
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directed evolution of Escherichia coli surface-displayed Vitreoscilla hemoglobin as an artificial metalloenzyme for the synthesis of 5-imino-1,2,4-thiadiazoles†
Yaning
Xu
a,
Fengxi
Li
 a,
Hanqing
Xie
a,
Yuyang
Liu
a,
Weiwei
Han
a,
Junhao
Wu
a,
Lei
Cheng
a,
Chunyu
Wang
b,
Zhengqiang
Li
*a and
Lei
Wang
*a
a,
Hanqing
Xie
a,
Yuyang
Liu
a,
Weiwei
Han
a,
Junhao
Wu
a,
Lei
Cheng
a,
Chunyu
Wang
b,
Zhengqiang
Li
*a and
Lei
Wang
*a
aKey Laboratory of Molecular Enzymology and Engineering of Ministry of Education, School of Life Sciences, Jilin University, Changchun 130023, P. R. China. E-mail: lzq@jlu.edu.cn; w_lei@jlu.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun 130023, P. R. China
First published on 25th April 2024
Abstract
Artificial metalloenzymes (ArMs) are constructed by anchoring organometallic catalysts to an evolvable protein scaffold. They present the advantages of both components and exhibit considerable potential for the in vivo catalysis of new-to-nature reactions. Herein, Escherichia coli surface-displayed Vitreoscilla hemoglobin (VHbSD-Co) that anchored the cobalt porphyrin cofactor instead of the original heme cofactor was used as an artificial thiourea oxidase (ATOase) to synthesize 5-imino-1,2,4-thiadiazoles. After two rounds of directed evolution using combinatorial active-site saturation test/iterative saturation mutagenesis (CAST/ISM) strategy, the evolved six-site mutation VHbSD-Co (6SM-VHbSD-Co) exhibited significant improvement in catalytic activity, with a broad substrate scope (31 examples) and high yields with whole cells. This study shows the potential of using VHb ArMs in new-to-nature reactions and demonstrates the applicability of E. coli surface-displayed methods to enhance catalytic properties through the substitution of porphyrin cofactors in hemoproteins in vivo.
Introduction
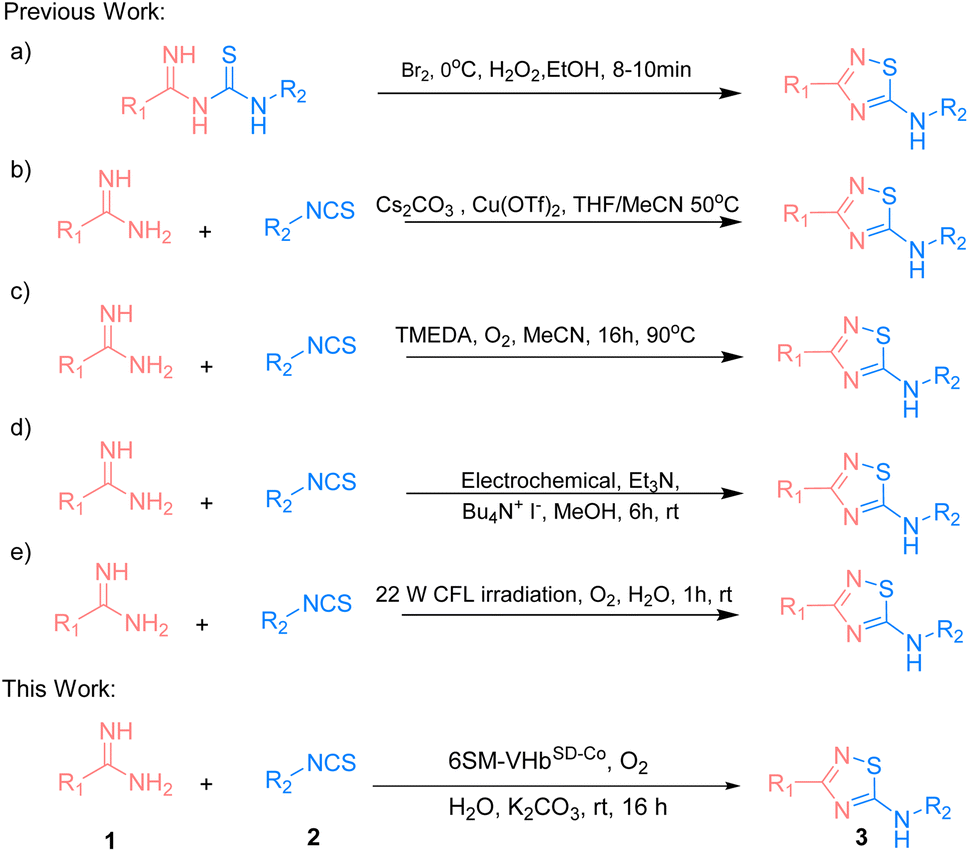
The 5-imino-1,2,4-thiadiazole motif is a unique unit in organic synthesis that can be easily transformed into a large number of functional groups.1,2 It also plays an important role in bioactive molecules.3–5 Existing methodologies focus on the intramolecular oxidative cyclization of imidoyl thiourea to form the corresponding 5-imino-1,2,4-thiadiazoles (Scheme 1a).6 In 1959, Tertiuk et al. proposed an intramolecular oxidative reaction from imidoyl thiourea by using Br2 as an oxidant at 0 °C for the synthesis of 5-imino-1,2,4-thiadiazoles.7 To date, several reports have been made on using stoichiometric oxidants (e.g., H2O2, I2 or hypervalent iodine) or transition metals for the synthesis of 5-imino-1,2,4-thiadiazoles via the oxidative cyclization of imidoyl thiourea. In 2014, Gong et al. presented Cu(OTf)2-catalyzed imidoyl thioureas in situ by amidine hydrochlorides with isothiocyanates for the synthesis of 5-imino-1,2,4-thiadiazoles via intramolecular N–S bond formation at 50 °C (Scheme 1b).8 In 2019, a strategy that used the tetramethylethylenediamine-mediated intramolecular oxidative synthesis of 5-imino-1,2,4-thiadiazoles in MeCN at 90 °C was proposed (Scheme 1c).9 In the succeeding years, Liu et al. explored an electrochemical method for the synthesis of 5-imino-1,2,4-thiadiazoles through intermolecular S–N coupling (Scheme 1d).10 Siddiqui et al. employed a visible light-promoted formation of the S–N bond for the synthesis of the 5-imino-1,2,4-thiadiazole motif in EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (4:1) at room temperature (Scheme 1e).11 However, some drawbacks exist, such as the use of high temperature, harmful organic solvents, or the limitation of the substrate scope. Therefore, exploring eco-friendly, green, and efficient strategies that have not yet been realized should be given importance when constructing these valuable and highly attractive 5-imino-1,2,4-thiadiazole structures.12–20
:H2O (4:1) at room temperature (Scheme 1e).11 However, some drawbacks exist, such as the use of high temperature, harmful organic solvents, or the limitation of the substrate scope. Therefore, exploring eco-friendly, green, and efficient strategies that have not yet been realized should be given importance when constructing these valuable and highly attractive 5-imino-1,2,4-thiadiazole structures.12–20
