
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of CT-coupled J-aggregation platform based on Aza-BODIPY for highly efficient phototherapy†
Shengmei
Wu‡
a,
Wenze
Zhang‡
b,
Chaoran
Li
b,
Zhigang
Ni
 a,
Weifeng
Chen
a,
Lizhi
Gai
*a,
Jiangwei
Tian
*b,
Zijian
Guo
c and
Hua
Lu
*a
a,
Weifeng
Chen
a,
Lizhi
Gai
*a,
Jiangwei
Tian
*b,
Zijian
Guo
c and
Hua
Lu
*a
aCollege of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, No. 2318, Yuhangtang Road, Hangzhou, 311121, P. R. China. E-mail: lizhigai@hznu.edu.cn; hualu@hznu.edu.cn
bState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 211198, P. R. China. E-mail: jwtian@cpu.edu.cn
cState Key Laboratory of Coordination Chemistry, Coordination Chemistry Institute, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China
First published on 27th February 2024
Abstract
Supramolecular engineering is exceptionally appealing in the design of functional materials, and J-aggregates resulting from noncovalent interactions offer intriguing features. However, building J-aggregation platforms remains a significant challenge. Herein, we report 3,5-dithienyl Aza-BODIPYs with a donor–acceptor–donor (D–A–D) architecture as the first charge transfer (CT)-coupled J-aggregation BODIPY-type platform. The core acceptor moieties in one molecule interact with donor units in neighboring molecules to generate slip-stacked packing motifs, resulting in CT-coupled J-aggregation with a redshifted wavelength up to 886 nm and an absorption tail over 1100 nm. The J-aggregates show significant photoacoustic signals and high photothermal conversion efficiency of 66%. The results obtained in vivo show that the J-aggregates have the potential to be used for tumor photothermal ablation and photoacoustic imaging. This study not only demonstrates Aza-BODIPY with D–A–D as a novel CT-coupled J-aggregation platform for NIR phototherapy materials but also motivates further study on the design of J-aggregation.
Introduction
With the advancement of optical diagnosis and nanomedicine, near-infrared organic dyes have demonstrated tremendous promise in the fields of disease diagnosis and phototherapy.1 Molecular engineering has been a widely used strategy to develop NIR dyes with desired properties.2 In addition to the molecular engineering strategy, supramolecular engineering offers an alternative approach for the design of NIR dyes by forming J-type aggregates.3 This strategy takes advantage of the exchange narrowing effect between chromophores, which can lead to a significant redshift in the absorption wavelength.4 J-aggregation has emerged as a convenient method to achieve NIR absorbance.4c However, the construction of J-aggregates is usually developed by serendipitous discovery, and the rational design of molecular structures to induce J-aggregation of organic dyes remains a challenge. Although there have been successful examples of J-aggregate formation, developing a systematic and predictable approach for designing J-aggregates is still an ongoing area of research.4b,d,5Classical J-aggregation refers to the accumulation of dye molecules in a slip-stacked manner along the direction of the dipole moment, with the slip angle of θ < 54.7°. This phenomenon leads to a narrow absorption peak in J-aggregation under the influence of the long-range Coulomb coupling. Typically, there is a wavelength shift of around 30 nm,6,7 which is limited to controlling the wavelength red shift. In addition, the narrow absorption peak (usually FWHM <40 nm) causes an unsatisfactory matching between the laser wavelength and absorption peak, making the selection of appropriate lasers for phototherapy difficult. Therefore, the development of J-aggregates with superior spectral characteristics has become an urgent matter. In D–A–D architectures, slip-stacked packing motifs are favored, where the central acceptor moiety in one molecule interacts with donor moieties in neighboring molecules (Fig. 1).4c,8 The particular arrangement of these interactions can result in different types of spectra with J-Coulomb dominance (conventional J-aggregation) or CT-coupled J-aggregation, the latter of which might even have an H-type J-Coulomb component.8,9 The mixing of multiple couplings results in band widening, which helps match lasers and is required for panchromatic light absorption. Currently, several CT-coupled J-aggregates based on the D–A–D architecture are limited to the squaraine system.10 The electron-deficient aza-BODIPY core is an ideal model to construct a D–A–D structure to form a slip-stacked packing motif.11 Recent studies have reported several aza-BODIPY dyes with conventional J-aggregation,12 in which main driving forces for aggregation are diverse, such as hydrogen bond,13 solvophobic interactions14 and π–π stacking interactions.15 Despite these advancements, there is still much to explore in the rational design and stabilization of J-aggregates. Herein, we present a general D–A–D scaffold incorporating thiophene units as donors16 into the 3,5-position of aza-BODIPY to construct CT-coupled J-aggregates. The resulting thiophene(D)–aza-BODIPY(A)–D scaffold exhibits hypsochromic and bathochromic absorption bands, corresponding to the mixing between intermolecular CT and Frenkel states due to pronounced D–A interaction. Moreover, the CT-coupled J-aggregates were demonstrated to be applicable to photoacoustic (PA) imaging-guided photothermal therapy (PTT).
