
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein painting for structural and binding site analysis via intracellular lysine reactivity profiling with o-phthalaldehyde†
Zhenxiang
Zheng
a,
Ya
Zeng
ab,
Kunjia
Lai
a,
Bin
Liao
a,
Pengfei
Li
a and
Chris Soon Heng
Tan
 *a
*a
aDepartment of Chemistry, College of Science, Southern University of Science and Technology, Shenzhen, Guangdong 518055, PR China. E-mail: christan@sustech.edu.cn
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon, Hong Kong, PR China
First published on 20th March 2024
Abstract
The three-dimensional structure and the molecular interaction of proteins determine their roles in many cellular processes. Chemical protein painting with protein mass spectrometry can identify changes in structural conformations and molecular interactions of proteins including their binding sites. Nevertheless, most current protein painting techniques identify protein targets and binding sites of drugs in vitro using a cell lysate or purified protein. Here, we tested 11 membrane-permeable lysine-reactive chemical probes for intracellular covalent labeling of endogenous proteins, which reveals ortho-phthalaldehyde (OPA) as the most reactive probe in the intracellular environment. An MS workflow and a new data analysis strategy termed RAPID (Reactive Amino acid Profiling by Inverse Detection) was developed to enhance detection sensitivity. RAPID with OPA successfully identified structural changes induced by the allosteric drug TEPP-46 on its target protein PKM2 and was applied to profile the conformation change of the proteome occurring in cells during thermal denaturation. The application of RAPID-OPA on cells treated with geldanamycin, selumetinib, and staurosporine successfully revealed their binding sites on target proteins. Thus, RAPID-OPA for cellular protein painting enables the identification of ligand-binding sites and detection of protein structural changes occurring in cells.
Introduction
Proteins perform cellular functions through their three-dimensional structures, local conformational features (e.g. catalytic sites), and the molecular interactions in which they are involved. Thus, investigating protein structural conformations, protein–ligand interaction, and protein–protein interaction is of great importance in cell biology and drug discovery. X-ray crystallography, nuclear magnetic resonance (NMR), and various spectroscopic techniques are commonly used to elucidate the structures of purified proteins in vitro, thus omitting the influence of the native cellular environment known to affect the protein structure, function and drug binding.1,2Proteomics methods based on mass spectrometry (MS), such as hydrogen–deuterium exchange (HDX),3 fast photochemical oxidation of proteins (FPOP),4–6 covalent protein painting (CPP),7–9 chemical cross-linking,10–13 and activity-based probe profiling (ABPP),14 have been applied to study the dynamic change of protein structures in cells but often with limited adaptability and sensitivity. Chemical cross-linking suffers from generally low reaction stoichiometry, restricting the methodology to profiling the most abundant proteins in the proteome, while reversible covalent labeling in HDX often compromises the detection of structural changes.15 FPOP requires an elaborate instrumental and experimental setup that hinders their popular adoption for global proteome study.16,17 In the ABPP strategy, designing and synthesizing probes without altering the drugs' activities or selectivity represent a very difficult and time-consuming process.18 Covalent protein painting (CPP), where solvent-accessible amino acids on proteins are covalently labeled with chemicals, is an accessible and promising chemical proteomics strategy for intracellular studies of protein structures and conformational changes.8 Chemically modified regions or specific amino acid sites constitute a “footprint” that is affected by the protein structure as well as interaction with proteins, metabolites and xenobiotics such as drugs. In particular, CPP with protein mass spectrometry does not require protein purification and can analyze thousands of proteins in parallel.
The amino acids that are often labeled in common chemical proteomics strategies are cysteine and lysine, of which lysine accounts for 6% of all residues in human proteins and is one of the most represented amino acids with an average of 30 lysine sites per protein.19 Lysine is present in many functional sites such as enzyme active sites,20 protein–protein interaction interfaces,21 and post-translational modification sites. Currently, the commonly used membrane-permeable probes for labeling or cross-linking primary amines are N-hydroxy succinimidyl ester (NHS ester), but they suffer from poor selectivity (also react with histidine, serine, and tyrosine), poor stability and low labeling efficiency under physiological conditions.22 Thus, there is a strong need for better probes and analysis strategies to analyze intracellular changes of the protein structure and interaction using CPP.
Here, we tested 11 membrane-permeable chemical probes revealing ortho-phthalaldehyde (OPA) with the highest reactivity intracellularly exceeding NHS esters, coherent with existing vitro studies showing that OPA is capable of amine reactions with peptides and proteins under physiological conditions.23 In particular, OPA has been used to modify proteins and peptides by intramolecular OPA–amine–thiol three-component processes for cyclization24,25 and OPA–amine two-component reactions for bioconjugation.26,27 Nevertheless, while OPA is the most reactive intracellular probe identified in this study, we observed that the number of OPA-labelled peptides identified is still limited for the practical use of CPP in the physiological intracellular context. To overcome this, we further develop a new data analysis strategy termed RAPID (Reactive Amino acid Profiling with Inverse Detection) and an optimized MS workflow that can be coupled with subsequent directed detection (e.g. parallel reaction monitoring) for identifying intracellular changes in the structures and molecular interactions of proteins. We show that RAPID-OPA can reveal global conformational changes of proteins occurring in cells during thermal denaturation as well as specific conformational changes induced in protein PKM2 by the allosteric inhibitor TEPP-46, and identified the binding sites of geldanamycin, selumetinib, and staurosporine on target proteins.
Results and discussion
Validation of the RAPID strategy
