
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating electron transfer and orbital interaction within metalloporphyrin-MOFs for highly sensitive NO2 sensing†
Er-Xia
Chen
ab,
Liang
He
 a,
Mei
Qiu
*c,
Yongfan
Zhang
d,
Yayong
Sun
a,
Wen-Hua
Li
a,
Jian-Ze
Xiao
a,
Jie
Chen
a,
Gang
Xu
*abe and
Qipu
Lin
*aef
a,
Mei
Qiu
*c,
Yongfan
Zhang
d,
Yayong
Sun
a,
Wen-Hua
Li
a,
Jian-Ze
Xiao
a,
Jie
Chen
a,
Gang
Xu
*abe and
Qipu
Lin
*aef
aState Key Laboratory of Structural Chemistry, Fujian Provincial Key Laboratory of Materials and Techniques toward Hydrogen Energy, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: gxu@fjirsm.ac.cn; linqipu@fjirsm.ac.cn
bFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, Fujian 350108, China
cCollege of Chemistry and Materials, Jiangxi Agricultural University, Nanchang, Jiangxi 330045, China. E-mail: qium@jxau.edu.cn
dCollege of Chemistry, Fuzhou University, Fuzhou, Fujian 350116, China
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
fState Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou, Fujian 350116, China
First published on 2nd April 2024
Abstract
The understanding of electron transfer pathways and orbital interactions between analytes and adsorption sites in gas-sensitive studies, especially at the atomic level, is currently limited. Herein, we have designed eight isoreticular catechol–metalloporphyrin scaffolds, FeTCP–M and InTCP–M (TCP = 5,10,15,20-tetrakis-catechol-porphyrin, M = Fe, Co, Ni and Zn) with adjustable charge transfer schemes in the coordination microenvironment and precise tuning of orbital interactions between analytes and adsorption sites, which can be used as models for exploring the influence of these factors on gas sensing. Our experimental findings indicate that the sensitivity and selectivity can be modulated using the type of metals in the metal–catechol chains (which regulate the electron transfer routes) and the metalloporphyrin rings (which fine-tune the orbital interactions between analytes and adsorption sites). Among the isostructures, InTCP–Co demonstrates the highest response and selectivity to NO2 under visible light irradiation, which could be attributed to the more favorable transfer pathway of charge carriers in the coordination microenvironment under visible light illumination, as well as the better electron spin state compatibility, higher orbital overlap and orbital symmetry matching between the N-2s2pz hybrid orbital of NO2 and the Co-3dz2 orbital of InTCP–Co.
Introduction
Understanding the atomic-scale sensing nature is essential for enhancing performance and gaining mechanistic insight into responses of analytes. Specifically, charge transfer during the gas sensing process and the interactions between analytes and adsorption sites at the atomic-level are fundamental factors in causing signal changes in gas induction.1,2 Although a variety of gas sensitive materials, such as semiconducting metal oxides,3–5 metal dichalcogenides,6,7 graphene-based hybrids,8,9 and conducting polymers,10,11 have attracted significant attention in the past few decades, their ambiguous structures or adsorption sites have limited the comprehension of the reaction processes between gas-sensitive materials and analytes at the atomic level, including electron transfer routes, electron spin states and orbital symmetry/overlap between adsorbates and adsorption sites, all of which have a microregulatory on gas-sensitive properties.As one of the emerging sensing materials, metal–organic frameworks (MOFs) might be promising platforms to address the issues because of their inherent features, such as well-defined and tailorable structures, accessible adsorption sites, high porosity and substantial surface areas.12–17 In line with our discovery in 2014 that ZIF-67 and [Co(im)2]n have chemiresistive behavior in detecting formaldehyde and trimethylamine,18,19 several chemiresistive sensors based on pure-MOFs have been developed.20–25 For instance, Kalidindi et al. utilized an amine-modified Zr-based MOF, NH2–UiO-66, to detect acidic gases (i.e., SO2, NO2 and CO2) at the parts-per-million (ppm) level.26 Dincă et al. investigated a two-dimensional (2D) MOF, Cu3(HITP)2 (HITP = 2,3,6,7,10,11-hexaiminotriphenylene), with high electrical conductivity and a detection limit of less than ppm (sub-ppm) for NH3 vapor.27 Zuo et al. synthesized a series of mixed-linker MOFs, which demonstrated adjustable sensing performance for volatile organic compounds (VOCs) by varying the ratio of mixed-linkers.28 In our research, we fabricated a chemiresistive heterostructured MOF-based sensor by integrating two types of MOF layer, achieving high selectivity and response to benzene at room temperature.29 Although MOFs have shown potential as sensitive materials for chemiresistive sensors, their interactions with gas (vapor) and the effect of charge transfer routes within MOFs during gas-sensing, particularly at the atomic-level, remain unclear. Consequently, it is still required to explore the influence of these factors on gas-sensitive properties by designing suitable MOFs.
The indirect (and direct) band gaps of typical semiconducting hematite (α-Fe2O3) and cubic indiumtrioxide (c-In2O3) are ∼2.1 eV (∼3.3 eV) and ∼2.8 eV (>3.5 eV), respectively (Fig. 1a–f).30–33 In the structures of α-Fe2O3 and c-In2O3, both Fe and In adopt six-coordination patterns to form 1D Fe3+–oxo or In3+–oxo chains. These chains are fused to produce 3D structures with the help of μ4-O moieties (Fig. 1a, b and d–f).34,35 Meanwhile, metalloporphyrins have gained much attention in the fields of molecular recognition, gas sensing and catalysis due to their open and adjustable metal sites in the porphyrin plane and excellent light absorption.36–39 Inspired by the difference in band gaps (especially the indirect band gaps) and structural features of α-Fe2O3/c-In2O3 and the characteristics of metalloporphyrins, assembling In3+/Fe3+–oxo chains and suitable metalloporphyrin ligands into MOFs might offer a platform for exploring the effect of electron transfer paths within MOFs and orbital interactions between analytes and adsorption sites during the gas response.
