
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphdiyne/metal oxide hybrid materials for efficient energy and environmental catalysis
Yuhua
Zhu
ac,
Shuhong
Zhang
a,
Xiaofeng
Qiu
a,
Quanguo
Hao
a,
Yan
Wu
a,
Zhu
Luo
*ab and
Yanbing
Guo
 *ab
*ab
aEngineering Research Center of Photoenergy Utilization for Pollution Control and Carbon Reduction, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan, Hubei 430082, China. E-mail: guoyanbing@mail.ccnu.edu.cn
bWuhan Institute of Photochemistry and Technology, 7 North Bingang Road, Wuhan, Hubei 430082, China
cSchool of Civil Engineering, Wuhan University, Wuhan 430072, China
First published on 6th March 2024
Abstract
Graphdiyne (GDY)-based materials, owing to their unique structure and tunable electronic properties, exhibit great potential in the fields of catalysis, energy, environmental science, and beyond. In particular, GDY/metal oxide hybrid materials (GDY/MOs) have attracted extensive attention in energy and environmental catalysis. The interaction between GDY and metal oxides can increase the number of intrinsic active sites, facilitate charge transfer, and regulate the adsorption and desorption of intermediate species. In this review, we summarize the structure, synthesis, advanced characterization, small molecule activation mechanism and applications of GDY/MOs in energy conversion and environmental remediation. The intrinsic structure–activity relationship and corresponding reaction mechanism are highlighted. In particular, the activation mechanisms of reactant molecules (H2O, O2, N2, etc.) on GDY/MOs are systemically discussed. Finally, we outline some new perspectives of opportunities and challenges in developing GDY/MOs for efficient energy and environmental catalysis.
1. Introduction
With the rapid development of human society and economy, the problems of energy crisis and environmental pollution have become increasingly prominent. Developing renewable energy sources and strengthening the control of pollutants are two effective strategies to solve the aforementioned problems.1–4 Catalysts with high efficiency and low cost play a crucial role in addressing challenges.5 Noble metal catalysts, including Pt, Pd, Ag, and Au, have been widely utilized due to their high catalytic performances.2,4 However, noble metal catalysts still suffer from limited availability, prohibitive cost, and relatively low stability, which prompt the exploration of alternative catalytic materials. Carbon-based materials are attracting growing attention due to their distinctive advantages in terms of low cost, large specific surface area, as well as tunable electronic structures,6–8 which exhibit potential applications in energy and environmental catalysis.Different from traditional inorganic carbon materials (graphene, carbon nanotubes, etc.) and organic polymerized carbon materials (carbon nitride, covalent organic frameworks, etc.), with single sp2 or sp3 hybridized states, graphdiyne (GDY), a rising-star carbon allotrope, comprising sp and sp2 hybridized carbon atoms, is an emerging two-dimensional (2D) planar carbon material.9–11 GDY possesses well dispersed electron-rich cavities and a large π-conjunction structure, which endows it with unique chemical and physical properties, including an adjustable intrinsic bandgap, fast charge transfer, excellent conductivity and so on. The electron mobility of GDY is considerably higher than that of common organic polymerized carbon materials, even up to several orders of magnitude.12–14 Moreover, the sp-hybridized carbon atoms and triangular cavities in the 2D GDY conjugated structure facilitate the formation of strong interfacial contacts with active components such as metal atoms and metal oxides.15,16 The fabrication of a wide range of unique GDY-based materials expands the applications of GDY in energy catalysis, environmental remediation, energy storage, electronics, life sciences, and other fields.17–20 In particular, GDY/MOs, through the interaction between GDY and metal oxides, enable the modulation of the electronic structure and increase the number of active sites, which improves the intrinsic activity, and enhances the stability.21–25 Therefore, the GDY/MOs exhibit unique advantages in heterogeneous catalysis.
Recently, many excellent and interesting research studies about GDY/MOs were published. However, a thorough overview of GDY/MOs for efficient energy and environmental catalysis has not been reported. This review summarizes the applications of GDY/MOs in the field of energy and environmental catalysis, emphasizing the uniqueness of the intrinsic relationship between the structure and performance and the corresponding mechanism. The structure, synthesis, characterization and applications of GDY/MOs are fully introduced. In particular, the activation behaviors on GDY/MOs for efficient energy and environmental catalysis, such as activation of H2O, O2 and N2, are systemically discussed. Finally, we comprehensively discuss the existing key challenges and prospects for fundamental and applied research of GDY/MOs in the field of energy and environmental remediation.
2. Typical structure, synthesis and characterization of GDY/MOs
2.1 Typical structure of GDY/MOs
Because of the abundant sp conjugated diyne bonds and unique triangular pore structure, GDY is widely used as a good carrier of MOs in the field of catalysis.11 Therefore, it is of great significance to study the unique structure of GDY/MOs. Herein, we summarize the structure of GDY/MOs as follows.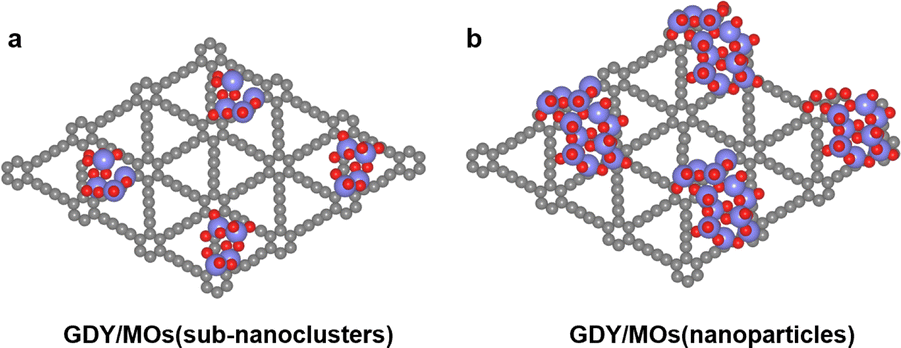 | ||
| Fig. 1 Schematic structure of GDY/MOs: (a) MOs as sub-nanoclusters; (b) MOs as nanoparticles. | ||
2.2 Synthesis of GDY/MOs
Metal oxides (MOs) have attracted wide attention in environmental and energy-related fields. In order to meet the different applications in these fields, it is of great significance to explore the controllable preparation and regulation of MOs. Recent studies have shown that the composite of carbon materials with MOs can modulate the structure of the MOs and thus enhance the catalytic activity and stability of MOs. Theoretical predictions demonstrated that the triangular cavities with the edges of sp-hybridized carbon atoms in the GDY would have a much stronger binding energy with many elements (for example, Li, K, and Ti) than the prevailing sp2-hybridized carbon materials.32–34 This particular interaction indicates that GDY may be an ideal material for interfacial modification on MOs. On this basis, the nanostructured NiCo2O4, TiO2, CoOx, MnO2, IrO2 and MoO3 connected with GDY have been successfully prepared for various potential applications.35–39 Therefore, it is of great significance to explore the synthesis of high quality GDY and GDY-based materials and reveal the relationship between their structures and properties. Here, this review systemically gives a summary and discussion of the synthesis methods of GDY/MOs.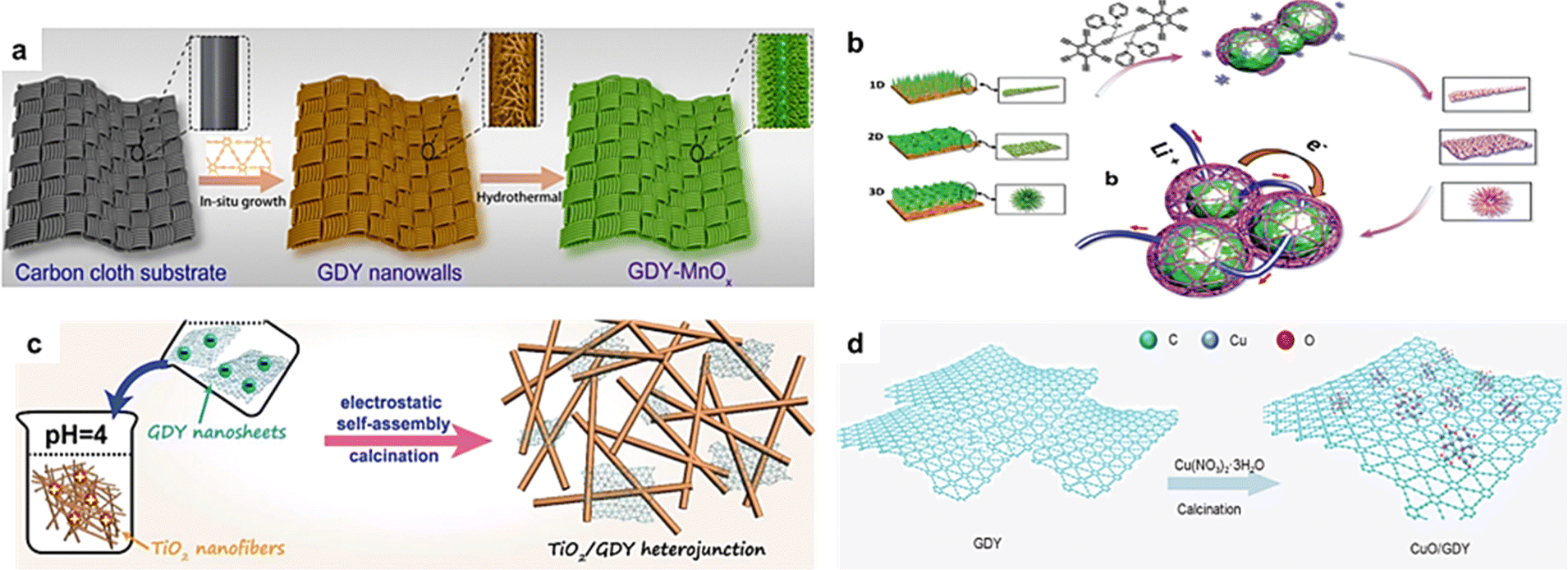
2.3 The advanced characterization of GDY/MOs
The structure of GDY/MOs plays a decisive role in catalytic performance. It can affect the quality of the GDY/MO surface and the rate of chemical reaction, change the position of active centers, binding energy, structure, electron distribution and charge transfer, and other properties. Therefore, appropriate structural characterization methods for GDY/MOs are essential. The reported characterization methods of GDY/MOs mainly involve two categories: image and spectroscopy. The characterization methods of images include scanning electron microscopy (SEM), transmission electron microscopy (TEM), atomic force microscopy (AFM), and scanning transmission electron microscopy (STEM). The spectroscopy characterization contains Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS) and so on. The image characterization is mainly used to describe the morphology characteristics of the GDY/MOs (particle size, elemental distribution, crystal structure, phase composition, structural defects, grain boundary structure and composition, etc.). Spectroscopy characterizes the structure of the catalyst (chemical composition, compositional changes, molecular valence, electronic configuration, interfacial interaction).2.3.1.1 SEM. SEM can be used to directly observe the surface structure of GDY/MOs, particle size, distribution, uniformity and agglomeration.
Combining with the energy spectrum, it can also be used to analyze their composition. For instance, Fig. 3a–c show the top SEM views of SnOx/GDY nanowalls.42 The top view demonstrated that vertical GDY nanowalls were in situ grown on the surface of the carbon cloth. The SEM images of GDY/MOs clearly describe the morphologies of GDY nanomaterials, including the height of the nanowalls, the size of nanoparticles and so on.
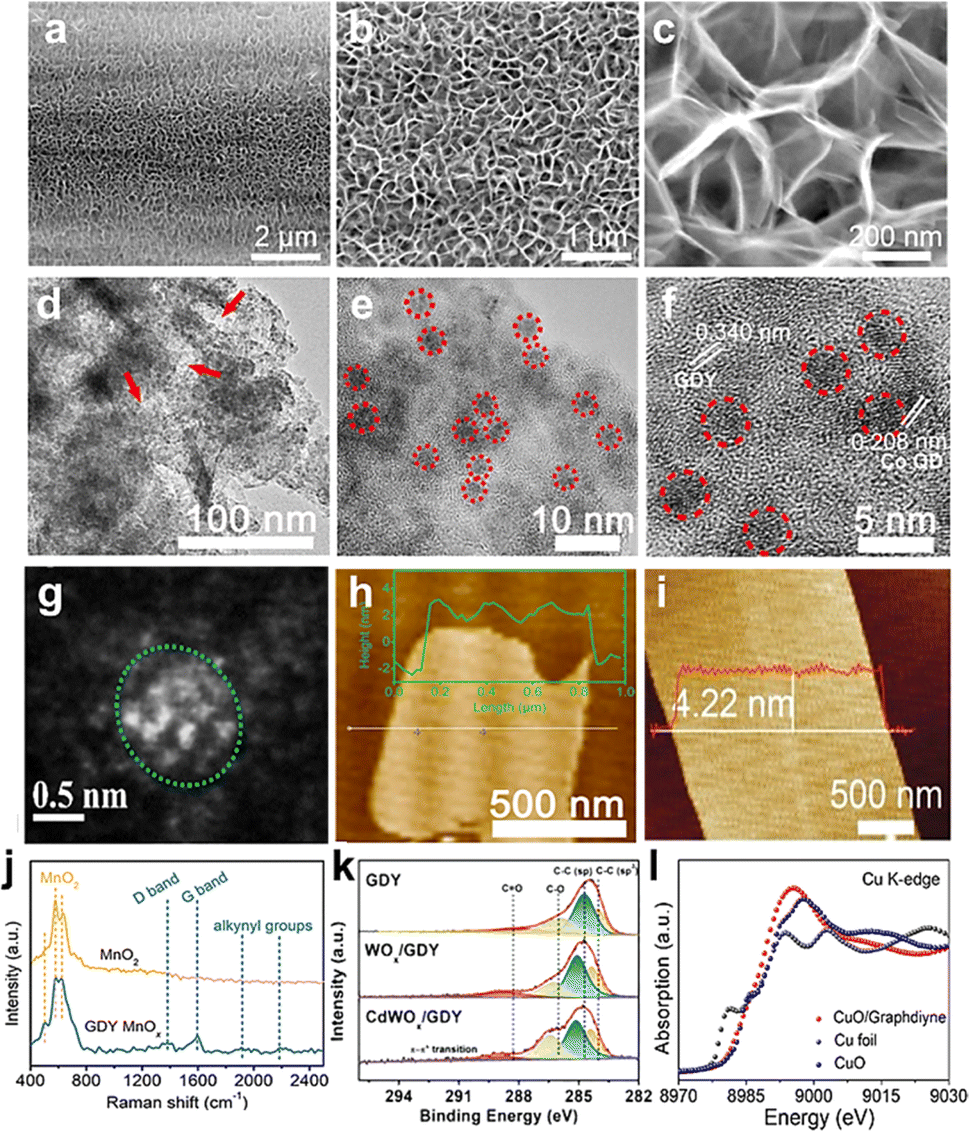 | ||
| Fig. 3 (a–c) SEM images of SnOx/GDY;42 (d and e) TEM images of GDY@CoOxQDs;37 (f) HRTEM image of GDY@CoOxQDs;37 (g) STEM images of GDY/CuO;29 (h) AFM images of GDY@CoOxQDs;37 (i) AFM image of IrOx/GDY;39 (j) Raman spectra of GDY/MnOx and MnO;38 (k) XPS spectra of WO3, WOx/GDY, and CdWOx/GDY;24 (l) XANES of CuO/GDY, CuO, and Cu foil.29 | ||
2.3.1.2 TEM. TEM and its associated electron diffraction, energy dispersive X-ray spectroscopy (EDX) and electron energy loss spectroscopy (EELS) are some of the most powerful methods for characterizing the morphology, crystal structure, composition and interfacial interaction of GDY/MOs. As shown in Fig. 3d–f,37 TEM and HRTEM images showed that Co quantum dots (CoOxQDs) were uniformly dispersed on the surface of porous GDY with the size of 3.7 nm. The CoOxQDs were grown on GDY nanowalls in the form of nanoparticles and the interfacial interaction may be present between GDY and CoOxOD. TEM is a commonly used method for characterizing GDY/MO morphology. However, this method is the result of local observation, so there are certain contingencies and statistical errors. The typical measurement range of TEM is 5 nm–500 μm. It is necessary to obtain the average particle size of nanoparticles through statistical analysis and measurement of particle size with a certain number of photos.