Lysine-reactive probes often react with proteins at low stoichiometry that necessitates enrichment to identify peptides containing the probe-modified lysine, while MS fragmentation could lead to probe cleavage generating daughter ions with an unpredictable mass-to-charge ratio (m/z).28 We hypothesized that these challenges can be circumvented by identifying lysine probes, preferably membrane-permeable, that interfere with the cleavage of proteins by trypsin at the C-terminus of arginine or lysine. This will lead to a decreased abundance of peptides that could have been generated otherwise by trypsin digestion of proteins. In addition, probe-modified lysine may not be amenable to further labelling in multiplexing MS analysis. Thus, by comparing unmodified peptides from control samples (without the probe) to probe-treated samples with an isobaric labelling MS strategy using TMT (Tandem Mass Tag) labelling reagents, low abundant probe-modified peptides could be inferred with increased sensitivity, while omitting tedious enrichment that increases experimental variability (Fig. 1A). Subsequently, a targeted search like multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM) can be used to validate and quantify the corresponding modified peptides with increased sensitivity. Cell intrinsic factors, such as changes in post-translation modification, structural conformation, and interactions of proteins with metabolites, drugs and other proteins, could lead to differentiated changes in lysine reactivity that can be explored with this analysis strategy termed RAPID (Reactive Amino Acid Profiling by Inverse Detection). Such a strategy together with the continued development of better intracellular amino acid reactive probes will expedite global structural and interaction analysis of proteins at various cellular states. To verify this strategy, we first tested 11 chemical probes, consisting of NHS esters and diformaldehydes, incubating them with intact cells followed by cell lysis, tryptic digestion and TMT-labelling for MS analysis. We observed that the sample treated with membrane-permeable OPA (Fig. 1B(b-1)) had the largest decrease in the abundance of peptides identified (Fig. 1B and C). We note that OPA contains two aldehyde groups that can be condensed with an amine by the Mannich reaction and/or reductive amination (Fig. 1D).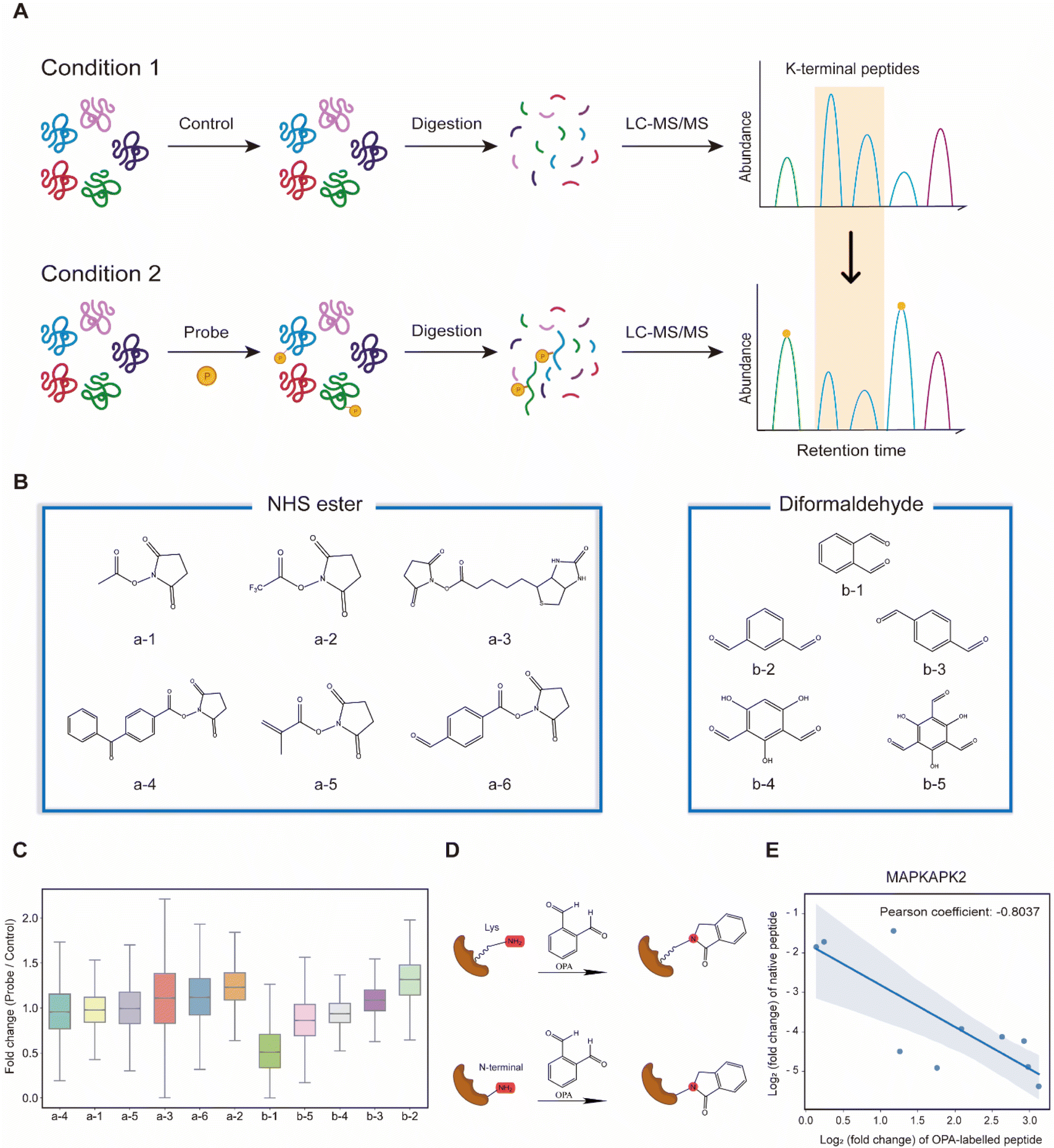 | ||
| Fig. 1 Proposal of the entire workflow for lysine reactivity profiling in complicated proteomes. (A) Principle of the RAPID strategy. (B) Chemical structure of lysine reactivity probes grouped into NHS esters and diformaldehydes. (C) Boxplot visualization of abundance changes for peptides in the HEK293T cell induced by each lysine reactivity probe. (D) Common strategies for peptide labeling via OPA. (E) Scatter plot shows the correlation in the abundance changes of OPA-labelled peptides and their native peptides in the MAPKAPK2 protein. | ||
The decrease in the abundance of native peptides identified could potentially arise from the direct interference of trypsin by OPA, and hence steps are incorporated in our workflow to remove excess OPA prior to enzymatic digestion of proteins (see the Methods).18 Nevertheless, to further validate that increase in OPA-labelled peptides leads to decreased abundance of native peptides, purified MAP kinase-activated protein kinase 2 (MAPKAPK2), which contains 30 lysine residues, was incubated with OPA followed by bottom-up MS analysis, where OPA is included as a variable lysine modification during the MS spectral search. Using a purified protein at a high concentration increases OPA reaction stoichiometry and the abundance of OPA-labelled peptides for MS detection. A total of 82 peptides were quantified, of which 19 peptides were OPA-labelled (Fig. S1A†). As anticipated, the native peptides of the identified OPA-labelled peptides showed a corresponding decrease in abundance (Fig. 1E, S1B and C†). These results strongly suggested that the intensity of OPA labelling can be inferred from changes in the abundance of native peptides.
Next, we hypothesized that the OPA-labelling reaction follows a second-order kinetic approach,29 with the modification rate increasing in a dose and time dependent manner. To systematically optimize the labelling conditions, we first incubated HEK293T cells with different OPA concentrations from 50 μM to 800 μM for a 15 min reaction (Fig. 2A). Overall, we observed decreasing abundance of native peptides with increasing OPA concentration from 100 μM onward (Fig. 2B). Next, we examined how the duration of incubation after different time points, namely 5, 10, and 15 min, affects labeling with 800 μM OPA. As anticipated, the abundance of native peptides decreases with time, where the greatest change was observed with 15 min of OPA incubation (Fig. 2C). We performed cell viability tests in the presence of 800 μM OPA at different incubation times (5 min, 15 min, and 30 min) and observed minimal change (<10%) up to 15 min (Fig. S2†). As OPA is known to react with cysteine also but at lower reactivity, we compared abundance change between native peptides with and without cysteine residues and observed a minimal difference (Fig. S3†). This together with the observed concentration- and time-dependent effect of OPA suggests that the decreased peptide abundance observed is largely due to the reaction of OPA with lysine and demonstrated that OPA could label proteins progressively under physiological conditions.
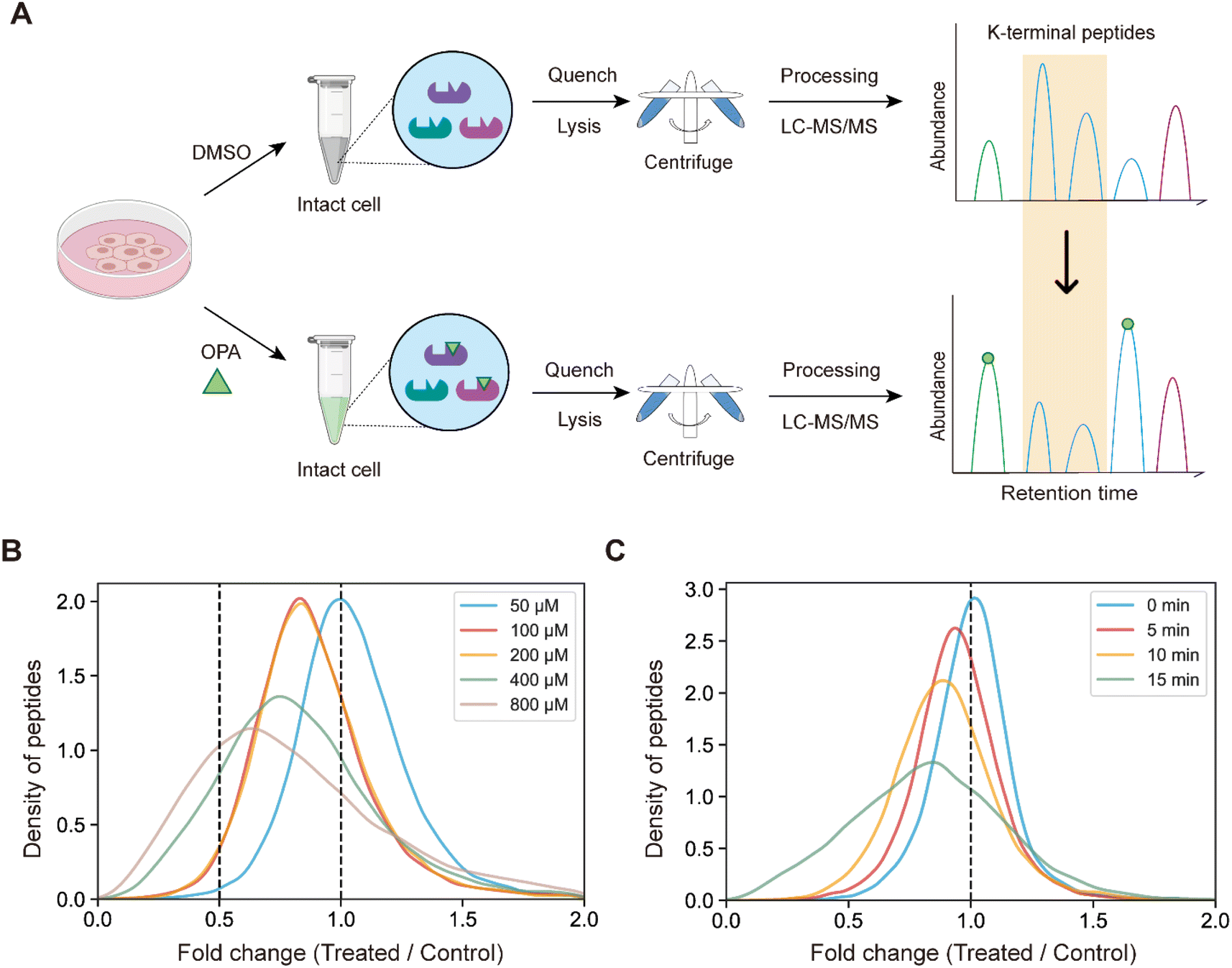 | ||
| Fig. 2 Experimental process and method optimization. (A) General workflow of RAPID-OPA. (B) Overall density plot of peptide abundance change with different concentrations of OPA. (C) Overall density plot of peptide abundance change with different labeling times of OPA. | ||
RAPID-OPA for monitoring the proteome-wide structural changes of proteins in cells
Overall, with 15 min of 800 μM OPA incubation in HEK293T cells, 8487 (13.47%) of the 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 005 native peptides across 6943 proteins identified exhibited a statistically significant (p-value < 0.05) decrease in abundance, while a spectral search incorporating variable OPA modification could only identify 673 OPA-labeled peptides (1.07%) (Fig. 3A). In comparison, lysine reactivity was better in HEK293T cell lysates with the same OPA concentration and duration, with 60.15% of the 52994 native peptides from 6057 proteins showing a statistically significant decrease in abundance (p-value < 0.05). The spectral search identified 2678 OPA-labeled peptides (5.05%) from the cell lysate, over four times higher than those identified from intracellular labelling (Fig. S4†). Next, we investigate the applicability of the RAPID-OPA workflow to study the proteome-wide intracellular conformational changes of protein structures. First, using the monomer structures of human proteins predicted by AlphaFold2, we observed that native peptides with the largest decrease in abundance after OPA labelling are over-represented in highly surface accessible lysine (Fig. 3B). Nevertheless, not all peptides with highly surface accessible lysine exhibit a marked decrease in abundance. As monomer structures of proteins were used in calculating the solvent accessible surface area (SASA), some of these peptides might be parts of the protein involved in interactions with the membrane, nucleic acids, and other proteins, which could also influence protein structural conformation not predicted by AlphaFold.30 We also observed that peptides with partially surface accessible lysine could exhibit a marked decrease in abundance, possibly due to the microenvironment of lysine with proximal amino acids that could influence its reactivity with OPA.21 Thus, changes in the structural conformation of the protein and lysine microenvironment could be reflected as differentiated changes in OPA labelling of lysine. To validate this, MS data from intact cells treated with OPA at three temperature points (namely 37 °C, 43 °C and 49 °C) were collected and analyzed (Fig. 3C). At these elevated temperatures, proteins are expected to at least partially unfold, exposing inner lysine for OPA labelling, which will result in less native peptides for MS detection (Fig. 3D). Indeed, we observed further decrease in native peptide abundance when cells are incubated with OPA at denaturing temperatures (43 °C and 49 °C) with more than 22068 (35.03%) and 29011 (46.05%) native peptides exhibiting a decrease in abundance compared to 8487 (13.47%) at 37 °C. Encouragingly, partially surface accessible lysine is enriched in peptides with the largest decrease in abundance (Fig. 3D, E and S5A–D†), confirming that RAPID-OPA can be used to detect the structural changes of proteins in cells.