 | ||
| Scheme 1 Strategies for synthesis of 5-imino-1,2,4-thiadiazole derivatives. | ||
Artificial metalloenzymes (ArMs) are an innovative type of biocatalysts that possess abiotic metal cofactors.21–26 They are promising tools for engineering new non-natural chemical transformations.27–33 Our group previously reported a Co(ppIX)-linked ArM that was designed based on Vitreoscilla hemoglobin (VHb).34,35 The cobalt porphyrin cofactor was incorporated into the cavity of VHb (VHbCo) in vivo by using a porphyrin synthesis-deficient Escherichia coli strain RP523,36 and a range of engineered VHbCo variants were found to catalyze intramolecular oxidative cyclization for the synthesis of 2-substituted benzoxazoles/benzothiazoles.
However, many metal cofactors are inhibited by cellular components and have limited access to the cytoplasm, indicating that the scaffold protein must be purified.37 These factors limit the throughput of genetic optimization schemes applied to ArMs and the applicability in vivo to expand natural metabolism. In recent decades, numerous studies have focused on making the assembly and screening of these biohybrid catalysts more convenient: (i) in vivo assembly of ArM with biosynthesized cobalt protoporphyrin IX (Co(ppIX)) under iron-limited, cobalt-rich growth conditions,38,39 (ii) in vivo assembly of ArM and cofactor with a system to transport the cofactor into the cytoplasm (e.g., recombinant production of heme proteins in E. coli strain Nissle 1917,40,41 ChuA42 and the Hug43 system for porphyrin transportation), and (iii) in vivo screening via cell surface display.37,44,45
To circumvent the limitation of the directed evolution of VHbCo based on our previous studies,34,35,46,47 we developed a new approach that uses E. coli surface-displayed method. We created a platform to display VHbCo on E. coli's outer membrane (VHbSD-Co) and established a whole-cell high-throughput screening strategy based on ultraviolet (UV) absorption for the in vivo directed evolution of VHbSD-Co with high catalytic reactivity for the synthesis of 5-imino-1,2,4-thiadiazoles. After two rounds of evolution, the best VHbSD-Co mutant exhibited excellent activity, producing thiadiazole products with broad substrate scopes, and it can be further evolved in different directions by adjusting the workflow. Our study provides a case for the systematic implementation and directed evolution of an ArM that can be applied to an in vivo non-natural reaction by using O2 as the oxidant and H2O as the solvent. Notably, this study is the first successful attempt to catalyze the synthesis of 1,2,4-thiadiazoles by using a biocatalyst, and it exhibits considerable potential for the further exploration of other non-natural reactions in vivo.
Result and discussion
Our study started with the reaction of benzamidine (1a) and phenyl isothiocyanate (2a) under aerobic conditions at room temperature to form the 5-imino-1,2,4-thiadiazole compound 3aa catalyzed by wild-type VHb. Apart from VHb, several hemeproteins and ArMs that were reported in our previous works were also evaluated (Table 1).34,35 The synthesis of 3aa could not proceed without a catalyst, as shown in entry 1. Cytochrome C, myoglobin, and horseradish peroxidase exhibited low reactivity, while hemoglobins catalyzed the formation of 3aa with better yields (entries 2–10). Among all of these hemeproteins, VHb presented the highest reactivity. Similar to the oxidation reactions reported in previous works, VHbCo demonstrated promising activity compared with wild-type VHb and VHbArM with Mn(ppIX) (VHbMn) and Zn(ppIX) (VHbZn) (entries 11–13). Therefore, VHbCo was selected to be the best biocatalyst for further study.|
|
|||
|---|---|---|---|
| Entry | Biocatalysts | Yieldb (%) | TON |
| a Reaction condition: benzamidines (1a, 50 mM), phenyl isothiocyanate (2a, 50 mM), catalyst (metalloporphyrin containing 0.05 mol%), K2CO3 30 mM, O2, PBS buffer (10 mM), stirred at rt for 12 h. b Determined by high-performance liquid chromatography (HPLC). | |||