2.3.1.3 STEM. STEM has the advantages of high resolution and sensitivity to chemical composition, and provides better visual images of GDY/MOs compared with TEM. Very recently, our group demonstrated that the neighboring sp-hybridized C and Cu sites on the interface of the sub-nanocluster CuO/GDY was the key structure to effectively modulate the O2 activation process in the bridging adsorption mode.29 The HAADF-STEM image (Fig. 3g) demonstrated the dark GDY and the relatively bright sub-nanocluster CuO. The particle size distribution of sub-nanocluster CuO from the HAADF-STEM image showed that the average particle size was around 0.82 nm. According to the HAADF-STEM image, the reconstruction model of the sub-nanocluster CuO suggested that the subnanocluster CuO was composed of 14 Cu atoms and 12 O atoms and there was interfacial chemical interaction between CuO and GDY. As an indispensable tool for in-depth study of GDY/MOs, STEM equipped with an aberration correction device can perform more precise and accurate structural characterization of GDY/MOs. With the introduction of the spherical error corrector, STEM with many analytical components can significantly improve the resolution to angstroms, and the ultra-high resolution can even clearly reveal the fine structure at the molecular level and atomic scale.
2.3.1.4 AFM. The AFM is capable of imaging samples in air or liquid and measuring the surface roughness and thickness of ultrathin 2D/quasi-2D GDY/MOs with high accuracy. As shown in Fig. 3h, the AFM images revealed that the GDY@CoOxQD nanosheet had thickness around 4.0 nm.37 Li et al. reported IrOx quantum dots synthesized by GDY-induced in situ controllable growth to enhance their activity in acidic electrolytes. The AFM images of IrOx/GDY showed a rough surface with numerous individual bright dots protruding (Fig. 3i),39 which implied the presence of QDs. In short, this characterization method is usually used to measure 3D surface topography and roughness of powder/film/bulk/fiber samples, nanosheet thickness, interphase thickness and so on. However, the use of AFM to characterize the interface structure of GDY/MOs is still rare.
2.3.2.1 Raman spectroscopy. Raman spectroscopy is an analytical method with scattering spectra to obtain information on molecular vibrations and rotations used for the structural characterization of GDY/MOs. In general, it can mainly be used to check the defects and bonding states of GDY. Fig. 3j shows the Raman spectra of MnO2 and GDY/MnOx. Compared with MnO2, the newly appeared peaks at 1382, 1589, 1912 and 2187 cm−1 corresponded to the D band, G band and alkynyl groups for GDY, respectively, which confirmed the successful synthesis of GDY/MnOx.38 After the GDY was connected with MnOx, the obvious shift of the G band and conjugated diyne link modes towards higher wave numbers can be observed, indicating the strong chemical interaction (charge transfer or chemical bonding) between GDY and metal species. Besides, MnOx/GDY showed a larger ID/IG ratio than pure GDY, which indicated the increase of the defect sites after the combination of GDY and MnOx. These results brought by the interactions between GDY and MnOx are all beneficial to the enhancement of the catalytic activity. In summary, Raman spectroscopy is very sensitive to molecular bonding and the structure of the sample, so each molecule or sample will have its own unique spectral “fingerprints”. These “fingerprints” can be used for chemical identification, morphology and phase, internal pressure/stress, and composition analysis. For GDY/MOs, Raman spectroscopy can not only show the unique alkynyl groups for GDY, but also supply some special structures of one kind of certain metal oxides.
2.3.2.2 XPS. XPS was mainly employed to check the existence of elements and measure the relative contents of GDY/MOs within the escape depths of photoelectrons in the near surface regions. Generally, information such as elemental composition, chemical state and molecular structure on the surface of the GDY/MOs can be obtained from the peak position and peak shape of the XPS. The content or concentration of the element on the surface of the samples can be obtained from the peak strength. Moreover, XPS can be used for verifying the interactions between GDY and MOs. As shown in Fig. 3k,24 the C 1s XPS spectra showed the characteristic peaks of sp-C, sp2-C, C–O, and C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for GDY, WOx/GDY, and CdWOx/GDY samples, demonstrating the robustness of GDY structures. Additionally, the sp-C for both WOx/GDY and CdWOx/GDY shifted to higher binding energies than that of GDY, implying the electron loss from sp-C to metal species. Compared with GDY, the newly appeared π–π* transition peak verified the interactions between GDY and WOx or CdWOx. Besides, the XPS results showed only the presence of W6+ in WO3, while the W 4f spectra of WOx/GDY and CdWOx/GDY all can be deconvoluted into two doublets corresponding to W6+ (35.8 eV for W 4f7/2 and 37.9 eV for W 4f5/2) and W5+ (35.3 eV for W 4f7/2 and 37.4 eV for W 4f5/2). These results indicated the presence of mixed-valent W species due to the charge transfer from sp-C in GDY to metal atoms. Therefore, XPS is a commonly used analytical technique to characterize the surface/interface composition of GDY/MOs, which is important for the investigation of the structure–performance relationship.
O for GDY, WOx/GDY, and CdWOx/GDY samples, demonstrating the robustness of GDY structures. Additionally, the sp-C for both WOx/GDY and CdWOx/GDY shifted to higher binding energies than that of GDY, implying the electron loss from sp-C to metal species. Compared with GDY, the newly appeared π–π* transition peak verified the interactions between GDY and WOx or CdWOx. Besides, the XPS results showed only the presence of W6+ in WO3, while the W 4f spectra of WOx/GDY and CdWOx/GDY all can be deconvoluted into two doublets corresponding to W6+ (35.8 eV for W 4f7/2 and 37.9 eV for W 4f5/2) and W5+ (35.3 eV for W 4f7/2 and 37.4 eV for W 4f5/2). These results indicated the presence of mixed-valent W species due to the charge transfer from sp-C in GDY to metal atoms. Therefore, XPS is a commonly used analytical technique to characterize the surface/interface composition of GDY/MOs, which is important for the investigation of the structure–performance relationship.
2.3.2.3 XAS. XAS is generally used to reflect the valence state of a single element, the coordination environment and the electronic structure of a single absorbing atom in GDY/MOs. For instance, Fig. 3l shows the X-ray absorption near edge structure (XANES) spectra at the Cu K-edge of the CuO/GDY catalysts, CuO, and Cu foil.29 Compared with Cu foil, the absorption edge of CuO/GDY shifted to a higher energy, close to that of CuO, indicating that the valence state of Cu in CuO/GDY was approximately +2. Besides, the results also indicated that there may be interfacial chemical bonds between CuO and GDY. In combination with XPS and Raman spectra, the structure and the interactions between GDY and MOs can be more accurately characterized.
3 Critical molecule activation behavior on GDY/MOs for efficient energy and environmental catalysis
In order to achieve efficient energy and environmental catalysis of GDY/MOs, the investigation of relevant reaction mechanisms is essential. Notably, the effective activation of important reactant molecules is the critical step towards efficient catalysis.43 Reactant molecules such as water (H2O), oxygen (O2), and nitrogen (N2) are important participants in environmental energy catalytic reactions, and their effective activation contributes to the improvement of GDY/MOs' catalytic performance.44,45 In the following, we will discuss in detail the activation behavior of these critical reactant molecules based on the GDY/MOs in the field of energy and environmental catalysis.3.1 Activation of H2O
H2O, as one of the most important compounds on the earth, widely exists in nature and plays an important role in heterogeneous catalytic chemical reactions. In the field of energy conversion, H2O is a clean reactant that can be activated and dissociated to generate hydrogen and oxygen, and other products.42,46 In the field of environmental remediation, H2O can act as an effective promoter or co-catalyst to enhance catalytic activity.43,47 GDY/MOs with a highly tunable interface structure due to the unique diacetylene bonds (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CC–) in GDY can provide more adsorption sites for H2O compared to other catalysts for H2O activation. The activation and dissociation of H2O molecule over GDY/MOs mainly produce hydrogen and hydroxyl species, which are the key species involved in catalytic reactions in the field of energy conversion and environmental remediation. There are two main mechanisms of H2O activation into reactive species (Fig. 4):48,49 one is the homolysis of water to produce hydroxyl radicals and hydrogen radicals; the other is the production of hydrogen protons and hydroxyl species by electrostatic forces or van der Waals forces.
C–CC–) in GDY can provide more adsorption sites for H2O compared to other catalysts for H2O activation. The activation and dissociation of H2O molecule over GDY/MOs mainly produce hydrogen and hydroxyl species, which are the key species involved in catalytic reactions in the field of energy conversion and environmental remediation. There are two main mechanisms of H2O activation into reactive species (Fig. 4):48,49 one is the homolysis of water to produce hydroxyl radicals and hydrogen radicals; the other is the production of hydrogen protons and hydroxyl species by electrostatic forces or van der Waals forces.
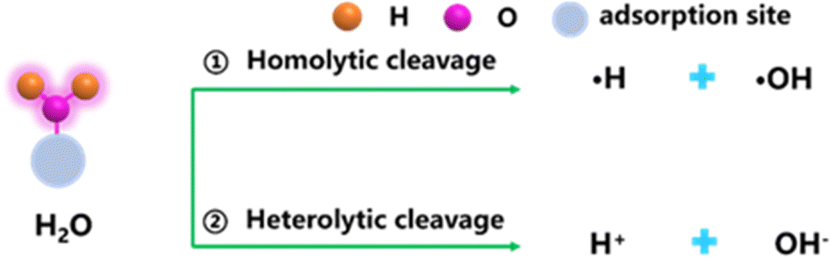 | ||
| Fig. 4 Two main mechanisms of H2O activation. | ||
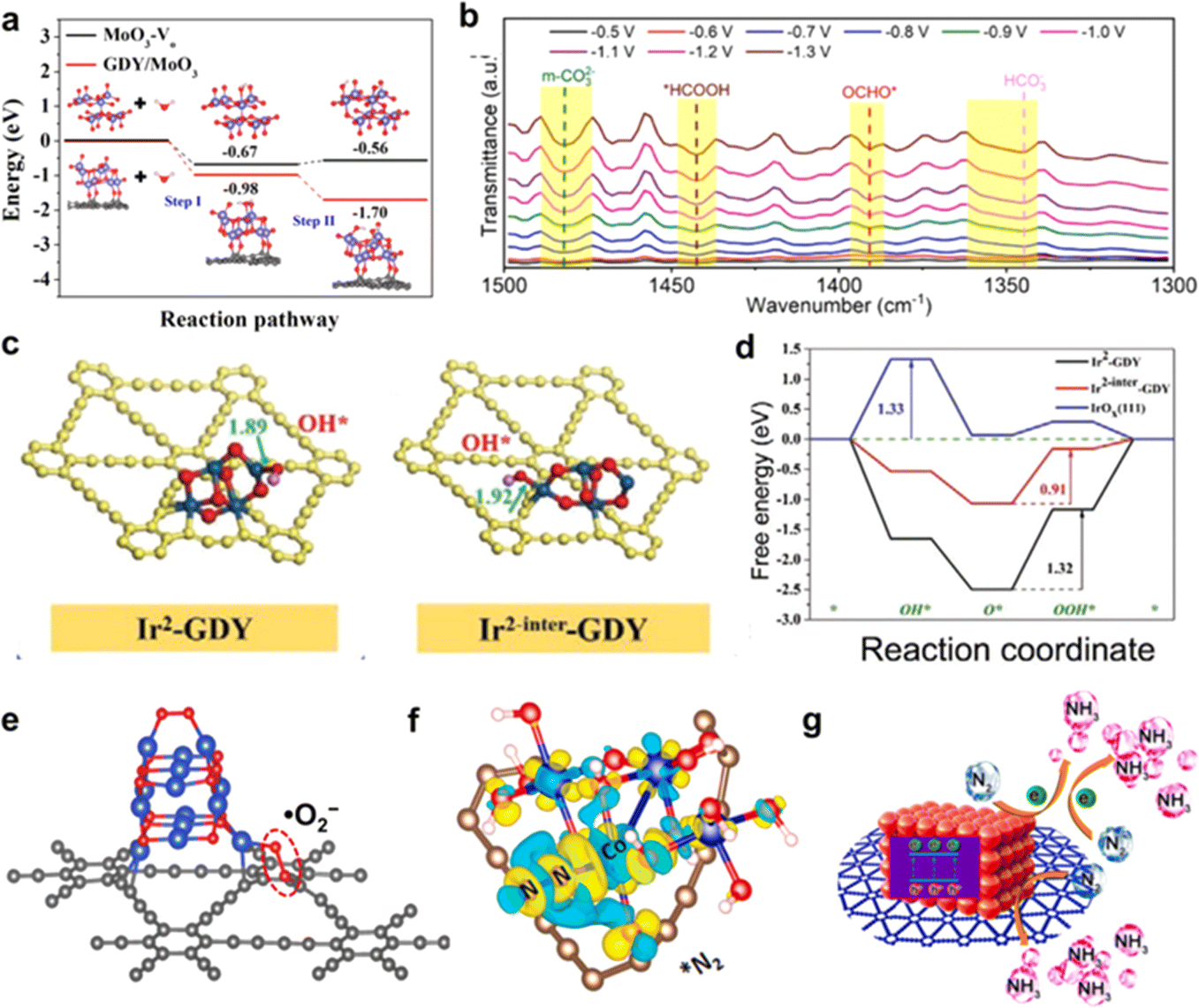 | ||
| Fig. 5 (a) Calculated energy diagram of H2O adsorption and H2O dissociation (inset: corresponding atomic structures of the initial state, H2O adsorption and water dissociation on MoO3-Vo and GDY/MoO3);22 (b) the operando in situ infrared spectra of SnOx/GDY at different potentials;31 (c) optimized adsorption structures of OH* on Ir2-GDY site and Ir2-inter site, respectively;39 (d) free energy diagrams of the OER on the Ir sites at equilibrium potential;39 (e) configuration of the O2 molecule adsorbed on the interface of CuO/GDY;29 (f) differential charge density distributions of (e) *N2 adsorbed on CoOxQDs/GDY;37 (g) schematic diagram of the ammonia production process on porous GDY@CoOxQDs.37 | ||
3.2 Activation of O2
Oxygen (O2) is an important gas that plays an indispensable role in oxidation reactions or generating chemical energy. However, the spin triplet state of oxygen is very stable and finds it difficult to directly participate in the chemical reaction. So, the issue of how to convert oxygen into reactive oxygen species (ROS, ·O2−, O22−, ·OH, O21, et al.) through chemical processes under mild conditions is still facing serious challenges. There are two main mechanisms of O2 activation into ROS (Fig. 6):29,51–54 energy transfer mechanism and electron transfer mechanism. In the energy transfer mechanism, the spin electron state of the spin-triplet state (3∑) O2 molecule is changed to 1O2via physical, chemical and biological methods. The electron transfer mechanism occurs through a continuous single-electron reduction process to sequentially form ·O2−, O22−, and ·OH.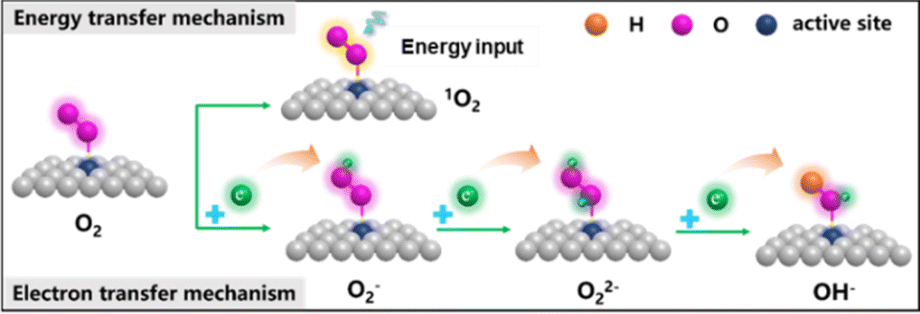 | ||
| Fig. 6 Two main mechanisms of O2 activation. | ||
The rapid activation of oxygen at the active site of the GDY/MOs to produce ROS is considered to be the main reaction pathway for degradation of antibacterial and aqueous/gaseous pollutants. However, there are few studies on the activation of oxygen by GDY/MOs. Zhang's group found that the generation ability of ROS (·O2−, ·OH, O21) and H2O2 was significantly enhanced after TiO2 nanofibers were incorporated into GDY.36 Hence TiO2/GDY showed superior photocatalytic antibacterial activities compared to TiO2. Moreover, oxygen activation plays an important role in the thermocatalytic degradation of gaseous pollutants. Recently, our group reported that the neighboring sp-hybridized C and Cu sites on sub-nanocluster CuO/GDY can effectively activate O2 to ·O2− in the bridging adsorption mode for CO oxidation (Fig. 5e).29 The unique sp-hybridized carbon in GDY could promote the activation of molecular oxygen to produce more ROS compared with the sp2-hybridized carbon in graphene, achieving excellent CO oxidation performance. In brief, GDY/MOs possess strong ability towards oxygen activation due to the interaction between GDY and MOs. And more GDY/MOs deserve to be designed and developed for energy and environmental applications.
3.3 Activation of N2
N2 activation at ambient temperature and pressure is a great challenge. Researchers have made great efforts to this end. The first step of N2 activation is the adsorption of N2 at the active site. Owing to the unique electronic structure of GDY, the efficient electron transfer between GDY and MOs in GDY/MOs can boost the adsorption/desorption of N2 and intermediate species. For example, GDY-induced iron vacancies in the ferroferric oxide/graphdiyne heterostructure (IVR-FO/GDY) promoted the generation of Fe vacancies in the crystalline structure and regulated the Fe valence state. The formation of iron vacancies activated the local O sites to transfer electron towards GDY, leading to GDY preserving the electron-rich feature, which facilitated electron transfer towards adsorbates. Thus, the N2 molecule was efficiently activated and could readily form *NNH. Moreover, IVR-FO/GDY with the iron vacancies promoted the transformation of key intermediate species (e.g., from *NH2NH to *NH2NH), resulting in impressive electrocatalytic nitrogen fixation ammonia performance. In addition, Li's group found that electrons could be transferred from the Co site to the π* orbital of N2 on GDY based quantum dots (GDY@CoOxQDs), which effectively weakened the NN bond facilitating the effective activation of NN bonds and the formation of the Co–N bond (Fig. 5f and g). CoOxQDs/GDY exhibited superior N2 adsorption and N2 activation abilities, leading to excellent photocatalytic performance for ammonia synthesis.37
4. Applications of GDY/MOs in catalysis
Based on the above discussion on the activation of reactant molecules by GDY/MOs, it can be seen that the interfacial interactions between GDY and MOs can effectively modulate the morphological and electronic structures of MOs, promoting the activation of reactant molecules (e.g., H2O, O2, N2, etc.) and regulating the adsorption of intermediate species, and thus the intrinsic activity of the catalyst was significantly enhanced. Besides, GDY is an emerging carbon material with a unique two-dimensional layered conjugated system, natural intrinsic band gap and ultra-high carrier mobility.9,55–60 Combining it with MOs can promote charge transfer and ion diffusion, and improve electrochemical reactivity and kinetics, which shows great performance advantages and application prospects in the fields of energy and environmental catalysis.4.1 Applications of GDY/MOs in energy catalysis
GDY/MOs demonstrate promising applications in energy catalysis, such as the hydrogen evolution reaction (HER),22 oxygen evolution reaction (OER),61 nitrogen reduction (NRR),62 and battery energy storage.63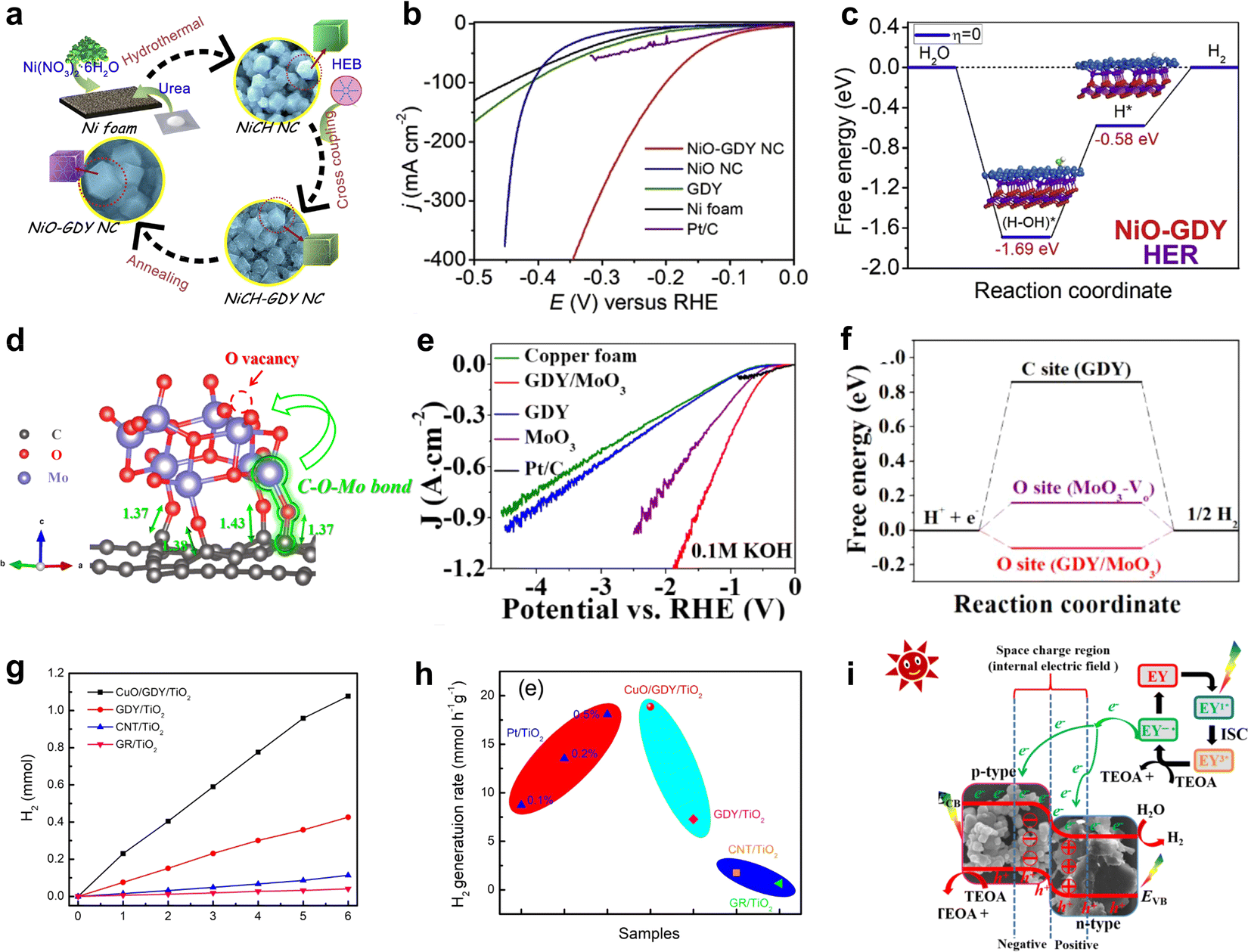 | ||
| Fig. 7 (a) The synthetic strategy for NiO-GDY;1 (b) polarization curves of NiO-GDY, NiO, GDY, Ni foam and Pt/C for the HER in 1.0 M KOH;1 (c) energy diagram for the HER on NiO-GDY;68 (d) C–O–Mo bonds at the interface between MoO3 and GDY;22 (e) polarization curves of the samples with high-current density in 0.1 M KOH;22 (f) the free energy of hydrogen adsorption (ΔGH*) on GDY, MoO3-Vo and GDY/MoO3;22 (g) H2 evolution of CuO/GDY/TiO2, CNT/TiO2 and GR/TiO2;71 (h) comparison of the H2 generation rate based on different photocatalysts;71 (i) photocatalytic hydrogen evolution mechanism of GO-15.21 | ||
GDY/MOs not only demonstrate excellent performance in electrochemical hydrogen evolution but also exhibit remarkable potential in photocatalytic hydrogen evolution.73 For example, CuO/GDY as a cocatalyst effectively promoted photogenerated carrier separation in TiO2 owing to the solid junction between GDY and CuO,71 facilitating the combination of hydrogen protons dissociated from water with electrons to produce hydrogen. Jin et al. reported a Co3O4/GDY catalyst (GO-15) with the p–n heterojunction, exhibiting a strong hydrogen evolution activity (2336.6 μmol g−1 h−1), which is 8.6 and 3.8 times higher than that of GDY and Co3O4.21 The coexistence of Co2+/Co3+ and a strong internal electric field in GO-15 greatly promoted charge separation and diversion of photon-generated carriers and accelerated the adsorption of eosin Y and hydrogen ions, which exhibited excellent photocatalytic H2 production performance.
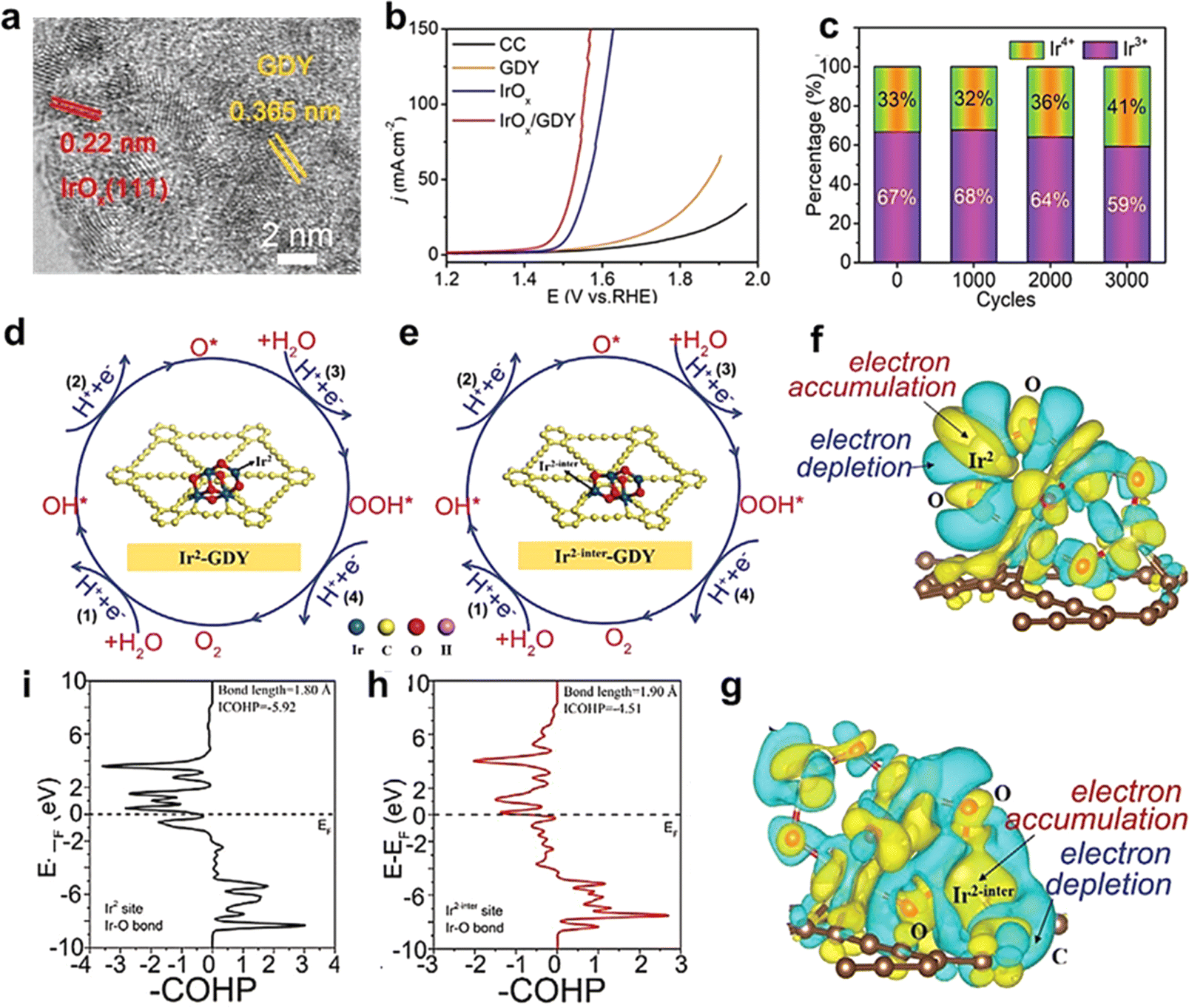 | ||
| Fig. 8 (a) High-resolution TEM images of IrOxQDs/GDY; (b) polarization curves of IrOxQDs/GDY, IrOx, GDY, and CC for the OER in 0.5 m H2SO4 at a scan rate of 2 mV s−1; (c) the percentage of Ir3+ and Ir4+ species in the catalyst after different cycles; four consecutive elementary electron steps on (d) non-heterogeneous and (e) heterogeneous interfacial iridium sites, respectively; differential charge density distribution of (f) non-heterogeneous interfacial iridium atom and (g) heterogeneous interfacial iridium atom, respectively; crystal orbital Hamilton populations (COHPs) of (i) Ir2 site (black line) and (h) Ir2-inter site.39 | ||
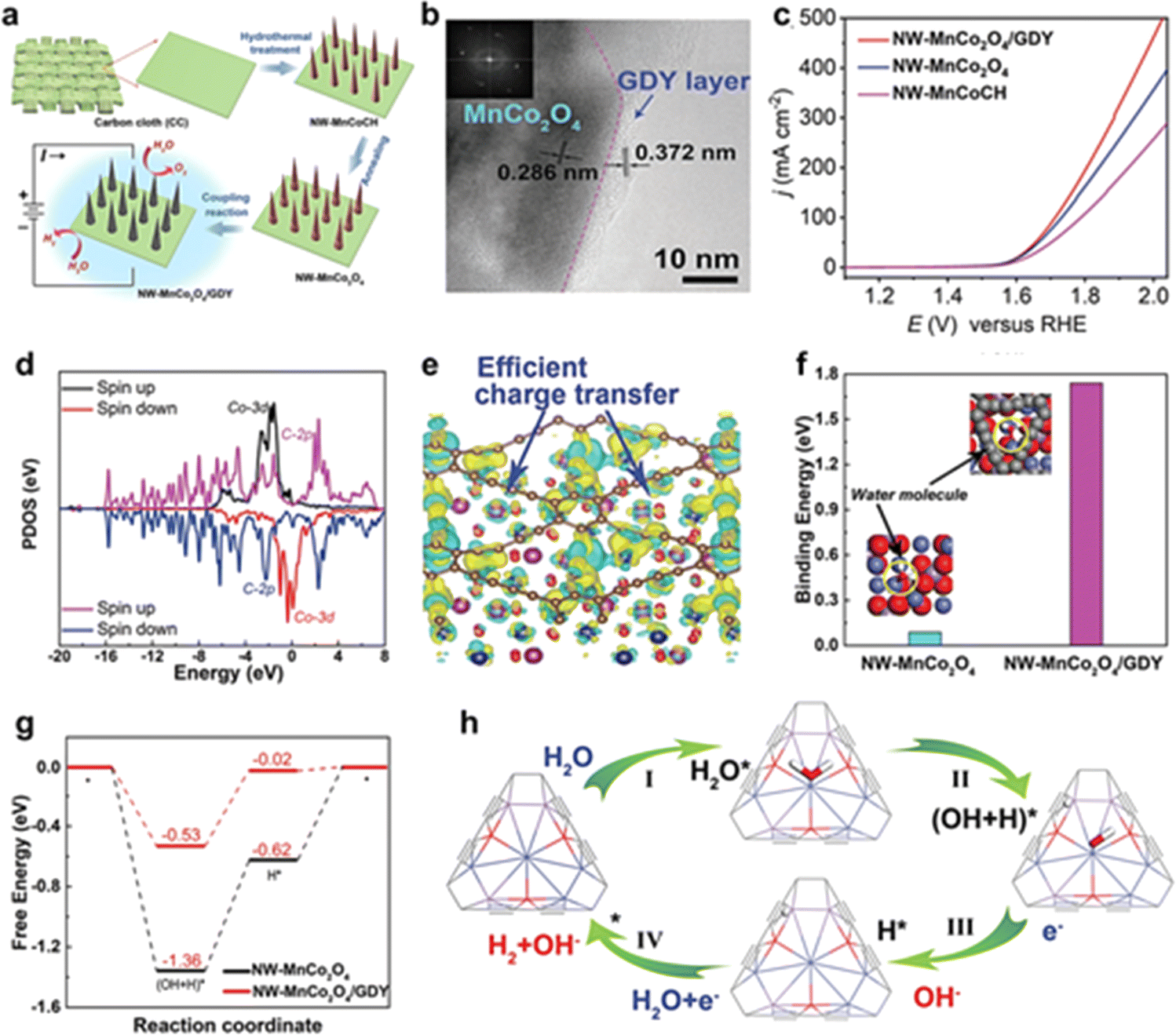 | ||
| Fig. 9 (a) The synthesis routes for the catalysts; (b) HRTEM, and SAED images of NW-MnCo2O4/GDY; (c) polarization curves of NW-MnCo2O4/GDY, NW-MnCo2O4, and NW-MnCoCH for the OER with a scan rate of 2 mV s−1; (d) the PDOS of Co and C atoms in the interface of NW-MnCo2O4/GDY; (e) the corresponding differential charge density distribution map of NW-MnCo2O4/GDY. Green and yellow areas represent the electron deletion and electron accumulation, respectively. The isosurface was set to 0.005 e bohr−3; (f) the binding energy of water molecules adsorbed on NW-MnCo2O4 and NW-MnCo2O4/GDY, respectively. (g) The free energy of H2O molecule dissociation and hydrogen evolution on NW-MnCo2O4 and NW-MnCo2O4/GDY, respectively; (h) schematic diagram of the proposed water splitting mechanism of NW-MnCo2O4/GDY.61 | ||