005 native peptides across 6943 proteins identified exhibited a statistically significant (p-value < 0.05) decrease in abundance, while a spectral search incorporating variable OPA modification could only identify 673 OPA-labeled peptides (1.07%) (Fig. 3A). In comparison, lysine reactivity was better in HEK293T cell lysates with the same OPA concentration and duration, with 60.15% of the 52994 native peptides from 6057 proteins showing a statistically significant decrease in abundance (p-value < 0.05). The spectral search identified 2678 OPA-labeled peptides (5.05%) from the cell lysate, over four times higher than those identified from intracellular labelling (Fig. S4†). Next, we investigate the applicability of the RAPID-OPA workflow to study the proteome-wide intracellular conformational changes of protein structures. First, using the monomer structures of human proteins predicted by AlphaFold2, we observed that native peptides with the largest decrease in abundance after OPA labelling are over-represented in highly surface accessible lysine (Fig. 3B). Nevertheless, not all peptides with highly surface accessible lysine exhibit a marked decrease in abundance. As monomer structures of proteins were used in calculating the solvent accessible surface area (SASA), some of these peptides might be parts of the protein involved in interactions with the membrane, nucleic acids, and other proteins, which could also influence protein structural conformation not predicted by AlphaFold.30 We also observed that peptides with partially surface accessible lysine could exhibit a marked decrease in abundance, possibly due to the microenvironment of lysine with proximal amino acids that could influence its reactivity with OPA.21 Thus, changes in the structural conformation of the protein and lysine microenvironment could be reflected as differentiated changes in OPA labelling of lysine. To validate this, MS data from intact cells treated with OPA at three temperature points (namely 37 °C, 43 °C and 49 °C) were collected and analyzed (Fig. 3C). At these elevated temperatures, proteins are expected to at least partially unfold, exposing inner lysine for OPA labelling, which will result in less native peptides for MS detection (Fig. 3D). Indeed, we observed further decrease in native peptide abundance when cells are incubated with OPA at denaturing temperatures (43 °C and 49 °C) with more than 22068 (35.03%) and 29011 (46.05%) native peptides exhibiting a decrease in abundance compared to 8487 (13.47%) at 37 °C. Encouragingly, partially surface accessible lysine is enriched in peptides with the largest decrease in abundance (Fig. 3D, E and S5A–D†), confirming that RAPID-OPA can be used to detect the structural changes of proteins in cells.
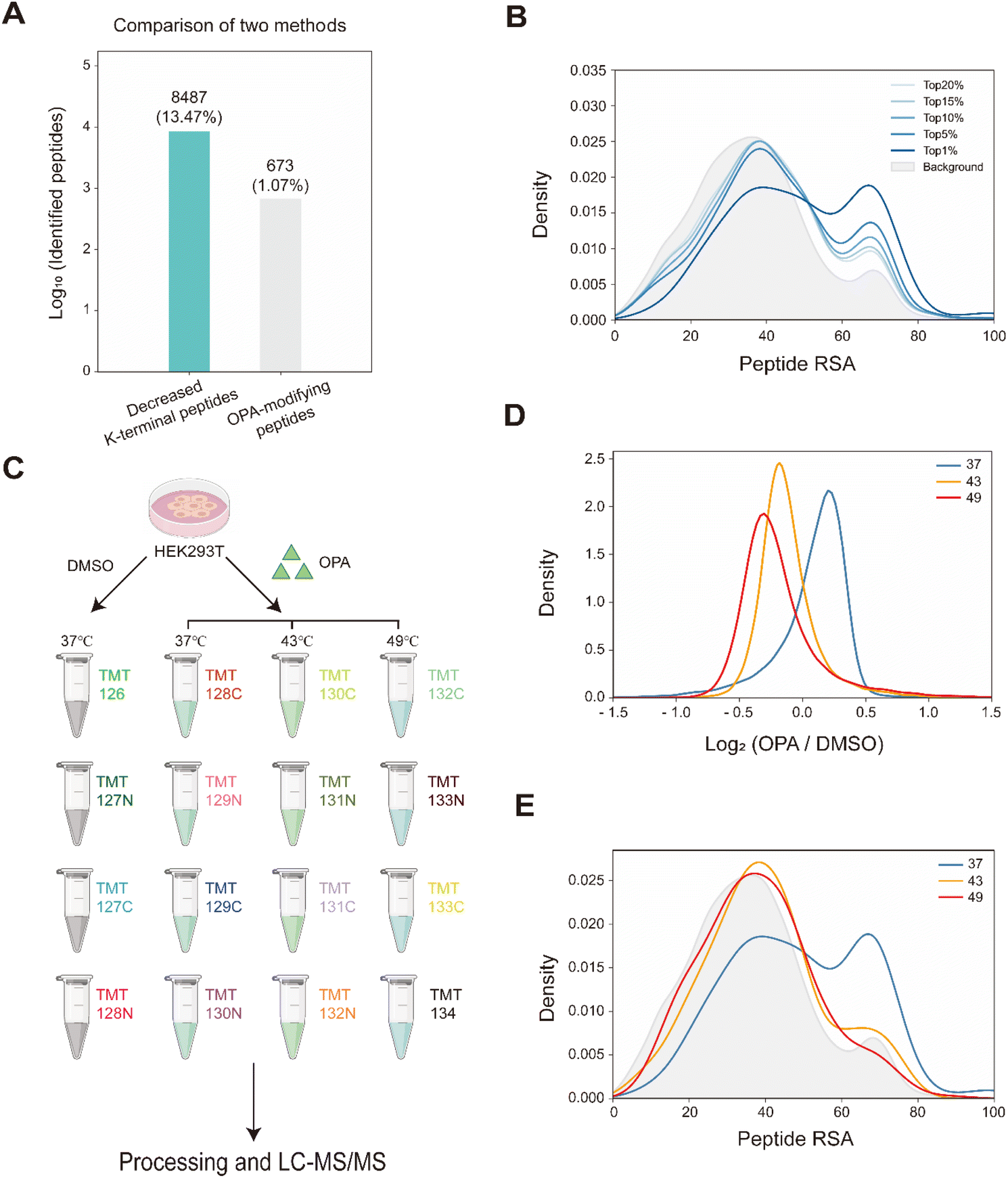 | ||
| Fig. 3 Monitoring the structural changes in proteomes. (A) Number of peptides with differentiated abundance identified in HEK293T cells. (B) RSA distribution of down-regulated peptides at 37 °C. (C) Schema of samples collected. HEK293T cells are treated with DMSO/OPA and heated at 3 temperatures for RAPID-OPA profiling. (D) Distribution of changes in native peptide abundance across different temperatures. (E) RSA distribution of the top 1% down-regulated peptides. | ||
Identifying protein structure conformation changes with RAPID-OPA
TEPP-46 binds and activate PKM2 (pyruvate kinase M2) by promoting the tetramerization of the protein.31 A recent study subjected a cell lysate to chemical labelling with isotope-coded formaldehyde (CD2O) to identify structural conformation changes induced in PKM2 by TEPP-46.32 To further validate the use of RAPID-OPA to identify conformational changes in protein structures directly in cells, HEK293T cells were treated with TEPP-46 for 20 min, followed by RAPID-OPA, where proteins were labelled with OPA before cell lysis and analyzed with MS in PRM mode (Fig. 4A). Four biological replicates were performed and processed for MS analysis with a single TMT set. Overall, we detected 30 native peptides of the PKM2 protein, of which 6 peptides exhibit a statistically significant decrease in abundance in samples incubated with OPA that were located near highly accessible areas or domains. However, in cells treated with TEPP-46, the decrease in abundance due to the formation of tetramers induced by the drug is significantly muted for the native peptide [K422].CCSGAIIVLTK433.