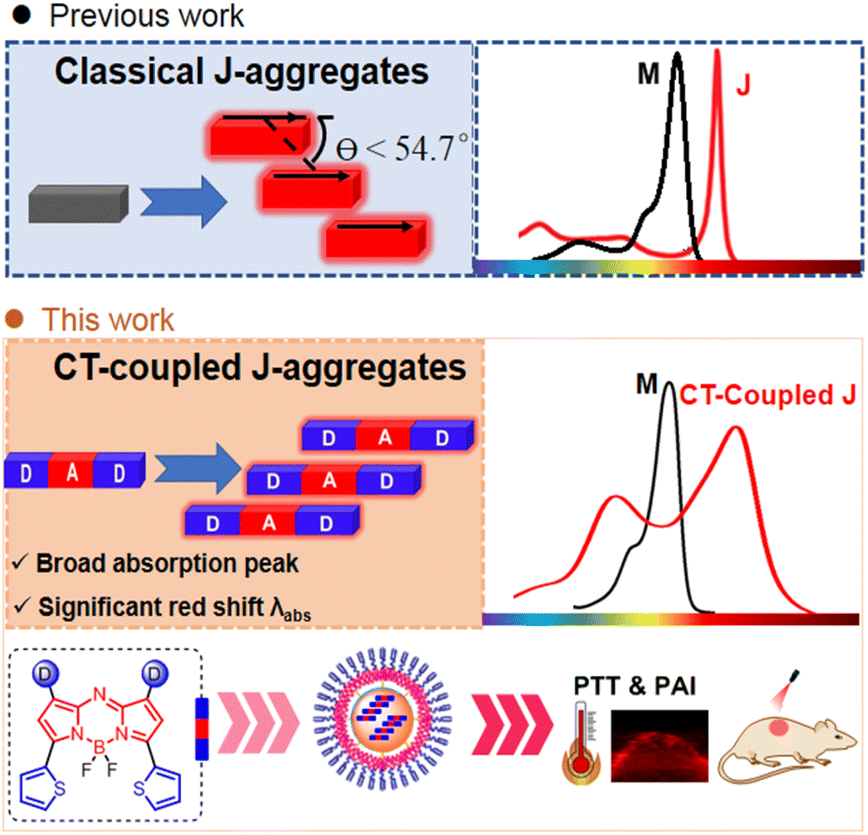 | ||
| Fig. 1 Creation of CT-coupled J-aggregates. Schematic illustration of classical J-aggregates and strategic design of CT-coupled J-aggregates in this study. | ||
Results and discussion
Molecular design
Slip-stacked packing motifs are highly favored in the D–A–D architecture. The aza-BODIPY core is an ideal acceptor due to its planar electron-deficient properties. The donor moiety at the 3,5-position plays a crucial role in forming the CT-coupled J-aggregates because the electronic effect at the 3,5-position contributes to HOMO and LUMO orbitals (Fig. S1 and Table S1†).17 The five-ring thiophene unit is a wonderful candidate with electron-rich nature. In addition, the angle between the five-ring thiophene (D) and aza-BODIPY core is smaller than that of the six-ring donor. This smaller angle facilitates increased conjugation and interaction between the donor and acceptor moieties, which is beneficial for the formation of the CT-coupled J-aggregates. Taking advantage of the unique properties of thiophenes, we developed a series of 3,5-dithienyl (D1)–aza-BODIPY(A)–D2 structures (Fig. 2A). These compounds are loaded with different electron-donor groups at the 1,7-position to achieve general CT-coupled J-aggregation. Additionally, D–A–A structures were designed to gain a deeper understanding of the aggregate characteristics.18 The synthesis of BDP1–5 adopts the classic Aza-BODIPY synthesis method.18a,19 The detailed synthesis process and characterization information are in the ESI.†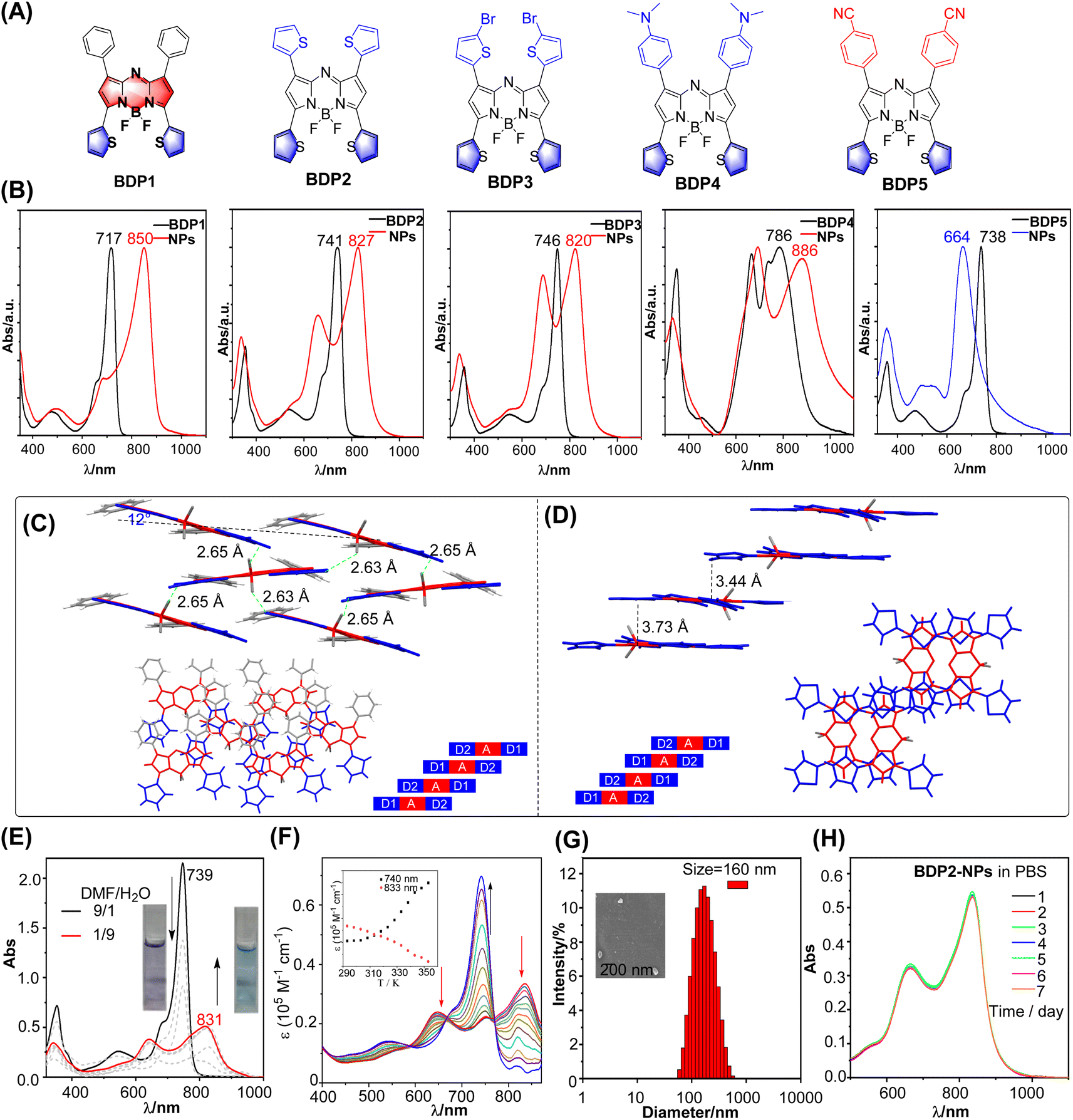 | ||
| Fig. 2 (A) Structure of BDP1–5. (B) Comparison of the absorption of BDP1–5 in THF and BDP1–5 NPs in water. (C and D) Molecular structure of BDP1 (left) and BDP2 (right) in the single crystal state with the F⋯H (thiophene) bond, packing motif in the single-crystal structure, and a schematic depiction of extended, antiparallelly aligned π-stacks, respectively. CCDC no. 877767 and 877768. Reproduced from ref. 18a with permission. (E) Absorption spectra of BDP2 in a DMF/water mixture with different water fractions. Inset: natural light image of BDP2 in DMF/water (1/9 and 9/1). (F) Temperature-dependent absorption spectra of BDP2 in a DMF/water (3/2) mixture from 293 to 353 K at a total concentration of 10 μM. (G) The size distribution of BDP2-NPs using DLS at 298 K. (H) Absorption spectra of BDP2-NPs in PBS for different days. | ||
Spectroscopic properties
We first investigated the spectroscopic properties of dichloromethane (Fig. 2B and Table S2†). BDP1–5 exhibited strong absorption spectra with a large molar extinction coefficient (3–7.5 × 104 M−1 cm−1) and maximum wavelength covering the 717–786 nm region through the modification of the substituent at the 1,7-position. The strong electron-donating N,N-dialkyl groups resulted in the splitting of the maximum absorption into two parallel peaks. This split can be attributed to the presence of local excitation (LE) and charge transfer (CT) transition states. Photobleaching resistance is a crucial property. The maximum absorption of BDP1–5 