| 1 | — | 0 | 0 |
| 2 | VHb | 38 | 760 |
| 3 | Myoglobin (equine skeletal muscle) | 20 | 400 |
| 4 | Hemoglobin (human) | 22 | 440 |
| 5 | Hemoglobin (rabbit) | 25 | 500 |
| 6 | Hemoglobin (bovine) | 26 | 520 |
| 7 | Cytochrome C (horse heart) | 15 | 300 |
| 8 | Cytochrome C (porcine heart) | 14 | 280 |
| 9 | Cytochrome C (bovine heart) | 20 | 400 |
| 10 | Horseradish peroxidase | 19 | 380 |
| 11 | VHbCo | 49 | 980 |
| 12 | VHbMn | 17 | 340 |
| 13 | VHbZn | Trace | Trace |

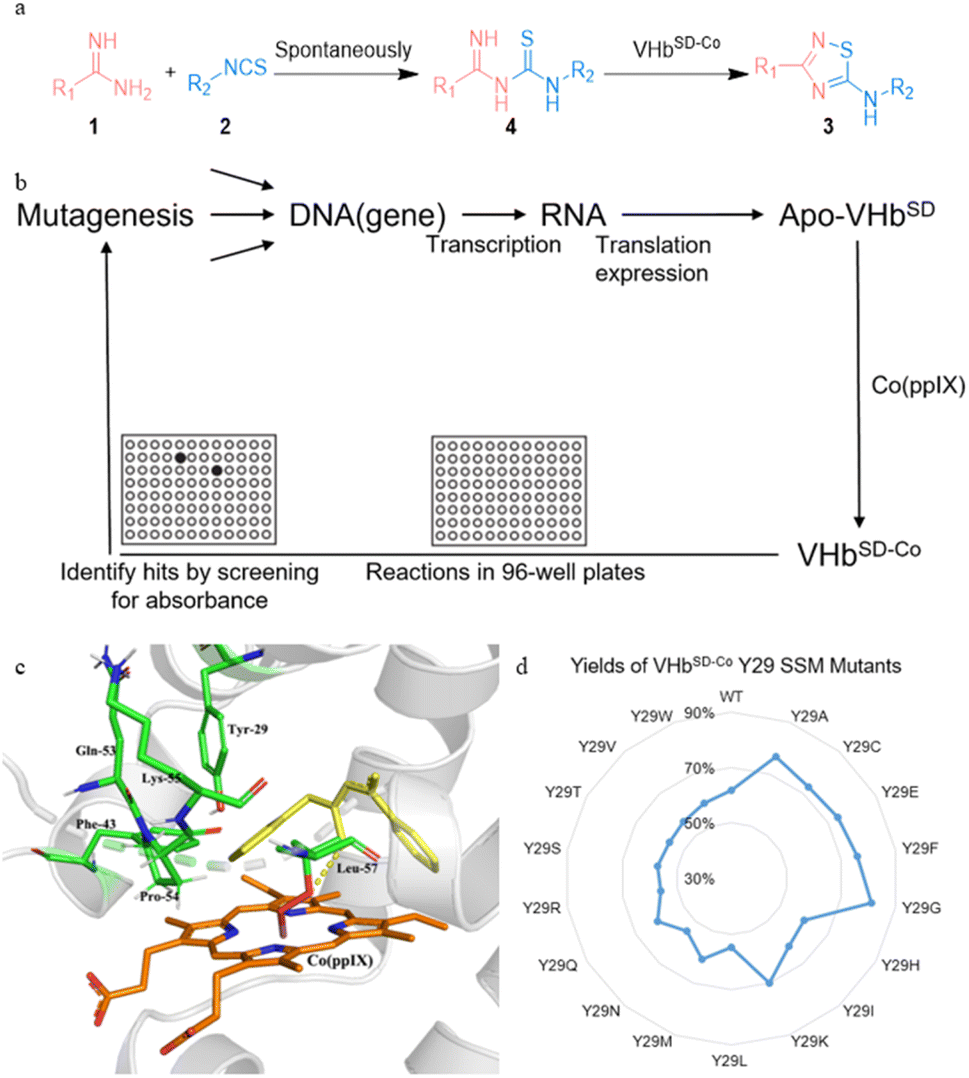
Considering that the intracellular assembly of VHbCo is inefficient for mutant screening and larger substrates experience difficulty in entering E. coli cells, we developed a VHbCo surface display platform on the outer membrane of E. coli by using Lpp–OmpA anchor. By fusing truncated E. coli lipoprotein Lpp, the first five β-strands of outer membrane protein OmpA, a Gly×5 linker, and VHb protein (with His×6 tag on the C-terminus), VHb was anchored onto the outer membrane. To demonstrate the successful construction of Lpp–OmpA–VHb fusion ArM (VHbSD-Co), E. coli cells were stained with a primary mouse-anti his-tag antibody after expression and labeled with a secondary fluorescent antibody, followed by fluorescence microscopy analysis (Fig. 1). Antibodies could not easily cross the membrane and stain the VHbCo protein expressed in the cytoplasm as we anticipated (Fig. 1b). The control experiment of E. coli cells without VHb protein revealed the fluorescence labeling of E. coli only in the presence of His×6 tag-labeled VHb displayed on the surface of the outer membrane (Fig. 1a and c). As expected, these results were consistent with previous reports of Lpp–OmpA-labeled proteins.37,44,45,48,49
 | ||
| Fig. 1 Fluorescence microscopy of immuno-stained E. coli cells. (a) E. coli cells without VHb, (b) E. coli cells expressing VHbCo in the cytoplasm, (c) E. coli cells expressing VHbSD-Co on the surface. Cells were labeled with a primary mouse anti-6xhistag-antibody followed by a fluorescently-labeled secondary goat-anti-mouse antibody. | ||
To test the catalytic activity of VHbSD-Co, the fusion protein was expressed in E. coli cells under anaerobic conditions. The cells were spun-down, and the pellets were resuspended in substrate buffer (1a 50 mM, 2a 50 mM, and K2CO3 30 mM). Then, the reaction mixture was incubated at room temperature for 12 h. The yield of the reaction was determined via high-performance liquid chromatography. As indicated in Table 2, purified fusion protein (entry 1) and cell lysis (entry 2) exhibited similar performance to VHbCo. The reaction catalyzed by VHbSD-Co on the whole cells demonstrated a noticeable enhancement in yield and turnover numbers (TON) (entry 3). To further demonstrate the feasibility of VHbSD-Co catalyzed in whole cells, a series of optimization experiments was carried out (Tables S1† and 2), and the results indicated that VHbSD scaffold, Co(ppIX) cofactor, O2, K2CO3 (for the neutralization of HCl in amidine salt), and aqueous solvent played essential roles in this biocatalysis method. The optimized conditions that led to a slight increase in yield and TON of 3aa (71%, 1420 TON) are as follows: when O2 is used as oxidant under aerobic conditions, VHbSD-Co catalyzes the synthesis of 3aa from 1a (50 mM) and 2a (50 mM) with K2CO3 (30 mM) as base in phosphate-buffered solution (PBS) (5% MeCN for the solubilization of substrates) for 16 h.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Yieldb (%) | TON |
| a Reaction condition: benzamidines (1a, 50 mM), phenyl isothiocyanate (2a, 50 mM), VHbSD-Co (Co(ppIX) containing 0.05 mol%, details were showed in ESI), K2CO3 30 mM, O2, PBS buffer (10 mM, 5% MeCN), stirred at rt for 16 h. b Determined by HPLC. c Without VHbSD. d Air instead of O2. e Without base. | ||||
| 1 | VHbSD-Co (purified) | K2CO3 | 53 | 1060 |
| 2 | VHbSD-Co (cell lysis) | K2CO3 | 47 | 940 |
| 3 | VHbSD-Co (whole cell) | K2CO3 | 71 | 1420 |
| 4 | VHbCo (whole cell) | K2CO3 | 25 | 500 |