[S] of the PKM2 protein suggesting that OPA-labelling of either the preceding Lys422 or the Lys433 within the peptide had been impeded (Fig. 4B and C). Both Lys422 and Lys433 are exposed in monomer PKM2 (Fig. 4E) but Lys422 is lysine found on the binding interface of tetramer PKM2 (Fig. 4D), with the RSA of Lys422 and Lys433 in tetrameric PKM2 less than 15% but over 90% in monomer PKM2 (Fig. 4E). As PKM2 changes from a monomer to a tetramer upon treatment with TEPP-46, this result further validated the use of RAPID-OPA for studying changes in the structural conformation and molecular interaction of proteins.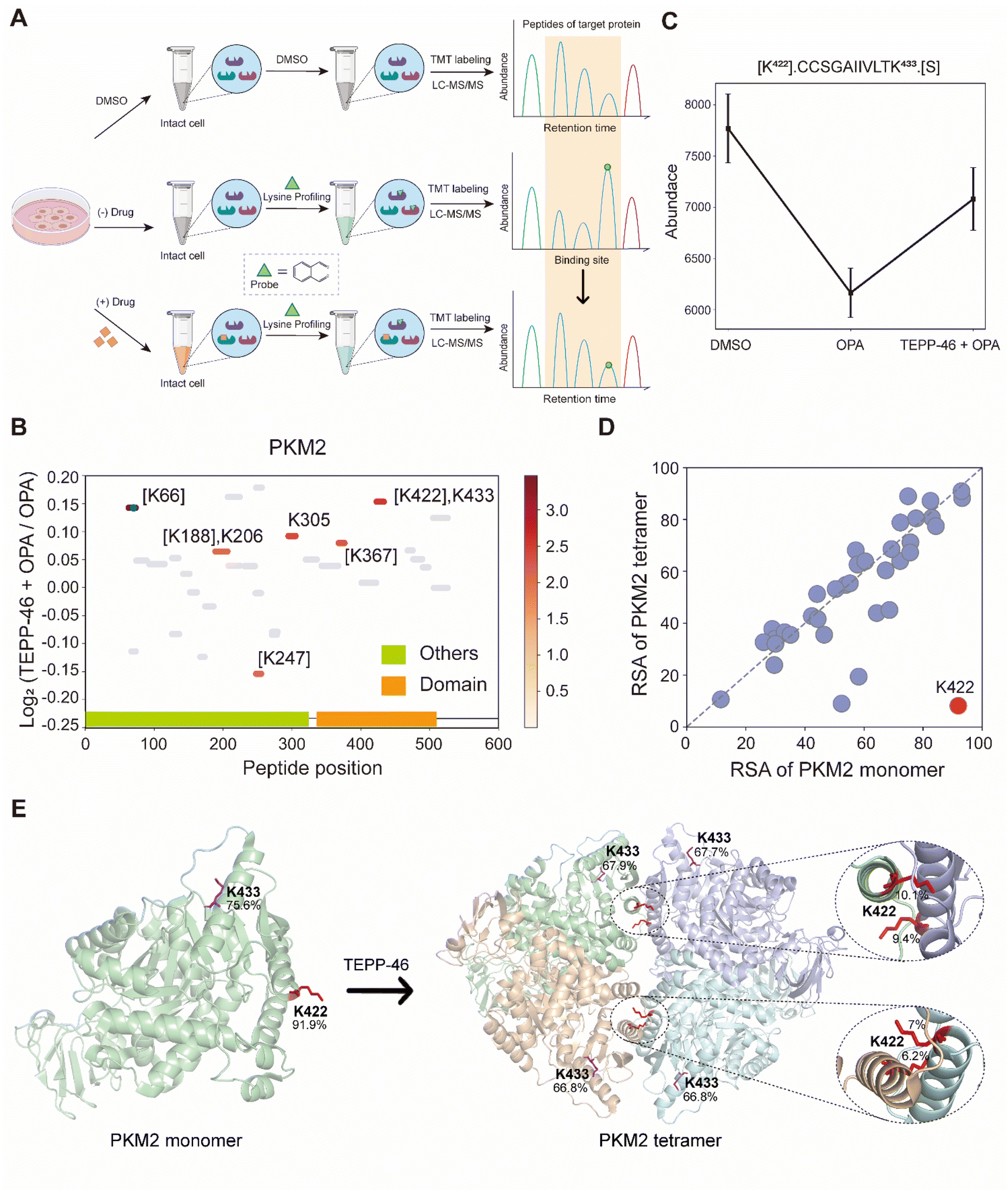 | ||
| Fig. 4 Interrogation of the allosteric proteins PKM2 induced by TEPP-46. (A) Schema of samples collected. HEK293T cells are treated with the drug before OPA. (B) Diagram showing the locations and labeling reactivity shifts of identified peptides in PKM2. (C) Abundance of peptides [K422].CCSGAIIVLTK433.[S] under different conditions. Data are presented as mean values ± s.d. (error bars). (D) Scatter plot showing the comparison of lysine RSA in monomer PKM2 and tetrameric PKM2. (E) RSA of K422 and K433 in tetrameric PKM2 and monomer PKM2. The four PKM2 monomers and K422 are represented in cartoon mode. The red square is the binding region of the PKM2 protein and the drug TEPP-46 (PDB ID:3BJF). | ||
Interrogation of the target proteins and binding sites of geldanamycin
Next, we used geldanamycin to assess applicability of RAPID-OPA to study the protein binding sites of drugs and chemicals. Geldanamycin (GA) is a natural heat shock protein 90 (Hsp90) inhibitor that specifically binds to ATP binding sites in the N-terminal domain of Hsp90.33 RAPID-OPA was applied on HEK293T cells treated with geldanamycin with good reproducibility in peptide quantification observed among four technical replicates performed (Fig. S6A–C†). The analysis identified 54 peptides and 59 peptides from known HSP90AA1 and HSP90AB1 target proteins of geldanamycin. Encouragingly, their corresponding OPA-labelled peptides were identified and exhibited significant changes in a reverse manner as anticipated. Specifically, the peptides with significant lysine residue shifts in reactivity were [R].ELISNSSDALDK58 OPAIR.[Y] and [R].YESLTDPSK69 OPALDSGK.[E] for HSP90AA1, and [R].ELISNASDALDK53 OPAIR.[Y] and [R].YESLTDPSK64 OPALDSGK.[E] and for HSP90AB1 (Fig. 5A and B), all of which were located within or near the known binding regions of geldanamycin in these proteins.18 For HSP90AA1, Lys58 was located directly in the interaction region, while Lys69 was located near the interaction region (Fig. 5C). The abundance of these OPA-labelled peptides is significantly lower in the drug-treated group suggesting that OPA-labelling of either the preceding Lys58, Lys69, Lys53 or Lys64 within the binding site has been hindered by the occupation of geldanamycin (Fig. 5D and E). In addition, both Lys100 of HSP90AA1 and Lys95 of HSP90AB1 were contained in native peptides, which were located in the domain with significant changes and are near the interaction region suggesting that conformational changes occurred after drug treatment (Fig. 5A–C). Thus, RAPID-OPA could also delineate the regions of ligand binding on target proteins.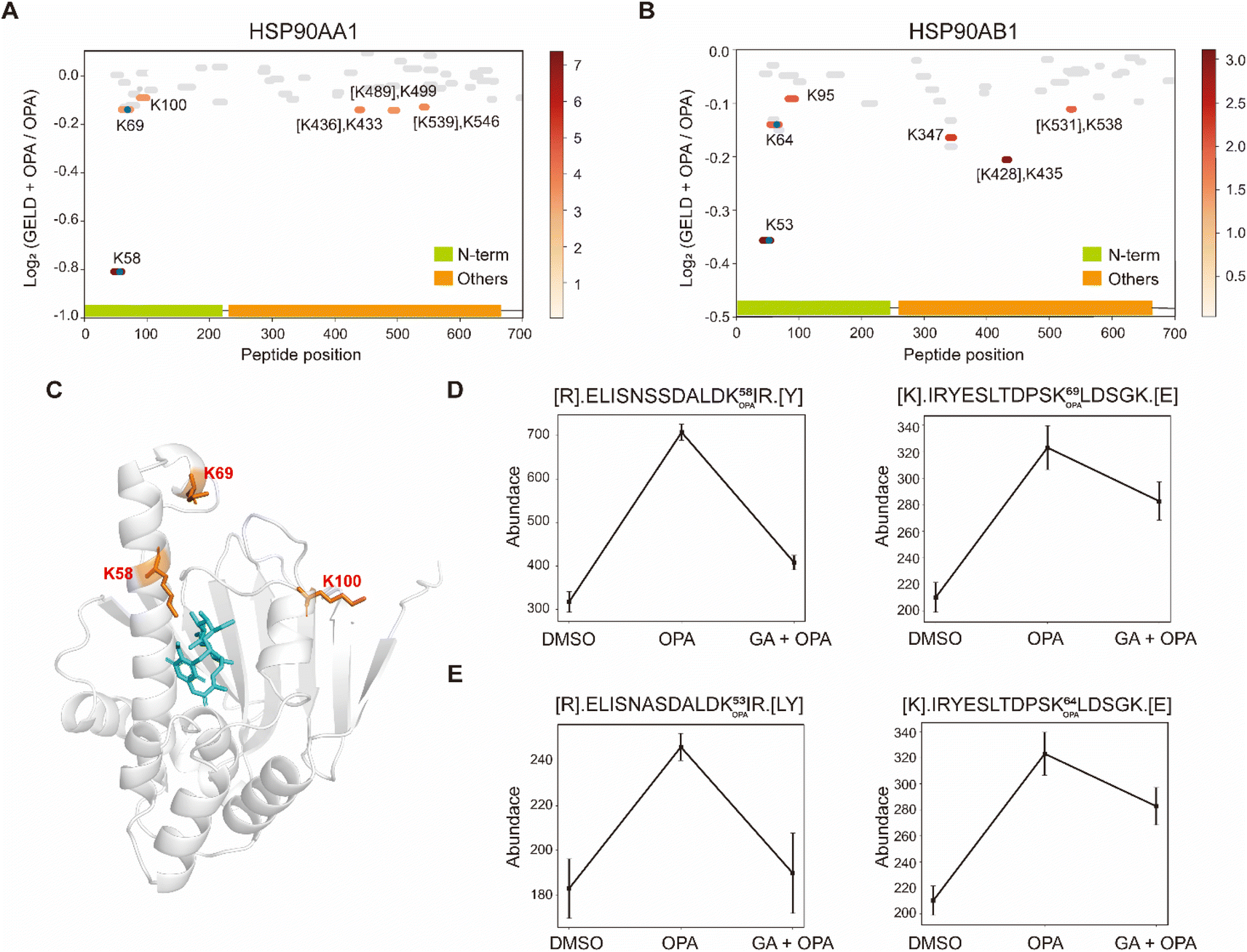 | ||
| Fig. 5 Interrogation of the target proteins and binding sites of geldanamycin. (A and B) Diagram showing the locations and labeling reactivity shifts of identified peptides in HSP90AA1 and HSP90AB1. The blue dot denotes OPA-label. (C) Co-crystallization result of HSP90AA1(PDB ID:1YET). (D) Abundance of peptides [R].ELISNSSDALDK58 OPAIR.[Y] and [K].IRYESLTDPSK69 OPALDSGK.[E] in HSP90AA1. (E) Abundance of peptides[R].ELISNASDALDK53 OPAIR.[Y] and [R].YESLTDPSK64 OPALDSGK.[E] in HSP90AB1. Data are presented as mean values ± s.d. (error bars). | ||
Interrogation of kinase inhibitors selumetinib and staurosporine with RAPID-OPA