exhibits minor change within 2%, whereas control indocyanine green (ICG) was significantly reduced after 30 min of irradiation (Fig. S2†), indicating their promising photobleaching resistance.We then investigated the aggregation behavior. The dyes were first dissolved in THF and later quickly injected into water containing DSPE-PEG2000 under stirring, leading to the formation of micellar nanoparticles (Fig. S3†). The UV/Vis spectra showed significant changes upon aggregation. D–A–D scaffolds BDP2–4 display broad absorption spectra (FWHM = 205 nm for BDP2, 209 nm for BDP3, and 298 nm for BDP4) with two distinguishable maxima with H-type JCoulomb and pronounced donor–acceptor interaction between monomers due to a significant HOMO–LUMO overlap (Fig. S1†). The energetically low-lying charge transfer (CT) states interact with Frenkel states, resulting in a mixing of electronic states. This mixing leads to the formation of optically allowed states at higher and lower energy compared to the monomer. In contrast, BDP1 with a weak donor at the 1,7-position exhibited a weak H-type JCoulomb and strong short-range coupling owing to wavefunction overlap. The substituent group greatly disturbed the type of aggregation. The 3,5-position phenyl aza-BODIPY displayed a minor absorptive change before and after aggregation, indicating that substituents at the 3,5-position have a significant effect on the formation of J-aggregation (Fig. S4†). In the case of the D–A–A structure, a blue-shifted and broad absorption band was observed, since it is prone to self-assembly into an extended 1d card-stack arrangement with pronounced Coulomb H-coupling (Fig. 2B).
Crystal packing can provide valuable insights into the aggregation behavior of molecules (Fig. 2C). According to the analysis of the X-ray structures of BDP1–2,18a they possess a rigid π system from B–F⋯H–C (thiophene) hydrogen interaction by inhibiting the rotation of thiophene. In the context of BDP1, it can be observed that the overlap between two parallel molecules is less pronounced, while there is a relatively higher degree of overlap with the neighboring molecules. This increase in overlap is primarily attributable to intermolecular interactions that arise between the electron-donating thiophene unit and non-parallel parent nucleus of the adjacent molecule, as well as between the thiophene unit and parent nucleus of a distinct molecule. Such intermolecular forces in turn facilitate the effective aggregation of the molecules. In the case of BDP2, it can be observed that the intermolecular spacing between parallel molecules is restricted to 3.73 and 3.44 Å, leading to an evident intermolecular π–π interaction.19 When examining the crystal packing from a top-down perspective, the presence of an electron-donating thiophene unit and another parallel electron-deficient core of the molecule is observed to play a significant role in this process. Overall, the crystal packing structure confirms the plausibility of utilizing a D–A–D strategy to facilitate the aggregation of molecules and intermolecular interactions.