| 5 | VHbSD (whole cell) | K2CO3 | 45 | 900 |
| 6c | Co(ppIX) and E.coli cells | K2CO3 | 9 | 180 |
| 7d | VHbSD-Co (whole cell) | K2CO3 | 27 | 540 |
| 8e | VHbSD-Co (whole cell) | — | 29 | 580 |
| 9 | VHbSD-Co (whole cell) | Na2CO3 | 43 | 860 |
| 10 | VHbSD-Co (whole cell) | NaHCO3 | 47 | 940 |
We also tested several previously reported VHbCo variants and mutants of axial His residue (Table S2†), but they did not perform better than the wild-type VHbCo due to the different steric hindrance effects of substrates. Therefore, we developed a high-throughput screening method to select VHbSD-Co variants as artificial thiourea oxidase (ATOase) with better catalytic activity. This method utilized the UV absorption discrepancy of N-(phenylcarbamothioyl)benzimidamide intermediate 4aa and product 3aa (Fig. S1† and 2a). Intermediate 4aa presented a characteristic absorption peak at 313 nm, while product 3aa had no UV absorption at this wavelength. In accordance with previously reported studies,50 substrates 1a and 2a formed intermediate 4aa spontaneously, enabling us to screen the VHbSD-Co variants based on the decrease in UV absorption with reaction in 96-well plates (Fig. 2b).
 | ||
| Fig. 2 High-throughput screening of whole-cell biocatalyst VHbSD-Co. | ||
To identify potential mutation sites, intermediate 4aa was docked into the structure of VHbSD-Co (with CoIII–superoxide intermediate) by using Autodock. The docking result indicated that Y29 residue on the B helix, and F43, Q53, P54, K55, and L57 residues on the loose loop region were closely related to the access of 4aa. Among them, Y29 was directly above the porphyrin plane and limited the exposure of intermediate 4aa to CoIII–superoxide intermediate. Therefore, a combinatorial active-site saturation test/iterative saturation mutagenesis (CAST/ISM) strategy51,52 was applied to evolve this ATOase, the six residues were divided into two groups (Y29 and F43/Q53/P54/K55/L57) according to the region they located. At first, a site saturation mutagenesis (SSM) library at Y29 was generated, and VHbSD-Co mutants performed significant changes compared with WT VHbSD-Co. Three better VHbSD-Co single mutants were identified in the surface-displayed whole-cell screening and the yield was reevaluated separately (yield of 3aa: VHbSD-Co Y29G, 81%; VHbSD-Co Y29A, 77%; VHbSD-Co Y29F, 76%). On the basis of the best single-site mutation VHbSD-Co Y29G (defined as 1SM-VHbSD-Co), a CAST library of F43, Q53, P54, K55, and L57 residues was created. After this high-throughput screening, several hits were identified (Table S3†) and the best six-site mutation, VHbSD-Co Y29G–F43P–Q53P–P54G–K55L–L57A (defined as 6SM-VHbSD-Co), was reevaluated and afforded an improved activity (yield of 3aa: 93%), and the SDS-PAGE verification was showed in Fig. S2.†
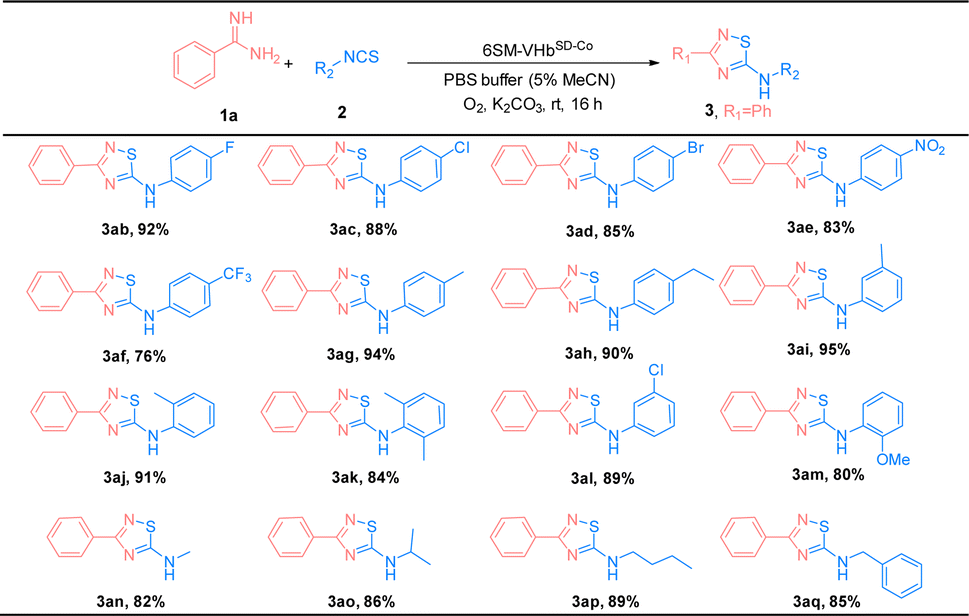
The substrate scope of amidines (1) and isothiocyanates (2) was explored using the best ATOase (6SM-VHbSD-Co), and the generality of this biocatalytic method for the synthesis of thiadiazoles was investigated. The results indicated that this biocatalyst was compatible with a range of amidines and isothiocyanates under standard conditions. As shown in Table 3, both electron-donating groups and electron-withdrawing groups on the phenyl groups of amidines (1) were applicable to the 6SM-VHbSD-Co (3ba–3la) and afforded excellent yields (85–96%). Benzamidine with a large spatial-resistance group showed a decrease on the reactivity, it might because the biphenyl benzamidine is a big substrate which is harder to access into the cavity of VHb, and also probably dispersed into the membrane due to the less water solubility (3ha). Meanwhile, small substituents on the phenyl group of substrates 1 do not lead to an increase in reactivity. This phenomenon may be due to the imperfect matching of intermediate 4 and the active cavity providing the worse catalytic performance based on the neighborhood and orientation effects of the enzyme. The yield of product 3ma–3oa presented varying enhancement by replacing 6SM-VHbSD-Co with 1SM-VHbSD-Co (the mutant with smaller substrate cavity).