Next, we applied RAPID-OPA to interrogate binding sites of the kinase inhibitor selumetinib on target proteins in HEK293T cells as described above in PRM mode. Selumetinib (SMT) is a selective, non-ATP-competitive, allosteric inhibitor of MEK1/2, which inhibits the RAS/RAF/MEK/ERK pathway. The analysis identified 11 peptides and 5 peptides from known target proteins MEK1 and MEK2, respectively. Among them, 4 and 1 significant peptides with lysine reactivity shifts of MEK1 and MEK2 respectively were located in the kinase domain (Fig. 6A and B). For MEK1, Lys97 was located directly in the drug–protein binding site, while other lysines with significant lysine shift were located near the interaction region (Fig. 6C). These results show that RAPID-OPA can be applied to study the effect of kinase inhibitors in the kinase domain in kinase proteins.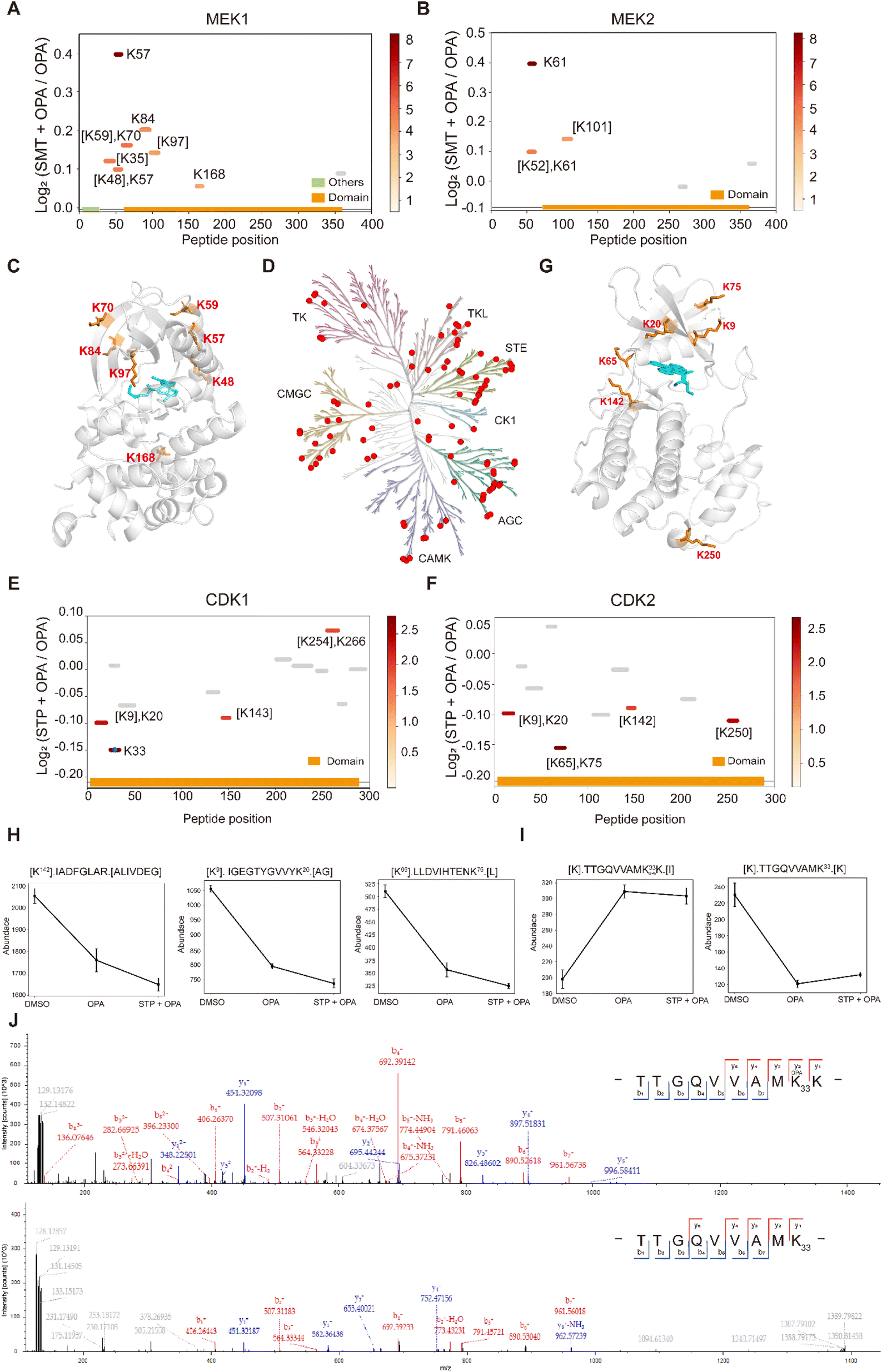 | ||
| Fig. 6 Interrogation of kinase inhibitors selumetinib and staurosporine with RAPID-OPA. (A and B) Diagram showing the locations and labeling reactivity shifts of identified peptides in MEK1 and MEK2. (C) Co-crystallization result of MEK1(PDB ID:7 M0T). (D) Distribution of the identified kinase proteins of staurosporine in HEK293T cells. (E and F) Diagram showing the locations and labeling reactivity shifts of identified peptides in CDK1 and CDK2. The blue dot denotes OPA-label. (G) Co-crystallization result of CDK2 (PDB ID:4ERW). (H) Abundance of peptides [K142].IADFGLAR.[A], [K9].IGEGTYGVVYK20.[A] and [K65].LLDVIHTENK75.[L] in CDK2. Data are presented as mean values ± s.d. (error bars). (I) Abundance of peptides [K].TTGQVVAMK33 OPAK.[I] and [K].TTGQVVAMK33.[K] in CDK1. Data are presented as mean values ± s.d. (error bars). (J) MS/MS result of peptides TTGQVVAMK33 OPAK (upper) and TTGQVVAMK33 (below). | ||
To further validate the feasibility of our method, we performed a proteome-wide scan for changes in lysine reactivity in HEK293T treated with staurosporine (STP), which is a potent, ATP-competitive, non-selective broad-spectrum inhibitor of protein kinases that binds to their ATP binding pockets. A total of 4173 peptides with a significant lysine reactivity shift were identified, with 183 (4.39%) peptides from 100 protein kinases and 467 (11.19%) peptides from 153 ATP-binding proteins (Fig. S7A–C†), which are over-represented among proteins with significance changes in peptide abundance (p-values < 0.05), consistent with the known binding proteins of the staurosporine (Fig. 6D). Of the peptides from kinases that showed significant changes in abundance, they were predominantly located in the kinase domain (N-lobe region) and ATP binding region of these kinase proteins (Fig. S6D†), further demonstrating the feasibility of our approach to identify drug targets and resolve binding sites at the proteome-wide level. For example, each of the four peptides of kinase proteins CDK1 and CDK2 with significant changes was located in the protein kinase domain and ATP binding region (Fig. 6E and F). Further analysis revealed that Lys9, Lys20, Lys65, Lys75 and Lys142 with significant shift changes were located within or near the known binding regions of staurosporine in the target protein CDK2 (Fig. 6G). Specifically, the abundance of peptides [K142].IADFGLAR.[A], [K9]. IGEGTYGVVYK20.[A] and [K65].LLDVIHTENK75.[L] which contain the above-mentioned lysines was lower in the drug-treated group, suggesting that these regions in the CDK2 protein might have undergone conformational change exposing these lysines further for OPA labelling (Fig. 6H). In addition, staurosporine also caused significant changes in the reactivity of some lysine residues in other structural domains of kinase proteins (Fig. S7D†). This indicates that the binding of staurosporine to the structural domain of these kinase may induce a conformational change. To confirm that the changes in peptide abundance arise from chemical modifications of OPA, targeted detection of their OPA-modified peptides was performed with PRM (Fig. S8 and 9†). For example, the abundance of the [K].TTGQVVAMK33.[K] native peptide of CDK1 is lower in the presence of OPA, while a directed search with PRM revealed its OPA-modified version [K].TTGQVVAM33 OPAK.[I] with a reverse trend (Fig. 6I). The MS/MS spectrum showed that OPA was site-specific at the lysine residue K33 at CDK1 (Fig. 6J).