To gain insight into the detailed aggregation state-related physiochemical properties, the absorption spectra of BDP2 under different DMF/water ratios were investigated, with the addition of water leading to a gradual decrease in the main absorption peak and appearance of a new absorption peak at 831 nm for BDP2 (Fig. 2E). Subsequently, the absorption peak of the monomer with maximum absorption of 739 nm increased on increasing the temperature from 293 K to 353 K, and this result verified that the self-assembly was reversible (Fig. 2F). We conducted an investigation into the aggregation mechanism of BDP2 employing a nucleation–elongation model.20 By analyzing the Vis-NIR spectral data and fitting resulting curves, we determined the fraction of aggregated molecules (αAgg), temperature (T), molar enthalpy (ΔHe), and elongation temperature (Te) (Fig. S5†). Our analysis revealed a molar enthalpy change of −18.2 kJ mol−1 and an elongation temperature of 360 K. Notably, the lower molar enthalpy indicates that BDP2 exhibits a higher aggregation propensity, suggesting its ease of aggregation. SEM was used to test its aggregates and confirmed that it formed a large number of the nanoparticles (Fig. S6a†).
Photothermal properties of BDP2-NPs
To ensure that BDP2 has good water solubility and stability, we performed a comparative characterization of the structure of the encapsulated nanoparticles. The hydrodynamic size of the BDP2-NPs measured by dynamic light scattering (DLS) was around 160 nm (Fig. 2G), which contributed to their accumulation at tumor sites through the enhanced permeability and retention effect,21 and their hydrodynamic diameters hardly changed after storage in a refrigerator at 4 °C for one week under normal physiological conditions (Fig. 2H, S6b and c†). While BDP2-NPs do not exhibit fluorescence emission due to the aggregation of free molecules in the nanoparticles, this property helps to improve their photothermal conversion ability, making them promising in photoacoustic/photothermal imaging and photothermal therapy.Based on the fact that BDP2-NPs exhibit negligible fluorescence emission in water, it is inferred that the excitation energy can be released through a non-radiative decay process.19a Therefore, we investigated whether the BDP2-NPs are an effective reagent in photothermal water media (Fig. 3). The temperature changes of the BDP2-NPs at different concentrations were monitored upon laser irradiation. Following 600 s of 808 nm laser irradiation at 0.96 W cm−2, the temperature difference increased by more than 25 °C for 30 μM BDP2-NPs. Positive correlations between the photothermal conversion efficiency of the BDP2-NPs and their concentration and laser power were found, indicating the controllable photothermal conversion behavior (Fig. 3a and b). Based on the photothermal effect and time constant,22 the photothermal conversion efficiency of the BDP2-NPs was calculated to be 66% (Fig. 3c). After five cycles of laser irradiation, the heating temperature of the BDP2-NPs remained stable at around 63 °C, indicating that the BDP2-NPs possess excellent photothermal properties (Fig. 3d). Subsequently, the temperature of the BDP2-NP solution increased from 25 °C to 56 °C after 5 min of laser irradiation, while only a minor temperature change was observed in the PBS reference (Fig. 3e). As compared to BDP2-NPs, BDP1-NPs possess similar PTT properties with a PCE value of 53% (Fig. S7†). Moreover, the BDP2-NPs produced the highest photoacoustic signal at 820 nm under 808 nm laser irradiation, indicating that the strong near-infrared absorption of the J-aggregate controlled the photoacoustic response.