 | ||
| Fig. 1 (a) The structure of α-Fe2O3; (b) the Fe–O chain in α-Fe2O3; (c) the schematic illustration of the indirect band gaps of α-Fe2O3 and c-In2O3; (d) the coordination configuration of Fe in α-Fe2O3 and In in c-In2O3; (e) the In–O chain in c-In2O3; (f) the structure of c-In2O3; (g) the 3D structure of FeTCP–M; (h) In/Fe–catechol coordination chains; (i) catechol–metalloporphyrin ligand; (j) the 3D structure of InTCP–M; all H atoms are omitted for clarity, M = Fe, Co, Ni and Zn. | ||
Under the guidance of theoretical simulation and calculation, we designed a series of isoreticular metalloporphyrinic metal–catechol (MPMC) scaffolds, FeTCP–M and InTCP–M, which feature In3+/Fe3+–oxo chains connected by catechol–metalloporphyrin arrays (Fig. 1g–j, where TCP = 5,10,15,20-tetrakis-catechol-porphyrin and M = Fe, Co, Ni, and Zn), to investigate the influence of electron transfer routes within MOFs and orbital interactions between analytes and adsorption sites on the overall chemiresistive sensing. Remarkably, we discovered that the sensing capability for NO2 can be efficiently tuned by regulating the metal types in both the metal–oxo chains and the porphyrin planes. Among these isomorphic scaffolds, InTCP–Co exhibited the highest response and selectivity toward NO2. This was attributed to several factors, including the impact of visible light absorption, the preferable electron transfer routes, a higher orbital overlap and suitable matching of the orbital symmetry and electron spin state between the N-2s2pz hybrid orbital of NO2 and the Co-3dz2 orbital of InTCP–Co.
Results and discussion
FeTCP–M and InTCP–M (M = Fe, Co, Ni, and Zn) were prepared using the solvothermal method, as detailed in the ESI (Sections S2 and S3†). Refinement of experimentally obtained high-resolution PXRD patterns (Fig. S1–S4†) established their isostructures.40 These structures consist of infinite In3+/Fe3+–oxo chains that are connected by eclipsed catechol–metalloporphyrin arrays (Fig. 1g–j). By manipulating the metal nodes in the metal–catechol rods, the energy band structures can be varied, which potentially influence electron transfer patterns and subsequently modulate the properties.23,27,40–44 Removal of the coordinated molecules in the axial direction leads to a planar tetragonal coordination geometry for the metals (Fe, Co, Ni and Zn) centered in the porphyrin rings (Fig. 1i). The different metal types in the porphyrin rings have distinct electron configurations, which could regulate the interactions between (In/Fe)TCP–M (M = Fe, Co, Ni and Zn) and analytes, thereby tuning the properties.45–47 Scanning electron microscopy (SEM) revealed a nanoplate-like grain morphology for (In/Fe)TCP–M (Fig. S5 and S6†). The microporous nature of (In/Fe)TCP–M was confirmed by N2 adsorption–desorption isotherms at 77 K (Fig. S7–S10†). The Brunauer–Emmett–Teller (BET) and Langmuir surface areas and pore size distribution are presented in Table S1.† As shown in Fig. S11 and S12,† the UV/vis diffuse reflectance spectra (DRS) of (In/Fe)TCP–M demonstrated a broad range of light absorption from 250 to 800 nm, indicating that they can easily generate photogenerated charge carriers under visible-light illumination.The well-defined chemical structure, regulated electronic structure, high porosity and gas uptake, and excellent visible light absorption make FeTCP–M and InTCP–M (M = Fe, Co, Ni, and Zn) suitable models for exploring the effect of charge transfer routes within MOFs and orbital interactions between adsorbates and adsorption sites on the chemiresistive sensing performance under visible light irradiation. Experimentally, FeTCP–M and InTCP–M (M = Fe, Co, Ni, and Zn) powders were individually coated on interdigital electrodes and used as sensitive devices (Fig. S13a†). The gas-sensing measurements were conducted using a custom-built instrument with a dynamic gas distribution system (Fig. S13b†), where the current of the devices in various gaseous analytes was monitored using a source meter (the details are in the Methods).
The real-time gas-sensing curves of FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn) showed different response values to 10 ppm NO2 at room temperature under visible-light irradiation (Fig. 2a). FeTCP–M (M = Fe, Co, Ni and Zn) demonstrated response values of 179%, 156%, 163% and 160%, respectively (Fig. 2a), indicating that the metal types in the porphyrin rings exerted no significant influence on the response values for FeTCP–M. In contrast, the response values of InTCP–M were substantially affected by the metal types in the porphyrin rings, resulting in response values of 426%, 937%, 187% and 172% for M = Fe, Co, Ni and Zn, respectively (Fig. 2a). Notably, InTCP–M demonstrated a higher response value to 10 ppm NO2 compared to FeTCP–M with identical metal types in the porphyrin rings, suggesting their different sensing mechanisms. Among FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn), InTCP–Co displayed the highest response value. Recycling experiments of the real-time sensing curves for 10 ppm NO2 were conducted to evaluate the stability and repeatability of (In/Fe)TCP–M. Their initial response values were maintained in six successive assays, demonstrating good stability and repeatability (Fig. 2b and S14–S20†).