In summary, the findings presented in this work demonstrate that lysine reactivity analysis using OPA coupled with the RAPID method could be used to reveal the drug-induced conformational changes of proteins and identify drug targets and their binding sites directly in cells toward obtaining data that better reflect the physiological state of cells tested. Nevertheless, the reaction of lysines with OPA could alter the structure of proteins, particularly after prolonged exposure, producing erroneous while obscuring true signals. Therefore, the method in its current stage has inherent false positives and false negatives, but the latter could be partially reduced by subjecting cells to drug treatment first before OPA incubation and by including appropriate control experiments. We envision that the development of cell-permeable probes with better and/or inducible reactivity could alleviate some of the caveats and when coupled with RAPID could expedite the routine intracellular study of protein structures and drug-binding sites.
Conclusions
In brief, we developed an intracellular chemical covalent labeling method based on lysine reactive shift coupled with a new data analysis strategy RAPID to analyze the intracellular conformational changes of proteins on a large scale. RAPID-OPA is able to detect global structural changes in proteins induced by elevated temperature. In addition, four drugs were used to benchmark the applicability of RAPID-OPA to study specific conformational changes and to identify ligand-binding sites. Thus, RAPID-OPA can be used to study intra-cellular protein conformational changes and for the identification of drug targets and their binding sites on proteins. We also envision that a data analysis strategy like RAPID could be readily combined with other reactive chemical probe tools to study the structural dynamics of proteomics and molecular interactions for various cellular processes and guide mechanistic studies of bioactive compounds.Experimental
Materials
TEPP-46, geldanamycin, selumetinib, and staurosporine were purchased from MedChem Express. All the compounds were purchased from Bidepharm (China). All the compounds were made up as solutions in DMSO (100×).Cell culture
HEK293T cells were cultured in DMEM medium (Gibco) supplemented with 10% FBS (PAN) and 1% penicillin–streptomycin (Gibco) at 37 °C and 5% CO2 in a humidified environment.In vivo covalent small molecules for lysine reactivity
HEK293T cells were treated with 800 μM individual probes or the corresponding concentration of the vehicle, dimethyl sulfoxide (DMSO), incubated at 37 °C for 15 min followed by addition of glycine solution to a final concentration of 2 mM with 10 min incubation to quench the reaction. Then, each mixture was subsequently subjected to a (2×) lysis buffer containing a final concentration of 100 mM HEPES (pH 7.5), 20 mM MgCl2, 10 mM β-glycerophosphate (sodium salt hydrate), 2 mM tris(2-carboxyethyl) phosphine hydrochloride (TCEP), 0.2 mM sodium orthovanadate, 0.2% (w/v) n-dodecyl β-D-maltoside (DDM), and EDTA-free protease inhibitor (Sigma-Aldrich, USA). The cell suspension was subjected to five times flash-freezing in liquid nitrogen and rapid thawing in water to facilitate cell lysis. After centrifugation at 21000g for 20 min at 4 °C, the supernatant was transferred to a new tube and the protein concentration was measured with a BCA assay kit (Thermo Fisher Scientific, USA).
RAPID-OPA analysis in purified proteins
MAP kinase-activated protein kinase 2 (MAPKAPK2) samples were prepared at 2 mg mL−1 with PBS (pH 7.4). Then, the MAPKAPK2 samples were subjected to labeling and quench steps as above. After centrifugation at 21000g for 20 min at 4 °C, the supernatant was transferred to a new tube and the protein concentration was measured with a BCA assay kit.
Optimization of RAPID-OPA labeling-time
HEK293T cells were treated with either 800 μM OPA or the corresponding concentration of DMSO, incubated at 37 °C for different durations (15 min, 10 min, 5 min, and 0 min) followed by addition of glycine solution to a final concentration of 2 mM with 10 min incubation to quench the reaction. The subsequent steps were the same as above.Optimization of RAPID-OPA reaction concentration
HEK293T cells were treated with either different concentrations (50 μM, 100 μM, 200 μM, 400 μM, and 800 μM) of OPA or the corresponding concentration of DMSO, incubated at 37 °C for 15 min. The subsequent steps were the same as above.Heat stimulation treatment
HEK293T cells were treated with either 800 μM OPA or the corresponding concentration of DMSO and heated in parallel in a PCR (VWR, Doppio Gradient) block for 15 min to three temperatures (37 °C, 43 °C, and 49 °C). The subsequent steps were the same as above.Profiling of lysine reactivity
HEK293T cells were treated with individual drugs (20 μM TEPP-46, selumetinib, and staurosporine and 100 μM geldanamycin) and incubated at 37 °C for 20 min. Next, the cells were incubated either with 800 μM OPA or the corresponding concentration of DMSO at 37 °C for 15 min. The subsequent steps were the same as above.In-solution digestion and TMT label
Each sample had 100 μg proteins in 60 μL 1× lysis buffer followed by reduction with 10 mM DTT in 95 °C for 10 min, and alkylated with 30 mM IAA for 30 min at room temperature. Then, protein was purified by methanol–chloroform precipitation (methanol/chloroform/water = 4:1:2). After being lyophilized to dryness, the protein pellet was resuspended in 8 M urea buffer and diluted to 2 M by adding 50 mM ammonium bicarbonate (ABC). Proteins were digested overnight at 37 °C with trypsin (Promega) and Lys-C (Wako, 125-05061) (trypsin/Lys-C/protein = 2:1:100 = w/w/w). The peptide concentration was measured with a Nanodrop (Thermo Fisher Scientific, USA). The samples of different compounds, RAPID-OPA labeling-times, and different concentrations with no need to label TMT were directly desalted with a C18 disk (3 M Empore, U.S.A.), followed by vacuum drying. The other tryptic digest samples were desalted with a C18 disk and labeled with the TMTpro reagent (Thermo Fisher Scientific, USA) followed by vacuum drying.