N triple bond (NN bonds up to 941 kJ mol−1).10 The Haber–Bosh reaction can efficiently convert nitrogen (N2) to NH3, but requires high temperature (300–500 °C) and pressure (150–200 atm), and is highly energy-consuming.37 It would be ideal if N2 conversion could be achieved at room temperature and atmospheric pressure. Electrocatalytic nitrogen fixation to ammonia (ENFA) has received widespread attention due to its advantages of low energy consumption, controllable reaction and green environmental protection. The efficient ENFA can efficiently convert the thermostable and chemical inert N2 to NH3 under ambient conditions.78 It has been reported that GDY has incomparable superiority over traditional carbon materials in the electrocatalysis field due to the infinite natural pores, evenly distributed surface charges, and high conductivity. It can effectively combine and regulate the morphology and electron structure of metal species, generating new active sites, facilitating electron transfer, and achieving excellent electrocatalytic activity.18,79,80 Huang and Li's groups utilized the advantageous properties of GDY in designing and fabricating an iron vacancy-rich ferroferric oxide/graphdiyne heterostructure (IVR-FO/GDY) for high-efficiency ENFA.62 The GDY-induced iron vacancies in IVR-FO/GDY promoted the generation of Fe vacancies in the crystalline structure and regulated the Fe valence state (Fig. 10), which facilitated electron transfer and boosted the efficient activation and adsorption/desorption of N2 and intermediate species. The ammonia yield (YNH3) was up to 60.88% and faradaic efficiency (FE) was up to 60.88%, leading to excellent ENFA performance. Different from ENFA, photocatalysis nitrogen fixation to ammonia (PNFA) uses the sustainable sunlight to drive NH3 synthesis from N2 and H2O at ambient temperature and pressure, which is a promising alternative for energy-consuming Haber–Bosh processes. The excellent hole transport properties of GDY endow GDY-based materials with natural advantages for photocatalysis.81,82 Li's group prepared cubic-like GDY@Fe3O4 (GDY@Fe-A) and rod-like GDY@Fe3O4 (GDY@Fe-B) for PNFA(Fig. 11).82 The coordination environment and valence states of Fe atoms were effectively regulated by GDY, which tuned the photocatalytic performance of the Fe3O4.62 The significant charge transfer between GDY and Fe-B promoted the increase of the OV number for GDY@Fe-B. Thus, GDY@Fe-B could generate more electron hole pairs under illumination, and effectively suppress the recombination of the generated electron–hole pairs, leading to effective nitrogen activation and high photocatalytic performance. GDY@Fe-B showed remarkable catalytic performance with an unprecedented YNH3 level of 1762.35 ± 153.71 μmol h−1 gcat−1 (the highest YNH3 could reach 1916.06 μmol h−1 gcat−1). The Li group also reported another high-performance graphdiyne/metal oxide hybrid material (GDY based quantum dots, GDY@CoOxQDs) for NH3 synthesis from N2 and H2O (Fig. 12).37 The GDY with acetylenic bonds, natural pores and high reduction ability contributed to in situ decoration and stabilization of Co quantum dots. The introduction of GDY and the coexistence of Co2+/Co3+ could facilitate electron transfer, which was beneficial for the adsorption and activation of N2. The electrons from the Co site were transferred to the π* orbital of N2 and formed a Co–N bond, leading to the enhancement of the PNFA performances. The YNH3 of GDY@CoOxQDs could reach 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 502 μmol h−1 gcat−1 in 46 repeated experiments, 100% selectivity, as well as a high long-term stability at room temperatures and pressures.
502 μmol h−1 gcat−1 in 46 repeated experiments, 100% selectivity, as well as a high long-term stability at room temperatures and pressures.
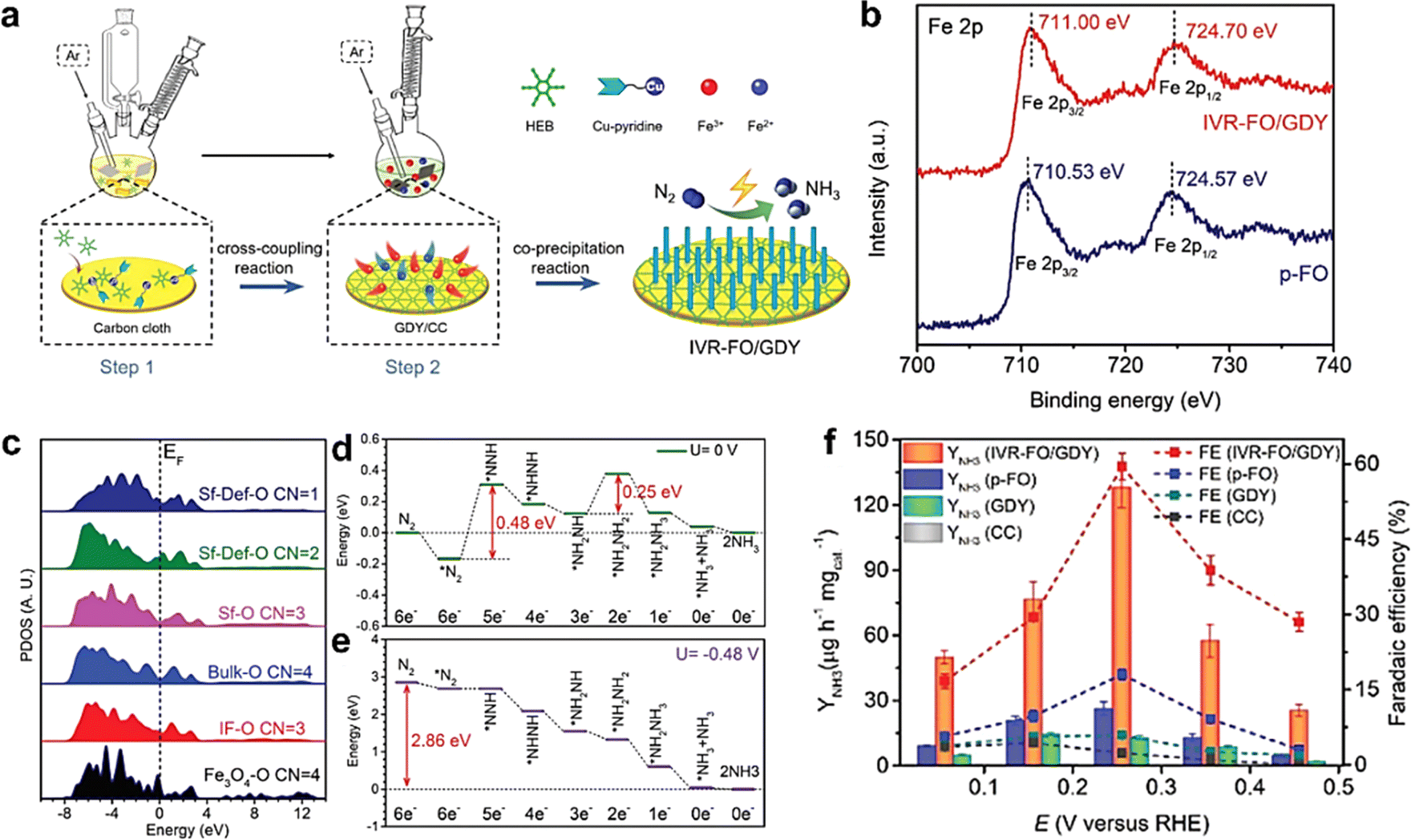 | ||
| Fig. 10 (a) The synthesis route of the IVR-FO/GDY catalyst; (b) Fe 2p XPS spectra of IVR-FO/GDY and p-FO; (c) the site-to-site PDOS of O-2p orbitals in IVR-FO/GDY; (d) energetic pathway of the electrocatalytic NRR on IVR-FO/GDY under U = 0 V; (e) energetic pathway of the electrocatalytic NRR on IVR-FO/GDY under U = −0.48 V; (f) YNH3 and FEs of the samples at different potentials in N2-saturated 0.1 m Na2SO4 (bars represent standard deviation).62 | ||
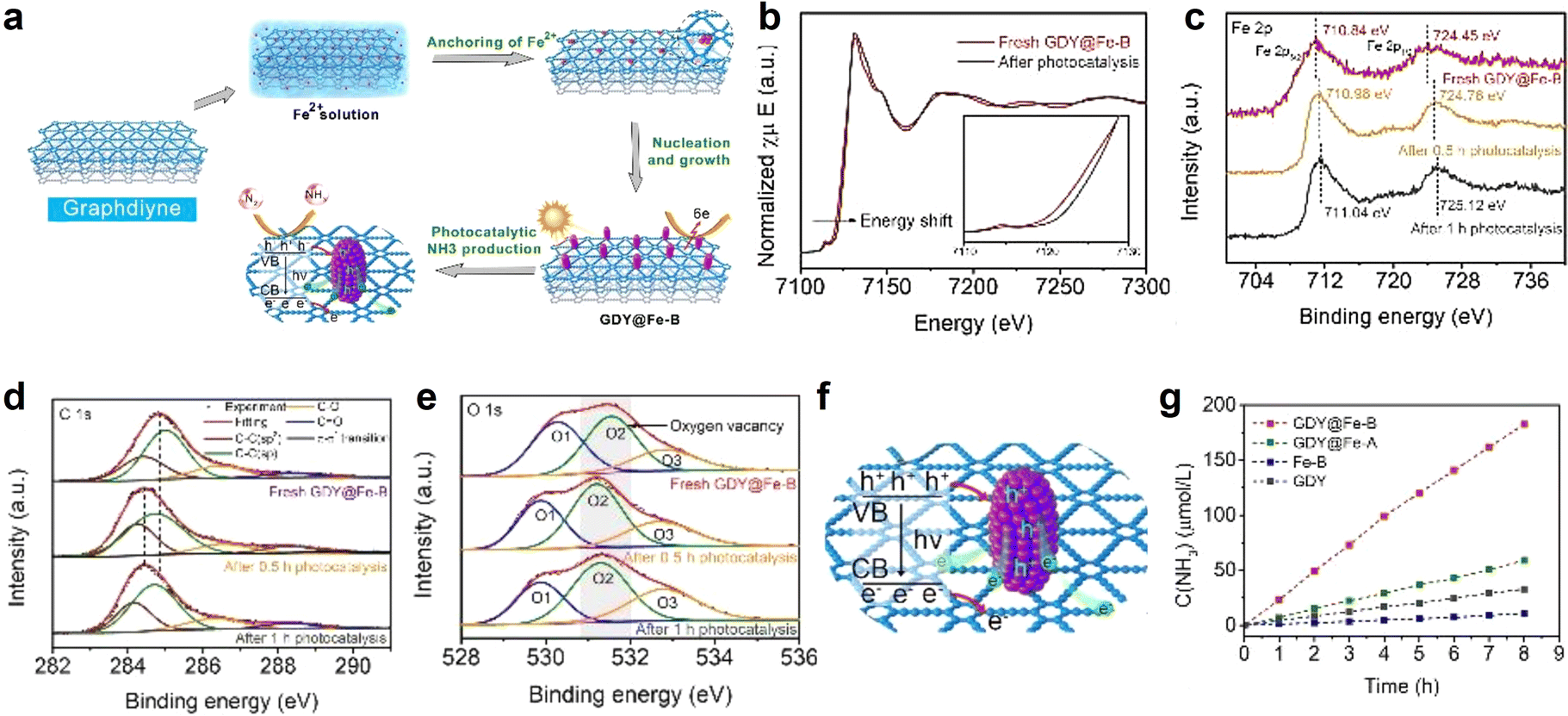 | ||
| Fig. 11 (a) Schematic illustration of the synthetic and reaction routes for GDY@Fe-B; (b) XANES spectra of GDY@Fe-B at the Fe K-edge after photocatalysis; (c) Fe 2p; (d) C 1s and (e) O 1s XPS spectra of GDY@Fe-B along with photocatalysis time; (f) schematic illustration of the mechanism of the photochemical nitrogen reduction reaction on GDY@Fe-B; (g) ammonia concentrations as the function of time for the catalysts under irradiation.82 | ||
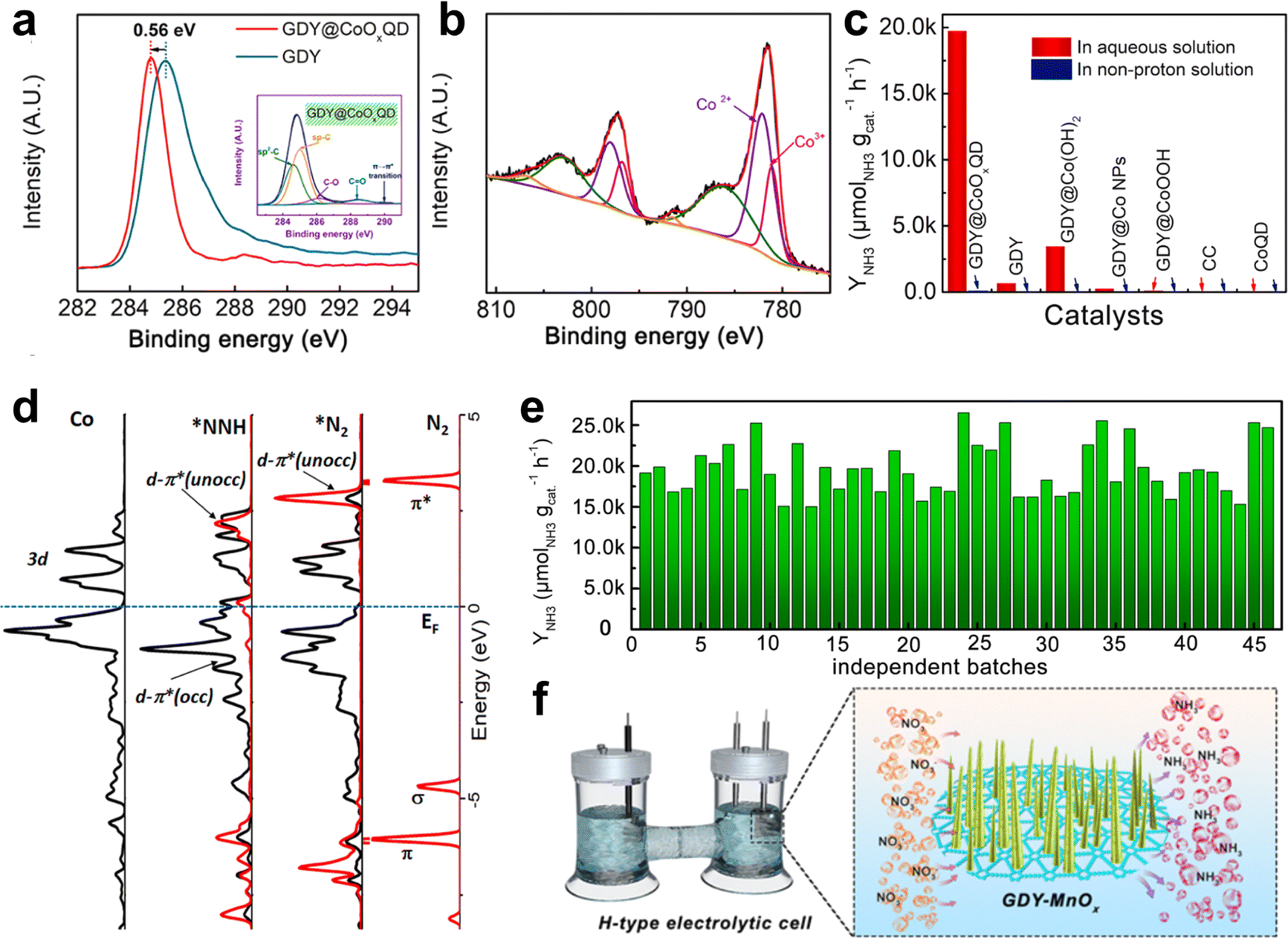 | ||
| Fig. 12 (a) C 1s XPS spectra of GDY@CoOxQDs and GDY. The inset is the deconvoluted C 1s XPS spectrum of GDY@CoOxQDs; (b) the deconvoluted Co 2p XPS spectra of GDY@CoOxQDs; (c) the NH3 yield rates of the samples in different solutions; (d) the orbital distributions and the corresponding differential charge density distributions of *N2 and *NNH adsorbed on CoOxQDs/GDY; (e) YNH3 of the GDY@CoOxQDs in independent experiments;37 (f) illustration of the NO3 to NH3 conversion in an H-type electrolytic cell.38 | ||
Besides, the electrocatalytic nitrate reduction reaction (NtRR) has shown great potential for efficient NH3 synthesis at room temperature and ambient pressure. Li's group reported the GDY–MnOx heterointerface for highly selective and active NH3 synthesis.38 It is found that the incomplete charge-transfer between GDY and Mn atoms at the interface structures lead to the formation of new active sites, increasing the number of active sites. The electrocatalytic NtRR test results showed that the GDY–MnOx exhibited excellent electrocatalytic performance for NH3 synthesis with a large NH3 yield of 463.4 μmol h−1 cm−2 and a high FE of 95.4% and long-term stability in 0.1 M KOH + 0.1 M NO3− aqueous electrolytes at room temperature and ambient pressure.
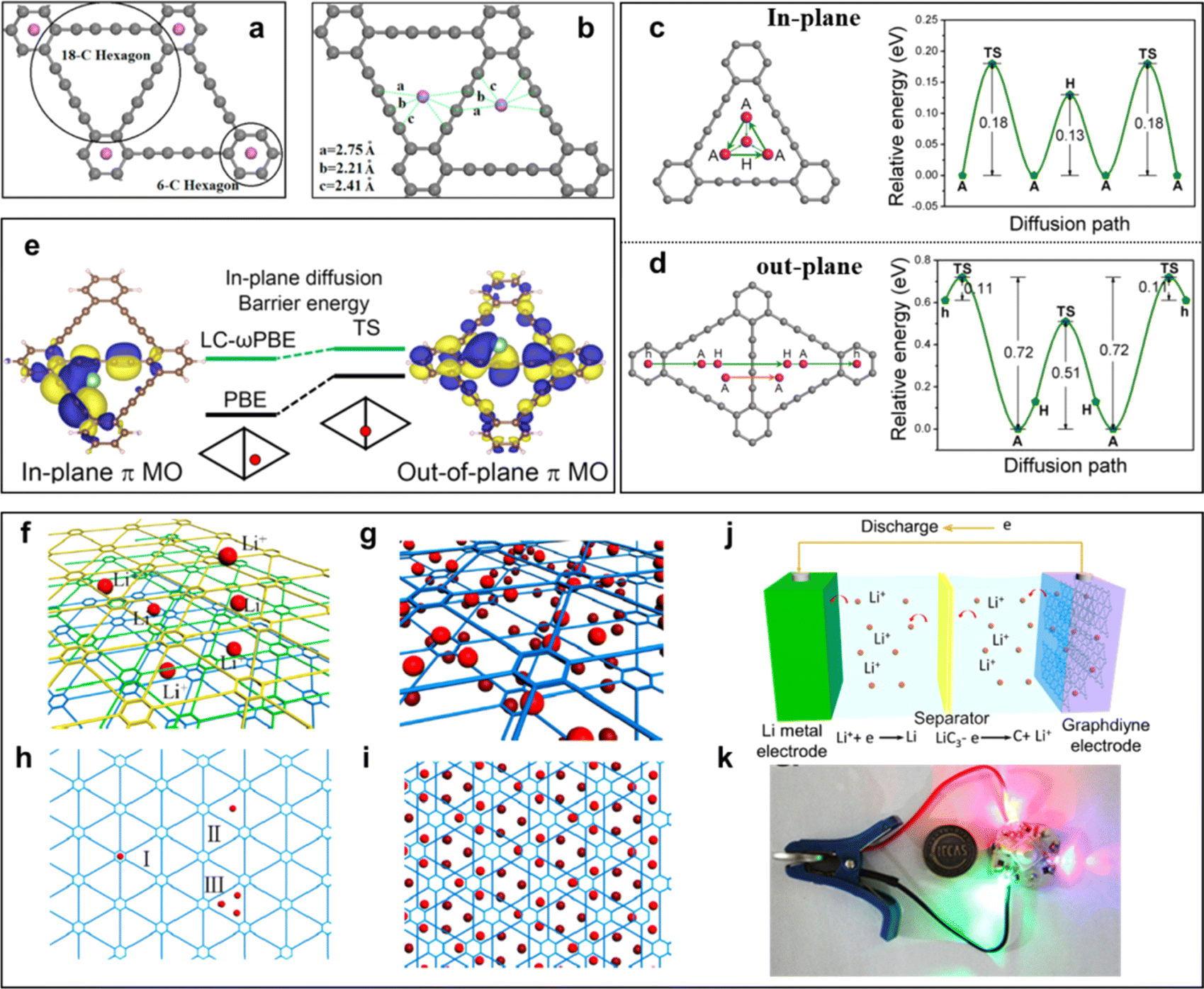 | ||
| Fig. 13 (a) Li in 6-C hexagon (two Li atoms are distributed on the two sides of the carbon network) and (b) Li in 18-C hexagon (both Li atoms are on the same side of the carbon network);83 (c) diffusion pathway of Li within one triangular-like pore and corresponding energy profile as a function of adsorption sites;84 (d) diffusion pathways of Li and corresponding energy profiles as a function of adsorption sites across a GDY layer; (f) Li-intercalated GDY; (g) three different sites for occupation by Li atoms in GDY; adsorption of Li atoms on (h and i) both sides;85 (j) representation of an assembled GDY-based battery; (k) photograph exhibition of the assembled full Li-ion battery and applied discharge for the lighting of an LED bulb. Li-storage of GDY in a Li-ion cell.86 | ||
The practical application of GDY as a high-efficiency lithium storage material in experiments was reported by Li et al. in 2015 (refs. 63 and 86) (Fig. 13j and k). Comparing experimental data with the theoretical value of surface capacity, the value of experimental results was larger than the theoretical capacity of monolayer GDY (Fig. 13h-I and h-II), but smaller than the monolayer GDY in Fig. 13h-III. This result indicated that Li storage in multilayer GDY occurred mainly through both the interlayer insertion/extraction way (Fig. 13f) and surface adsorption/desorption method on the surface of the GDY films (Fig. 13g and i). Based on the above calculation research, the intrinsic advantages make GDY an ideal candidate for constructing electrode materials. Furthermore, GDY/MOs can improve interfacial ion and electron conductivities, and enhance the interfacial stability to realize the construction of high-efficiency electrodes. In 2018, the Huang group first reported the GDY/MOs, TiO2@GDY, with superior electrochemical performances for lithium-ion batteries.87 The results indicated that TiO2@GDY showed a high reversible capacity of 432.4 mA h g−1 after 300 cycles at a current density of 1 A g−1. It was about 3 times that of TiO2. There were two main reasons why the TiO2@GDY was so effective. One was the heterojunction interfaces (Fig. 14a), the built-in electric field could facilitate Li-ion diffusion from GDY to TiO2, and different work functions brought about electron transfer from TiO2 (∼5.70 eV) to GDY (∼4.52 eV). The same mechanism of heterojunction interfaces of MnO2/GDY (Fig. 14d) was also reported by Huang's group after 2 years.88 The research indicated that about 200 cycles later, the irreversible capacity was maintained at 450 mA h g−1 while the MnO2 was just about 100 mA h g−1 (Fig. 14e). The vacancies can also significantly affect the performance of GDY/MOs. It not only provided more ion storage sites, but also facilitated the ion diffusion and improved the electrochemical reactivity and kinetics. For example, the local electric field produced by the oxygen-vacancies can further enhance the electrochemical performances of TiO2@GDY (Fig. 14b and c). Recently, the dual-vacancy strategy for the Cu0.95V2O5@GDYcatalyst (Fig. 14f) was firstly reported by Li's group to enhance the fast-charging capability of lithium-ion batteries.89 Compared to single VCu or Vo vacancy conditions, the lowest Li-ion migration energy barrier was achieved when dual-VCu and Vo were both present in the Cu0.95V2O5@GDY catalyst (Fig. 14h). The Cu0.95V2O5@GDY anodes with a built-in electric field (E) pointing from GDY to Cu0.95V2O5 (Fig. 14g) showed high capacities of 120.3 mA h g−1 for 20000 cycles at extremely high current densities of 10 Ag−1 (Fig. 14i). Besides the oxygen vacancy, the cationic vacancies also play a key role in modulating the electronic structure of electrode materials. For example, iron cationic vacancies (IV) of the GDY/ferroferric oxide heterostructure (IV-GDY-FO) were reported by Li's Group.90 The charge transfer from FO to GDY could induce high valence Fe and exist stably with Fe vacancy (Fig. 14l). Fe vacancies can decrease diffusion energy barrier (Fig. 14j) and improve adsorption energies (Fig. 14k), and accelerate the kinetic process to realize speedy charge transfer and ion diffusion.
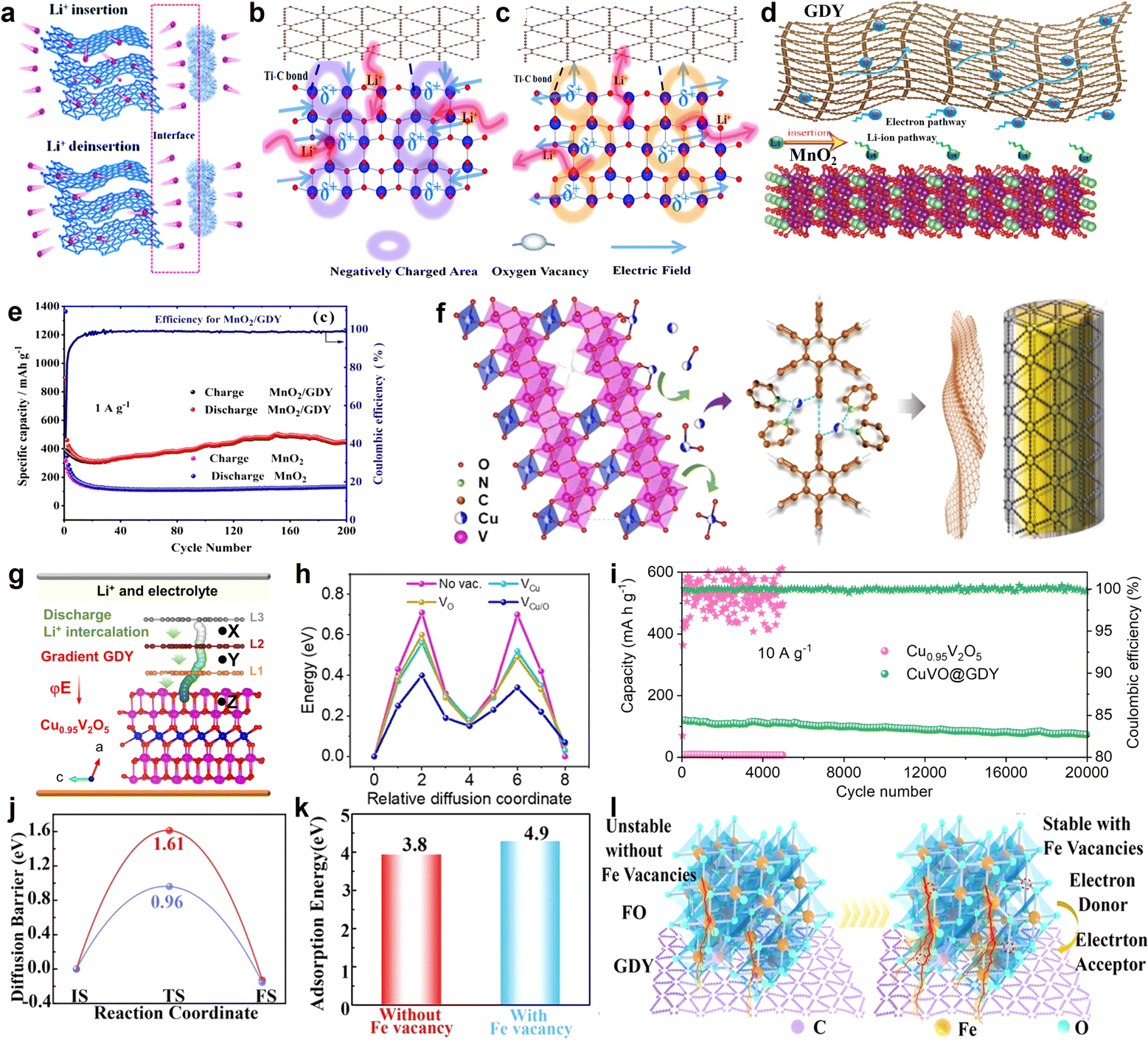 | ||
| Fig. 14 (a) An interfacial built-in electric field (E) between TiO2 and GDY, which facilitates charge transfer across the interface; (b) and (c) schematic illustration of the potential mechanism behind the improved electrochemical performances due to the oxygen vacancy derived local built-in electric field;87 (d) hybrid nanostructured MnO2 nanowire/GDY and the electron/Li-ion pathway;88 (e) cycling performance at 1 A g−1 of MnO2/GDY, MnO2 electrodes;88 (f) Cu ions migrate out of the Cu0.95V2O5 bulk phase to form VCu/O;89 (g) illustration of the Li (green atoms) diffusion pathway from gradient GDY to Cu0.95V2O5via a built-in electric field (E); (h) the Li-ion migration energy barrier in the Cu0.95V2O5 bulk phase;89 (i) cycling performance of Cu0.95V2O5 and CuVO@GDY at a high current density of 10 A g−1;89 (j) diffusion barrier without Fe vacancies and with Fe vacancies;90 (k) Li adsorption energy without Fe vacancies and with Fe vacancies;90 (l) the scheme of formation of Fe vacancy in the presence of GDY.90 | ||
4.2 Applications of GDY/MOs in environmental remediation
C–CC–) in GDY could promote the activation of molecular oxygen to produce more ROS (Fig. 15b, ·O2−). Furthermore, the neighboring sp-hybridized C is more favorable to promote the rapid dissociation of carbonate than sp2-hybridized C, which facilitates the low-temperature CO oxidation. CuO/GDY can readily convert 50% CO at around 133 °C, which is 34 °C lower than that for CuO/graphene (Fig. 15c).
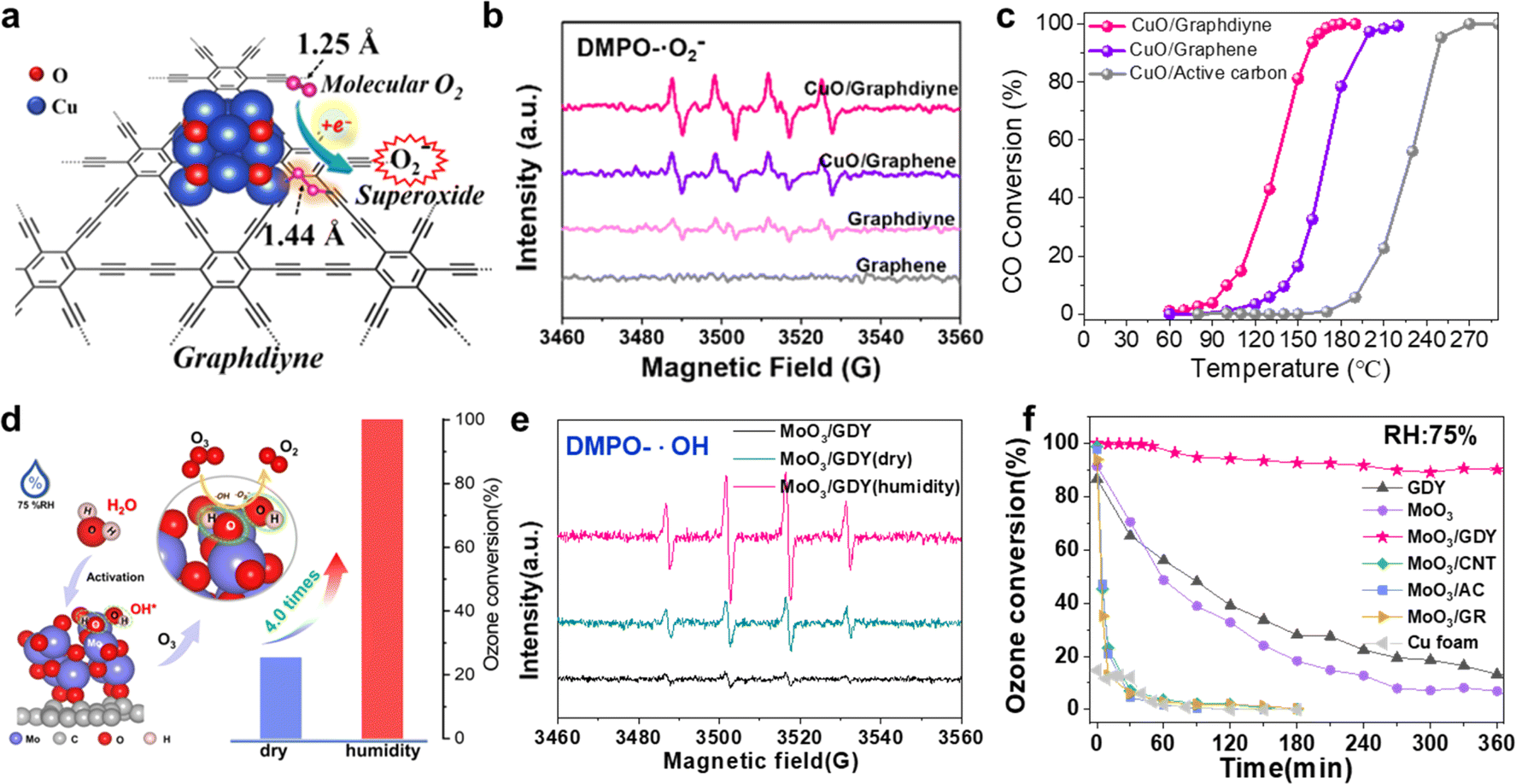 | ||
| Fig. 15 (a) Neighboring sp-hybridized carbon-involved O2 activation on the interface of sub-nanocluster CuO/GDY; (b) EPR spectra of CuO/GDY and GDY for ˙·O2− in DMPO spin trap solution; (c) light-off curves for CO oxidation over CuO/GDY, CuO/graphene, and CuO/active carbon samples [reaction conditions: 1% CO + 5% O2/N2 balanced, total flow rate of 25 mL min−1, and WHSV = 10000 mL (g−1 h−1)];29 (d) rapid O3 decomposition over water-activated monolithic MoO3/GDY nanowalls under high humidity; (e) EPR spectra of pristine MoO3/GDY, O3-exposed MoO3/GDY under dry and humid conditions for ˙OH in DMPO spin trap solution; (f) the O3 conversion of GDY, MoO3, MoO3/GDY, MoO3/CNTs, MoO3/AC, MoO3/GR and Cu foam under humid conditions (75% RH).8 | ||
Moreover, our group has also made some progress in ozone (O3) removal. O3, as a greenhouse gas and secondary atmospheric pollutant, has caused serious harm to the ecosystem and human health. Therefore, efficient and rapid removal of O3 is of great significance to the protection of environmental safety and human health. Recently, our group has reported a three-dimensional monolithic MoO3/GDY catalyst with a C–O–Mo bond. The sp-C in GDY could donate electrons to MoO3 through the C–O–Mo bond, which facilitated the activation of water to hydroxyl species (Fig. 15d), serving as the new active sites (MoO3/GDY-2OH) for rapid and efficient O3 decomposition under high humidity (RH = 75%).8 And further, EPR spectroscopic experiments show that the MoO3/GDY catalyst can accelerate O3 decomposition and produce more active oxygen species (·OH and ·O2−) under humid conditions (Fig. 15e). As shown in Fig. 15f, MoO3/GDY showed the best O3 decomposition activity under high humidity conditions (75% RH) than control samples: Cu foam and GDY, MoO3 and other carbon-based MoO3 materials (MoO3/CNTs, MoO3/AC, MoO3/GR). This study demonstrates the feasibility of water-promoted O3 decomposition on GDY/MOs and provides new insights on the design of water activation for heterogeneous catalysis in a high-humidity atmosphere.
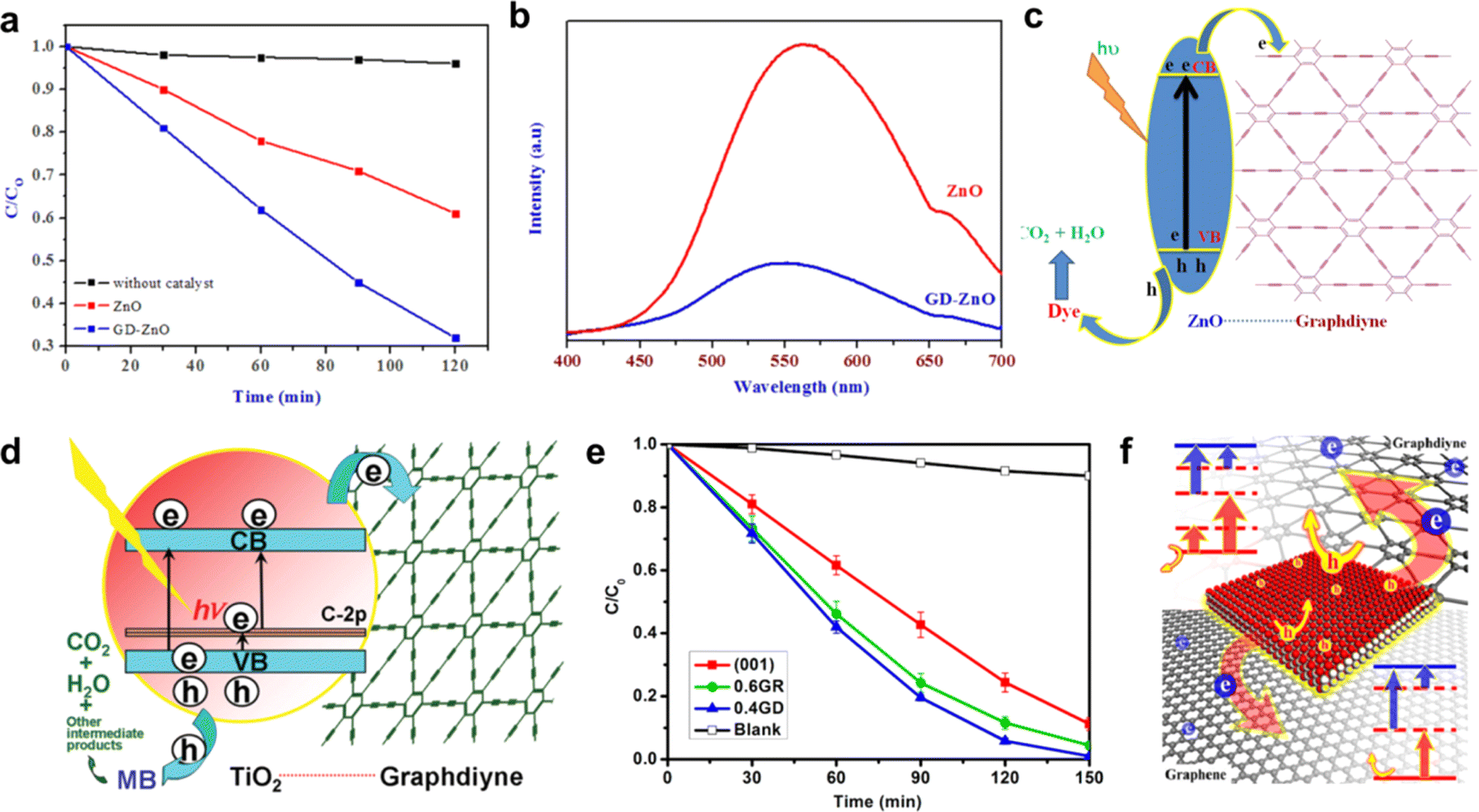 | ||
| Fig. 16 (a) Photocatalytic performance of GDY–ZnO nanohybrids and ZnO nanoparticles in the degradation of methylene blue;93 (b) photoluminescence spectra of ZnO and GDY–ZnO nanohybrids;93 (c) schematic representation of GD-ZnO nanohybrid photocatalysis;93 (d) schematic structure of P25-GD and tentative processes of the photodegradation of methylene blue (MB) over P25-GDY;94 (e) photocatalytic degradation of MB over TiO2(001), TiO2(001)GDY, TiO2(001)GR composites, and the blank experiment; (f) schematic of the photocatalytic properties of TiO2-graphdiyne, and TiO2-graphene composites.95 | ||
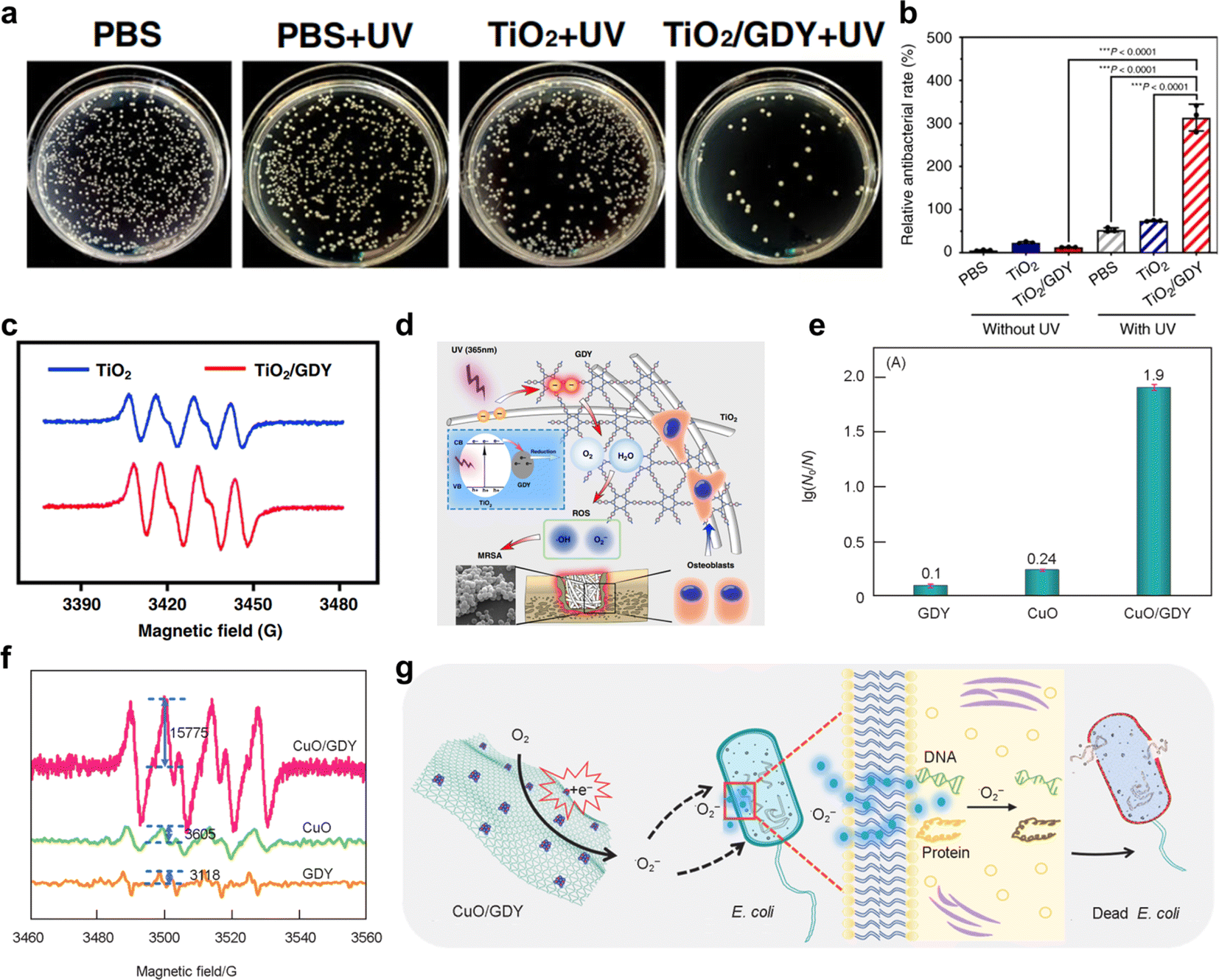 | ||
| Fig. 17 (a)Photographs and quantitative analysis of the bacterial colonies of the infected femurs treated with TiO2, TiO2/GDY, or PBS as a control, with UV irradiation (365 nm, 2 W cm−1, 5 min);36 (b) semiquantification of the live/dead staining for the ratio of PI- and calcein AM-stained areas;36 (c) ESR spectra of TiO2, TiO2/GDY for ˙O2−;36 (d) schematic of the TiO2/GDY in orthopedic implant infection, reactive oxygen species are produced in the process;36 (e) E. coli inactivation efficiency of GDY, CuO and CuO/GDY;25 (f) ESR spectra of GDY, CuO and CuO/GDY for ·O2−;25 (g) illustration of the possible antibacterial mechanisms of CuO/GDY nanomaterials through oxidative stress.25 | ||
4.3 CO2 reduction reaction
The depletion of fossil fuels and continued emissions of CO2 have resulted in a growing energy and environmental crisis. The CO2 reduction into chemical fuels is recognized as a potential solution to these issues.100–105 At present, CO2 reduction reactions (CO2RR) mainly face the following challenges: (1) the CO2 molecule is difficult to adsorb and activate; (2) hydrogen evolution reactions that compete with CO2 reduction reactions should be effectively inhibited; (3) the stability of the catalyst needs to be improved; (4) the selectivity of reduction products is difficult to control and the mechanism of the CO2 reduction reaction needs further study. To date, several attempts have been made to solve these problems.Recently, Yu et al. successfully synthesized TiO2 nanofibers supported by the GDY cocatalyst (Fig. 18a) through an electrostatic self-assembly method for photocatalytic CO2 reduction.106 The first calculations and in situ XPS tests showed that delocalized electrons in GDY can be hybridized with empty orbitals in TiO2, forming an internal electric field (IEF) at the interfaces of TiO2/GDY (Fig. 18b). The unique structure of IFE, combined with the photothermal effect of GDY, can enhance charge separation, directed electron transfer, CO2 adsorption and activation (Fig. 18c–e), thus significantly improving the efficiency and selectivity of CO2 photoreduction. Li et al. developed a GDY/SnOx electrocatalyst with a novel heterostructure,31 achieving highly selective reduction of CO2 to formates at high current density. The results showed that GDY has advantages in effectively regulating the valence state of SnOx, increasing the charge transfer ability and the number of active sites.