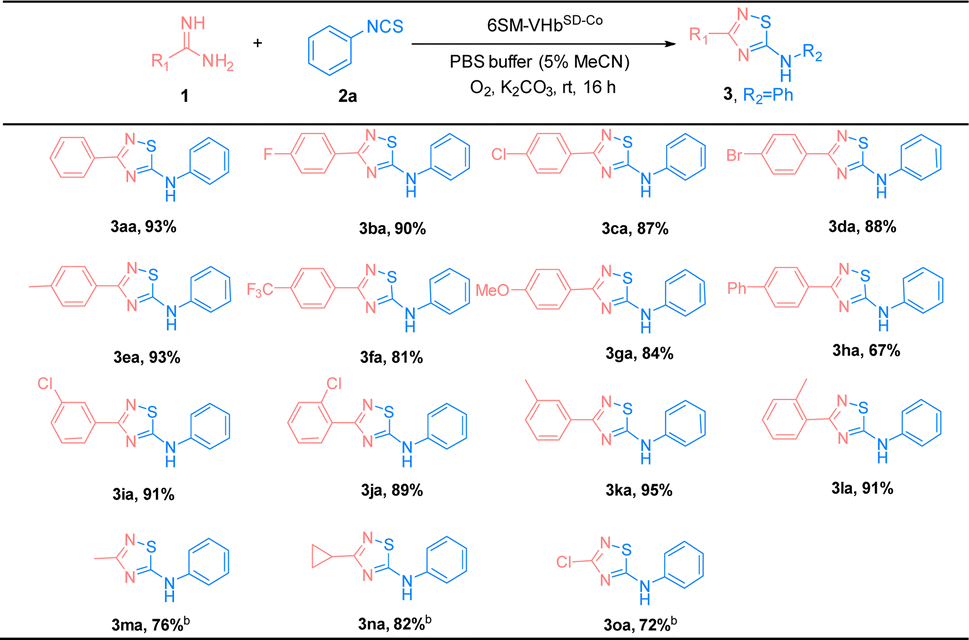
Thereafter, the scope of isothiocyanates was investigated and demonstrated a similar pattern to amidines (Table 4). The steric effects influenced the overall yield of thiadiazoles as demonstrated by the ortho- and para-substituted phenyl isothiocyanates producing decreased yields of products, compared with their meta-substituted counterparts (3ca, 3ia, and 3ja; 3ea, 3ka, and 3la). Encouraged by the successful results, we focused on the further application of this evolved ATOase. A preparation-scale reaction (0.5 mmol) for product 3aa was performed and afforded a satisfactory yield (95%).
| a Reaction condition: benzamidines (1a, 0.5 mmol), phenyl isothiocyanate (2a, 0.5 mmol), 6SM-VHbSD-Co (Co(ppIX) containing 0.05 mol%, details were showed in ESI), K2CO3 0.3 mmol, O2, PBS buffer (10 mM, 5% MeCN), stirred at rt for 16 h. Isolated yield. |
|---|
|
To clarify the mechanism of this reaction, some control experiments were performed under specific conditions (Table 5). The results indicated that without ATOase 6SM-VHbSD-Co, substrates 1a and 2a coupled and converted into intermediate 4aa spontaneously (entry 1). With the addition of ATOase, intermediate 4aa afforded 3aa in 98% yield (entry 2). When the reaction was conducted in the presence of radical scavenger 2,2,6,6-tetramethylpiperdine-1-oxide (TEMPO), the yield of 3aa decreased dramatically, implying the involvement of a radical pathway in this ATOase-catalyzed reaction (entry 3). The control experiments entries 4 and 5 demonstrated the essential role of the O2 and Co(ppIX) cofactor of ATOase.
| Entry | Conditions | Yieldb (%) | |
|---|---|---|---|
| a Standard reaction condition: benzamidines (1a, 50 mM), phenyl isothiocyanate (2a, 50 mM), VHbSD-Co (Co(ppIX) containing 0.05 mol%, details were showed in ESI), K2CO3 30 mM, O2, PBS buffer (10 mM, 5% MeCN), stirred at rt for 16 h. b Determined by HPLC. | |||
| 1 | Without VHbSD-Co | 4aa | 86 |
| 2 | Using 4aa as substrate | 3aa | 98 |
| 3 | TEMPO (3 equiv.) | 3aa | Trace |
| 4 | N2 atmosphere | 3aa | Trace |
| 5 | VHbSD without Co(ppIX) | 3aa | Trace |
On the basis of the previous literature10,53,54 and our earlier study on VHbCo-catalyzed aerobic oxidation,34,35 the mechanism of this reaction was proposed as shown in Scheme 2. First, amidines (1) and isothiocyanates (2) were coupled spontaneously to generate thiourea intermediate 4. Under aerobic conditions, the Co(ppIX) cofactor interacted with O2 to generate a CoIII–superoxide intermediate, which acidified 4 to form a thiyl radical I through an electron transfer process and converted to a base itself, and then thiyl radical I transformed to intermediate II after proton transfer. Next, the nucleophilic attack on the sulfur atom by imino nitrogen led to cyclization to form intermediate III, which was accompanied by a proton transfer to CoIII–superoxide intermediate and generated a peroxo anion. After proton exchange with solvent, III converted into 5-imino-1,2,4-thiadiazole product 3 and H2O2 was generated simultaneously which could also facilitate this reaction (the generation of H2O2 was shown in Fig. S3†).
 | ||
| Scheme 2 Proposed mechanism of VHbSD-Co-catalyzed synthesis of 1,2,4-thiadiazole. | ||
To further explain the reactivity enhancement and catalytic mechanism of VHbSD-Co, thiourea intermediate (4aa) was docked into the active cavity of WT-VHbSD-Co, 1SM-VHbSD-Co and 6SM-VHbSD-Co by using the AutoDock Vina tool55 in Chimera.56 On the basis of the highest scored structures of the docking, Fig. 3 depicts the interactions of intermediate 4aa with VHbSD-Co, 1SM-VHbSD-Co and 6SM-VHbSD-Co (the structure of 1SM-VHbSD-Co and 6SM-VHbSD-Co were constructed by homology modeling strategy using SWISS-MODEL57). In particular, intermediate 4aa was accessed in a hydrophobic pocket in 6SM-VHbSD-Co and well-accommodated into the active center by van der Waals contacts between proximate residues (F28, G29, F33, P43, D44, L55 and A58) and the thiourea intermediate, residue G54 stabilized intermediate by forming a hydrogen bond with the imino nitrogen on the intermediate, meanwhile, pi–alkyl interactions (L32, A57 and V98) and amide-pi stacked interactions (P53) also contributes to the approach of the intermediate (a 2D diagram showed by Discovery Studio Visualizer 4.0 (ref. 58) in Fig. S4†). The sulfur atom on intermediate 4aa maintains a slightly far distance from the oxygen atom on CoIII–superoxide intermediate in WT-VHbSD-Co (Fig. 3a), and the mutation of six amino acids with shorter side chains (Gly, Ala) and rigid side chains (Pro) adjusts the conformation of the flexible loop region (CD spectrum of WT-VHbSD-Co and 6SM-VHbSD-Co were showed in Fig. S5†), reduces spatial resistance to the intermediate, and brings the distance between intermediate 4aa and the oxygen atom in the CoIII–superoxide intermediate closer in mutant 6SM-VHbSD-Co (Fig. 3c).
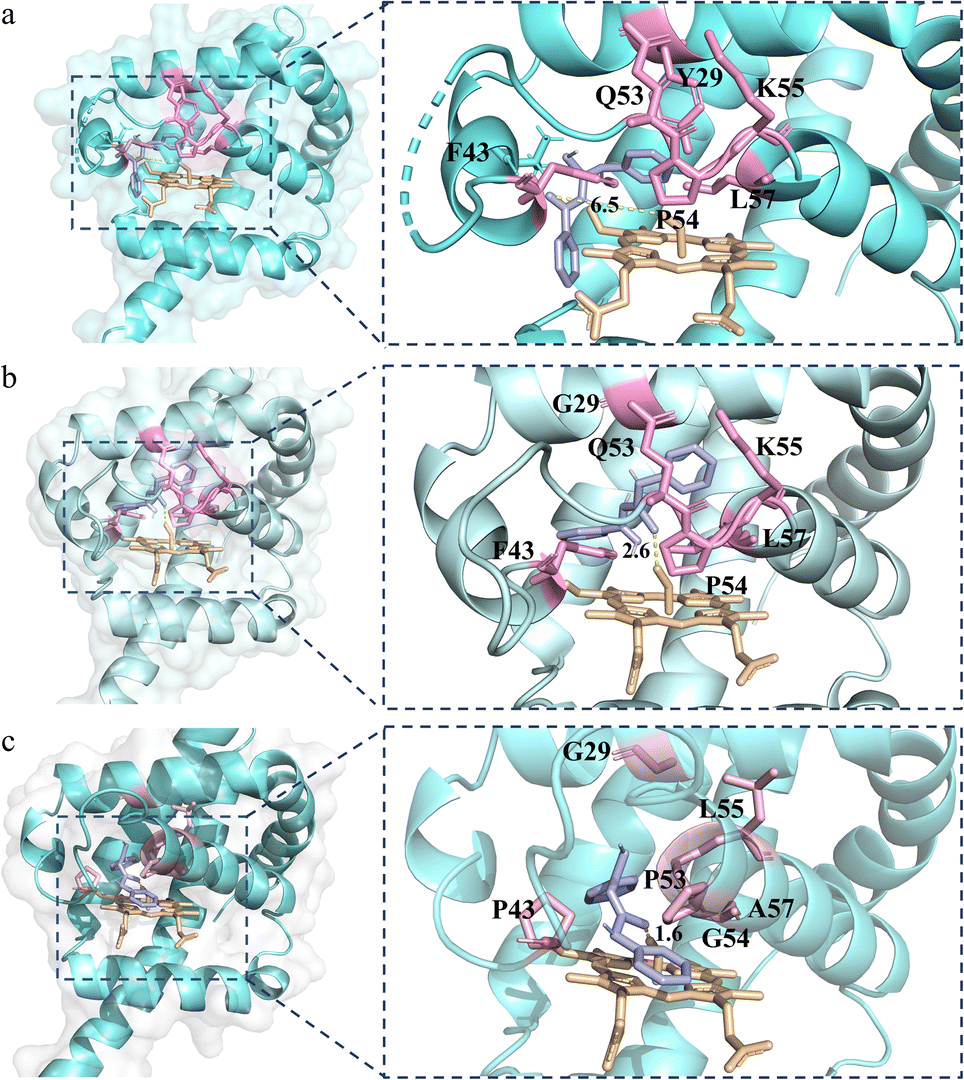 | ||
| Fig. 3 Structure model of thiourea intermediate 4aa (light purple) in the active site of VHbSD-Co. (a) Structure of WT-VHbSD-Co in complex with 4aa. (b) Structure of 1SM-VHbSD-Co in complex with 4aa. (c) Structure of 6SM-VHbSD-Co in complex with 4aa. | ||
Conclusions
We evolved an artificial thiourea oxidase based on the porphyrin substitution of VHb. The oxidase catalyzed amidines (1) and isothiocyanates (2) to form 5-imino-1,2,4-thiadiazoles (3). To simplify the protocol of ArM construction and the screening of directed evolution, we implemented an E. coli surface-displayed method by using Lpp–OmpA–VHb fusion protein. Through a CAST/ISM evolution of Y29, F43, Q53, P54, K55, and L57 residues, we obtained a six-site mutation 6SM-VHbSD-Co (Y29G–F43P–Q53P–P54G–K55L–L57A) with improved ATOase activity (up to 96% yield and 1920 TON, 2.5-fold increased versus WT VHb) and broad substrate scopes (31 examples). The molecular docking of ATOase and thiourea intermediate 4 further indicated the rationale of the reactivity enhancement. We expect that this surface-displayed method can be extended to streamline the construction and directed evolution of ArMs for new-to-nature reactions and in vivo synthetic biology explorations.Data availability
All the data supporting this article have been included in ESI.†Author contributions
Yaning Xu: conceptualization, investigation, writing – original draft. Fengxi Li: methodology, investigation. Hanqing Xie: investigation. Yuyang Liu & Weiwei Han: docking simulation. Junhao Wu: investigation. Lei Cheng: investigation. Chunyu Wang: NMR investigation. Zhengqiang Li: conceptualization, supervision, project administration, resources. Lei Wang: conceptualization, supervision, project administration, resources, writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the Science and Technology Development Program of Jilin Province (No. 20230101135JC) and the Graduate Innovation Fund of Jilin University (No. 2023CX044).Notes and references
- E. R. Devi, R. Sreenivasulu, M. V. B. Rao, K. P. Rao, R. V. Nadh and M. Sireesha, Russ. J. Gen. Chem., 2021, 91, 1105–1111 CrossRef CAS.
- B. M. Sahoo, B. K. Banik, A. Tiwari, V. Tiwari and M. K. Mahapatra, Curr. Organocatal., 2023, 10, 237–249 CrossRef CAS.
- G. F. Makhaeva, N. V. Kovaleva, N. P. Boltneva, S. V. Lushchekina, E. V. Rudakova, T. S. Stupina, A. A. Terentiev, I. V. Serkov, A. N. Proshin, E. V. Radchenko, V. A. Palyulin, S. O. Bachurin and R. J. Richardson, Bioorg. Chem., 2020, 96, 103387 CrossRef PubMed.
- V. Palomo, D. I. Perez, C. Perez, J. A. Morales-Garcia, I. Soteras, S. Alonso-Gil, A. Encinas, A. Castro, N. E. Campillo, A. Perez-Castillo, C. Gil and A. Martinez, J. Med. Chem., 2012, 55, 1645–1661 CrossRef CAS PubMed.
- M. Promzeleva, T. Volkova, A. Proshin, O. Siluykov, A. Mazur, P. Tolstoy, S. Ivanov, F. Kamilov and I. Terekhova, ACS Biomater. Sci. Eng., 2018, 4, 491–501 CrossRef CAS PubMed.
- L. Rubab, A. Anum, S. A. Al-Hussain, A. Irfan, S. Ahmad, S. Ullah, A. A. Al-Mutairi and M. E. A. Zaki, Catalysts, 2022, 12, 1329 CrossRef CAS.
- F. Kurzer and W. Tertiuk, J. Chem. Soc., 1959, 2851–2857, 10.1039/JR9590002851.
- H.-Y. Kim, S. H. Kwak, G.-H. Lee and Y.-D. Gong, Tetrahedron, 2014, 70, 8737–8743 CrossRef CAS.
- Z. Yang, T. Cao, S. Liu, A. Li, K. Liu, T. Yang and C. Zhou, New J. Chem., 2019, 43, 6465–6468 RSC.
- Z. Yang, J. Zhang, L. Hu, L. Li, K. Liu, T. Yang and C. Zhou, J. Org. Chem., 2020, 85, 3358–3363 CrossRef CAS PubMed.
- A. Verma, A. Srivastava, S. K. Tiwari, N. Yadav, M. D. Ansari, V. B. Yadav, H. Sagir and I. R. Siddiqui, J. Heterocycl. Chem., 2020, 57, 3493–3498 CrossRef CAS.
- S. Chauhan, P. Verma, A. Mishra and V. Srivastava, Chem. Heterocycl. Compd., 2020, 56, 123–126 CrossRef CAS.
- J. Y. Chen, M. Selvaraju, Y. T. Lin, S. Dhole, C. Y. Lin and C. M. Sun, J. Org. Chem., 2020, 85, 5570–5579 CrossRef CAS PubMed.
- J. Yuan, Q. Xia, W. Zhu, C. Wu, B. Wang, B. Liu, X. Yang, Y. Xu and H. Xu, ChemPhotoChem, 2020, 4, 445–450 CrossRef CAS.
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed.
- P. Kamboj and V. Tyagi, Green Chem., 2024, 26, 1990–1999 RSC.
- P. Kamboj and V. Tyagi, Tetrahedron, 2023, 148, 133679 CrossRef CAS.
- C. Mateo, O. Abian, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2000, 26, 509–515 CrossRef CAS PubMed.
- S. Peirce, J. J. Virgen-Ortíz, V. G. Tacias-Pascacio, N. Rueda, R. Bartolome-Cabrero, L. Fernandez-Lopez, M. E. Russo, A. Marzocchella and R. Fernandez-Lafuente, RSC Adv., 2016, 6, 61707–61715 RSC.
- K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107–122 CrossRef CAS PubMed.
- Y. L. Deng, S. Dwaraknath, W. O. Ouyang, C. J. Matsumoto, S. Ouchida and Y. Lu, Angew. Chem., Int. Ed., 2023, 62, e202215719 CrossRef CAS PubMed.
- S. Kato, A. Onoda, U. Schwaneberg and T. Hayashi, J. Am. Chem. Soc., 2023, 145, 8285–8290 CrossRef CAS PubMed.
- Y. Okamoto, T. Mabuchi, K. Nakane, A. Ueno and S. Sato, ACS Catal., 2023, 13, 4134–4141 CrossRef CAS.
- Y. Gu, B. J. Bloomer, Z. Liu, R. Chen, D. S. Clark and J. F. Hartwig, Angew. Chem., Int. Ed., 2021, 61, e202110519 CrossRef PubMed.
- E. J. Moore, V. Steck, P. Bajaj and R. Fasan, J. Org. Chem., 2018, 83, 7480–7490 CrossRef CAS PubMed.
- R. Mao, D. M. Taylor, D. J. Wackelin, T. Rogge, S. J. Wu, K. M. Sicinski, K. N. Houk and F. H. Arnold, Nat. Synth., 2024, 3, 256–264 CrossRef.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- U. Markel, D. F. Sauer, J. Schiffels, J. Okuda and U. Schwaneberg, Angew. Chem., Int. Ed., 2019, 58, 4454–4464 CrossRef CAS PubMed.
- I. Drienovská, L. Alonso-Cotchico, P. Vidossich, A. Lledós, J. D. Maréchal and G. Roelfes, Chem. Sci., 2017, 8, 7228–7235 RSC.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC.
- Y. Jiang, C. Wang, N. Ma, J. Chen, C. Liu, F. Wang, J. Xu and Z. Cong, Catal. Sci. Technol., 2020, 10, 1219–1223 RSC.
- J. Chen, F. Kong, N. Ma, P. Zhao, C. Liu, X. Wang and Z. Cong, ACS Catal., 2019, 9, 7350–7355 CrossRef CAS.
- P. Zhao, J. Chen, N. Ma, J. Chen, X. Qin, C. Liu, F. Yao, L. Yao, L. Jin and Z. Cong, Chem. Sci., 2021, 12, 6307–6314 RSC.
- F. Li, Y. Xu, Y. Xu, J. Ma, H. Xie, H. Yang, W. Han, C. Wang, Z. Li and L. Wang, Org. Chem. Front., 2023, 10, 3509–3514 RSC.
- Y. Xu, F. Li, N. Zhao, J. Su, C. Wang, C. Wang, Z. Li and L. Wang, Green Chem., 2021, 23, 8047–8052 RSC.
- J. J. Woodward, N. I. Martin and M. A. Marletta, Nat. Methods, 2007, 4, 43–45 CrossRef CAS PubMed.
- T. Heinisch, F. Schwizer, B. Garabedian, E. Csibra, M. Jeschek, J. Vallapurackal, V. B. Pinheiro, P. Marlière, S. Panke and T. R. Ward, Chem. Sci., 2018, 9, 5383–5388 RSC.
- L. J. Perkins, B. R. Weaver, A. R. Buller and J. N. Burstyn, De novo biosynthesis of a nonnatural cobalt porphyrin cofactor in E. coli and incorporation into hemoproteins, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017625118 CrossRef CAS PubMed.
- B. R. Weaver, L. J. Perkins, F. O. Fernandez Candelaria, J. N. Burstyn and A. R. Buller, Molecular Determinants of Efficient Cobalt-Substituted Hemoprotein Production in E. coli, ACS Synth. Biol., 2023, 12, 3669–3679 CrossRef CAS PubMed.
- Z. N. Liu, J. Huang, Y. Gu, D. S. Clark, A. Mukhopadhyay, J. D. Keasling and J. F. Hartwig, J. Am. Chem. Soc., 2022, 144, 883–890 CrossRef CAS PubMed.
- K. Fiege, C. J. Querebillo, P. Hildebrandt and N. Frankenberg-Dinkel, Biochemistry, 2018, 57, 2747–2755 CrossRef CAS PubMed.
- V. S. Lelyveld, E. Brustad, F. H. Arnold and A. Jasanoff, J. Am. Chem. Soc., 2011, 133, 649–651 CrossRef CAS PubMed.
- Y. Guo, G. Guo, X. Mao, W. Zhang, J. Xiao, W. Tong, T. Liu, B. Xiao, X. Liu, Y. Feng and Q. Zou, BMC Microbiol., 2008, 8, 226 CrossRef PubMed.
- A. Baiyoumy, J. Vallapurackal, F. Schwizer, T. Heinisch, T. Kardashliev, M. Held, S. Panke and T. R. Ward, ACS Catal., 2021, 11, 10705–10712 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- F. Li, Y. Xu, Y. Xu, H. Xie, J. Wu, C. Wang, Z. Li, Z. Wang and L. Wang, Org. Lett., 2023, 25, 7115–7119 CrossRef CAS PubMed.
- H. Xie, F. Li, Y. Xu, C. Wang, Y. Xu, J. Wu, Z. Li, Z. Wang and L. Wang, Green Chem., 2023, 25, 6853–6858 RSC.
- G. Georgiou, D. L. Stephens, C. Stathopoulos, H. L. Poetschke, J. Mendenhall and C. F. Earhart, Protein Eng., Des. Sel., 1996, 9, 239–247 CrossRef CAS PubMed.
- J. A. Francisco, C. F. Earhart and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2713–2717 CrossRef CAS PubMed.
- B. Wang, Y. Meng, Y. Zhou, L. Ren, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2017, 82, 5898–5903 CrossRef CAS PubMed.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed.
- T. Naqvi, A. C. Warden, N. French, E. Sugrue, P. D. Carr, C. J. Jackson and C. Scott, PLoS One, 2014, 9, e94177 CrossRef PubMed.
- Z. Q. Xiong, Q. H. Zhong, S. R. Sheng and J. M. Chen, Arkivoc, 2021, 340–348, DOI:10.24820/ark.5550190.p011.577.
- L. Yang, L. Song, S. Tang, L. Li, H. Li, B. Yuan and G. Yang, Eur. J. Org Chem., 2019, 2019, 1281–1285 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- A. S. Inc., Accelrys, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00005f |
| This journal is © The Royal Society of Chemistry 2024 |