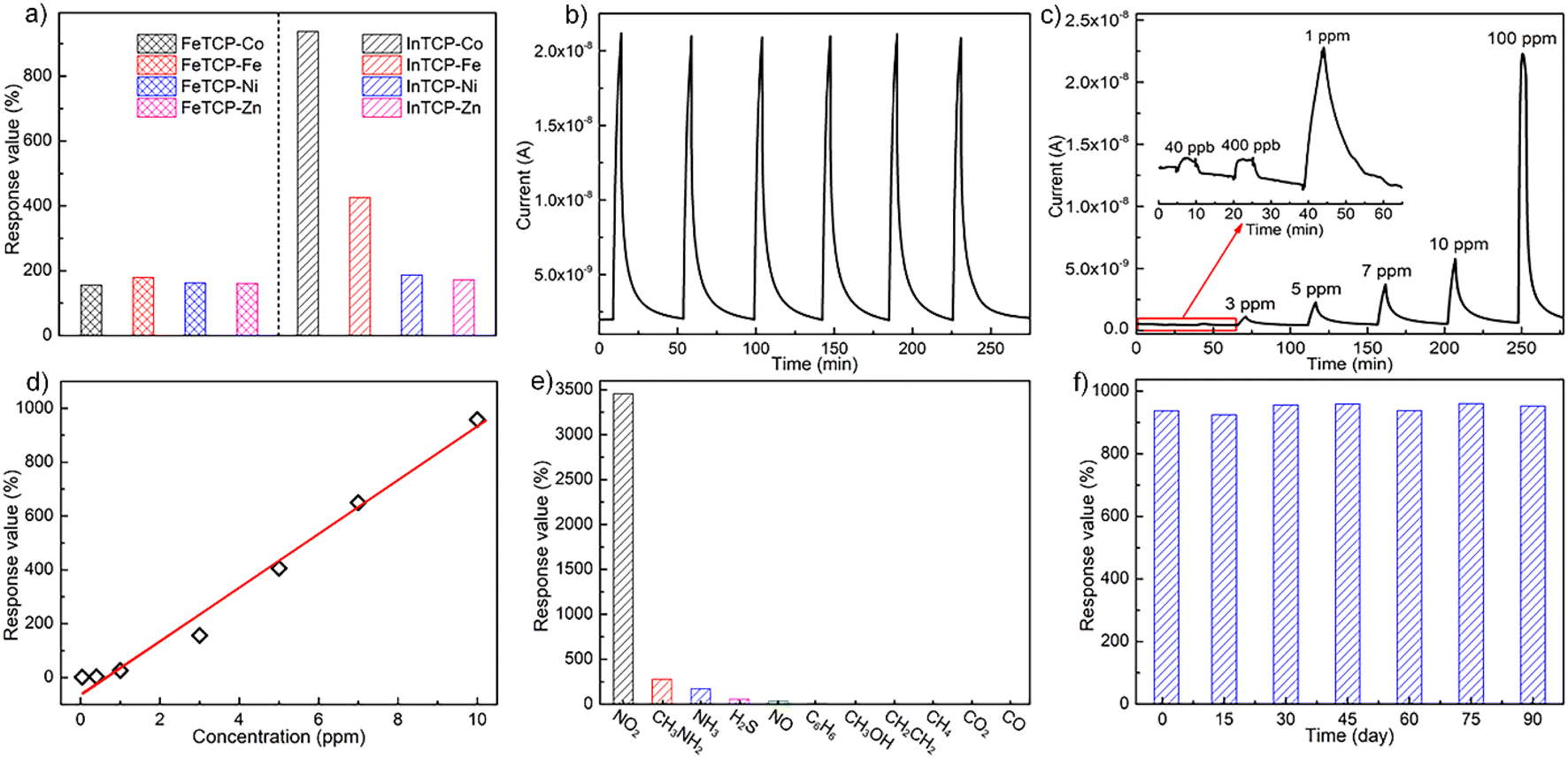 | ||
| Fig. 2 (a) The response values of FeTCP–M and InTCP–M sensors (M = Fe, Co, Ni and Zn) to 10 ppm NO2; (b) the dynamic response–recovery curve of InTCP–Co to 10 ppm NO2 with six consecutive cycles; (c) the dynamic response–recovery curve of InTCP–Co to NO2 at different concentrations; (d) the plot of response value-concentration of InTCP–Co to NO2; (e) the response values of InTCP–Co to different interfering gases; (f) the stability of InTCP–Co to 10 ppm NO2 in 90 days. | ||
NO2 concentration (40 ppb to 100 ppm) dependent responses of InTCP–Co are presented in Fig. 2c, where InTCP–Co exhibits excellent response/recovery performance over a wide range of NO2 concentrations. The corresponding plot of response vs. concentration of InTCP–Co towards NO2 displays a good linear relationship (R2 = 0.982, Fig. 2d) within the range of 40 ppb to 10 ppm. Fig. S21† shows that InTCP–Co has a response time of 3.8 min and a recovery time of 13.1 min when exposed to 10 ppm NO2. The relatively long recovery time may be caused by the relatively strong affinity between NO2 and InTCP–Co. To assess the anti-interference performance of the InTCP–Co sensor in practical applications, the selectivity of InTCP–Co was further evaluated by examining the response of the sensor to various interfering gases at 100 ppm, including ammonia (NH3), methylamine (CH3NH2), carbon dioxide (CO2), carbon monoxide (CO), nitric oxide (NO), hydrogen sulfide (H2S), methane (CH4), ethylene (C2H4), methanol (CH3OH), and benzene (C6H6). As shown in Fig. 2e, compared with NO2, the response values of the interfering gases were negligible. Further experiments showed that InTCP–Co could still distinguish NO2 when NO2 was mixed with other gases (Fig. S22†), confirming that InTCP–Co possesses excellent selectivity.
Additional reference experiments were conducted at different temperatures without visible light irradiation. The response of InTCP–Co to 10 ppm NO2 was notably ineffective (Fig. S23–S28†), suggesting that visible photo-induced charge carriers can improve gas sensing properties, whereas temperature has a negligible effect on gas sensitivity. When the catechol–metalloporphyrin ligand (3,4-TDHPP–Co) was used as the sensing material, the response value to 10 ppm NO2 was only 53% under the same operating conditions (Fig. S29 and S30†). This finding indicates that assembling 3,4-TDHPP–Co into the porous MOFs can effectively enhance gas sensitivity. Moreover, after three months, the original response value of InTCP–Co towards 10 ppm NO2 was retained as demonstrated in Fig. 2f. The PXRD patterns in Fig. S31† also illustrate the structural integrity of InTCP–Co and its excellent long-term stability as a sensor. These features indicate that InTCP–Co is a good sensing material for NO2 at room temperature and under visible light irradiation (Table S2†).
To unveil the impact of substitutable metal species in the porphyrin rings and 1D coordination nodes on gas sensing properties, we resorted to DFT calculations. The optimal configurations for NO2 adsorption on FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn) are presented in Fig. S39 and S40.† NO2 molecules are adsorbed on the metals in the porphyrin rings in a V-shape configuration. It can be seen from the charge difference density maps (Fig. 3a and S41–S44†) that the charge density distributions are different between NO2 and the adsorption sites of FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn), indicating distinct interactions between NO2 and (In/Fe)TCP–M (M = Fe, Co, Ni and Zn). We further confirmed the different interplay between NO2 and (In/Fe)TCP–M (M = Fe, Co, Ni and Zn) by N 1s XPS spectroscopy (Fig. 3b and S32–S38†). After NO2 capture, two new peaks appear at approximately 405.2 and 406.5 eV, respectively, when compared to (In/Fe)TCP–M (M = Fe, Co and Ni) prior to the NO2 response (Fig. 3b, S32, S33 and S35–S37†). The peak at a lower bonding energy (∼405.2 eV) was assigned to NO2− species and the peak at a higher bonding energy (∼406.5 eV) was ascribed to NO3− species or NO2 (N2O4) chemisorbed on (In/Fe)TCP–M (M = Fe, Co and Ni) in the molecular form,48–50 which is consistent with in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFT, Fig. 3c). The emerging bands at 1207 and 1480 cm−1 belong to NO2− species.51–53 The bands at 1218, 1248 and 1306 cm−1 are assigned to NO3− species.51,54 However, no obvious changes were observed in the N 1s XPS spectra of (In/Fe)TCP–Zn before and after NO2 sorption (Fig. S34 and S38†), suggesting that the interactions between NO2 and (In/Fe)TCP–Zn were weaker than those between NO2 and (In/Fe)TCP–M (M = Fe, Co and Ni).
 | ||
| Fig. 3 (a) The charge difference density map of NO2 affined on InTCP–Co (the other parts of the structure of InTCP–Co have been omitted for clarity); (b) the N 1s XPS spectra of InTCP–Co before and after the NO2 response; (c) the in situ DRIFT spectra of InTCP–Co in a 100 ppm NO2 atmosphere. | ||
To further explore the different interactions between NO2 and (In/Fe)TCP–M (M = Fe, Co, Ni and Zn), we analyzed the density of states (DOS) of the MPMC scaffolds before and after NO2 capture. As shown in Fig. 4a, there are no significant changes in the total density of states (TDOS) near the Fermi level of FeTCP–M (M = Fe, Co, Ni and Zn) before and after NO2 sorption, indicating the weak orbital interactions between NO2 and the main orbital compositions near the Fermi level. However, the TDOS of InTCP–M (M = Fe, Co, Ni and Zn) show marked differences before and after NO2 sorption when changing the metal types in porphyrin rings (Fig. 4b). The TDOS of InTCP–Zn remain essentially unchanged, indicating weak orbital interaction between NO2 and the major orbital compositions near the Fermi level of InTCP–Zn. This might be mainly attributed to the full occupation of Zn-3d orbitals. The changes in the TDOS are substantial for InTCP–M (M = Fe, Co, Ni) before and after NO2 capture, especially around the Fermi level, indicating that the orbital interactions between NO2 and the dominant orbital components near the Fermi level of InTCP–M (M = Fe, Co, Ni) are stronger.
 | ||
| Fig. 4 The TDOS of (a) FeTCP–M and (b) InTCP–M before and after the NO2 response (M = Fe, Co, Ni and Zn); (c) the TDOS and PDOS of InTCP–Co before and after the NO2 response; (d) the PDOS of NO2 and Co-4s, Co-3p, Co-3d orbitals of InTCP–Co after the NO2 response; (e) the PDOS of Co-4s, Co-3p and Co-3d orbitals of InTCP–Co before the NO2 response; (f) the PDOS of electron spin states in the Co-3dz2 orbital of InTCP–Co. | ||
The projected density of states (PDOS) of InTCP–M after NO2 adsorption demonstrates that the orbital interactions between NO2 and InTCP–M mainly derive from the sp2 hybrid orbital of NO2 and the M-3dz2 orbitals of InTCP–M (M = Fe, Co and Ni, Fig. 4c, d and S45–S48†). Further analysis shows that the M-3dz2 orbitals interacting with NO2 stem from the beta states (M = Fe, Co and Ni, Fig. 4e, f, S45 and S46†). The crystal orbital overlap population (COOP) is widely used to understand the bonding and antibonding contributions of interacting components and its integrated value (ICOOP) could reflect the strength of the interaction.55,56 It can be seen from Table S3† that the interaction between beta states of electron spin in M-3dz2 and NO2 follows the order: InTCP–Co > InTCP–Fe > InTCP–Ni > InTCP–Zn, in line with the sensitive response to NO2 under visible light irradiation. Compared with visible light illumination, the temperature does not significantly affect the sensitive response to NO2 for InTCP–Co. The reason may be that visible light can conduce to produce photogenerated electrons, which can then jump into the unoccupied Co-3dz2 orbitals, thereby enhancing the orbital interaction between NO2 and InTCP–Co to improve the sensitive response to NO2. But photoelectrons could not be produced under the dark conditions or by heating. Therefore, the visible-light-driven sensitive response to NO2 is higher than that in the dark or at different temperatures. Although the PDOS of FeTCP–M after NO2 adsorption also manifests that the orbital interactions between NO2 and FeTCP–M originate from the beta state of electron spin in the M-3dz2 orbitals (M = Fe, Co, and Ni, Fig. S51–S54†), the ICOOP values of the beta state of electron spin in M-3dz2 orbitals and NO2 are different (Table S3†). It can be seen from the PDOS of FeTCP–M (M = Fe, Co, Ni and Zn, Fig. S49 and S50†) that the photoelectrons primarily transfer to the Fe–oxo chains. Hence, the sensitive response to NO2 with the same concentrations does not exhibit a notable difference under the visible-light excitation for FeTCP–M (M = Fe, Co, Ni and Zn). This indicates that the degree of orbital overlap, the electron spin state in the orbital, the electron transfer routes in the microenvironment within a MOF and visible light irradiation have a significant influence on the sensitive response to NO2.
To reveal the selectivity of InTCP–Co to various analytes, we conducted calculations to determine the optimal adsorption configurations for ten typical interfering gases on InTCP–Co (Fig. S55 and S56†). It can be seen that, C6H6, CH4 and CO2 were not adsorbed on InTCP–Co in the optimal adsorption configuration, suggesting that the interactions between InTCP–Co and C6H6/CH4/CO2 are weak individually. It was further confirmed by the adsorption energies (Eads) and the DOS of InTCP–Co after C6H6, CH4 and CO2 interaction (Table S4 and Fig. S57–S61†). The weak affinities between InTCP–Co and C6H6/CH4/CO2 cannot cause changes in the structure or the local electron/hole concentrations in InTCP–Co. Therefore, InTCP–Co shows negligible response values for C6H6/CH4/CO2. Except for C6H6, CH4 and CO2, the other seven typical interfering gases could be adsorbed on InTCP–Co. However, their adsorption energies are diverse, indicating that the interactions between these analytes and InTCP–Co are different. Among all of the measured analytes, InTCP–Co possesses the most negative energy (−3.08 eV) for the NO2 response, indicating that the interaction between NO2 and InTCP–Co is stronger than that of other analytes measured. The DOS of InTCP–Co after adsorbing diverse analytes (NO2, CH3NH2, NH3, H2S, NO, CH3OH, CH2CH2 and CO) have changed differently (Fig. 4c, d and S62–S68†), which further indicates the distinctive orbital interactions between diverse analytes and InTCP–Co.
To investigate the inherent essence of the different orbital interactions between the adsorbed analytes and Co in the porphyrin ring of InTCP–Co, further analyses have been performed using the PDOS associated with the crystal orbital Hamilton populations (COHP)57–59 and natural bond orbital (NBO).60,61 It is well known that there are unpaired electrons in free NO2 and NO molecules. The electrons in CH3NH2, NH3, CO, H2S, CH3OH, and C2H4 in their free states are all paired. As shown in Fig. 5a, the redistributed PDOS of N in NO2 and Co in InTCP–Co are overlapped and changed upon trapping NO2 into InTCP–Co, demonstrating that orbital hybridization interaction occurs between Co in InTCP–Co and N in NO2. To further disclose the origin of the orbital hybridization between N in NO2 and Co in InTCP–Co, we examined the COHP curve of Co–N. The COHP curve clearly indicates that the orbital hybridization is driven by the Co-3dz2 and N-2s2pz hybrid orbitals (Fig. 5a) which display a bonding character, indicating the presence of a relatively strong orbital interaction between N in NO2 and Co in InTCP–Co. Additionally, the NBO analysis is employed to further visualize the orbital hybridization interaction. As shown in Fig. 5b, the symmetrical N-2s2pz hybrid orbital occupied by one electron in NO2 hybridizes with the Co-3dz2 orbital filled with one electron (photo-induced electron) to form a relatively stable Co–N bond, generating one σ bonding orbital occupied by paired electrons and one empty antibonding orbital (σ*). The formation of this relatively stable Co–N bond leads to changes in the coordination configuration of Co and the Co-3d orbital energy level splitting (Fig. S69†), which may cause tiny alterations in the structure or the local electron/hole concentrations in InTCP–Co. Therefore, InTCP–Co exhibits a response to NO2.
 | ||
| Fig. 5 (a) The PDOS patterns of N in NO2 in the free state, Co in InTCP–Co and N in NO2 after the NO2 response, and the COHP curve of the Co–N bond between Co in InTCP–Co and N in NO2 after the NO2 response; (b) the dominant interaction and the energy levels of the scalar relativistic Kohn–Sham molecular orbitals of InTCP–Co after NO2 adsorption with correlation with the orbitals from Co-3dz2 and NO2; (c) the PDOS patterns of N in NH3 in the free state, Co in InTCP–Co and N in NH3 after the NH3 response, and the COHP curve of the Co–N bond between Co in InTCP–Co and N in NH3 after the NH3 response; (d) the major interaction and the energy levels of the scalar relativistic Kohn–Sham molecular orbitals of InTCP–Co after the NH3 response with correlation with the orbitals from Co-3dz2 and NH3; (e) the PDOS patterns of N in NO in the free state, Co in InTCP–Co and N in NO after the NO response, and the COHP curve of the Co–N bond between Co in InTCP–Co and N in NO after the NO response; (f) the main interaction and energy levels of the scalar relativistic Kohn–Sham molecular orbitals of InTCP–Co after the NO response with correlation with the orbitals from Co-3dz2 and NO; the electron in Co-3dz2 orbital derives from photogenerated electrons in (b), (d) and (f). | ||
As a representative analyte with paired electrons in its free states, NH3 was chosen as a comparison to probe the orbital hybridization interaction with InTCP–Co. As shown in Fig. 5c, although hybridization between Co-3dz2 and N-2s2pz hybrid orbitals is observed, the interaction is weak due to the antibonding characteristics of the majority of the 3dz2–2s2pz hybrid in the COHP curve. In addition, the Fermi level falls in the antibonding region of the 3dz2–2s2pz hybrid orbitals according to the COHP analysis. This is mainly caused by the fact that the N-2s2pz hybrid orbital occupied by two electrons (lone pair electrons) in NH3 hybridizes with the Co-3dz2 orbital with one electron (light-generated electron) to form one σ bonding orbital occupied by paired electrons and one σ antibonding orbital (σ*) occupied by one electron (Fig. 5d). In comparison to NO2, the weak interaction between NH3 and InTCP–Co is likely to cause minute changes in the structure or the local electron/hole concentrations in InTCP–Co. Consequently, InTCP–Co exhibits low response values for the measured analytes with all paired electrons in their molecules in their free states.
Despite the presence of an unpaired electron in NO in its free state, it occupies the πpx antibonding orbital 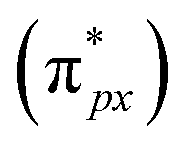 . Notoriously, the
. Notoriously, the  orbital is antisymmetric, which mismatches with the symmetrical Co-3dz2 orbital in InTCP–Co (Fig. 5e and f). As a result, the orbital hybridization interaction between the symmetrical Co-3dz2 orbital and
orbital is antisymmetric, which mismatches with the symmetrical Co-3dz2 orbital in InTCP–Co (Fig. 5e and f). As a result, the orbital hybridization interaction between the symmetrical Co-3dz2 orbital and  orbital is extremely weak. Although the symmetry of the
orbital is extremely weak. Although the symmetry of the 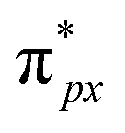 orbital matches the degenerate Co-3dxz and Co-3dyz orbitals, the orbital hybridization interaction between
orbital matches the degenerate Co-3dxz and Co-3dyz orbitals, the orbital hybridization interaction between  and Co-3dxz/Co-3dyz is also weak. This is due to the fact that the Co-3dxz and Co-3dyz orbitals each are occupied by two electrons, which leads to an unstable Co–N bond accompanied by a
and Co-3dxz/Co-3dyz is also weak. This is due to the fact that the Co-3dxz and Co-3dyz orbitals each are occupied by two electrons, which leads to an unstable Co–N bond accompanied by a  orbital occupied by one electron (Fig. 5e and f). Similarly, the Co-3dz2 orbital occupied by one electron (photogenerated electron) also forms an unstable Co–N bond with the σ bonding orbital (σpz–pz) or σ antibonding orbital
orbital occupied by one electron (Fig. 5e and f). Similarly, the Co-3dz2 orbital occupied by one electron (photogenerated electron) also forms an unstable Co–N bond with the σ bonding orbital (σpz–pz) or σ antibonding orbital 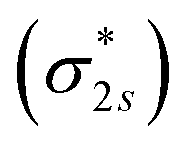 . In brief, the orbital interaction between NO and InTCP–Co is weak. Compared with NO2, NO does not cause significant changes in the structure or local electron/hole concentrations, resulting in a lower response value to NO than that to NO2 in InTCP–Co.
. In brief, the orbital interaction between NO and InTCP–Co is weak. Compared with NO2, NO does not cause significant changes in the structure or local electron/hole concentrations, resulting in a lower response value to NO than that to NO2 in InTCP–Co.
Based on the experimental results and theoretical calculations, a potential mechanism is proposed to explain the exceptional selective sensitivity of InTCP–Co to NO2 under visible light irradiation. Specifically, when InTCP–Co sensors were exposed to the NO2 atmosphere, NO2 was adsorbed on the Co site of InTCP–Co in a V-shaped fashion. The N-2s2pz hybrid orbital (filled by one unpaired electron) in NO2 hybridizes with the Co-3dz2 orbital (occupied by one photogenerated electron with the beta spin state) to form a relatively stable Co–N bond, leading to a change in the coordination configuration of Co and the splitting of the Co-3d orbital energy level. These changes might cause alterations in the structure or local electron/hole concentrations in InTCP–Co, resulting in a conductivity variation and generating a sensing response to NO2. When NO2 is removed from the test environment, the amount of NO2 attached to InTCP–Co gradually decreases, which could revert the coordination configuration of Co and the splitting of the Co-3d orbital energy levels to their original states, then causing the local electron/hole concentrations in InTCP–Co to return to their original states. Thereby, the current recovers. With the assistance of visible light irradiation, the photogenerated electrons can transition to the unoccupied Co-3dz2 orbitals, enhancing the orbital interaction between NO2 and InTCP–Co. Therefore, compared to the conditions without light, the response value to NO2 could be improved under visible light illumination. Among the examined combinations of FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn), InTCP–Co exhibits a higher response value and excellent selectivity to NO2, which is primarily attributed to superior electron spin state matching, increased orbital overlap, improved orbital symmetry compatibility between the N-2s2pz hybrid orbital of NO2 and Co-3dz2 orbital, more favourable electron transfer paths and the promotion of the photoelectrons.
Conclusions
In summary, we have fabricated a series of reversible chemiresistive gas sensors using MOFs featuring metalloporphyrin facets aligned by a periodic array of In/Fe–oxo pillars, FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn). The response of the catechol–metalloporphyrin scaffolds to NO2 has been investigated under visible light excitation, and we found that it could be modulated by varying the metal species individually coordinated in porphyrin centers and with catechol groups. Among them, InTCP–Co reveals the highest response and selectivity toward NO2 under visible light illumination, with an experimental detection limit as low as 40 ppb. Both the experimental results and theoretical calculations reveal that the excellent response performance of InTCP–Co to NO2 could be ascribed to multiple factors, including the better matching of electron spin state and orbital symmetry, the higher orbital overlap between the N-2s2pz hybrid orbital of NO2 and Co-3dz2 orbital, and the more favorable electron transfer pathway in InTCP–Co. This work has provided a new perspective for the design of high-performance chemiresistive gas sensor by regulating the transfer route of charge carriers, electron spin states, orbital overlap and orbital symmetry of analytes and sensitive substances.Methods
Syntheses of FeTCP–M (M = Fe, Co, Ni and Zn)40
FeCl2 (0.075 mmol, 0.0095 g), 3,4-TDHPP–M (0.075 mmol, 0.0597 g, 0.0599 g, 0.0599 g, and 0.0604 g for M = Fe, Co, Ni and Zn, respectively), N,N-dimethylformamide (DMF, 3.0 mL), water (0.5 mL) and methanol (0.5 mL) were mixed in a 23 mL Teflon-lined stainless steel container and then heated to 140 °C for 4 days. After cooling to room temperature, brown crystal samples were obtained after washing with methanol and drying, named FeTCP–M (M = Fe, Co, Ni and Zn), respectively.Syntheses of InTCP–M (M = Fe, Co, Ni and Zn)40
In(NO3)3·4.5H2O (0.075 mmol, 0.0286 g), 3,4-TDHPP–M (0.075 mmol, 0.0597 g, 0.0599 g, 0.0599 g, and 0.0604 g for M = Fe, Co, Ni and Zn, respectively), water (1.0 mL) and DMF (3.0 mL) were mixed in a 23 mL Teflon-lined stainless steel container and then heated to 140 °C for 4 days. After cooling to room temperature, brown crystal samples were obtained after washing with methanol and drying, named InTCP–M (M = Fe, Co, Ni and Zn), respectively.Fabrication of sensor devices
The ground sample was mixed with moderate isopropanol to form paste. Then the paste was coated on the interdigital electrodes of the sensor substrates (Fig. S13a†). Then the coated sensors were placed in a vacuum oven at 60 °C for 12 h to remove isopropanol.Gas sensing test
The gas sensing characterization was carried out on a home-made system (Fig. S13b†) which was reported in the previous work.62 In the gas sensing test, the light source was a 300 W Xe lamp with a 420 nm cutoff filter and the light intensity can be modulated by adjusting the current. The current was 12 mA and the distance between the gas sensor and light source was 8 cm in this work. The current was recorded in real-time using a source meter (Keithley 2602B) and the bias voltage of the device was 5 V during the gas sensing test. The gas (air and the target gas are diluted in the air and NO is diluted in N2 because NO is easily oxidized in the air) flow rate was controlled at 600 mL min−1 which was monitored using mass flow controllers (CS-200C, Beijing Sevenstar Qualiflow Electronic Equipment Manufacturing Co. Ltd, China). The analytical gas was introduced into a quartz tube and the concentration was obtained by controlling the flow ratio of analytical gas and air. The response value (R) of the target gas is defined as:where Ig is the current of the fabricated sensor in the target gas and I0 is the current of the fabricated sensor in the air. The response or recovery time is expressed as the time required to reach 90% of the maximum current value change.

In situ diffuse reflectance infrared Fourier transform (DRIFT) spectrum
A Nicolet 6700 FT-IR spectrometer with an MCT/A detector cooled using liquid nitrogen was employed to record the DRIFTS spectra. The background of DRIFTS spectra was obtained with InTCP–Co in synthetic air (79% nitrogen, 21% oxygen). The time-resolved DRIFTS spectra of NO2 adsorption (100 ppm NO2 in synthetic air) were recorded under the same conditions as the background test. All spectra were collected with a resolution of 4 cm−1 by scanning 128 times.Density functional theory (DFT)-based calculations
The DFT calculations and structure optimization of FeTCP–M and InTCP–M (M = Fe, Co, Ni and Zn) were carried out using the Vienna ab initio simulation package (VASP)63,64 based on the projected augmented wave (PAW) pseudopotential method.65–67 The generalized gradient approximation (GGA) of the exchange-correlation functional with the Perdew–Burke–Ernzerhof (PBE) functional was introduced.68 The Brillouin-zone integration was sampled using a (1 × 1 × 1) Monkhorst–Pack mesh69 and the kinetic energy cutoff for the plane-wave expansion was set to 400 eV in the simulations. The K-points were improved in the calculations of the crystal orbital Hamilton population (COHP). All of the atoms in the structures were fully relaxed to the ground state. The vdW-DF method was employed to solve the spin-polarized effect and the effect of dispersion. The energy convergence changes and maximum force for geometric optimization were set to 0.02 eV Å−1 and 10−6 eV, respectively. A (1 × 1 × 1) unit cell as a model was constructed in the DFT calculations. The adsorption energies (Eads) of different analytes on InTCP–Co are defined as Eads = EA+B − EA − EB, where EA+B, EA, and EB are the total DFT energies of analytes adsorbed on InTCP–Co, InTCP–Co and isolated analyte molecules, respectively.Author contributions
E.-X. C. and Q. L. conceived and designed the idea. E.-X. C. carried out the experiments and characterization, L. H. provided help for modifying pictures in the article, M. Q. performed DFT calculations and analysis, Y. Z. provided resources for DFT, Y. S. performed gas adsorption measurements, W.-H. L., J.-Z. X. and J. C. provided help for gas sensing measurements, G. X. provided resources for gas sensing measurements, and E.-X. C., G. X. and Q. L. wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (22201283, 22325109, 22171263, 62227815, 22269009, and 21773030), the National Key Research and Development Project (2022YFA1503900), the National Science Foundation of Fujian Province (2022J05090, 2022T3008, and 2021J02017), the Scientific Research and Equipment Development Project of CAS (YJKYQ20210024), Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZR138), the Open Project Program of the State Key Laboratory of Photocatalysis on Energy and Environment (SKLPEE-KF202204), Fuzhou University and the Self-Deployment Project Research Program of Haixi Institutes, Chinese Academy of Sciences (CXZX-2022-GH09).Notes and references
- D. B. Cao, X. Liu, J. P. Lewis, W. Guo and X. Wen, Angew. Chem., Int. Ed., 2022, 61, e202202751 CrossRef CAS PubMed.
- Q. K. Li, X. F. Li, G. Zhang and J. Jiang, J. Am. Chem. Soc., 2018, 140, 15149–15152 CrossRef CAS PubMed.
- L.-Y. Zhu, L.-X. Ou, L.-W. Mao, X.-Y. Wu, Y.-P. Liu and H.-L. Lu, Nano-Micro Lett., 2023, 15, 89 CrossRef CAS PubMed.
- X. Yang, Y. Deng, H. Yang, Y. Liao, X. Cheng, Y. Zou, L. Wu and Y. Deng, Adv. Sci., 2022, 10, e2204810 CrossRef PubMed.
- O. Lupan, N. Ababii, D. Santos-Carballal, M.-I. Terasa, N. Magariu, D. Zappa, E. Comini, T. Pauporté, L. Siebert, F. Faupel, A. Vahl, S. Hansen, N. H. de Leeuw and R. Adelung, Nano Energy, 2021, 88, 106241 CrossRef CAS.
- Y. Kim, S. Lee, J. G. Song, K. Y. Ko, W. J. Woo, S. W. Lee, M. Park, H. Lee, Z. Lee, H. Choi, W.-H. Kim, J. Park and H. Kim, Adv. Funct. Mater., 2020, 30, 2003360 CrossRef CAS.
- X. Geng, D. Liu, C. C. Hewa-Rahinduwage, S. L. Brock and L. Luo, Acc. Chem. Res., 2023, 56, 1087–1096 CrossRef CAS PubMed.
- G. Shao, O. Ovsianytskyi, M. F. Bekheet and A. Gurlo, Chem. Commun., 2020, 56, 450–453 RSC.
- H. Lim, H. Kwon, H. Kang, J. E. Jang and H.-J. Kwon, Nat. Commun., 2023, 14, 3114 CrossRef CAS PubMed.
- X. Liu, W. Zheng, R. Kumar, M. Kumar and J. Zhang, Coord. Chem. Rev., 2022, 462, 214517 CrossRef CAS.
- C. Zhu, Y. Xu, T. Zhou, L. Liu, Q. Chen, B. Gao and T. Zhang, Sens. Actuators, B, 2022, 365, 131928 CrossRef CAS.
- K. Wang, Y. Li, L.-H. Xie, X. Li and J.-R. Li, Chem. Soc. Rev., 2022, 51, 6417–6441 RSC.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- Z. Ji, T. Li and O. M. Yaghi, Science, 2020, 369, 674–780 CrossRef CAS PubMed.
- H. Lyu, O. I. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 2387–2396 CrossRef CAS PubMed.
- W. Zhang, J. Wu, W. Shi, P. Qin, W. Lang, X. Zhang, Z. Gu, H. Li, Y. Fan, Y. Shen, S. Zhang, Z. Liu, Y. Fu, W. Zhang and F. Huo, Adv. Mater., 2023, 35, e2303216 CrossRef PubMed.
- L. He, Y.-J. Guo, Y.-H. Xiao, E.-X. Chen, M.-B. Luo, Z.-H. Li and Q. Lin, CCS Chem., 2022, 4, 2286–2293 CrossRef CAS.
- E.-X. Chen, H. R. Fu, R. Lin, Y. X. Tan and J. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 22871–22875 CrossRef CAS PubMed.
- E.-X. Chen, H. Yang and J. Zhang, Inorg. Chem., 2014, 53, 5411–5413 CrossRef CAS PubMed.
- H.-Y. Li, S. N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC.
- M.-S. Yao, W.-H. Li and G. Xu, Coord. Chem. Rev., 2021, 426, 213479 CrossRef CAS.
- W.-T. Koo, J.-S. Jang and I.-D. Kim, Chem, 2019, 5, 1938–1963 CAS.
- Z. Meng, A. Aykanat and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 2046–2053 CrossRef CAS PubMed.
- H.-Z. Li, Y. Pan, Q. Li, Q. Lin, D. Lin, F. Wang, G. Xu and J. Zhang, J. Mater. Chem. A, 2023, 11, 965–971 RSC.
- Y.-M. Jo, Y. K. Jo, J.-H. Lee, H. W. Jang, I. S. Hwang and D. J. Yoo, Adv. Mater., 2023, 35, e2206842 CrossRef PubMed.
- M. E. DMello, N. G. Sundaram, A. Singh, A. K. Singh and S. B. Kalidindi, Chem. Commun., 2019, 55, 349–352 RSC.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- X.-C. Zhou, C. Liu, J. Su, Y.-F. Liu, Z. Mu, Y. Sun, Z.-M. Yang, S. Yuan, M. Ding and J.-L. Zuo, Angew. Chem., Int. Ed., 2023, 62, e202211850 CrossRef CAS PubMed.
- M.-S. Yao, J.-W. Xiu, Q.-Q. Huang, W.-H. Li, W.-W. Wu, A.-Q. Wu, L.-A. Cao, W.-H. Deng, G.-E. Wang and G. Xu, Angew. Chem., Int. Ed., 2019, 58, 14915–14919 CrossRef CAS PubMed.
- T. K. Townsend, E. M. Sabio, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2011, 4, 4270–4275 RSC.
- S. M. Majhi, S. T. Navale, A. Mirzaei, H. W. Kim and S. S. Kim, Inorg. Chem. Front., 2023, 10, 3428–3467 RSC.
- R. Tian, Z. Gao, G. Chen, H. Guan, C. Dong and E. Comini, Sens. Actuators, B, 2023, 383, 133584 CrossRef CAS.
- D. Wang, C. Han, C. Zheng, H. Fang, D. Xu and H. Zhao, Sens. Actuators, B, 2023, 387, 133833 CrossRef CAS.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- N. Pattanayak, P. Panda and S. Parida, Ceram. Int., 2022, 48, 7636–7642 CrossRef CAS.
- R. Singh and A. Mukherjee, ACS Catal., 2019, 9, 3604–3617 CrossRef CAS.
- Z. W. Hao, M. M. Dong, R. Q. Zhang, C. K. Wang and X. X. Fu, Phys. Chem. Chem. Phys., 2021, 23, 11852–11862 RSC.
- H. Lee, H. Park, D. Y. Ryu and W. D. Jang, Chem. Soc. Rev., 2023, 52, 1947–1974 RSC.
- Y.-X. Li, J. Li, H.-B. Zeng, X.-J. Zhang, S. Cosnier and D. Shan, Anal. Chem., 2023, 95, 3493–3498 CrossRef CAS PubMed.
- E.-X. Chen, M. Qiu, Y.-F. Zhang, L. He, Y.-Y. Sun, H.-L. Zheng, X. Wu, J. Zhang and Q. Lin, Angew. Chem., Int. Ed., 2022, 61, e202111622 CrossRef CAS PubMed.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780–13783 CrossRef CAS PubMed.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed.
- Y. S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed.
- K. Lee, W. C. Isley, A. L. Dzubak, P. Verma, S. J. Stoneburner, L. C. Lin, J. D. Howe, E. D. Bloch, D. A. Reed, M. R. Hudson, C. M. Brown, J. R. Long, J. B. Neaton, B. Smit, C. J. Cramer, D. G. Truhlar and L. Gagliardi, J. Am. Chem. Soc., 2014, 136, 698–704 CrossRef CAS PubMed.
- H. Nishihara, M. Ohwada, T. Kamimura, M. Nishimura, H. Tanaka, S. Hiraide, M. T. Miyahara, K. Ariga, Q. Ji, J. Maruyama and F. Tani, Chem. Commun., 2018, 54, 7822–7825 RSC.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- J. Ding, X. Guan, J. Lv, X. Chen, Y. Zhang, H. Li, D. Zhang, S. Qiu, H. L. Jiang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 3248–3254 CrossRef CAS PubMed.
- E. Ozensoy, C. H. F. Peden and J. Szanyi, J. Phys. Chem. B, 2005, 109, 15977–15984 CrossRef CAS PubMed.
- B. Sun, H. Lv, Z. Liu, J. Wang, X. Bai, Y. Zhang, J. Chen, K. Kan and K. Shi, J. Mater. Chem. A, 2021, 9, 6335–6344 RSC.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS.
- Q. Zhao, B. Chen, J. Li, X. Wang, M. Crocker and C. Shi, Appl. Catal., B, 2020, 277, 119215 CrossRef CAS.
- J. Abdul Nasir, J. Guan, T. W. Keal, A. W. Desmoutier, Y. Lu, A. M. Beale, C. R. A. Catlow and A. A. Sokol, J. Am. Chem. Soc., 2023, 145, 247–259 CrossRef CAS PubMed.
- F. Wang, P. Wang, T. Lan, Y. Shen, W. Ren and D. Zhang, ACS Catal., 2022, 12, 7622–7632 CrossRef CAS.
- M. Diaz-Lopez, S. A. Guda and Y. Joly, J. Phys. Chem. A, 2020, 124, 6111–6118 CrossRef CAS PubMed.
- P. C. Müller, C. Ertural, J. Hempelmann and R. Dronskowski, J. Phys. Chem. C, 2021, 125, 7959–7970 CrossRef.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Blöchl, J. Phys. Chem. Lett., 1993, 97, 8617–8624 CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- B. D. Dunnington and J. R. Schmidt, J. Chem. Theory Comput., 2012, 8, 1902–1911 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- M.-S. Yao, W.-X. Tang, G.-E. Wang, B. Nath and G. Xu, Adv. Mater., 2016, 28, 5229–5234 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06909e |
| This journal is © The Royal Society of Chemistry 2024 |