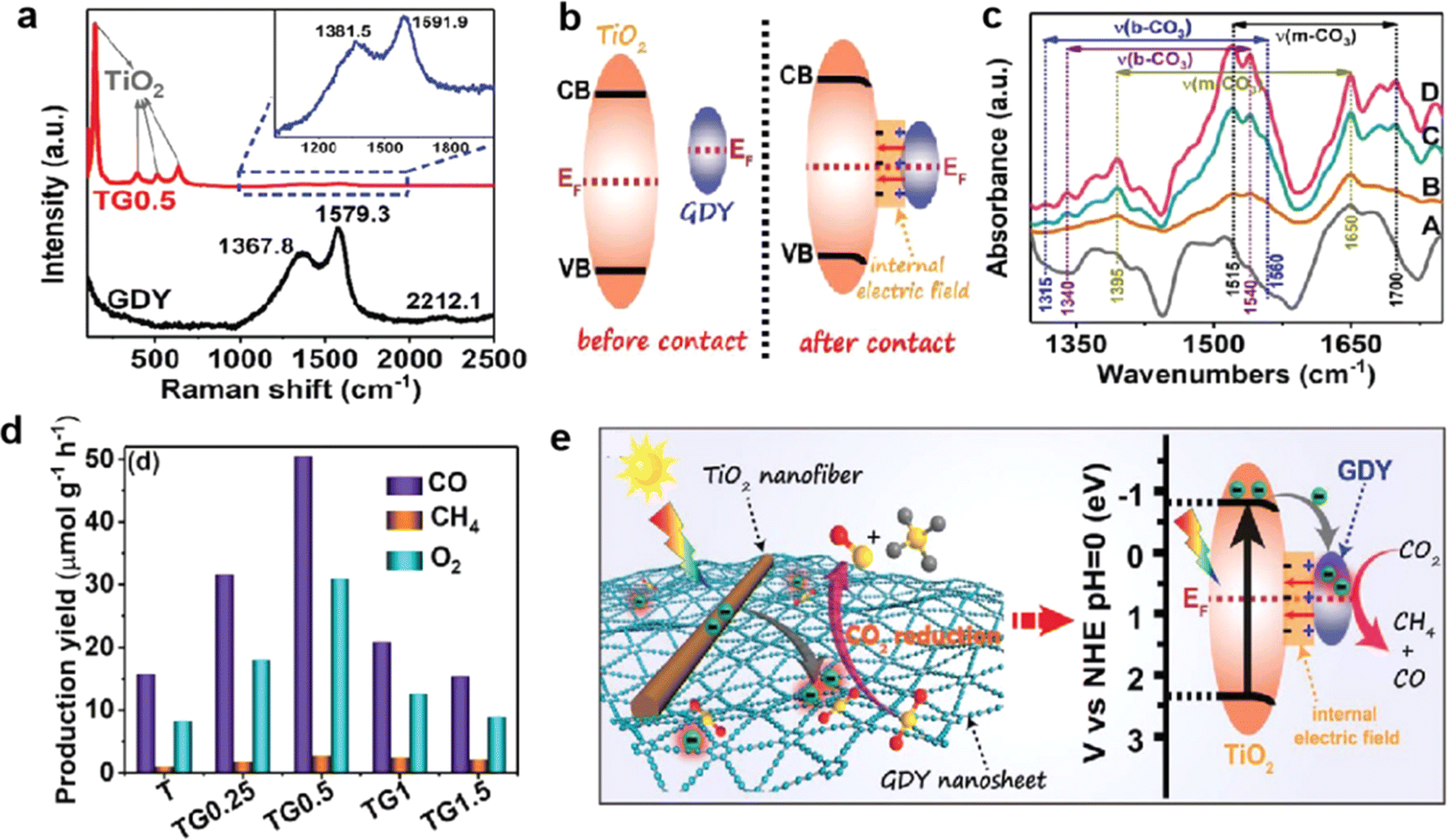 | ||
| Fig. 18 2 after adsorption of CO2/H2O for 60 min in dark (A) and TiO2/GDY after adsorption of CO2/H2O for 20 (B), 40 (C), and 60 (D) min in dark; (a) Raman spectra of GDY and TiO2/GDY; (b) schematic diagram for the electron transfer and the formation of IEF between TiO2 and GDY upon their contact; (c) in situ DRIFTS spectra of TiO2 after adsorption of CO2/H2O for 60 min in dark (A) and TiO2/GDY after adsorption of CO2/H2O for 20 (B), 40 (C), and 60 (D) min in dark; (d) photocatalytic activities of CO2 reduction over the samples; (e) schematic illustration of the TiO2/GDY heterojunction: internal electric field-induced charge transfer and separation under UV-visible light irradiation for CO2 photoreduction.106 | ||
The maximum reaction selectivity was 99.5% and the formate yield was 323.42 μmol h−1 cm−2 at a high current density of 112 mA cm−2 and an applied voltage of −0.9 V vs. RHE. From the operando in situ infrared (IR) measurements, an upward peak at 1390 cm−1 indicated that the OCHO* species was the key intermediate in the formation of formate. And the appearance of *CO (1956 and 2012 cm−1) explained the later emergence of CO. Combined with these results, they further proposed that the formation of OCHO* was an exothermic process while the formation of *COOH was endothermic conversely. Therefore, to generate formate, the limiting potential is determined by the OCHO* hydrogenation step. In conclusion, the conversion efficiency and the selectivity of target products can be improved by studying the mechanism of the CO2RR deeply and designing efficient catalysts. In addition, photocatalytic conversion of CO2 into energy-rich products is also an ideal strategy to pursue renewable energy;103–105 GDY/MOs possess excellent CO2 activation and charge separation/transfer ability and may be a potential photocatalytic CO2 conversion material.
5. Conclusions and perspectives
In this review, we systematically summarized the structure, synthesis and advanced characterization methods of GDY/MOs and their applications in energy conversion and environmental remediation (HER, OER, NRR, CO2RR, air pollution control, liquid organic pollution control, antibacterial activity, etc.). In particular, the detailed structure–performance relationship, activation behaviors (activation of H2O, O2, CO2 and N2), intermediate species and corresponding mechanisms for energy and environmental catalysis were discussed.The unique sp-hybridized carbon atoms and triangular cavities in the GDY-conjugated structure facilitate the formation of strong interfacial contacts with metal oxide. The interaction between metal oxide and GDY endows GDY/MOs with fast charge transfer at the interface, promoting the adsorption and activation of reactant molecules (H2O, N2), and regulating the adsorption and desorption of active intermediates, which exhibits excellent performance in water electrolysis (HER and OER) and NRR. GDY as the carrier can disperse MOs uniformly and induce the formation of metal vacancy which could regulate the charge distribution around vacancies and adjacent atoms, facilitating electronic transportation, enlarging the lithium-ion diffusion, and thus displaying distinguished battery energy storage. The interfacial chemical bonds formed between sp-hybridized C and MOs endow GDY/MOs with the ability to activate oxygen to form ·O2− species, enabling GDY to be used as an outstanding catalyst for low temperature CO oxidation and antimicrobial performance. The heterogeneous interfaces formed by high charge mobility GDY and MOs could generate more electron hole pairs under illumination, and effectively suppress the recombination of the generated electron–hole pairs, leading to excellent photocatalytic performance for carbon dioxide reduction, nitrogen reduction and degradation of organic pollutants such as methylene blue and rhodamine. Therefore, we believe that further exploration of GDY/MOs will further reveal their intrinsic properties and pave the way for practical applications in energy and environment-related fields.
The above unique advantages enable GDY/MOs to show excellent performance and great application potential in energy conversion and environmental remediation. Although many advances have been made in the synthesis and application of GDY/MOs for energy and environmental catalysis, the research and efficient utilization of GDY/MOs are still in the development stage and further improvements are still desired. Based on the current perspectives, it is worth carrying out more in-depth explorations into the synthesis, properties and practical applications in future research.
(i) The research of GDY/MOs is mainly focused on basic research. It is essential to explore the service life of the GDY/MOs and expand the practical applications in the fields of energy conversion and environmental remediation: (1) exploring low-cost, large-scale, uniform and controllable synthesis of GDY/MOs; (2) assembling catalysts into device realizable applications, such as a complete set of devices directly capable of seawater electrolysis and a complete set of air purification device.
(ii) At present, the photocatalytic water splitting efficiency of GDY/MOs still needs to be improved. We can effectively choose GDY/MOs by DFT calculations and machine learning, and further enhance the photocatalytic water splitting performance by modulating the interfacial structure of GDY and suitable MOs.
(iii) Current research on GDY/MOs in the field of energy is mainly focused on water splitting and NH3 synthesis. However, GDY can effectively transfer electrons to form strong interactions with MOs, which can promote the adsorption and activation of small molecules, and may have a greater application prospect in fuel cells and syngas preparation.
(iv) Besides CO and O3, other gaseous pollutants also threaten the environment and human health, such as volatile organic compounds (VOCs). The strong interaction between acetylenic links in GDY and MOs can effectively activate O2 and H2O to generate ·O2−, ·OH, which has great potential for the oxidation of VOCs. Moreover, the catalytic mechanism of GDY/MOs for VOCs deserves in-depth and comprehensive exploration.
(v) Emerging pollutants (EPs) are a class of chemical pollutants that pose potential or substantial threats to human health and ecological environment. EPs mainly include pharmaceuticals and personal care products, endocrine disrupting chemicals, antibiotics, persistent organic pollutants, disinfection by-products and other industrial chemicals, etc. The GDY/MOs with high specific surface area and strong activation ability for organic pollutants can benefit the adsorption and elimination of some EPs.
(vi) Currently, the selectivity of the CO2RR and NRR on GDY/MOs could reach nearly 100%, but its conversion rate is still very low, and it is necessary to continue to design and synthesize high-performance GDY/MO materials.
(vii) The relationship between the structures, properties, and performance of the GDY/MOs, active intermediates and mechanisms need to be thoroughly revealed and understood at the molecular and atomic levels through machine learning, and advanced in situ and ex situ techniques. At present, the research of GDY/MOs in energy and environmental catalysis is in its initial stage, there is still much room for improvement.
Undoubtedly, GDY/MOs are a key material for the development of high-performance and new technologies in the fields of energy conversion and environmental remediation. It is believed that GDY/MOs are capable of delivering incredible performance appropriate for the applications including but not limited to energy and environmental aspects. We are looking forward to a better tomorrow for GDY/MO research.
Author contributions
Y. Z., S. Z., Y. W. and Y. G. conceived the idea. Z. L. and Y. G supervised the project. The manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant number 22076060, 22376075), Natural Science Foundation for Distinguished Young Scholars of Hubei province (grant number 2021CFA085), Natural Science Foundation of Hubei Province, China (grant number 2022CFB321), The Recruitment Program of Global Young Experts start-up funds, The Program of Introducing Talents of Discipline to Universities of China (111 program, B17019), Longyan Major Science and Technology Project (2022LYF1006), and Knowledge Innovation Program of Wuhan Basic Research (2022010801020289).References
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- B. Song, M. Chen, G. Zeng, J. Gong, M. Shen, W. Xiong, C. Zhou, X. Tang, Y. Yang and W. Wang, J. Hazard. Mater., 2020, 398, 122957 CrossRef CAS PubMed.
- M. Li, N. Han, X. Zhang, S. Wang, M. Jiang, A. Bokhari, W. Zhang, M. Race, Z. Shen, R. Chen, M. Mubashir, K. S. Khoo, S. S. Teo and P. L. Show, Environ. Res., 2022, 205, 544 Search PubMed.
- Y. Zhu, Y. Yao, Z. Luo, C. Pan, J. Yang, Y. Fang, H. Deng, C. Liu, Q. Tan, F. Liu and Y. Guo, Molecules, 2019, 25, 18 CrossRef PubMed.
- Z. Zuo, D. Wang, J. Zhang, F. Lu and Y. Li, Adv. Mater., 2018, 31, 1803762 CrossRef.
- G. Zhu, W. Zhu, Y. Lou, J. Ma, W. Yao, R. Zong and Y. Zhu, Nat. Commun., 2021, 12, 4152 CrossRef CAS.
- Q. Hao, Z. Li, Y. Shi, R. Li, Y. Li, S. Ouyang, H. Yuan and T. Zhang, Nano Energy, 2022, 102, 107723 CrossRef CAS.
- Y. Zhu, L. Yang, J. Ma, Y. Fang, J. Yang, X. Chen, J. Zheng, S. Zhang, W. Chen, C. Pan, B. Zhang, X. Qiu, Z. Luo, J. Wang and Y. Guo, Angew. Chem., Int. Ed., 2023, e202309158 CAS.
- B. Li, C. Lai, M. Zhang, G. Zeng, S. Liu, D. Huang, L. Qin, X. Liu, H. Yi, F. Xu, N. An and L. Chen, Adv. Energy Mater., 2020, 10, 2000177 CrossRef CAS.
- X. Zheng, S. Chen, J. Li, H. Wu, C. Zhang, D. Zhang, X. Chen, Y. Gao, F. He, L. Hui, H. Liu, T. Jiu, N. Wang, G. Li, J. Xu, Y. Xue, C. Huang, C. Chen, Y. Guo, T. B. Lu, D. Wang, L. Mao, J. Zhang, Y. Zhang, L. Chi, W. Guo, X. H. Bu, H. Zhang, L. Dai, Y. Zhao and Y. Li, ACS Nano, 2023, 17, 14309–14346 CrossRef CAS.
- Y. Fang, Y. Liu, L. Qi, Y. Xue and Y. Li, Chem. Soc. Rev., 2022, 51, 2681–2709 RSC.
- S. Chai, X. Chen, X. Zhang, Y. Fang, R. S. Sprick and X. Chen, Environ. Sci.: Nano, 2022, 9, 2464–2469 RSC.
- M. Wang, M. Wang, H. H. Lin, M. Ballabio, H. Zhong, M. Bonn, S. Zhou, T. Heine, E. Canovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2020, 142, 21622–21627 CrossRef CAS.
- Y. Fang, Y. Hou, X. Fu and X. Wang, Chem. Rev., 2022, 122, 4204–4256 CrossRef CAS.
- X. Gao, H. Liu, D. Wang and J. Zhang, Chem. Soc. Rev., 2019, 48, 908–936 RSC.
- J. Liu, Q. Ma, Z. Huang, G. Liu and H. Zhang, Adv. Mater., 2019, 31, e1800696 CrossRef.
- X. Chen, X. Jiang and N. Yang, Small, 2022, 18, e2201135 CrossRef PubMed.
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1460 CrossRef.
- C. Pan, C. Wang, Y. Fang, Y. Zhu, H. Deng and Y. Guo, Environ. Sci.: Nano, 2021, 8, 1863–1885 RSC.
- J. Liu, C. Chen and Y. Zhao, Adv. Mater., 2019, 31, e1804386 CrossRef.
- Y. Cao, X. Hao, X. Guo, K. Wang, G. Wang and Z. Jin, Ind. Eng. Chem. Fundam., 2021, 60, 18397–18407 CrossRef CAS.
- Y. Yao, Y. Zhu, C. Pan, C. Wang, S. Hu, W. Xiao, X. Chi, Y. Fang, J. Yang, H. Deng, S. Xiao, J. Li, Z. Luo and Y. Guo, J. Am. Chem. Soc., 2021, 143, 8720–8730 CrossRef CAS.
- J. Tang, M. Zhao, X. Cai, L. Liu, X. Li and T. Jiu, Chem. Res. Chin. Univ., 2021, 37, 1309–1316 CrossRef CAS.
- X. Luan, Z. Zheng, S. Zhao, Y. Xue and Y. Li, Adv. Funct. Mater., 2022, 32, 2202843 CrossRef CAS.
- C. Pan, X. Liu, X. Zhang, F. Mao, P. Xu, Y. Zhu, H. Deng, Z. Luo, H. Sun, L. Zhang and Y. Guo, Chem. Res. Chin. Univ., 2021, 37, 1341–1347 CrossRef CAS.
- S. Zhang and X. Wang, Acc. Mater. Res., 2022, 3, 1285–1298 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS.
- M. Peng, C. Dong, R. Gao, D. Xiao, H. Liu and D. Ma, ACS Cent. Sci., 2021, 7, 262–273 CrossRef CAS PubMed.
- C. Pan, C. Wang, X. Zhao, P. Xu, F. Mao, J. Yang, Y. Zhu, R. Yu, S. Xiao, Y. Fang, H. Deng, Z. Luo, J. Wu, J. Li, S. Liu, S. Xiao, L. Zhang and Y. Guo, J. Am. Chem. Soc., 2022, 144, 4942–4951 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- X. Chen, X. Zheng, C. Zhang, D. Zhang, Y. Gao, S. Chen, Y. Xue and Y. Li, Nano Energy, 2023, 114, 108622 CrossRef CAS.
- S. C. Shekar and R. S. Swathi, J. Phys. Chem. C, 2015, 119, 8912–8923 CrossRef CAS.
- Z. Chen, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2017, 121, 3463–3468 CrossRef CAS.
- J. He, S. Y. Ma, P. Zhou, C. X. Zhang, C. He and L. Z. Sun, J. Phys. Chem. C, 2012, 116, 26313–26321 CrossRef CAS.
- Z. Jin, X. Wang, X. Hao, G. Wang, X. Guo and K. Wang, 2D Mater., 2022, 9, 025014 CrossRef.
- R. Wang, M. Shi, F. Xu, Y. Qiu, P. Zhang, K. Shen, Q. Zhao, J. Yu and Y. Zhang, Nat. Commun., 2020, 11, 4465 CrossRef CAS PubMed.
- Y. Liu, Y. Xue, L. Hui, H. Yu, Y. Fang, F. He and Y. Li, Nano Energy, 2021, 89, 106333 CrossRef CAS.
- X. Luan, L. Qi, Z. Zheng, S. Zhao, Y. Gao, Y. Xue and Y. Li, Chem. Commun., 2023, 59, 7611–7614 RSC.
- Z. Wang, Z. Zheng, Y. Xue, F. He and Y. Li, Adv. Energy Mater., 2021, 11, 2101138 CrossRef CAS.
- J. Zhou, X. Gao, R. Liu, Z. Xie, J. Yang, S. Zhang, G. Zhang, H. Liu, Y. Li, J. Zhang and Z. Liu, J. Am. Chem. Soc., 2015, 137, 7596–7599 CrossRef CAS.
- F. Wang, Z. Zuo, L. Li, F. He, F. Lu and Y. Li, Adv. Mater., 2019, 31, 1806272 CrossRef PubMed.
- Z. Luo, R. Miao, T. D. Huan, I. M. Mosa, A. S. Poyraz, W. Zhong, J. E. Cloud, D. A. Kriz, S. Thanneeru, J. He, Y. Zhang, R. Ramprasad and S. L. Suib, Adv. Energy Mater., 2016, 6, 1600528 Search PubMed.
- Q. Wei, C. Yu, X. Song, Y. Zhong, L. Ni, Y. Ren, W. Guo, J. Yu and J. Qiu, J. Am. Chem. Soc., 2021, 143, 6071–6078 CrossRef CAS PubMed.
- X. Zhang, F. Bi, Z. Zhu, Y. Yang, S. Zhao, J. Chen, X. Lv, Y. Wang, J. Xu and N. Liu, Appl. Catal., B, 2021, 297, 120393 CrossRef CAS.
- S. Chen, Z. Zhang, W. Jiang, S. Zhang, J. Zhu, L. Wang, H. Ou, S. Zaman, L. Tan, P. Zhu, E. Zhang, P. Jiang, Y. Su, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 12807–12815 CrossRef CAS PubMed.
- X. Niu, Q. Tang, B. He and P. Yang, Electrochim. Acta, 2016, 208, 180–187 CrossRef CAS.
- J. Saavedra, C. J. Pursell and B. D. Chandler, J. Am. Chem. Soc., 2018, 140, 3712–3723 CrossRef CAS.
- J. Zhang, C. Muck-Lichtenfeld and A. Studer, Nature, 2023, 619, 506–513 CrossRef CAS.
- Y. Wang, Y. Qu, B. Qu, L. Bai, Y. Liu, Z. D. Yang, W. Zhang, L. Jing and H. Fu, Adv. Mater., 2021, 33, e2105482 CrossRef.
- L. Xiang, L. Zhang, J. Shao, F. Lin, Z. Wang, B. Yan and G. Chen, J. Hazard. Mater., 2023, 441, 129997 CrossRef CAS.
- H. Gu, X. Liu, X. Liu, C. Ling, K. Wei, G. Zhan, Y. Guo and L. Zhang, Nat. Commun., 2021, 12, 5422 CrossRef CAS PubMed.
- L. Kang, B. Wang, Q. Bing, M. Zalibera, R. Buchel, R. Xu, Q. Wang, Y. Liu, D. Gianolio, C. C. Tang, E. K. Gibson, M. Danaie, C. Allen, K. Wu, S. Marlow, L. D. Sun, Q. He, S. Guan, A. Savitsky, J. J. Velasco-Velez, J. Callison, C. W. M. Kay, S. E. Pratsinis, W. Lubitz, J. Y. Liu and F. R. Wang, Nat. Commun., 2020, 11, 4008 CrossRef CAS.
- X. P. Zhang, A. Chandra, Y. M. Lee, R. Cao, K. Ray and W. Nam, Chem. Soc. Rev., 2021, 50, 4804–4811 RSC.
- Y. Mao, P. Wang, L. Li, Z. Chen, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2020, 59, 3685–3690 CrossRef CAS PubMed.
- Y. Du, W. Zhou, J. Gao, X. Pan and Y. Li, Acc. Chem. Res., 2020, 53, 459–469 CrossRef CAS.
- M. Li, Q. Lv, W. Si, Z. Hou and C. Huang, Angew. Chem., Int. Ed., 2022, 61, e202208238 CrossRef CAS.
- Y. Guo, J. Liu, Q. Yang, L. Ma, Y. Zhao, Z. Huang, X. Li, B. Dong, X. Z. Fu and C. Zhi, Small, 2020, 16, e1907341 CrossRef.
- Q. Lv, W. Si, J. He, L. Sun, C. Zhang, N. Wang, Z. Yang, X. Li, X. Wang, W. Deng, Y. Long, C. Huang and Y. Li, Nat. Commun., 2018, 9, 3376 CrossRef PubMed.
- C. Huang, Y. Li, N. Wang, Y. Xue, Z. Zuo, H. Liu and Y. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS PubMed.
- T. Lu, X. Hu, J. He, R. Li, J. Gao, Q. Lv, Z. Yang, S. Cui and C. Huang, Nano Energy, 2021, 85, 106024 CrossRef CAS.
- L. Qi, Z. Zheng, C. Xing, Z. Wang, X. Luan, Y. Xue, F. He and Y. Li, Adv. Funct. Mater., 2022, 32, 2107179 CrossRef CAS.
- Y. Fang, Y. Xue, L. Hui, H. Yu, C. Zhang, B. Huang and Y. Li, Adv. Sci. (Weinh), 2022, 9, e2102721 CrossRef.
- C. Huang, S. Zhang, H. Liu, Y. Li, G. Cui and Y. Li, Nano Energy, 2015, 11, 481–489 CrossRef CAS.
- A. Pei, R. Xie, Y. Zhang, Y. Feng, W. Wang, S. Zhang, Z. Huang, L. Zhu, G. Chai, Z. Yang, Q. Gao, H. Ye, C. Shang, B. H. Chen and Z. Guo, Energy Environ. Sci., 2023, 16, 1035–1048 RSC.
- Y. Yao, Z. Jin, Y. Chen, Z. Gao, J. Yan, H. Liu, J. Wang, Y. Li and S. Liu, Carbon, 2018, 129, 228–235 CrossRef CAS.
- R. Samanta, B. K. Manna, R. Trivedi, B. Chakraborty and S. Barman, Chem. Sci., 2024, 15, 364–378 RSC.
- Q. Liu, Z. Yan, J. Gao, H. Fan, M. Li and E. Wang, Chem. Sci., 2023, 14, 11830–11839 RSC.
- H. Yu, Y. Xue, L. Hui, F. He, C. Zhang, Y. Liu, Y. Fang, C. Xing, Y. Li, H. Liu and Y. Li, Nano Energy, 2019, 64, 103928 CrossRef CAS.
- C. Zhang, Y. Xue, L. Hui, Y. Fang, Y. Liu and Y. Li, Mater. Chem. Front., 2021, 5, 5305–5311 RSC.
- L. Hui, Y. Xue, B. Huang, H. Yu, C. Zhang, D. Zhang, D. Jia, Y. Zhao, Y. Li, H. Liu and Y. Li, Nat. Commun., 2018, 9, 5309 CrossRef CAS.
- J. Li, X. Han, D. Wang, L. Zhu, M. H. Ha-Thi, T. Pino, J. Arbiol, L. Z. Wu and M. Nawfal Ghazzal, Angew. Chem., Int. Ed., 2022, 61, e202210242 CrossRef.
- Y. Gao, Y. Xue, F. He and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206946119 CrossRef CAS PubMed.
- Q. Hao, Y. Song, H. Ji, Z. Mo, X. She, J. Deng, T. Muhmood, X. Wu, S. Yuan, H. Xu and H. Li, Appl. Surf. Sci., 2018, 459, 845–852 CrossRef CAS.
- H. Y. Lin, Z. X. Lou, Y. Ding, X. Li, F. Mao, H. Y. Yuan, P. F. Liu and H. G. Yang, Small Methods, 2022, 6, e2201130 CrossRef PubMed.
- J. Li, X. Gao, L. Zhu, M. N. Ghazzal, J. Zhang, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2020, 13, 1326–1346 RSC.
- X. Luan and Y. Xue, Chem. Res. Chin. Univ., 2021, 37, 1268–1274 CrossRef CAS.
- M. A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS.
- H. Yu, L. Hui, Y. Xue, Y. Liu, Y. Fang, C. Xing, C. Zhang, D. Zhang, X. Chen, Y. Du, Z. Wang, Y. Gao, B. Huang and Y. Li, Nano Energy, 2020, 72, 104667 CrossRef CAS.
- X. Wang, X. Hu, L. Zheng, Q. Lv, J. He, X. Li, R. Li, T. Lu and C. Huang, Nano Energy, 2023, 117, 108919 CrossRef CAS.
- X. Gao, J. Li, R. Du, J. Zhou, M.-Y. Huang, R. Liu, J. Li, Z. Xie, L.-Z. Wu, Z. Liu and J. Zhang, Adv. Mater., 2017, 29, 1605308 CrossRef PubMed.
- Y. Fang, Y. Xue, L. Hui, H. Yu and Y. Li, Angew. Chem., Int. Ed., 2020, 60, 3170–3174 Search PubMed.
- C. Sun and D. J. Searles, J. Phys. Chem. C, 2012, 116, 26222–26226 CrossRef CAS.
- H. Zhang, Y. Xia, H. Bu, X. Wang, M. Zhang, Y. Luo and M. Zhao, J. Appl. Phys., 2013, 113, 044309 Search PubMed.
- J. Kim, S. Kang, J. Lim and W. Y. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 2677–2683 CrossRef CAS PubMed.
- S. Zhang, H. Liu, C. Huang, G. Cui and Y. Li, Chem. Commun., 2015, 51, 1834–1837 RSC.
- Z. Lin, G. Liu, Y. Zheng, Y. Lin and Z. Huang, J. Mater. Chem. A, 2018, 6, 22655–22661 RSC.
- Y. Lin, H. Kang, M. Liang, X. Ye, J. Li, Q. Feng, Y. Zheng and Z. Huang, Appl. Surf. Sci., 2020, 526 Search PubMed.
- F. Wang, J. An, H. Shen, Z. Wang, G. Li and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202216397 CrossRef CAS.
- M. Luo, J. Yang, X. Li, M. Eguchi, Y. Yamauchi and Z.-L. Wang, Chem. Sci., 2023, 14, 3400–3414 RSC.
- Q. Peng, J. Crean, L. Han, S. Liu, X. Wen, S. De and A. Dearden, Nanotechnol., Sci. Appl., 2014, 7, 1–29 CAS.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2011, 112, 1555–1614 CrossRef PubMed.
- S. Thangavel, K. Krishnamoorthy, V. Krishnaswamy, N. Raju, S. J. Kim and G. Venugopal, J. Phys. Chem. C, 2015, 119, 22057–22065 CrossRef CAS.
- S. Wang, L. Yi, J. E. Halpert, X. Lai, Y. Liu, H. Cao, R. Yu, D. Wang and Y. Li, Small, 2011, 8, 265–271 CrossRef PubMed.
- N. Yang, Y. Liu, H. Wen, Z. Tang, H. Zhao, Y. Li and D. Wang, ACS Nano, 2013, 7, 1504–1512 CrossRef CAS PubMed.
- Q. Bai, H. Luo, S. Shi, S. Liu, L. Wang, F. Du, Z. Yang, Z. Zhu and N. Sui, J. Colloid Interface Sci., 2022, 613, 376–383 CrossRef CAS.
- G. Liu, Z. Hu, X. Chen, W. Li, Y. Wu, Z. Liu, L. Miao, Z. Luo, J. Wang and Y. Guo, J. Hazard. Mater., 2023, 465, 133269 CrossRef PubMed.
- C. Pan, B. Zhang, T. Pan, H. Huang, S. Song, X. Cai, Y. Wang, H. Sun, Z. Luo, L. Zhang and Y. Guo, Environ. Sci.: Nano, 2023, 10, 3072–3083 RSC.
- C. Pan, H. Shen, G. Liu, X. Zhang, X. Liu, H. Liu, P. Xu, W. Chen, Y. Tian, H. Deng, H. Sun, J. Wang, Z. Luo, L. Zhang and Y. Guo, ACS Appl. Nano Mater., 2022, 5, 10980–10990 CrossRef CAS.
- S. Sorcar, Y. Hwang, C. A. Grimes and S.-I. In, Mater. Today, 2017, 20, 507–515 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- Z. Zeng, Y. Yan, J. Chen, P. Zan, Q. Tian and P. Chen, Adv. Funct. Mater., 2018, 29, 1806500 CrossRef.
- Y. Wang, Y. Liu, L. Tan, X. Lin, Y. Fang, X. F. Lu, Y. Hou, G. Zhang and S. Wang, J. Mater. Chem., 2023, 11, 26804–26811 RSC.
- B. Su, M. Zheng, W. Lin, X. F. Lu, D. Luan, S. Wang and X. W. Lou, Adv. Energy Mater., 2023, 13, 2203290 CrossRef CAS.
- B. Su, Y. Kong, S. Wang, S. Zuo, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415–27423 CrossRef CAS.
- F. Xu, K. Meng, B. Zhu, H. Liu, J. Xu and J. Yu, Adv. Funct. Mater., 2019, 29, 1904256 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |