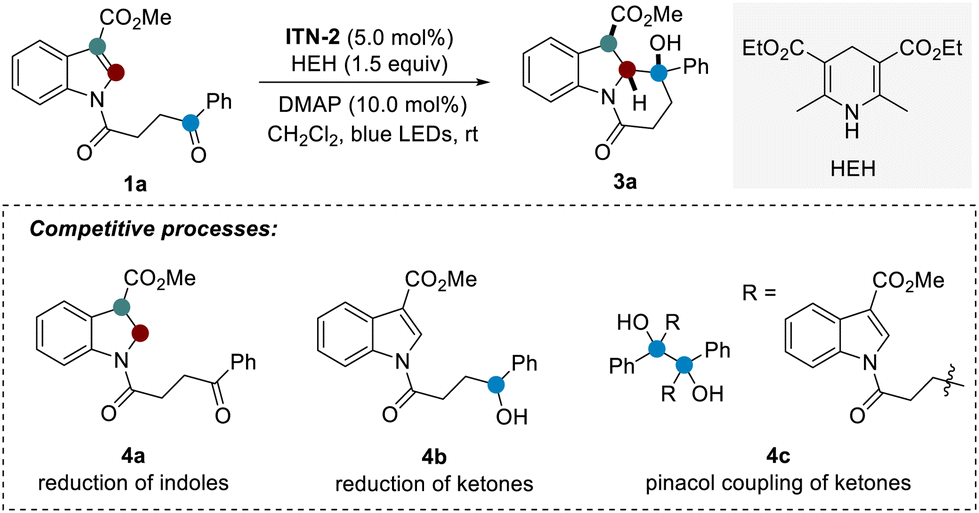
Then, we continued to investigate the intramolecular dearomatization of indoles with ketones (Table 1). In fact, this process remains problematic because of energetic barriers caused by aromaticity and competition between dearomatization and reduction or pinacol C–C coupling (1a → 4a–4c). The formation of multiple stereocenters also faces the challenge of diastereoselective control. With N-acylated indole derivative 1a as the model substrate, our optimization revealed that the use of ITN-2 as the photocatalyst, 4-dimethylaminopyridine (DMAP) as the additive, and Hantzsch ester (HEH) as the reductant in dichloromethane led to the formation of expected dearomatized product 3a in 90% yield with excellent diastereoselectivity under blue LED irradiation at room temperature (entry 1). Notably, the presence of a catalytic amount of DMAP significantly improved the diastereoselectivity, likely a result of thermodynamic control (entry 2). Consistently, ITN-1 and ITN-3 afforded inferior efficiency (entries 3 and 4). Changing dichloromethane to other commonly used solvents such as acetone, CH3CN, or DMSO resulted in dramatically decreased yields (8–37%) for the desired product 3a (entries 5–7), demonstrating the important role of solvents. When DIPEA was used as the external reductant instead, the dearomatization reaction also occurred well, albeit with a lower yield of 3a (entry 8). Lastly, the control experiments indicated that photocatalyst ITN-2, reductant HEH, and visible light are all indispensable (entries 9–11).
|
|
|||
|---|---|---|---|
| Entry | Variations from standard conditions | Yieldb (%) | Drc |
| a Reaction conditions: a solution of ITN-2 (2.6 mg, 5.0 mol%), 1a (0.1 mmol, 1.0 equiv.), HEH (0.15 mmol, 1.5 equiv.), and DMAP (1.2 mg, 10.0 mol%) in CH2Cl2 (1.0 mL, 0.1 M) was irradiated by blue LEDs (30 W) at room temperature under a nitrogen atmosphere for 24 h. b Determined by 1H NMR yield using CH2Br2 as an internal standard. c Ratios of diastereoisomers were determined by crude 1H NMR analysis. d Isolated yield. n.d. = not determined. DMAP = 4-dimethylaminopyridine. DIPEA = N,N-diisopropylethylamine. | |||
| 1 | None | 91 (90)d | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | No DMAP | 91 | 1.7:1 |
| 3 | ITN-1 instead of ITN-2 | 0 | n.d. |
| 4 | ITN-3 instead of ITN-2 | 59 | >20:1 |
| 5 | Acetone instead of CH2Cl2 | 37 | 16:1 |
| 6 | CH3CN instead of CH2Cl2 | 13 | n.d. |
| 7 | DMSO instead of CH2Cl2 | 8 | n.d. |
| 8 | DIPEA instead of HEH | 45 | >20:1 |
| 9 | No ITN-2 | 0 | n.d. |
| 10 | No HEH | 0 | n.d. |
| 11 | No light | 0 | n.d. |
With the optimized conditions in hand, we explored the scope of photocatalytic intramolecular dearomatization of indole derivatives with ketones (Scheme 4). It was observed that the aromatic ketones bearing either electron-withdrawing (e.g. 4-Cl, 4-F) or electron-donating [e.g. 4-Me, 4-Et, 4-nPr, 4-iPr, 4-tBu, 4-OMe, 2,4-(Me)2, 2,5-(Me)2, 3,4-(Me)2, 3,4-(OMe)2] groups on the arene skeletons were well accommodated. In all cases, highly functionalized benzannulated indolizidines possessing three consecutive stereocenters were furnished in good to excellent yields with remarkable diastereoselectivity (3a–3n: 75–98% yields, 18:1 → >20:1 dr). Similarly, the structure and relative configuration of 3k were confirmed by X-ray crystallographic analysis.14 In addition, indole derivatives tethered with naphthyl and thienyl ketones were compatible with this transformation (3o–3p: 85–87% yields, >20:1 dr). Changing the substituents at the C-3 position of indoles to ethyl ester or acetyl groups also proved feasible (3q–3r: 73–85% yields, >20:1 dr). Furthermore, the reactions of substrates 1s–1u containing varied substituents (–CN, –F, –Me) on the indole skeletons occurred smoothly, delivering the desired indolizidines 3s–3u in good yields with extraordinary diastereoselectivity (86–96% yields, 18:1 → >20:1 dr).
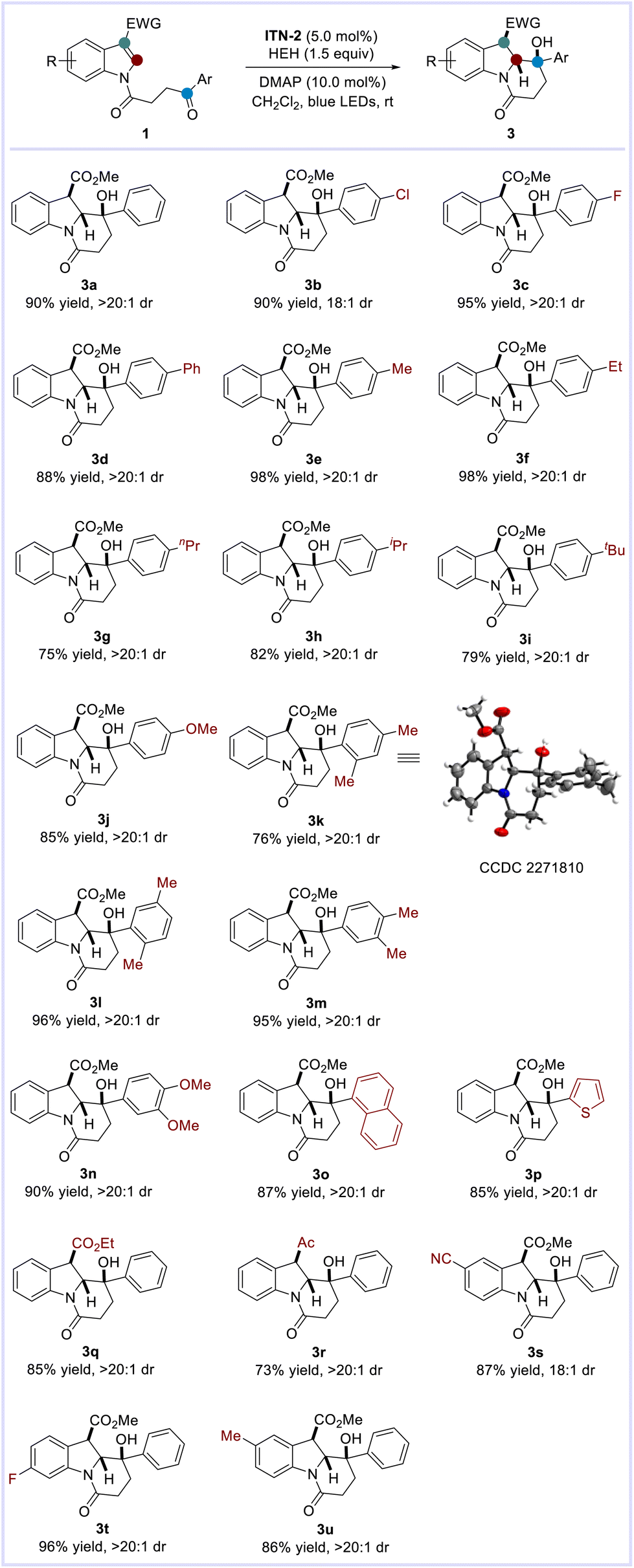 | ||
| Scheme 4 Substrate scope for dearomatization.a,b aReaction conditions: a solution of ITN-2 (5.2 mg, 5.0 mol%), 1 (0.2 mmol, 1.0 equiv.), HEH (0.3 mmol, 1.5 equiv.), and DMAP (2.4 mg, 10.0 mol%) in CH2Cl2 (2.0 mL, 0.1 M) was irradiated by blue LEDs (30 W) at room temperature under a nitrogen atmosphere for 12–48 h. bIsolated yield. Ratios of diastereoisomers were determined by crude 1H NMR analysis. | ||
In order to demonstrate the synthetic practicality, gram-scale syntheses were carried out (Scheme 5). With ITN-2 as the photocatalyst, the C–H functionalization and dearomatization reactions of substrate 1a on 4.47 mmol scale were selectively achieved under the optimized reaction conditions, yielding polycyclic indole 2a and indoline derivative 3a in 85% yield (1.28 g) and 91% yield (1.36 g), respectively.
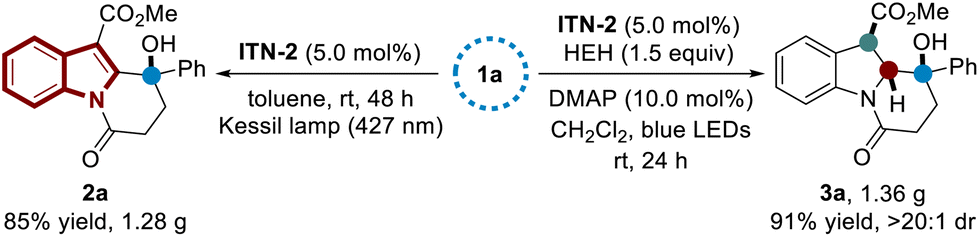 | ||
| Scheme 5 Gram-scale reaction. | ||
Several experiments were performed to probe the possible mechanism (Scheme 6A). First, both reactions were subjected to ultraviolet light irradiation (365 nm) without the addition of ITN-2. It was observed neither of the reactions proceeded, thus excluding the operation of the energy transfer mechanism. Upon the addition of TEMPO (2,2,6,6-tetramethylpiperidine N-oxyl), the expected transformations were completely inhibited, which suggested the involvement of radicals. Accordingly, the plausible mechanism for photocatalytic C–H functionalization and dearomatization was proposed as exemplified by the reactions of 1a (Scheme 6B).
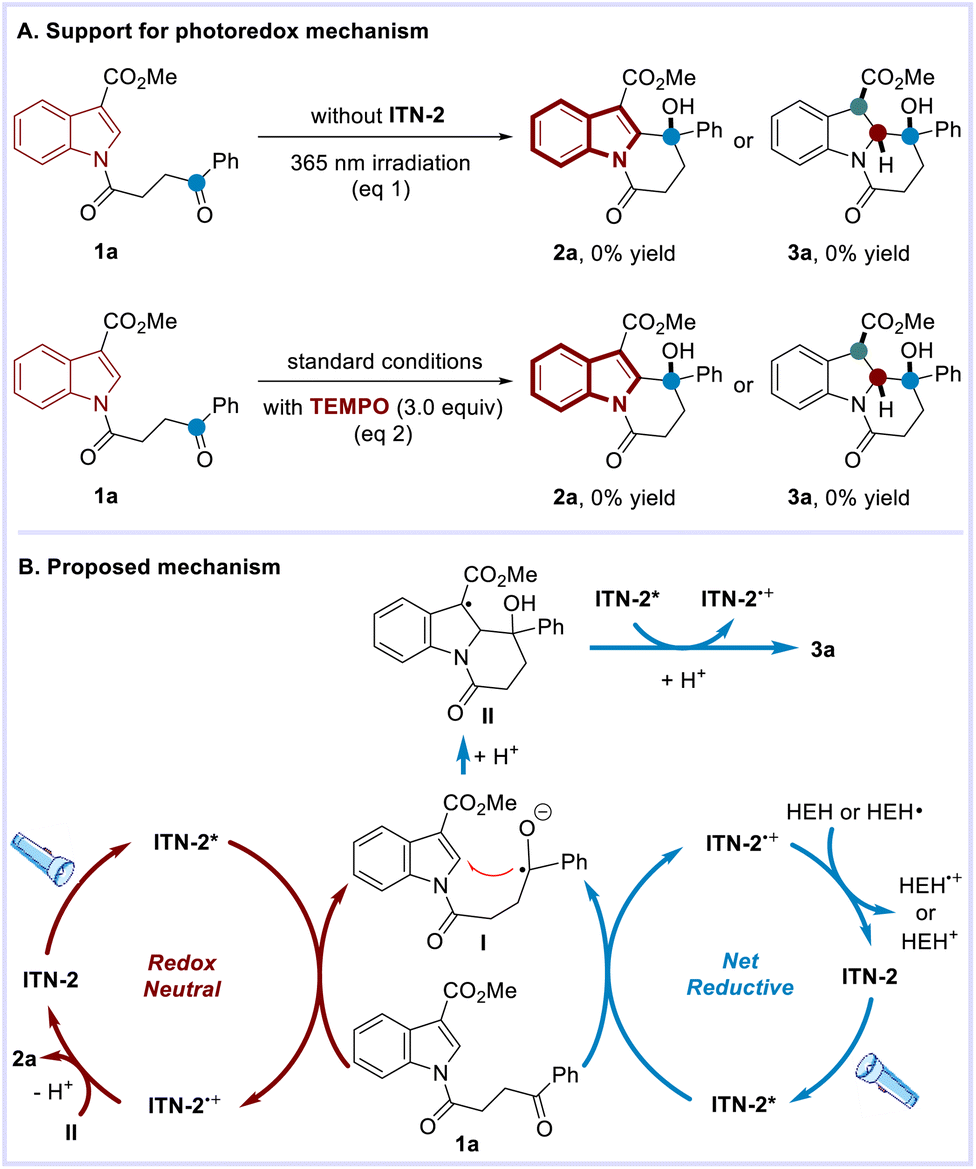 | ||
| Scheme 6 Proposed mechanism. | ||
Irradiation of ITN-2 affords its excited state. Oxidative quenching of ITN-2* by indole derivative 1a generates ITN-2˙+ and the corresponding ketyl radical (I), which is supposed to undergo addition to electron-deficient indole. After protonation, intermediate II proceeds diversely in the two transformations. In the C–H functionalization reaction, single-electron oxidation of II by ITN-2˙+ delivers aromatization product 2a upon the release of a proton and completes the catalytic cycle. With respect to the dearomatization reaction, single-electron reduction of II by excited state ITN-2* occurs instead, yielding the benzannulated indolizidine 3a after accepting a proton. ITN-2˙+ can be further converted back to ITN-2 in the presence of an external reductant. While C–H functionalization is a redox-neutral transformation, dearomatization is a net-reductive process requiring a stoichiometric external reductant to turn over the photocatalytic cycle.
Conclusions
In summary, we have developed a photocatalytic protocol to engineer C–H functionalization and dearomatization processes selectively. Crucial to the strategy are the different redox events promoted by a new organic photocatalyst – isoazatruxene ITN-2. Under redox-neutral conditions, intramolecular C–H functionalization is directed, providing polycyclic indoles bearing tertiary alcohols in good yields. Under net-reductive conditions, reductive dearomatization of indole derivatives with ketones can be realized instead, in which the corresponding benzannulated indolizidines possessing three consecutive stereocenters are furnished with remarkable diastereoselectivity. We anticipate this study will stimulate the development of novel photocatalysts for previously challenging transformations.Data availability
All experimental data is available in the ESI.†Author contributions
X. Z. conceived the project. B. A., H. C., J.-L. C., and F. L. performed the experiments under S.-L. Y. and X. Z.'s supervision. C. Z. performed the DFT calculations. B. A., C. Z., S.-L. Y., and X. Z. cowrote the paper.Conflicts of interest
A patent has been filed on the use of isoazatruxenes and their derivatives in photocatalysis.Acknowledgements
We thank NSFC (22071024, 22271047), and the Natural Science Foundation of Fujian Province (2021J06020) for generous financial support.Notes and references
- For selected reviews, see: (a) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS PubMed; (b) A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2019, 48, 4401–4423 RSC; (c) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7558–7598 CrossRef CAS PubMed; (d) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed; (e) P. Kumar, P. J. Nagtilak and M. Kapur, New J. Chem., 2021, 45, 13692–13746 RSC; (f) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (g) S. Brunen, B. Mitschke, M. Leutzsch and B. List, J. Am. Chem. Soc., 2023, 145, 15708–15713 CrossRef CAS.
- For selected reviews, see: (a) P. W. Rabideau and Z. Marcinow, Org. React., 1992, 42, 1–334 CAS; (b) R. M. Moriarty and O. M. Prakash, Org. React., 2001, 57, 327–415 CAS; (c) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed; (d) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed; (e) L. M. Repka and S. E. Reisman, J. Org. Chem., 2013, 78, 12314–12320 CrossRef CAS PubMed; (f) S. P. Roche, J.-J. Y. Tendoung and B. Tréguier, Tetrahedron, 2015, 71, 3549–3591 CrossRef CAS; (g) M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem.–Eur. J., 2016, 22, 2856–2881 CrossRef CAS PubMed; (h) R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816–9849 CrossRef CAS PubMed; (i) M. Okumura and D. Sarlah, Synlett, 2018, 29, 845–855 CrossRef CAS; (j) J. Bariwal, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3831–3848 RSC; (k) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC; (l) G. Huang and B. Yin, Adv. Synth. Catal., 2019, 361, 405–425 CrossRef CAS; (m) F.-T. Sheng, J.-Y. Wang, W. Tan, Y.-C. Zhang and F. Shi, Org. Chem. Front., 2020, 7, 3967–3998 RSC; (n) M. Okumura and D. Sarlah, Eur. J. Org Chem., 2020, 2020, 1259–1273 CrossRef CAS PubMed; (o) Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Chem. Soc. Rev., 2022, 51, 2145–2170 RSC.
- For selected examples, see: (a) A. Taheri, B. Lai, C. Cheng and Y. Gu, Green Chem., 2015, 17, 812–816 RSC; (b) S. P. Pitre, J. C. Scaiano and T. P. Yoon, ACS Catal., 2017, 7, 6440–6444 CrossRef CAS PubMed; (c) K. Wu, Y. Du and T. Wang, Org. Lett., 2017, 19, 5669–5672 CrossRef CAS PubMed; (d) D. Alpers, M. Gallhof, J. Witt, F. Hoffmann and M. Brasholz, Angew. Chem., Int. Ed., 2017, 56, 1402–1406 CrossRef CAS PubMed; (e) D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Chem. Sci., 2018, 9, 9012–9017 RSC; (f) E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394–3402 CrossRef CAS PubMed; (g) F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183–2194 CrossRef CAS; (h) J. Yang, D.-W. Ji, Y.-C. Hu, X.-T. Min, X. Zhou and Q.-A. Chen, Chem. Sci., 2019, 10, 9560–9564 RSC; (i) Y.-Z. Cheng, Q.-R. Zhao, X. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 18069–18074 CrossRef CAS; (j) M. Zhu, C. Zheng, X. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2636–2644 CrossRef CAS PubMed; (k) M. Zhu, X.-L. Huang, H. Xu, X. Zhang, C. Zheng and S.-L. You, CCS Chem., 2020, 3, 652–664 CrossRef; (l) W.-J. Zhou, Z.-H. Wang, L.-L. Liao, Y.-X. Jiang, K.-G. Cao, T. Ju, Y. Li, G.-M. Cao and D.-G. Yu, Nat. Commun., 2020, 11, 3263 CrossRef CAS PubMed; (m) H. Li, L. Guo, X. Feng, L. Huo, S. Zhu and L. Chu, Chem. Sci., 2020, 11, 4904–4910 RSC; (n) J. Ma, F. Schäfers, C. Daniliuc, K. Bergander, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 9639–9645 CrossRef CAS PubMed; (o) M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. K. Gupta, N. A. Meanwell, A. Mathur and T. G. M. Dhar, J. Am. Chem. Soc., 2020, 142, 3094–3103 CrossRef CAS PubMed; (p) K. Sun, A. Shi, Y. Liu, X. Chen, P. Xiang, X. Wang, L. Qu and B. Yu, Chem. Sci., 2022, 13, 5659–5666 RSC; (q) E. Georgiou, D. Spinnato, K. Chen, P. Melchiorre and K. Muñiz, Chem. Sci., 2022, 13, 8060–8064 RSC; (r) X. Chang, F. Zhang, S. Zhu, Z. Yang, X. Feng and Y. Liu, Nat. Commun., 2023, 14, 3876 CrossRef CAS PubMed; (s) X. Lv, Y.-N. Qi, J. Wang, X. Zhao and Z. Jiang, Org. Lett., 2023, 25, 3114–3119 CrossRef CAS PubMed; (t) T.-T. Song, Y.-K. Mei, Y. Liu, X.-Y. Wang, S.-Y. Guo, D.-W. Ji, B. Wan, W. Yuan and Q.-A. Chen, Angew. Chem., Int. Ed., 2023, e202314304 Search PubMed; (u) J. Qin, Z. Zhang, Y. Lu, S. Zhu and L. Chu, Chem. Sci., 2023, 14, 12143–12151 RSC.
- For selected examples, see: (a) G. Tan, Q. You, J. Lan and J. You, Angew. Chem., Int. Ed., 2018, 57, 6309–6313 CrossRef CAS PubMed; (b) Y. Dong, A. W. Schuppe, B. K. Mai, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2022, 144, 5985–5995 CrossRef CAS PubMed; (c) X. Li, L. Hu, S. Ma, H. Yu, G. Lu and T. Xu, ACS Catal., 2023, 13, 4873–4881 CrossRef CAS.
- For selected reviews, see: (a) Y. Yao, M. Alami, A. Hamze and O. Provot, Org. Biomol. Chem., 2021, 19, 3509–3526 RSC; (b) S. M. Umer, M. Solangi, K. M. Khan and R. S. Z. Saleem, Molecules, 2022, 27, 7586 CrossRef CAS PubMed; (c) D. P. Zlotos, Y. M. Mandour and A. A. Jensen, Nat. Prod. Rep., 2022, 39, 1910–1937 RSC ; for selected examples, see: ; (d) R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, Tetrahedron, 1963, 19, 247–288 CrossRef CAS; (e) L. Randriambola, J.-C. Quirion, C. Kan-Fan and H.-P. Husson, Tetrahedron Lett., 1987, 28, 2123–2126 CrossRef CAS; (f) P. J. Stephens, J.-J. Pan, F. J. Devlin, M. Urbanová and J. Hájíček, J. Org. Chem., 2007, 72, 2508–2524 CrossRef CAS PubMed; (g) X. Zhang and J. C. Anderson, Angew. Chem., Int. Ed., 2019, 58, 18040–18045 CrossRef CAS PubMed; (h) S. Al-Rashed, A. Baker, S. S. Ahmad, A. Syed, A. H. Bahkali, A. M. Elgorban and M. S. Khan, Bioorg. Chem., 2021, 107, 104626 CrossRef CAS PubMed.
- (a) S.-T. Chen and J.-M. Fang, J. Org. Chem., 1997, 62, 4349–4357 CrossRef CAS PubMed; (b) D. Ameena and T. J. Snape, Med. Chem. Commun., 2013, 4, 893–907 RSC.
- For selected reviews, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248–1254 RSC; (e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (f) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (g) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (h) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed; (i) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (j) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (k) A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2019, 48, 4401–4423 RSC; (l) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed; (m) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC; (n) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (o) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Sci. China: Chem., 2019, 62, 24–57 CrossRef CAS; (p) A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed; (q) Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS; (r) M. Latrache and N. Hoffmann, Chem. Soc. Rev., 2021, 50, 7418–7435 RSC; (s) J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC; (t) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC; (u) G.-Q. Xu and P.-F. Xu, Chem. Commun., 2021, 57, 12914–12935 RSC; (v) A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed; (w) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed; (x) Y. Sakakibara and K. Murakami, ACS Catal., 2022, 12, 1857–1878 CrossRef CAS; (y) T. Bortolato, S. Cuadros, G. Simionato and L. Dell'Amico, Chem. Commun., 2022, 58, 1263–1283 RSC; (z) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (a a) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed; (a b) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (a c) D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed.
- For selected reviews, see: (a) K. N. Lee and M.-Y. Ngai, Chem. Commun., 2017, 53, 13093–13112 RSC; (b) Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949–2961 CrossRef CAS PubMed; (c) Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC ; for selected examples, see: ; (d) S. Gross and H.-U. Reissig, Org. Lett., 2003, 5, 4305–4307 CrossRef CAS PubMed; (e) K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS PubMed; (f) Z. Qu, T. Tian, Y. Tan, X. Ji, G.-J. Deng and H. Huang, Green Chem., 2022, 24, 7403–7409 RSC; (g) C. N. Rao and H.-U. Reissig, Eur. J. Org Chem., 2022, e202200264 CrossRef CAS; (h) V. J. Mayerhofer, M. Lippolis and C. J. Teskey, Angew. Chem., Int. Ed., 2023, 63, e202314870 CrossRef PubMed; (i) W.-Y. Zhang, H.-C. Wang, Y. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2023, 145, 10314–10321 CrossRef CAS PubMed; (j) M. Wang, C. Zhang, C. Ci, H. Jiang, P. H. Dixneuf and M. Zhang, J. Am. Chem. Soc., 2023, 145, 10967–10973 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- For selected examples, see: (a) A. Call, C. Casadevall, F. Acuña-Parés, A. Casitas and J. Lloret-Fillol, Chem. Sci., 2017, 8, 4739–4749 RSC; (b) J. Cai, Y. Li, Z. Ye, W. Wang, Y. Lin and L. Gong, Green Synth. Catal., 2023, 4, 253–257 CrossRef CAS.
- For selected examples, see: (a) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed; (b) K. Li, Q. Wan, C. Yang, X.-Y. Chang, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 14129–14133 CrossRef CAS PubMed; (c) A. Caron, É. Morin and S. K. Collins, ACS Catal., 2019, 9, 9458–9464 CrossRef CAS; (d) N. Noto, Y. Hyodo, M. Yoshizawa, T. Koike and M. Akita, ACS Catal., 2020, 10, 14283–14289 CrossRef CAS; (e) M. Liu, L. Tan, R. T. Rashid, Y. Cen, S. Cheng, G. Botton, Z. Mi and C.-J. Li, Chem. Sci., 2020, 11, 7864–7870 RSC; (f) T. Luo, L. Li, Y. Chen, J. An, C. Liu, Z. Yan, J. H. Carter, X. Han, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, C. C. Tang, M. Schröder and S. Yang, Nat. Commun., 2021, 12, 3583 CrossRef CAS PubMed; (g) H. Wang, J.-P. Qu and Y.-B. Kang, Org. Lett., 2021, 23, 2900–2903 CrossRef CAS PubMed; (h) F. Lan, C.-S. Liu, C. Zhou, X. Huang, J.-Y. Wu and X. Zhang, J. Mater. Chem. A, 2022, 10, 16578–16584 RSC; (i) S. Kundu, L. Roy and M. S. Maji, Org. Lett., 2022, 24, 9001–9006 CrossRef CAS PubMed; (j) Y. Yan, G. Li, J. Ma, C. Wang, J. Xiao and D. Xue, Green Chem., 2023, 25, 4129–4136 RSC.
- For selected reviews, see: (a) R. Ali and S. Alvi, Tetrahedron, 2020, 76, 131345 CrossRef CAS; (b) C. Zhou, B. An, F. Lan and X. Zhang, Chem. Commun., 2023, 59, 13245–13257 RSC.
- (a) C. Ruiz, E. M. García-Frutos, D. A. da Silva Filho, J. T. López Navarrete, M. C. R. Delgado and B. Gómez-Lor, J. Phys. Chem. C, 2014, 118, 5470–5477 CrossRef CAS; (b) T. Deng, W. Yan, X. Liu, G. Hu, W. Xiao, S. Mao, J. Lin, Y. Jiao and Y. Jin, Org. Lett., 2022, 24, 1502–1506 CrossRef CAS PubMed; (c) R. Gao, B. An, C. Zhou and X. Zhang, Molecules, 2022, 27, 8722 CrossRef CAS PubMed.
- See the ESI† for more details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and copies of NMR spectra. CCDC 2271792, 2271797, 2271798, 2271810 and 2309597. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00120f |
| This journal is © The Royal Society of Chemistry 2024 |