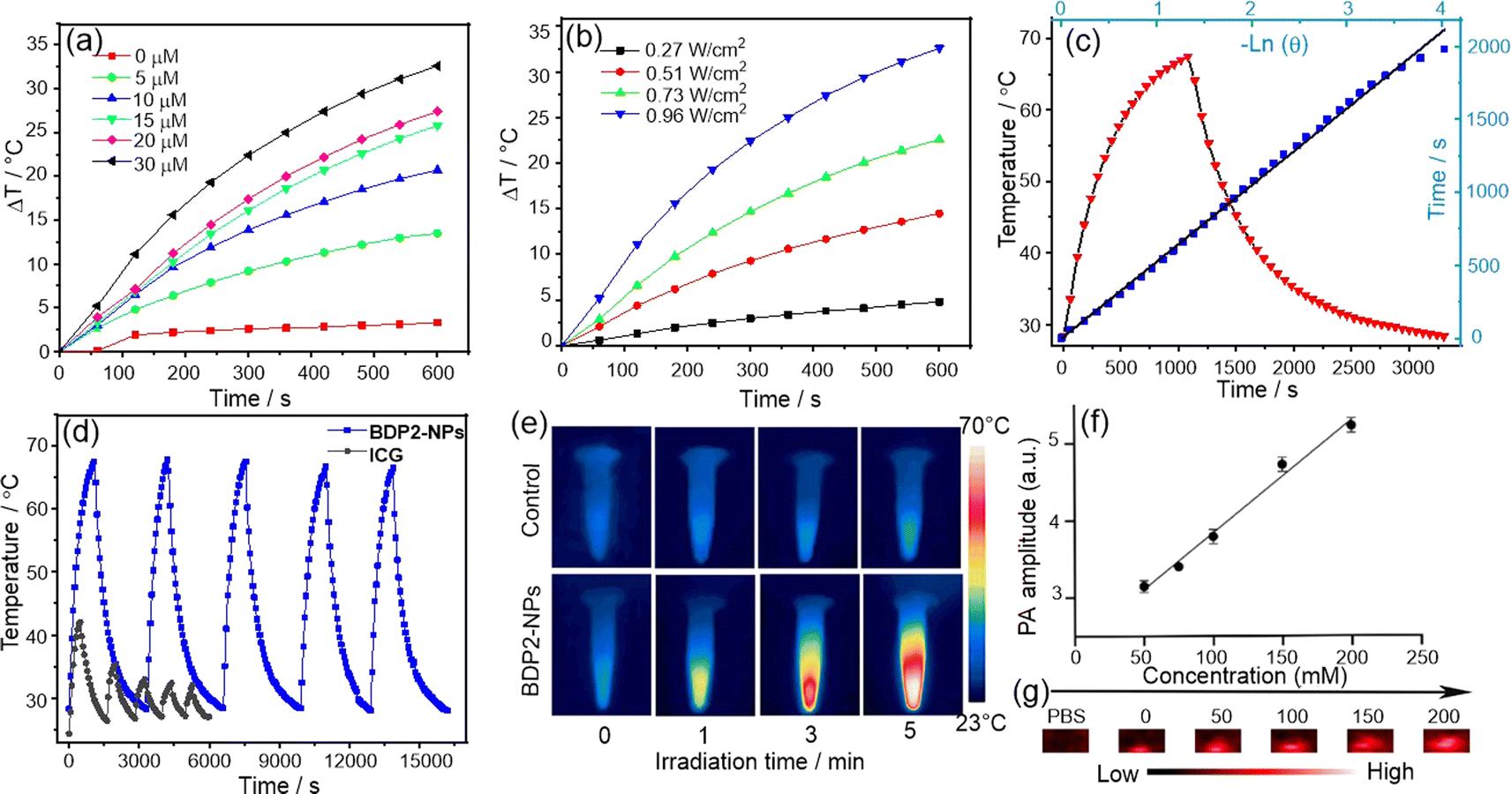 | ||
| Fig. 3 (a) Heating curves of the BDP2-NPs at various concentrations upon laser irradiation (0.96 W cm−2); (b) heating curves of the BDP2-NPs (30 μM) at increasing power density; (c) heating curves of the BDP2-NPs (30 μM) during a laser-on and off switching process, with the inset showing the related linear cooling time data versus −ln(θ). η = 66%. (d) Heating and cooling curves of the BDP2-NPs (30 μM) upon five times laser irradiation (0.96 W cm−2), with ICG (30 μM) as the control. (e) Photothermal imaging of the BDP2-NPs at different irradiation times. (f) Photoacoustic signals at 820 nm for different concentrations of the BDP2-NPs; data are presented as mean ± SD (n = 3). (g) Photoacoustic signals showing a proportional relationship with the BDP2-NP concentration. | ||
The photoacoustic signal intensity displayed a linear correlation with concentration along with a correlation coefficient of 0.9808 (Fig. 3f and g). In addition, neither the BDP1-NPs nor BDP2-NPs were able to generate singlet oxygen, suggesting that this type of dye aggregates to produce a specific photothermal reagent (Fig. S8†). Thus, the remarkable photoacoustic signal of the NPs meets the requirements of in vivo imaging and could be utilized in future in vivo PTT experiments.
Phototoxicity and biocompatibility
Afterward, we examined the photothermal therapy (PTT) efficacy of the BDP2-NPs in living cells. The classical MTT assay was utilized to study the biocompatibility and phototoxicity of the BDP2-NPs in Hepa 1-6 cells and HepG2 cells. The results (Fig. 4a) show that under normal conditions without a laser, the cell viability rate of the BDP2-NPs from 0 to 100 μM was almost always higher than 80%, suggesting their excellent biocompatibility. In contrast, under laser irradiation at lower power (0.1 W cm−2), the rate of cell viability decreased significantly through the photothermal effect. In addition, fluorescence microscopy imaging was performed using Calcein AM/pyridine iodide (PI) dual staining to investigate the effect of the BDP2-NPs on cell survival (Fig. 4b). As the laser (0.2 W cm−2) irradiation time increased, the red fluorescence (PI) gradually intensified, while the green fluorescence (Calcein AM) diminished, indicating the successful occurrence of excellent photothermal therapy (PTT). Flow cytometry analysis demonstrated that even at low laser power, after 8 min of laser (0.1 W cm−2) irradiation, cell apoptosis could be induced by the photothermal effect effectively (Fig. S9†). These experiments certified that the BDP2-NPs possessed sufficient phototoxicity and biocompatibility for follow-up PTT.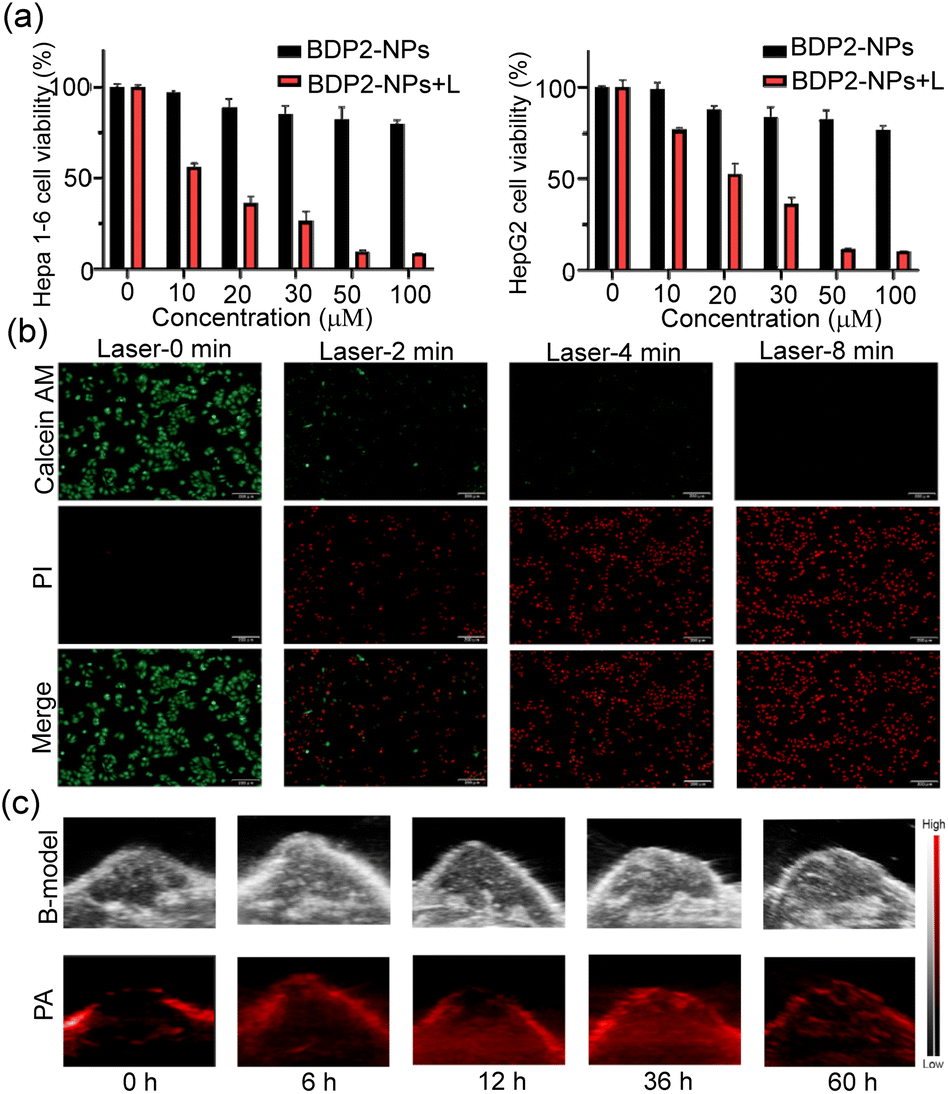 | ||