Tip-based fractionation of TMT-labelled peptides
The fractionation on the C18 membrane is performed by the acetonitrile concentration fractionation method, called a tip-based manual fractionation.34 C18 is filled in a 200 μL pipette tip, and the number of C18 is equal to the total amount of peptides to be fractionated. The C18 device was activated with methanol and 80% (v/v) ACN and 0.5% (v/v) acetic acid, followed by 1% (v/v) FA and 5 mM ammonium formate to wash off the excess methanol solution. The TMT-labelled samples were diluted with 1% (v/v) FA and loaded onto a C18 tip. The peptide adsorbed on the C18 membrane is gradually eluted and collected according to 24 stepwise increases of ACN in 5 mM ammonium formate at pH 10. (3%, 5%, 7%, 9%, 11%, 13%, 15%, 17%, 19%, 21%, 23%, 24%, 26%, 28%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, and 80%) collected in a round robin manner across 6 fractions. Accordingly, the concentrations of acetonitrile for each fraction are (3%, 15%, 26%, and 50%), (5%, 17%, 28%, and 55%), (7%, 19%, 30%, and 60%), (9%, 21%, 35%, and 65%), (11%, 23%, 40%, and 70%), and (13%, 24%, 45%, 80%). Finally, the six fractions were lyophilized to dryness and redissolved in 0.1% (v/v) formic acid in water for nano-LC-MS/MS analysis.LFQ-RAPID analysis
Samples that were not labeled with TMT were subjected to analysis using a Q-Exactive instrument coupled with an Easy-nLC 1200 system (Thermo Fisher Scientific). The analytical column, consisting of an integrated spray tip (100 μm i.d. × 20 cm), was packed with 1.9 μm/120 Å ReproSil-Pur C18 beads (Dr Maisch GmbH) for peptide separation at a flow rate of 250 nL min−1. The samples were directly loaded onto the column with buffer A [0.1% (v/v) FA in water]. An 80 min gradient separation was performed as follows: 4–8% buffer B [90% (v/v) ACN in buffer A] over 2 min, 8–25% buffer B for 50 min, 25–40% buffer B for 10 min, and 40–97% buffer B over 2 min, followed by 16 min wash with 97% buffer B. Peptides were detected on a Q-Exactive instrument, utilizing full MS scans on an Orbitrap mass analyzer with a resolution of 70000 and top 10 MS2 scans at a resolution of 35000. HCD fragmentation was performed with a normalized collision energy (NCE) of 32, and a dynamic exclusion time of 30 seconds was applied. Raw files were searched using Proteome Discoverer (PD) software (version 2.4, Thermo Fisher Scientific) against the human proteome fasta database (Uniprot, 20376 entries, downloaded on May 03, 2022). The maximum missed cleavage for trypsin digestion was set to 2. The mass tolerance for peptide precursors was 10 ppm and the mass tolerance for fragment ions was 0.02 Da. OPA modification (+116.1062 Da, K), oxidation (+15.995 Da, M), and deamidation (+0.984 Da, N, Q) were selected as variable modifications. FDR control for the protein and peptide is 1% at the strict level and 5% at the relaxed level.
Multiplexed LC-RAPID analysis
TMT-labeled samples were diluted with 0.1% FA prior to separation on a 20 cm × 100 μm EASY-Spray C18 LC column with a 135 min gradient on an UltiMate 3000 HPLC system (Thermo Fisher Scientific). The mobile phase was solvent A (0.5% acetic acid in water) and solvent B (80% ACN, 0.5% acetic acid in water). MS data were acquired with an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific): MS1 scan resolution was 60000 with the m/z range of 350–1200, the AGC target set as the standard and the maximum injection time of 45 ms. MS2 scan was performed at first mass (m/z) 110 and the Orbitrap resolution was 30000 using turbo TMT with HCD collision energies at 38. Raw data were also searched by PD, adding TMT pro modification (+304.207 Da) as variable modifications and others setups were the same.
TMT-PRM analysis
For the PRM method, the LC-MS/MS instruments and chromatographic setups were identical to the multiplexed LC-RAPID method. The PRM scan mode, MS scan mode, was set at the maximum injection time of 50 ms, and the AGC target was set as custom at the resolution of 60000. MS2 scan mode was set at the maximum injection time of 100 ms, AGC target set as custom and an isolation window of 0.7 m/z at the resolution of 30000. Then 38% normalized collision energy was set for HCD fragmentation. Targeted PKM2, MEK and CDK1 precursor transitions and their corresponding m/z were extracted from the previous DDA discovery run and put into the PRM method as an inclusion list. Raw data were also searched by PD and the setups were the same as above.
RSA prediction
Solvent accessibility prediction was performed using GetArea35 (http://curie.utmb.edu/getarea.html). All human protein structures were downloaded from AlphaFold2 and imported into GetArea to get the accessibility profiles for each residue. The RSA value is the ratio of real side-chain SASA to average SASA in the tripeptide Gly–X–Gly in an ensemble of 30 random conformations. In this study, we calculated the peptide RSA value, the average RSA value of all amino acids in the peptide, which considered the solvent accessibility of the entire peptide and focused on the local solvent accessibility of the lysine site(s). For any predicted protein structure by AlphaFold2, all residues were given the per-residue confidence scores (pLDDT) values, with values greater than 70 indicating confident results.Data analysis
All statistical and bioinformatics analyses were performed in the Python language (version 3.10) or R language (version 4.3.0). The R package limma was employed to identify differential peptides under different conditions and peptides with a p-value < 0.05 were considered as significantly changed.For the analysis of OPA-labelled and native peptides detected in the proteomics dataset, any contaminant peptides and null signal peptides were removed, and only unique peptides are retained after deduplication by sequence. Then, all lysine residues were identified and matched to GetArea-predicted RSA and AlphaFold2 pLDDT values by the UniProt accession ID and sequence, and the low-confidence protein structures (pLDDT value <70) were not included for structure analysis.
For the analysis and visualization of target protein structures, the co-crystallized protein–ligand structures were downloaded from the Protein Data Bank (PDB) (https://www.rcsb.org/). PyMOL (version 2.5.4) was used to show the protein structure or the interaction model of the protein and ligand, and the identified lysine site(s) by RAPID-OPA were annotated.
Data availability
All the raw MS data have been uploaded onto the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD046150.36,37Author contributions
C. T. conceived and led the project. Z. Z. and Y. Z. analyzed the data. Y. Z. and B. L. wrote the algorithms. Z. Z. and K. L. performed the biological experiments. P. L. suggested and provided the compounds. Z. Z., Y. Z. and C. T. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by grants from the Shenzhen Innovation of Science and Technology Commission (No. JCY20200109140814408), the National Key Research and Development Program of China (No. 2021YFA1302603), and the National Natural Science Foundation of China (No. 22074060 and 22150610470) awarded to Chris Soon Heng Tan. The figures are created with MedPeer (https://medpeer.cn).Notes and references
- H. Ji, X. Lu, S. Zhao, Q. Wang, B. Liao, L. G. Bauer, K. V. M. Huber, R. Luo, R. Tian and C. S. H. Tan, Target deconvolution with matrix-augmented pooling strategy reveals cell-specific drug-protein interactions, Cell Chem. Biol., 2023, 30, 1478–1487 CrossRef CAS PubMed.
- J. D. Vasta, A. Michaud, C. A. Zimprich, M. T. Beck, M. R. Swiatnicki, H. Zegzouti, M. R. Thomas, J. Wilkinson, J. A. Crapster and M. B. Robers, Protomer selectivity of type II RAF inhibitors within the RAS/RAF complex, Cell Chem. Biol., 2023, 30, 1354–1365 CrossRef CAS PubMed.
- M. Kaldmae, A. Leppert, G. Chen, M. Sarr, C. Sahin, K. Nordling, N. Kronqvist, M. Gonzalvo-Ulla, N. Fritz, A. Abelein, S. Laiotan, H. Biverstal, H. Jornvall, D. P. Lane, A. Rising, J. Johansson and M. Landreh, High intracellular stability of the spidroin N-terminal domain in spite of abundant amyloidogenic segments revealed by in-cell hydrogen/deuterium exchange mass spectrometry, FEBS J., 2020, 287, 2823–2833 CrossRef PubMed.
- J. A. Espino, V. S. Mali and L. M. Jones, In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells, Anal. Chem., 2015, 87, 7971–7978 CrossRef CAS PubMed.
- A. Rinas, V. S. Mali, J. A. Espino and L. M. Jones, Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry, Anal. Chem., 2016, 88, 10052–10058 CrossRef CAS PubMed.
- U. Kaur, D. T. Johnson and L. M. Jones, Validation of the Applicability of In-Cell Fast Photochemical Oxidation of Proteins across Multiple Eukaryotic Cell Lines, J. Am. Soc. Mass Spectrom., 2020, 31, 1372–1379 CrossRef CAS PubMed.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- C. Bamberger, S. Pankow, S. Martinez-Bartolome, M. Ma, J. Diedrich, R. A. Rissman and J. R. Yates 3rd, Protein Footprinting via Covalent Protein Painting Reveals Structural Changes of the Proteome in Alzheimer's Disease, J. Proteome Res., 2021, 20, 2762–2771 CrossRef CAS PubMed.
- D. Cox, A. R. Ormsby, G. E. Reid and D. M. Hatters, Protein painting reveals pervasive remodeling of conserved proteostasis machinery in response to pharmacological stimuli, npj Syst. Biol. Appl., 2022, 8, 46 CrossRef CAS PubMed.
- Y. Yang, H. P. Song, D. He, S. Zhang, S. Z. Dai, S. X. Lin, R. Meng, C. Wang and P. R. Chen, Genetically encoded protein photocrosslinker with a transferable mass spectrometry-identifiable label, Nat. Commun., 2016, 7, 12299 CrossRef CAS PubMed.
- B. Yang, S. B. Tang, C. Ma, S. T. Li, G. C. Shao, B. B. Dang, W. F. DeGrado, M. Q. Dong, P. G. Wang, S. Ding and L. Wang, Spontaneous and specific chemical cross-linking in live cells to capture and identify protein interactions, Nat. Commun., 2017, 8, 2240 CrossRef PubMed.
- W. Hu, Y. Yuan, C. H. Wang, H. T. Tian, A. D. Guo, H. J. Nie, H. Hu, M. J. Tan, Z. Tang and X. H. Chen, Genetically Encoded Residue-Selective Photo-Crosslinker to Capture Protein-Protein Interactions in Living Cells, Chem, 2019, 5, 2955–2968 CAS.
- Z. Li, D. Wang, L. Li, S. Pan, Z. Na, C. Y. Tan and S. Q. Yao, "Minimalist" cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling, J. Am. Chem. Soc., 2014, 136, 9990–9998 CrossRef CAS PubMed.
- M. R. Lazear, J. R. Remsberg, M. G. Jaeger, K. Rothamel, H. L. Her, K. E. DeMeester, E. Njomen, S. J. Hogg, J. Rahman, L. R. Whitby, S. J. Won, M. A. Schafroth, D. Ogasawara, M. Yokoyama, G. L. Lindsey, H. X. Li, J. Germain, S. Barbas, J. Vaughan, T. W. Hanigan, V. F. Vartabedian, C. J. Reinhardt, M. M. Dix, S. J. Koo, I. Heo, J. R. Teijaro, G. M. Simon, B. Ghosh, O. Abdel-Wahab, K. Ahn, A. Saghatelian, B. Melillo, S. L. Schreiber, G. W. Yeo and B. F. Cravatt, Proteomic discovery of chemical probes that perturb protein complexes in human cells, Mol. Cell, 2023, 83, 1725–1742 CrossRef CAS PubMed.
- X. R. Liu, M. M. Zhang and M. L. Gross, Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications, Chem. Rev., 2020, 120, 4355–4454 CrossRef CAS PubMed.
- G. H. Xu and M. R. Chance, Hydroxyl radical-mediated modification of proteins as probes for structural proteomics, Chem. Rev., 2007, 107, 3514–3543 CrossRef CAS PubMed.
- X. R. Liu, D. L. Rempel and M. L. Gross, Protein higher-order-structure determination by fast photochemical oxidation of proteins and mass spectrometry analysis, Nat. Protoc., 2020, 15, 3942–3970 CrossRef CAS PubMed.
- C. Ruan, J. Zhou, Z. Li, K. Li, Z. Fang, X. Zhang and M. Ye, Proteome-Wide Deconvolution of Drug Targets and Binding Sites by Lysine Reactivity Profiling, Anal. Chem., 2022, 94, 3352–3359 CAS.
- S. M. Hacker, K. M. Backus, M. R. Lazear, S. Forli, B. E. Correia and B. F. Cravatt, Global profiling of lysine reactivity and ligandability in the human proteome, Nat. Chem., 2017, 9, 1181–1190 CrossRef CAS PubMed.
- G. Akçay, M. A. Belmonte, B. Aquila, C. Chuaqui, A. W. Hird, M. L. Lamb, P. B. Rawlins, N. Su, S. Tentarelli, N. P. Grimster and Q. B. Su, Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain, Nat. Chem. Biol., 2016, 12, 931–936 CrossRef PubMed.
- Z. Liu, W. Zhang, B. Sun, Y. Ma, M. He, Y. Pan and F. Wang, Probing conformational hotspots for the recognition and intervention of protein complexes by lysine reactivity profiling, Chem. Sci., 2020, 12, 1451–1457 RSC.
- C. L. Tung, C. T. Wong, E. Y. Fung and X. Li, Traceless and Chemoselective Amine Bioconjugation via Phthalimidine Formation in Native Protein Modification, Org. Lett., 2016, 18, 2600–2603 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, H. Liu, H. Y. Chow, R. Tian, Y. M. Eva Fung and X. Li, OPA-Based Bifunctional Linker for Protein Labeling and Profiling, Biochemistry, 2020, 59, 175–178 CrossRef CAS PubMed.
- C. H. P. Cheung, T. H. Chong, T. Wei, H. Liu and X. Li, Guanidine Additive Enabled Intermolecular ortho-Phthalaldehyde-Amine-Thiol Three-Component Reactions for Modular Constructions, Angew Chem. Int. Ed. Engl., 2023, 62, e202217150 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhang, C. T. T. Wong and X. Li, Chemoselective Peptide Cyclization and Bicyclization Directly on Unprotected Peptides, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef CAS PubMed.
- B. Li, L. Wang, X. Chen, X. Chu, H. Tang, J. Zhang, G. He, L. Li and G. Chen, Extendable stapling of unprotected peptides by crosslinking two amines with o-phthalaldehyde, Nat. Commun., 2022, 13, 311 CrossRef CAS PubMed.
- X. Chu, B. Li, H. Y. Liu, X. Sun, X. Yang, G. He, C. Zhou, W. Xuan, S. L. Liu and G. Chen, Bioconjugation via Hetero-Selective Clamping of Two Different Amines with ortho-Phthalaldehyde, Angew Chem. Int. Ed. Engl., 2023, 62, e202212199 CrossRef CAS PubMed.
- D. A. Polasky, D. J. Geiszler, F. C. Yu, K. Li, G. C. Teo and A. I. Nesvizhskii, MSFragger-Labile: A Flexible Method to Improve Labile PTM Analysis in Proteomics, Mol. Cell. Proteomics, 2023, 22, 100538 CrossRef CAS PubMed.
- Y. P. Zhou and R. W. Vachet, Covalent Labeling with Isotopically Encoded Reagents for Faster Structural Analysis of Proteins by Mass Spectrometry, Anal. Chem., 2013, 85, 9664–9670 CrossRef CAS PubMed.
- N. B. Herrington, D. Stein, Y. C. Li, G. Pandey and A. Schlessinger, Exploring the Druggable Conformational Space of Protein Kinases Using AI-Generated Structures, bioRxiv, 2023, preprint, DOI:10.1101/2023.08.31.555779.
- D. Anastasiou, Y. Yu, W. J. Israelsen, J. K. Jiang, M. B. Boxer, B. S. Hong, W. Tempel, S. Dimov, M. Shen, A. Jha, H. Yang, K. R. Mattaini, C. M. Metallo, B. P. Fiske, K. D. Courtney, S. Malstrom, T. M. Khan, C. Kung, A. P. Skoumbourdis, H. Veith, N. Southall, M. J. Walsh, K. R. Brimacombe, W. Leister, S. Y. Lunt, Z. R. Johnson, K. E. Yen, K. Kunii, S. M. Davidson, H. R. Christofk, C. P. Austin, J. Inglese, M. H. Harris, J. M. Asara, G. Stephanopoulos, F. G. Salituro, S. Jin, L. Dang, D. S. Auld, H. W. Park, L. C. Cantley, C. J. Thomas and M. G. Vander Heiden, Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis, Nat. Chem. Biol., 2012, 8, 839–847 CrossRef CAS PubMed.
- Y. Tian, N. Wan, H. Zhang, C. Shao, M. Ding, Q. Bao, H. Hu, H. Sun, C. Liu, K. Zhou, S. Chen, G. Wang, H. Ye and H. Hao, Chemoproteomic mapping of the glycolytic targetome in cancer cells, Nat. Chem. Biol., 2023, 19, 1480–1491 CrossRef CAS PubMed.
- R. R. A. Kitson and C. J. Moody, Learning from Nature: Advances in Geldanamycin- and Radicicol-Based Inhibitors of Hsp90, J. Org. Chem., 2013, 78, 5117–5141 CrossRef CAS PubMed.
- X. Lu, B. Liao, S. Y. Sun, Y. H. Mao, Q. Wu, R. J. Tian and C. S. H. Tan, Scaled-Down Thermal Profiling and Coaggregation Analysis of the Proteome for Drug Target and Protein Interaction Analysis, Anal. Chem., 2023, 95, 13844–13854 CrossRef CAS PubMed.
- R. Fraczkiewicz and W. Braun, Exact and Efficient Analytical Calculation of the Accessible Surface Areas and Their Gradients for Macromolecules, J. Comput. Chem., 1998, 19, 319–333 CrossRef CAS.
- J. Ma, T. Chen, S. F. Wu, C. Y. Yang, M. Z. Bai, K. X. Shu, K. L. Li, G. Q. Zhang, Z. Jin, F. C. He, H. Hermjakob and Y. P. Zhu, iProX: an integrated proteome resource, Nucleic Acids Res., 2019, 47, D1211–D1217 CrossRef PubMed.
- T. Chen, J. Ma, Y. Liu, Z. G. Chen, N. Xiao, Y. T. Lu, Y. J. Fu, C. Y. Yang, M. S. Li, S. F. Wu, X. Wang, D. S. Li, F. C. He, H. Hermjakob and Y. P. Zhu, iProX in 2021: connecting proteomics data sharing with big data, Nucleic Acids Res., 2022, 50, D1522–D1527 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00032c |
| This journal is © The Royal Society of Chemistry 2024 |