| Fig. 4 (a) Cell viability of Hepa1-6 and HepG2 cells incubated with the BDP2-NPs at various concentrations in the dark and after laser irradiation (808 nm, 0.1 W cm−2, 10 min); data are presented as mean ± SD (n = 5). (b) Photothermal induced apoptosis imaging by fluorescence microscopy. Scale bar: 200 μm. (c) Representative images of the ultrasound (US) and photoacoustic imaging of the BDP2-NPs in Hepa1-6 tumor-bearing mice. | ||
BDP2-NP in vivo tumor PA imaging
The feasibility of the BDP2-NPs for in vivo PA imaging was then investigated. A weak PA signal was observed before BDP2-NP injection (0 h), attributed to the deoxygenated and oxyhemoglobin.23 First, 100 μL of 100 μM the BDP2-NPs was injected into Hepa 1-6 tumor-bearing C57BL/6 mice through the tail vein, and PA images of tumors were recorded at different times (Fig. 4c and S10†). The photoacoustic signal of the BDP2-NPs in the tumors gradually increased after injection and reached a peak at 36 h, suggesting that the BDP2-NPs have good stability and significant tumor accumulation in blood circulation. These results indicated that BDP2-NPs can be used for photoacoustic imaging of in vivo tumors to precisely guide laser irradiation for photothermal therapy.BDP2-NP in vivo tumor photothermal therapy
Encouraged by the great PA properties and outstanding tumor cell damage capability of the BDP2-NPsin vitro and in vivo, PA image-guided in vivo PTT was studied in Hepa 1-6 tumor-bearing C57BL/6 mice (Fig. 5). The tumor area of the mice was irradiated for 5 min using an 808 nm laser (0.2 W cm−2) at 36 h after the tail vein injection of the BDP2-NPs. As shown in Fig. 5a, with the increase of the irradiation time, the temperature of the tumor area in the BDP2-NP group significantly increased to approximately 50 °C; however, the temperature in the control group which was injected with PBS did not change notably. To investigate the apoptosis of cancer cells induced by PTT in vivo, mouse tumor tissues were harvested for hematoxylin and eosin (H&E) staining and TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining after 24 h of photothermal treatment (Fig. 5c). In the BDP2-NPs + laser group (BDP2-NPs + L), massive destruction and apoptosis of tumor tissues were observed compared to the other groups. After laser irradiation, the tumor volume (Fig. 5b and S11†) and body weight (Fig. S12†) of the mice were recorded for 15 consecutive days, and the tumor volume of the BDP2-NPs + L group was significantly reduced, which has statistical significance compared with the other groups. However, there was no significant change in body weight, indicating that the photothermal effect of the BDP2-NPs could effectively inhibit the growth of tumors in mice without toxicity (Fig. S12†). To further verify the biocompatibility of BDP2, different groups were injected with 100 μL of BDP2-NPs/PBS through the tail vein. After 36 h, the laser group adopted same irradiation conditions as before. Then, 48 h after the end of the laser irradiation, the mice were sacrificed, serum was taken to detect ALT, AST, BUN, and CREA, and organs were harvested for H&E staining analysis (Fig. S13†). The results show that BDP2-NPs itself has no obvious toxicity toward mice, and the photothermal effect of the BDP2-NPs will not have adverse effects on mice. All these results indicate that the BDP2-NPs successfully inhibit tumor proliferation, show excellent PTT effect, and possess significant advantages in tumor localization and treatment at the in vivo level.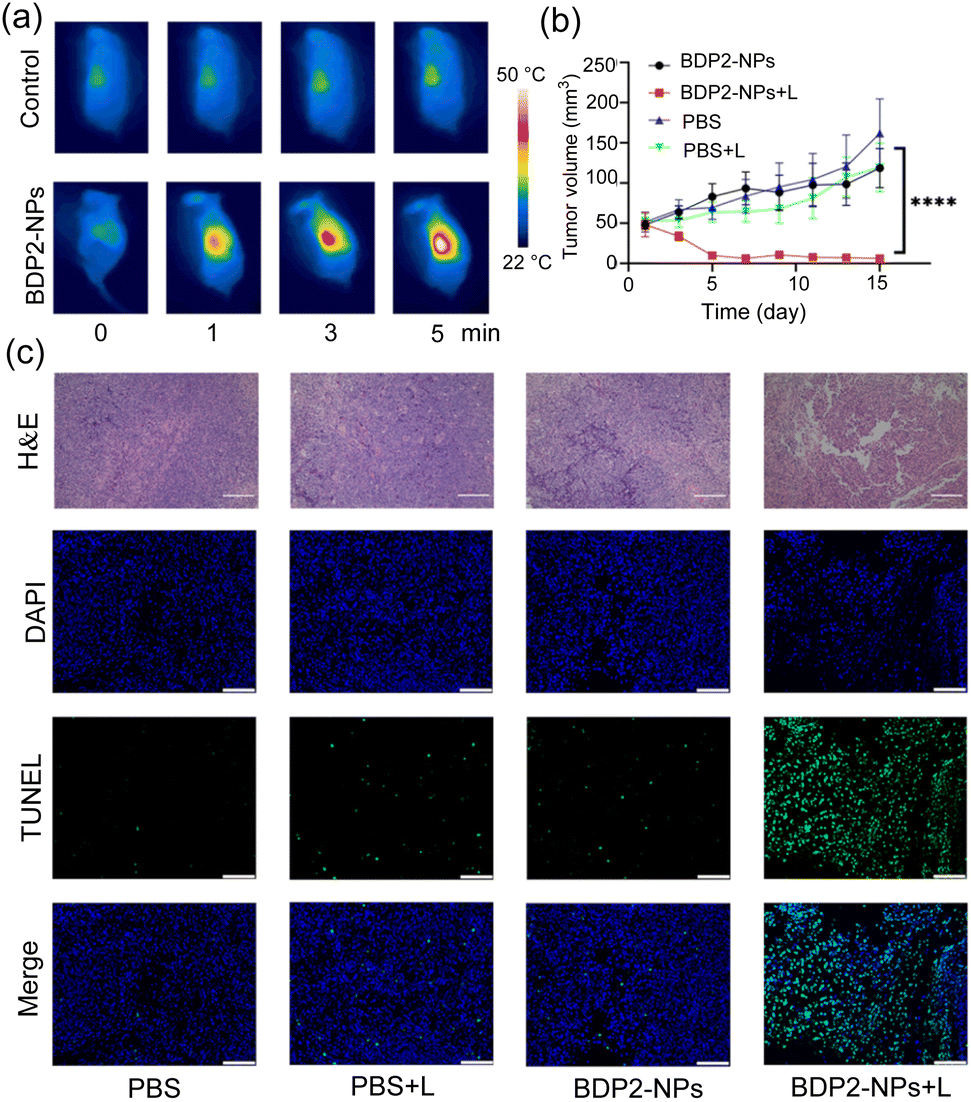 | ||
| Fig. 5 (a) Representative thermographic images. (b) Tumor growth curves of various groups after different treatments; data are presented as mean ± SD (n = 4 mice). ****P < 0.0001. (c) H&E (scale bar: 200 μm) and TUNEL (scale bar: 20 μm) staining images in the tumor area of each group after various treatments. | ||
Conclusions
We have rationally designed a molecular platform based on 3,5-dithienyl-Aza-BODIPY and investigated strategies for accurately constructing aggregates using substituents with different electronic effects. Photophysical property investigations and single-crystal structure analysis have demonstrated that interactions within and between D–A–D structures significantly contribute to the formation of J aggregates. Additionally, the 3,5-position thiophene unit plays a crucial role in this aggregation process. By employing a D–A–D architecture, we have achieved CT-coupled J-aggregation based on a BODIPY chromophore for the first time. The CT-coupled J-aggregates showed excellent biocompatibility, NIR PA imaging capability for tumor diagnosis, and high PTT performance in in vitro and in vivo studies. Our research provides valuable insights into the rational design of a CT-coupled J-aggregation platform, paving the way for exploring organic NIR photothermal agents for biophotonics applications.Ethical statement
All animal experiments were conducted in accordance with the approved agreement of the Institutional Animal Ethics Committee of China Pharmaceutical University (approval code: 2022-12-021). Furthermore, all laboratory animal procedures strictly adhered to the guidelines outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.Data availability
The ESI† includes all experimental details, including TD-DFT calculations, spectroscopic measurements, synthesis and characterization of all products reported in this study. NMR spectra of all products are included as well.Author contributions
Conceptualization: S. W., L. G. and H. L; methodology: S. W. and W. Z.; investigation: C. L. and W. C.; visualization: Z. N. and H. L.; supervision: L. G., J. T. and H. L.; writing—original draft: H. L., J. T. and L. G.; writing—review & editing: H. L., J. T. and Z. G. All authors have been involved in checking the data, and have approval the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21801057, 21871072, and 21775166) for the financial support. Theoretical calculations were carried out at the Computational Centre for Molecular Design of Organosilicon Compounds, Hangzhou Normal University.Notes and references
- (a) Z. Zeng, S. S. Liew, X. Wei and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 26454–26475 CrossRef CAS PubMed; (b) H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- (a) X. Hu, Z. Chen, H. Ao, Q. Fan, Z. Yang and W. Huang, Adv. Opt. Mater., 2022, 10, 2201067 CrossRef CAS; (b) H. Gao, X. Zhi, F. Wu, Y. Zhao, F. Cai, P. Li and Z. Shen, Angew. Chem., Int. Ed., 2023, 62, e202309208 CrossRef CAS PubMed; (c) L. Gai, R. Zhang, X. Shi, Z. Ni, S. Wang, J. L. Zhang, H. Lu and Z. Guo, Chem. Sci., 2023, 14, 1434 RSC.
- S. Xu, H.-W. Liu, S.-Y. Huan, L. Yuan and X.-B. Zhang, Mater. Chem. Front., 2021, 5, 1076–1089 RSC.
- (a) L. Zhao, X. Ren and X. Yan, CCS Chem., 2021, 3, 678–693 CrossRef; (b) N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed; (c) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed; (d) J. H. Kim, T. Schembri, D. Bialas, M. Stolte and F. Würthner, Adv. Mater., 2022, 34, 2104678 CrossRef CAS PubMed.
- (a) Y. Tian, D. Yin and L. Yan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2023, 15, e1831 CAS; (b) J. Heo, D. P. Murale, H. Y. Yoon, V. Arun, S. Choi, E. Kim, J.-S. Lee and S. Kim, Aggregate, 2022, 3, e159 CrossRef CAS.
- C. Spitz, J. Knoester, A. Ouart and S. Daehne, Chem. Phys., 2002, 275, 271–284 CrossRef CAS.
- C. A. Steinbeck, M. Ernst, B. H. Meier and B. F. Chmelka, J. Phys. Chem. C, 2008, 112, 2565–2573 CrossRef CAS.
- N. J. Hestand, C. Zheng, A. R. Penmetcha, B. Cona, J. A. Cody, F. C. Spano and C. J. Collison, J. Phys. Chem. C, 2015, 119, 18964–18974 CrossRef CAS.
- (a) N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS; (b) N. J. Hestand and F. C. Spano, Chem. Phys., 2016, 481, 262–271 CrossRef CAS.
- (a) C.-A. Shen, D. Bialas, M. Hecht, V. Stepanenko, K. Sugiyasu and F. Würthner, Angew. Chem., Int. Ed., 2021, 60, 11949–11958 CrossRef CAS PubMed; (b) A. Kaczmarek-Kedziera and D. Kędziera, Theor. Chem. Acc., 2016, 135, 214 Search PubMed; (c) K. Ilina, W. M. MacCuaig, M. Laramie, J. N. Jeouty, L. R. McNally and M. Henary, Bioconjugate Chem., 2020, 31, 194–213 CrossRef CAS PubMed.
- (a) H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC; (b) N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS; (c) W. Sheng, F. Lv, B. Tang, E. Hao and L. Jiao, Chin. Chem. Lett., 2019, 30, 1825–1833 CrossRef CAS; (d) Z. Shi, X. Han, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020, 49, 7533–7567 RSC.
- Z. Chen and Z. Chen, Org. Chem. Front., 2023, 10, 2581–2602 RSC.
- L. Jing, Y. Zhang, X. Ren and Z. Chen, Angew. Chem., Int. Ed. Engl., 2021, 38, 10–19 CAS.
- (a) M. H. Y. Cheng, K. M. Harmatys, D. M. Charron, J. Chen and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 13394–13399 CrossRef CAS; (b) B. Lei, H. Pan, Y. Zhang, X.-K. Ren and Z. Chen, Org. Biomol. Chem., 2021, 19, 6108–6114 RSC; (c) D. Tian, H. Pan, Y. Zhang, X.-K. Ren and Z. Chen, Tetrahedron Lett., 2021, 76, 153216 CrossRef CAS.
- (a) Y. Tian, D. Yin, Q. Cheng, H. Dang, C. Teng and L. Yan, J. Mater. Chem. B, 2022, 10, 1650–1662 RSC; (b) X. Guo, J. Yang, M. Li, F. Zhang, W. Bu, H. Li, Q. Wu, D. Yin, L. Jiao and E. Hao, Angew. Chem., Int. Ed., 2022, 61, e202211081 CrossRef CAS PubMed; (c) W. Sheng, Z. Wang, E. Hao and L. Jiao, Chin. Chem. Lett., 2021, 32, 1249–1252 CrossRef CAS.
- C. Gustafsson, H. Shirani, P. Leira, D. R. Rehn, M. Linares, K. P. R. Nilsson, P. Norman and M. Lindgren, ChemPhysChem, 2021, 22, 323–335 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- (a) X. Zhang, H. Yu and Y. Xiao, J. Org. Chem., 2012, 77, 669–673 CrossRef CAS ; CCDC no. 877767–877768. Reproduced with permission.; (b) R. Gresser, H. Hartmann, M. Wrackmeyer, K. Leo and M. Riede, Tetrahedron, 2011, 67, 7148–7155 CrossRef CAS; (c) A. E. Pogonin, A. Y. Shagurin, M. A. Savenkova, F. Y. Telegin, Y. S. Marfin and A. S. Vashurin, Molecules, 2020, 25, 5361 CrossRef CAS PubMed.
- (a) Y. Ge and D. F. O'Shea, Chem. Soc. Rev., 2016, 45, 3846–3864 RSC; (b) G. Kubheka, J. Mack, T. Nyokong and Z. Shen, Molecules, 2020, 25, 3689 CrossRef CAS PubMed; (c) Y. Wang, D. Zhang, K. Xiong, R. Shang and X.-D. Jiang, Chin. Chem. Lett., 2022, 33, 115–122 CrossRef CAS.
- (a) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS; (b) Z. Chen, Y. Liu, W. Wagner, V. Stepanenko, X. Ren, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 5729–5733 CrossRef CAS; (c) X. Wang, Z. Jiang, Z. Liang, T. Wang, Y. Chen and Z. Liu, Sci. Adv., 2022, 8, eadd5660 CrossRef CAS PubMed.
- (a) E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed; (b) J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS PubMed.
- (a) X. Song, L. Feng, C. Liang, K. Yang and Z. Liu, Nano Lett., 2016, 16, 6145–6153 CrossRef CAS PubMed; (b) J. Jo, C. H. Lee, R. Kopelman and X. Wang, Nat. Commun., 2017, 8, 471 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06976a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |