
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA guide to troubleshooting metal sacrificial anodes for organic electrosynthesis
Skyler D.
Ware
 a,
Wendy
Zhang
a,
Weiyang
Guan
b,
Song
Lin
b and
Kimberly A.
See
*a
a,
Wendy
Zhang
a,
Weiyang
Guan
b,
Song
Lin
b and
Kimberly A.
See
*a
aDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: ksee@caltech.edu
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA
First published on 6th March 2024
Abstract
The development of reductive electrosynthetic reactions is often enabled by the oxidation of a sacrificial metal anode, which charge-balances the reductive reaction of interest occurring at the cathode. The metal oxidation is frequently assumed to be straightforward and innocent relative to the chemistry of interest, but several processes can interfere with ideal sacrificial anode behavior, thereby limiting the success of reductive electrosynthetic reactions. These issues are compounded by a lack of reported observations and characterization of the anodes themselves, even when a failure at the anode is observed. Here, we weave lessons from electrochemistry, interfacial characterization, and organic synthesis to share strategies for overcoming issues related to sacrificial anodes in electrosynthesis. We highlight common but underexplored challenges with sacrificial anodes that cause reactions to fail, including detrimental side reactions between the anode or its cations and the components of the organic reaction, passivation of the anode surface by an insulating native surface film, accumulation of insulating byproducts at the anode surface during the reaction, and competitive reduction of sacrificial metal cations at the cathode. For each case, we propose experiments to diagnose and characterize the anode and explore troubleshooting strategies to overcome the challenge. We conclude by highlighting open questions in the field of sacrificial-anode-driven electrosynthesis and by indicating alternatives to traditional sacrificial anodes that could streamline reaction optimization.
 Skyler D. Ware | Skyler Ware received her BS in chemistry from The Ohio State University in 2018. In 2023, she earned her PhD in chemistry from Caltech under the supervision of Prof. Kimberly See. Her research focuses on electrochemical interfaces and non-aqueous electrolytes in energy storage systems and organic electrosynthesis. |
 Wendy Zhang | Wendy Zhang received her BS and MS in chemistry from the College of William and Mary in 2015 and 2016 under the supervision of Prof. William McNamara. In 2023, she earned her PhD in chemistry from Caltech under the supervision of Prof. Kimberly See. Her research in the See lab focuses on understanding and manipulating electrode interfaces to improve the metal sacrificial anode performance for organic electrosynthesis. |
 Weiyang Guan | Weiyang Guan was born in Zhejiang, China. After obtaining her bachelor's degree from the University of Wisconsin-Madison in 2019, she became interested in electrochemistry and joined the Song Lin group at Cornell University for further studies. Her PhD research is focused on applying reductive electrochemistry for strong bonds activation, especially in the generation of reactive intermediates for downstream transformations. |
 Song Lin | Song Lin grew up in Tianjin, China. After earning a BS degree from Peking University in 2008, he pursued graduate studies at Harvard University working under the direction of Professor Eric Jacobsen. He then carried out postdoctoral studies with Professor Chris Chang at UC Berkeley. In the summer of 2016, Song moved to Ithaca to start his independent career. He was promoted to Associate Professor with tenure in July 2021 and then to Full Professor in January 2023. He is currently an Associate Editor for Organic Letters and serves on the Editorial Advisory Board of Synlett, Chem, and Chemistry–A European Journal as well as the Scientific Advisory Board of Snapdragon Chemistry. The Lin Laboratory's research lies at the interface of electrochemistry and organic chemistry, with a main objective of using fundamental principles of electrochemistry and radical chemistry to discover new organic transformations and uncover new reaction mechanisms. |
 Kimberly A. See | Kimberly See is an Assistant Professor of Chemistry in the Division of Chemistry and Chemical Engineering at Caltech. She was born and raised in Colorado and received her BS in Chemistry from the Colorado School of Mines. Kim earned her PhD in Chemistry at the University of California, Santa Barbara where she worked with Prof. Ram Seshadri and Galen Stucky. Kim was awarded the St. Elmo Brady Future Faculty Postdoctoral Fellowship at the University of Illinois at Urbana-Champaign and worked with Prof. Andrew Gewirth in the Department of Chemistry. Now, her group at Caltech studies electrochemical systems with sustainability applications, ranging from next-generation battery chemistries to developing electrolytes and electrodes for green organic electrosynthesis. Her group studies both interfaces and bulk materials properties in electrochemical devices and how they change as a result of charge transfer reactions. |
1. Introduction
Organic electrosynthesis has undergone a major revival in the past several years.1–12 The rapidly growing field enables new reactivity that is not achievable with traditional synthetic methods.13,14 Furthermore, the use of electrons as reagents offers routes to greener and safer synthetic conditions, potentially eliminating the need for harsh or hazardous chemicals.11 Electrosynthesis also enables greater selectivity in synthetic reactions via the ability to fine-tune either the applied potential to promote specific reduction or oxidation reactions, or the applied current to select for the most kinetically labile reactions.15 Electrochemical experiments offer a handle by which to probe the reaction mechanism, enabling greater mechanistic insight than would be achievable with chemical reductants.16–18Though an attractive goal of organic electrosynthesis is to develop paired electrolyses in which reactions at both the anode and the cathode contribute to value-added products,1,2,19–23 individual half reactions must be well understood if they are to be combined into a larger process. In reductive electrosynthesis, these half reactions frequently rely on charge balancing via the oxidation of a sacrificial anode, typically Mg, Al, Zn, or Fe.24 Sacrificial anodes have enabled the use of electrochemistry in a variety of organic reactions.7–10,22,24–26 During electrolysis, the metal electrode is oxidized, releasing metal cations into solution as shown in Fig. 1. The cations are often thought to be inert to the reductive chemistry of interest. In some situations, the cations do participate in the reaction of interest; in these cases the anode is not truly sacrificial and plays a more substantial role.27–29
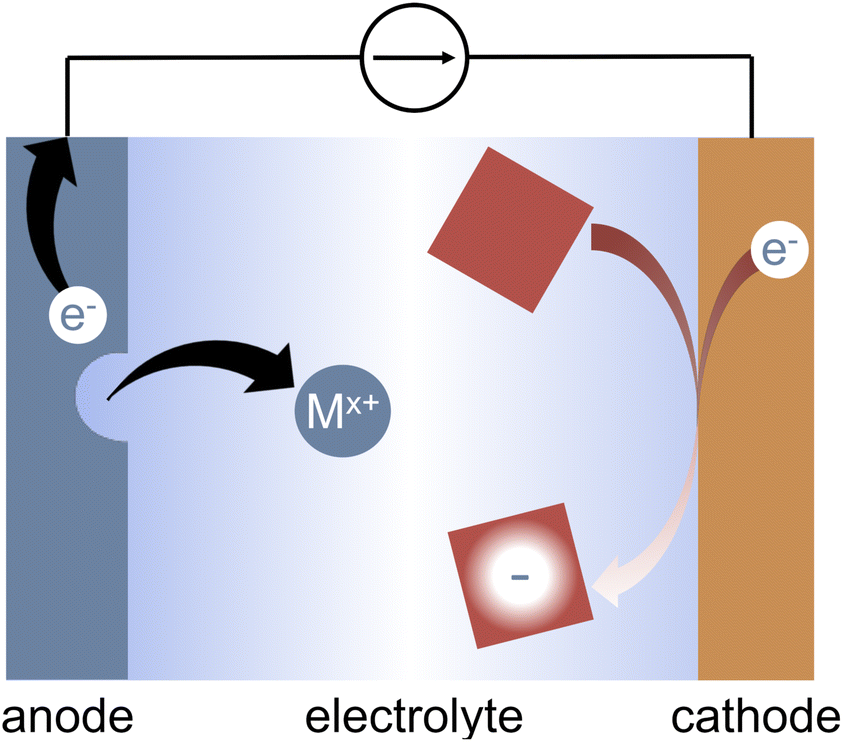 | ||
| Fig. 1 Sacrificial metal anodes enable reductive electrosynthesis by charge-balancing the target reductive reactions at the cathode. During a reductive reaction, the metal sacrificial anode is oxidized, releasing metal cations into solution. | ||
In an ideal case, electrolysis with sacrificial anodes offers several benefits during early stages of reaction development. First, the oxidation reaction itself is typically straightforward and occurs at a known, constant potential. The oxidation of the metal anode is sufficient to charge balance the reductive reaction of interest, thereby preventing unwanted oxidation of substrates or additives in the solution. The addition of metal cations into solution over the course of the reaction is less likely to limit the scope of the reductive reaction than the use of other chemicals as sacrificial reductants.21 Furthermore, metal electrodes are generally inexpensive and easy to store, and they provide a less hazardous alternative to commonly used sacrificial reductants.30
The ideal sacrificial anode should not limit or interfere with the reductive reaction of interest, either through direct interactions with reaction components or through its electrochemical performance. In particular, four major criteria must be satisfied to ensure that the reaction is not limited by the sacrificial anode:
(1) Both the metal anode and the cations generated during electrolysis should not degrade any electrolyte components or reagents used in the reductive reaction.
(2) Any inherent reactivity between the metal and the electrolyte solution should not form an insulating surface film or prohibit oxidation of the anode.
(3) The anode should permit metal stripping throughout the reaction, meaning that it must not be passivated by products or byproducts formed over the course of the electrolysis.
(4) Metal cations generated from anodic oxidation should not undergo competitive reduction at the cathode.
These four criteria are outlined in Fig. 2 and serve as a basis for the outline of our discussion.
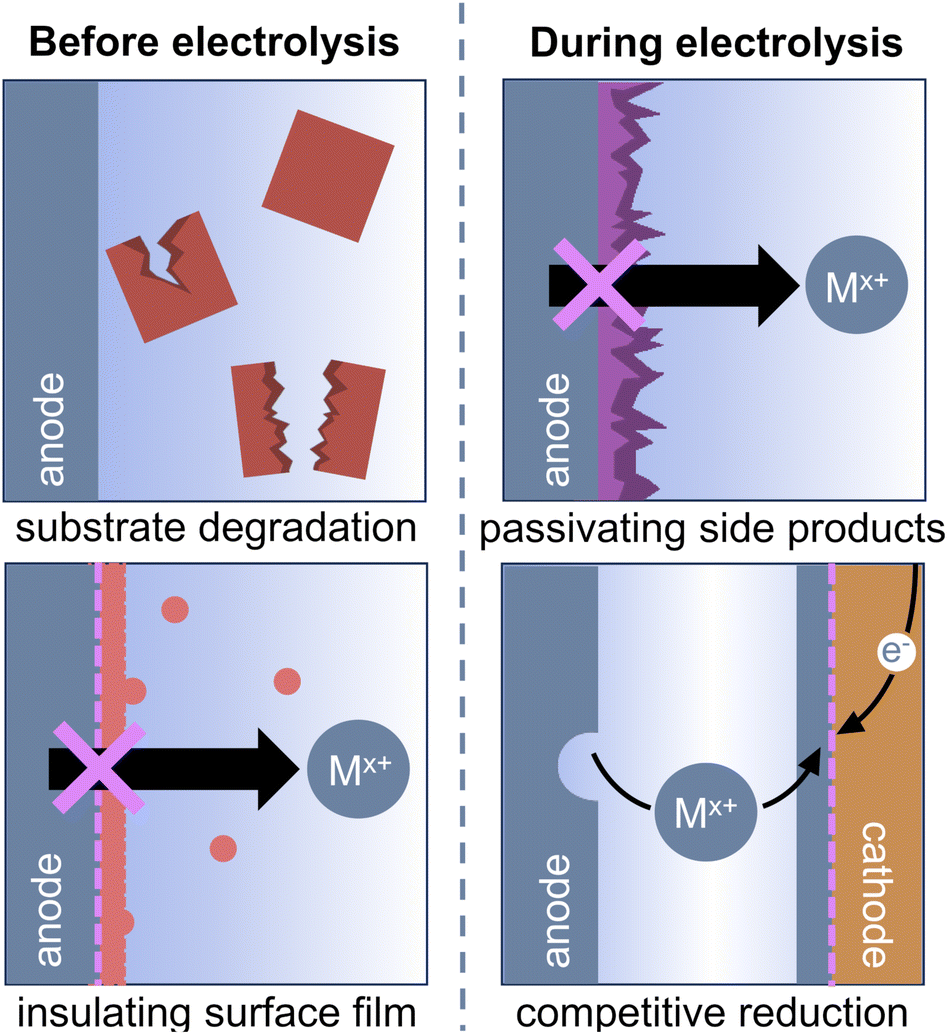 | ||
| Fig. 2 Sacrificial anodes can deviate from ideal behavior and limit the reductive reaction of interest, even under conditions that would normally be compatible with said reaction. Chemical reactions between the electrode and substrate, formation of an insulating surface film on the anode, anode passivation by byproducts formed during the reductive reaction, and competitive plating of cations from the sacrificial anode at the cathode can all prevent ideal anode performance. | ||
Though many reductive electrosynthetic reactions assume that each of these sacrificial anode criteria are met, in reality several processes can interfere with these assumptions and prevent ideal sacrificial anode behavior. The surface chemistry at metal electrodes is extremely sensitive to the chemicals present in the reaction solution, a fact that has been well studied in the analytical electrochemistry community. In addition to the electrode materials, the success of an electrosynthetic reaction depends on a host of factors that must be optimized, including chemical parameters such as the solvent, reactants, and additives, as well as electrochemical parameters including the supporting electrolyte and the magnitude of the applied potential or current.26,27,31–36 Changes to any of these parameters can induce nonideal behavior at the sacrificial anode, leading to failed reactions, low yields, hazardous short-circuits, and/or extreme voltages that exceed the compliance limits of the potentiostat and stop the reaction early. Thus, the performance of the anode can impose limits on the available reaction conditions, eliminating chemical space that would otherwise be compatible with the reaction of interest.
The issues associated with sacrificial anode performance are compounded by a lack of reported observations of the anodes themselves.37,38 Many electrosynthetic works simply select the sacrificial anode that provides the highest yield in optimization experiments without investigating the anodic chemistry, even when a failure at the anode is observed. Not only does this strategy limit the range of compatible conditions for the reductive reaction, it also ignores potential mechanistic contributions from the cations generated during sacrificial anode oxidation. Fortunately, problems at the anode can often be diagnosed and rectified with a few brief experiments, leading to higher product yields and an expanded set of compatible reaction conditions.
Understanding and characterizing processes at the anode requires knowledge of both electrochemical and surface characterization techniques that can interrogate the chemistry at the electrode. Here, we integrate lessons from electrochemistry, interface characterization, and organic synthesis to elucidate strategies for overcoming issues related to sacrificial anodes. We highlight four common but underexplored challenges that cause reactions to fail. For each, we suggest electrochemical and surface characterization experiments to diagnose the problem, and we present experiment design strategies for troubleshooting sacrificial anodes. The fundamentals of electrochemical techniques in organic synthesis, as well as classes of synthetic reactions enabled by sacrificial anodes, have been reviewed extensively elsewhere and will not be covered here.15–17,24–27,39–42 We conclude with an outlook for the future of this multidisciplinary field, including open challenges related to sacrificial anode optimization and potential new directions for developing reductive half reactions with minimal interference from anodic processes. We hope that the strategies presented here will improve the process of screening and optimizing electrosynthetic reactions, expand the chemical space in which reductive reactions can occur, enable more robust yields from reductive electrosyntheses, and encourage collaborations at the intersection of electrochemistry, organic synthesis, and interface chemistry.
2. Side reactions
As in all synthetic procedures, side reactions between reagents must be considered when optimizing a reaction. In the case of electrosynthesis, care must be taken to avoid side reactions between components of the electrochemical system as well as between the substrates and additives common to traditional organic synthesis. In particular, reactions between any combination of the electrodes, solvent, supporting electrolyte, substrates, and intermediates must be considered. Even if not directly related to the reductive chemistry, chemical reactions between the sacrificial anode and any other component of the electrolyte solution can lead to low yields of the desired product.2.1 Examples of side reactions with the anode
In many cases, reactions that occur at the sacrificial anode are chemical reductions of a reactant by a strongly electropositive anode, such as Mg. The canonical example of such a detrimental side reaction is the reaction between Mg metal and organic halides (R–X) to form Grignard reagents, RMgX, and related compounds in solution.43,44 Given that organic halides are frequently used as substrates for reductive electrosynthesis, such side reactions are likely problematic during the optimization stage of many methodologies. It should be noted that the formation of most Grignard reagents with non-activated Mg requires an induction period, in which the native MgO surface layer is removed from the Mg source to expose reactive Mg, but the length of this induction period depends on the other compounds in solution as well as any pretreatment steps, such as polishing the Mg metal. In electrochemical systems, the anode is usually chemically or mechanically polished to ensure effective Mg stripping, but the solution composition will be reaction-dependent. In addition to affecting the substrates and mechanistic pathway, the formation of Grignard reagents can contribute to the growth of a high-impedance passivation layer at the Mg anode,45,46 which will be discussed in more detail in Section 4. In all cases, the electrolyte should be checked for potential Grignard-forming conditions before a Mg sacrificial anode is employed.The formation of Grignard reagents is not the only undesired side reaction that can occur at sacrificial anodes. In addition to alkyl halides, Mg can also react with esters and ketones. Condon et al. observed that in an attempted alkylation of decyl trichloroacetate with triethylborane, the ester group was reduced when a Mg sacrificial anode was used, leading to the formation of decanol as an undesired side product. The researchers noted that when a Mg or Zn sacrificial anode was employed, the electrolysis time was shortened from the expected 1 h to 30 min, suggesting that some chemical reduction of the substrate occurred at the anode. No chemical reduction was observed when an Al or Fe anode was employed.47 In the electrochemical allylation of carbonyl compounds, Durandetti et al. observed side reactions between carbonyl species and a Mg sacrificial anode resulting from enolization of the ketone substrate. The authors attribute this reactivity to the reducing power of Mg; similar side reactions were not observed when a Zn anode was used in place of Mg.48 Mg can chemically reduce a variety of substrates, including activated alkenes, pyridine derivatives, and cyanoarenes (Fig. 3), which could lead to undesired byproducts in an electrochemically driven reaction.49–52 Zn can chemically reduce carbonyl halides, potentially causing similar issues.53 Substrates that can be chemically reduced by a mildly reducing metal can often also be reduced by more strongly reducing metals, necessitating the careful choice of sacrificial anodes in an electroreductive reaction. The choice of solvent can introduce potential side reactions as well. Saboureau et al. observed overconsumption of a Mg sacrificial anode during electrolyses carried out in dimethylformamide (DMF) solvent, which was traced back to chemical corrosion of the Mg anode via reductive decomposition of the solvent. Zn and Al sacrificial anodes were not subject to the same corrosion reaction.54 Furthermore, Zn sacrificial anodes can chemically reduce Ni and Pd catalysts for reductive coupling reactions, possibly complicating reaction optimization and mechanistic studies.55,56
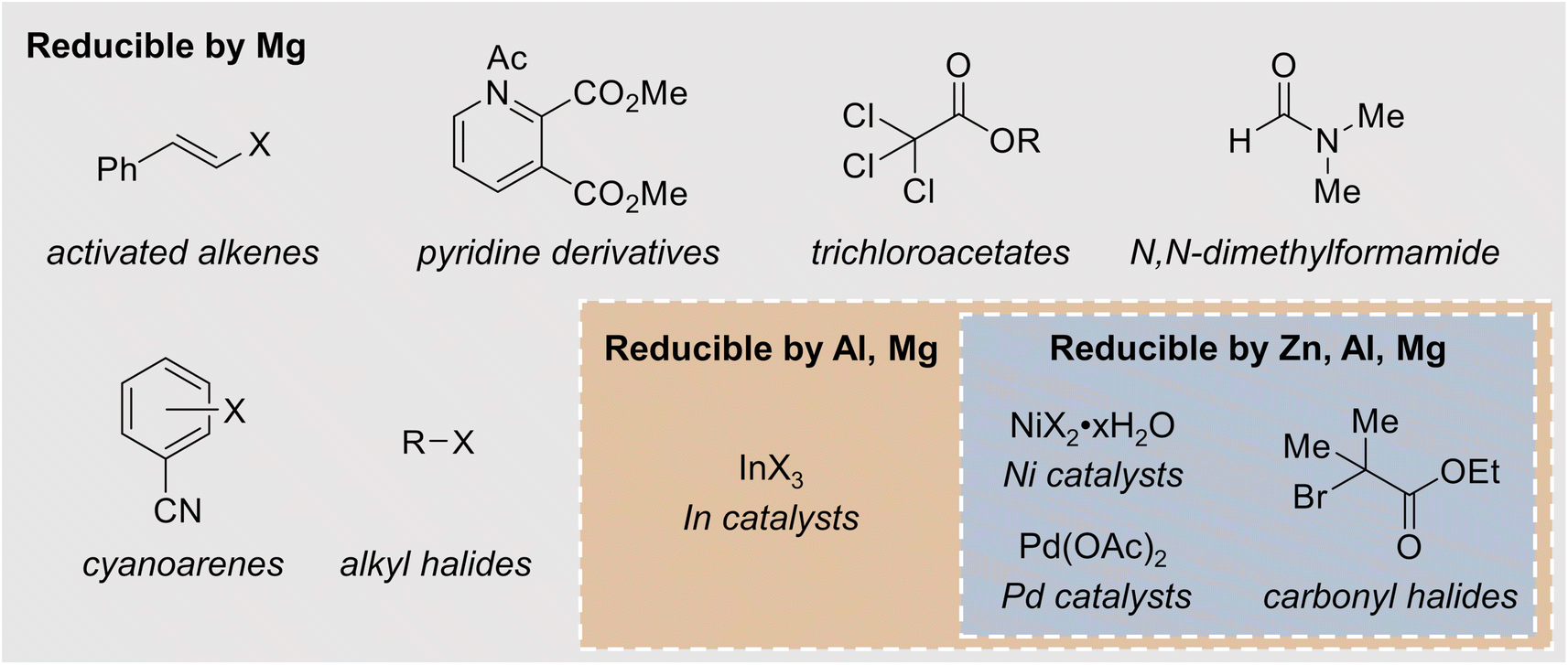 | ||
| Fig. 3 Examples of substrates and catalysts that can be chemically reduced by various metal sacrificial anodes. | ||
2.2 Examples of side reactions with anodically generated cations
In addition to side reactions between the metal anode and reaction components in solution, the cations generated during oxidation of the anode can also interfere with the reductive reaction. Nedelec et al. demonstrated that the nature of the cation generated from the sacrificial anode dictates the extent of electrochemically driven cross-coupling products of alkyl halides.35 Introduction of metal cations and salts via oxidation of the anode can also lead to diminished yields. For example, Peters et al. observed that anode-derived Mg salts limited the success of an electrochemical Birch reaction. Addition of Mg salts directly to the electrolyte reduced yields of the desired diene from 74% to 30%. In cases when the solution was not stirred during electrolysis, smaller anode–cathode distances were correlated with lower yields, suggesting that metal salts generated at the anode negatively impacted yields.57 During electrochemical atom transfer radical polymerization (ATRP) reactions catalyzed by Cu–amine complexes, anodically generated Al3+ can hinder the catalyst's reactivity, providing a competitive coordination pathway for the amine ligands, and the Al sacrificial anode can chemically reduce the Cu catalyst to Cu(0).58–602.3 Symptoms of side reactions with the anode
As detailed above, there are several indicators of side reactions between the sacrificial anode and another component of the system. Similarly to traditional chemical reactions, high conversion of the starting material but low yields of the desired product serve as key markers of background reactivity. Side products may appear in 1H-NMR spectra of the post-electrolysis solution, either as discrete and identifiable species or as an intractable mixture formed from decomposition of the substrate, additives, or solvent. In certain cases, degradation of the electrolyte solution may be visible through a color change or the formation of precipitates.2.4 Diagnosing the problem
A few experiments can be undertaken to determine whether the formation of an undesired side product is related to the sacrificial anode. Control experiments without electricity, such as placing the anode in the electrolyte solution without applying a current or potential bias, can indicate whether chemical reactions between the anode and electrolyte are occurring. Perhaps the simplest electrochemical experiment is to exchange the anode for a different metal. A less reducing metal, such as Zn, is less likely to undergo detrimental side reactions with electrolyte components, but the nature of any observed side reactions will be solution-dependent and must be optimized as such. However, if a specific sacrificial anode must be used, or if an understanding of the side reaction is important to mechanistic development, further electrochemical characterization can be carried out. A divided cell is an excellent tool that can be employed to decouple any observed reductive decomposition from reactions occurring at the cathode. Saboureau et al. adopted this approach when determining the nature of the Mg corrosion reaction in DMF during the electrosynthesis of carboxylic acids from organic halides. A Mg anode was fitted to the anodic compartment, which was then charged with an organic halide (4-chloro-trifluoromethylbenzene) and several supporting electrolytes in succession. The cathodic reaction was the straightforward electrochemical reduction of 1,2-dibromoethane, an easily reducible species that serves as an efficient counter reaction. Analysis of the anodic compartment after electrolysis indicated that the aromatic halide was not consumed at the anode and that only DMF had degraded.54 This set of divided cell experiments conclusively demonstrated the nature of the side reaction between anodically polarized Mg and DMF.2.5 Troubleshooting the problem
First, we discuss solutions to the anode chemically reducing components in the electrolyte. A simple solution would be to use a less reducing metal; however, another metal could introduce new side reactions or different challenges. Alternatively, a divided cell can be used to carry out the electrolysis itself, not just in a diagnostic role, to prevent crossover from the cathodic chamber into the anodic chamber. A more complicated but exciting solution would be to generate a surface film on the anode metal that conducts the corresponding metal cation but is electronically insulating, preventing the metal from reacting directly with the organic substrate via electron transfer. The film would be akin to the solid-electrolyte interphase (SEI) formed on anodes in Li-ion batteries; however, such a strategy has not been pursued in the context of sacrificial anodes and will thus be discussed in the Summary and future outlook section.Next, we discuss issues of reactivity between the metal cation generated at the anode and components in the solution. A divided cell can again be used to prevent the generated cation from reacting with anything in the reaction mixture. If an undivided cell is required, a strongly coordinating binding agent could sequester the generated cation, provided that the bound complex does not react further with the electrolyte or passivate the anode.
It is important to acknowledge that not all reactions between the sacrificial anodes and the electrolyte solution are deleterious. The cations and salts produced by oxidation of the anode can participate in the reductive reaction, stabilize products formed at the cathode, or function as products in their own right. For example, Mellah and coworkers have explored the use of a samarium sacrificial anode to directly generate Sm(II) reagents for C–C bond formation in solution.61–63 The Sm metal anode is oxidized to Sm2+, and various Sm2+ salts including SmCl2, SmBr2, SmI2, and Sm(OTf)2 (OTf = triflate, CF3SO−3) were formed in situ through the addition of nBu4X salts.63 Cations or salts formed from oxidation of the anode can also function as in situ-generated reactants participating in the reduction reaction. Lu et al. demonstrated low product yields from an electroreductive radical silylation reaction when a divided cell was used, in part because anodically generated Mg2+ is proposed to participate in the overall reaction mechanism.64 Manabe et al. also demonstrated such reactivity in the electrochemical reduction of triphenylphosphine oxide to triphenylphosphine. AlCl3, generated via oxidation of an Al sacrificial anode, facilitated selective cleavage of the P–O bond.65 Gosmini et al. demonstrated that the presence of Fe(II) salts, formed from pre-electrolysis of a sacrificial Fe anode, was necessary to enable electrochemical cross-coupling of aryl halides and 2-chloropyrimidine; the same is true for the electroreductive coupling of aldehydes or ketones with gem-dichloro complexes.66,67 Fe(II) salts were also found to facilitate the electrochemical cyclopropanation reaction of halogenated compounds and activated olefins.68 Even if the generated cations do not participate directly in the reaction, they can stabilize products or species of interest formed during the reductive reaction. For example, Mg2+ and Al3+ cations coordinate to carboxylate anions formed as the products of electrocarboxylation reactions.25,69,70 This coordination stabilizes the carboxylate and forms a precipitate, which can easily be extracted from the organic solution. Similarly, Zn2+ cations can stabilize intermediates formed during the reduction of quinolines.71 Furthermore, metal anodes can chemically reduce species in solution to form reagents that are otherwise difficult to access. For example, Hilt and Smolko observed the chemical reduction of In(III) to In(0) at an Al anode. The In(0) was then oxidized at the anode to form In(I), a catalyst for the allylation of aldehydes, ketones, and esters.72 The metal anode can also influence the selectivity of the reaction. In an electrochemical thiolation reaction via cross-electrophile coupling of alkyl bromides with functionalized thiosulfonates, Ang et al. determined that the cross-coupling reaction only proceeds when a Mg sacrificial anode is used. Attempts to run the reaction with a Zn, Fe, or Cu anode resulted in homocoupling of the thiosulfonate to form diphenyldisulfide as the sole product after 3 h electrolysis.73
3. Anode passivation by inherent metal reactivity
Reactions between the sacrificial anode and reaction components can affect the electrochemical behavior of the anode itself, in addition to altering the electrolyte solution. Decomposition of certain electrolyte components can form ionically insulating surface layers on the sacrificial anode, prohibiting further contact between the electrolyte and the metal and preventing oxidation of the anode. Many anode materials — particularly electropositive metals such as Mg and Al — contain native oxide layers that similarly limit efficient metal oxidation.3.1 Examples of passivation by native oxides
The native surface oxide layers that form on Mg and Al develop before exposure to the electrolyte solution. While the oxide layer can be beneficial in preventing undesired chemical reactivity between the anode and substrate before electrolysis, in many cases the surface film is ionically insulating and does not permit oxidation or dissolution of the anode material.74–79 Solid-state conductivity of multivalent cations is very difficult due to their high charge density and large size.80 Once the oxides are formed, they are very thermodynamically stable, which is obvious from their position on the Ellingham diagram. In fact, MgO and Al2O3 are two of the most thermodynamically stable oxides relative to the corresponding metal.81 Mg is so oxophilic that even if it is sputtered in ultra-high vacuum, the surface is still covered by MgO.82 The surface film thus limits the electrochemical reaction by preventing the anodic stripping from charge-balancing the cathodic reaction at low overpotentials. The oxide layer forms whenever Mg or Al is in contact with air or moisture.74,83 As such, a rigorous electrode polishing procedure is required to remove the passivating oxide film. Polishing the anode has the added benefit of removing any impurities or oxidized products remaining on the anode from previous reactions. If the anode has been used in prior reactions, macroscale deposits of oxidized product can be removed via sonication, electropolishing, or an acid rinse.57,84 The electrode should then be mechanically polished under an inert atmosphere using a razor blade, fine-grit sandpaper, or a rotary tool to minimize growth of the oxide surface film.Even if the sacrificial anode is rigorously polished, electropositive metals can still form an insulating surface film when placed in contact with organic electrolytes. Even with rigorous drying procedures, electrolytes can still contain ppb to ppm amounts of trace water that will react with the metal. Additionally, solvent decomposition or reactions between the anode and the supporting electrolyte can form insulating surface films that prevent oxidation of the anode.64,65,84,85 Importantly, these insulating surface films may not be immediately apparent or visible to the naked eye. Even an electrode that appears shiny and metallic with no obvious corrosion could experience difficulties with anodic oxidation, preventing the cathodic reaction from proceeding to completion and limiting reaction scale-up.65,86
3.2 Examples of passivation by supporting electrolyte
Reactions between the anode material and the supporting electrolyte are particularly insidious as these are two components that are optimized independently but are in fact codependent. Selecting either an anode material or a supporting electrolyte too early in the optimization process can lead to inadvertent exclusion of compatible reaction conditions if the only factor limiting the cathodic reaction is the non-obvious evolution of an insulating surface film on the anode. Fortunately, the formation of such surface films has been studied extensively in the battery community. Several supporting electrolytes commonly used in organic electrosynthesis have already been screened for metal deposition and stripping for use in battery applications, and many are found to be incompatible with Mg and Al stripping.65,87–89 In particular, fluoride-containing electrolytes — including those containing BF4−, PF6−, AsF6−, or others with anions that can hydrolyze in the presence of trace water to form HF — have been shown to form ionically insulating MFx-rich surface films on Mg and Al.65,87 Other electrolytes, including those containing CF3SO3−, (CF3SO2)2N− (bistriflimide or TFSI), or ClO4−, can also interfere with Mg anodes.87–89 Using supporting electrolytes containing any of the aforementioned anions with a Mg or Al sacrificial anode is likely to lead to passivation of the anode and unsatisfactory electrochemical performance.3.3 Symptoms of passivation by inherent reactivity
Several physical and electrochemical signatures could indicate formation of an insulating surface film on the anode. During an electrolysis experiment, a sharp increase in the overall cell voltage might be observed, especially at the start of the reaction. Depending on the instrument used, the voltage may increase until the compliance limit of the device is reached, causing the reaction to stop prematurely. In many cases, the insulating surface film is not readily apparent and no visible electrode fouling is observed because the surface films can be very thin.3.4 Diagnosing the problem
The first step in determining if a reaction fails due to the formation of an insulating surface film on the anode is to search the literature for prior reports of reactivity between the anode and the solvent or supporting electrolyte. For Mg anodes in particular, research on SEIs in Mg metal batteries is a good starting point for exploring inherent reactivity between the anode and electrolyte.87,89,90 If no prior reports of passivation exist, several electrochemical experiments can point to the existence of an ionically insulating surface film. Linear sweep voltammetry (LSV), in which the sacrificial anode is the working electrode and the potential is swept anodically, can indicate whether metal stripping is observed at the expected potential. Fig. 4a shows an example of an LSV experiment designed to assess metal stripping at an Al electrode.91 When a TBABF4 supporting electrolyte in tetrahydrofuran (THF) is used, the oxidative current density is extremely low and no Al oxidation is observed due to the formation of a passivating film resulting from a chemical reaction between Al and the electrolyte. The current density also decreases upon subsequent scans, indicating that Al stripping becomes more difficult due to the growth of the passivating film. It should be noted that a separate reference electrode is required for such experiments to eliminate confounding effects that may arise from the possible passivation of the counter electrode in a two-electrode cell. Several reference electrodes suitable for use in nonaqueous systems have been developed.92–95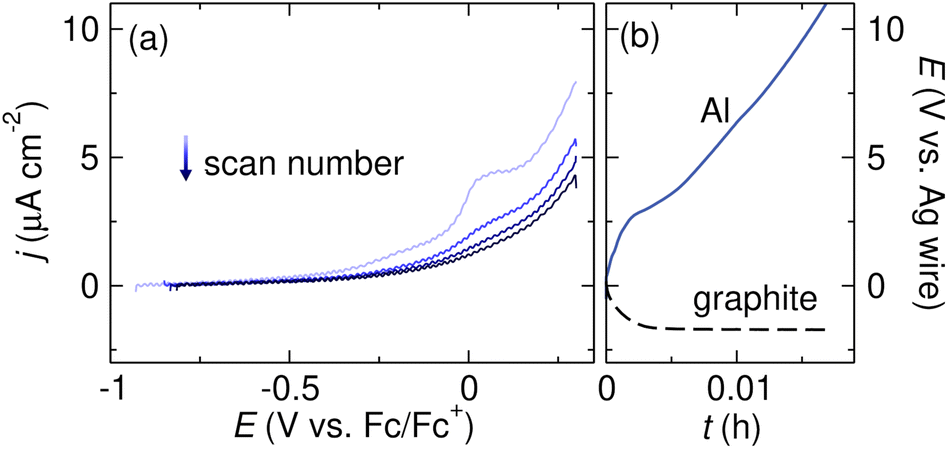 | ||
| Fig. 4 (a) Linear sweep voltammograms of an Al sacrificial anode in THF with 0.1 M TBABF4 supporting electrolyte. The voltammograms were collected at 5 mV s−1 scan rate with 85% iR compensation. (b) Voltage profiles of Al and graphite electrodes during galvanostatic Al stripping in THF with 0.5 M TBABF4 supporting electrolyte. The Al stripping experiment was conducted with tBuBr as a sacrificial reductant. Adapted from ref. 91 with permission from the Royal Society of Chemistry. | ||
The potential at the anode during a constant current electrolysis can also be monitored to confirm that the high voltage observed in an electrolysis experiment is related to processes at the anode and not the cathode. Fig. 4b shows the potentials of an Al anode and a graphite cathode during reduction of tBuBr. The potential at the cathode is steady and constant, suggesting that the reduction proceeds as expected. However, the voltage at the Al anode increases to >10 V within a few seconds and quickly reaches the compliance limit of the potentiostat. This sharp polarization and extreme overpotential at the anode indicate that the oxidation reaction does not proceed smoothly, likely due to the presence of a passivating surface film.91 Monitoring both the cathode and anode voltage during electrolysis requires the use of a nonaqueous reference electrode and a potentiostat equipped with the hardware necessary to record both the working and counter electrode potentials.
Electrochemical impedance spectroscopy (EIS) may indicate changes in the anode surface resistance over the course of a reaction. To diagnose an ionically insulating surface film, an EIS experiment in a two-electrode cell using the electrolyte of interest and with both electrodes made from the sacrificial anode metal will provide the resistance associated with oxidizing the anode; a notably high resistance (more than a few kΩ) suggests that an insulating surface film has formed, preventing metal stripping and limiting the anode's performance in the reductive reaction.96 Note, however, that a low resistance does not necessarily mean that no surface film has formed, as an electronically conductive but ionically insulating surface film can exhibit low impedance.
If the nature of the reaction that causes surface passivation is not known, various surface characterization techniques can pinpoint the electrolyte component that reacts with the anode. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) provide information about the morphology of the anode after reaction and a spatially resolved elemental distribution map, respectively. These techniques require high vacuum and thus will only probe solid products at the surface. Therefore, if elements exclusively present in the electrolyte are observed, then we can assume the electrolyte has reacted with the metal.97 For a more in-depth understanding of the surface reaction, X-ray photoelectron spectroscopy (XPS) provides information about the chemical environment of each element within the top 5–10 nm of the surface film.91,98 XPS is useful for determining the identity of the surface species and for characterizing extremely thin surface films, such as the oxide layers formed when Mg and Al are exposed to oxidants.
3.5 Troubleshooting the problem
Research in the fields of corrosion science and energy storage have demonstrated that passivating metal oxide layers on Mg or Al can be removed by adding halide salts to the electrolyte solution. In an oxidizing environment, chloride ions migrate through the oxide film that forms on pure Al metal, breaking down the oxide layer and forming “pits” in the metal; corrosion by such pitting reactions has been well studied and is not limited to chlorides.99–104 Chloride ions have also been shown to break down non-oxide insulating surface films on Al. Manabe et al. demonstrated that an insulating AlF3 surface film which formed when Al was in contact with PF6−-containing electrolyte could be removed by introducing TBACl as a co-supporting electrolyte, along with the chelating amine tetramethylethylenediamine (TMEDA) to promote Al stripping.65Bromide additives can also break down the insulating surface film on Al anodes. Zhang et al. observed that adding TBABr as a co-supporting electrolyte enabled Al oxidation in electrolytes that would otherwise passivate the metal (Fig. 5a). The current densities observed in the LSV with TBABr co-supporting electrolyte are much higher than those observed without TBABr (shown in Fig. 4a). The current density also steadily increases upon subsequent scans, suggesting that surface passivation does not limit Al stripping. In a follow-up electrolysis experiment, the Al anode potential remained low and constant for several hours (Fig. 5b), suggesting that Br− contributes to the formation of an ionically conductive surface film on Al. The beneficial effect of additives is not limited to bromides in this case; Cl−-, Br−-, and I−-containing additives all enabled Al oxidation.91
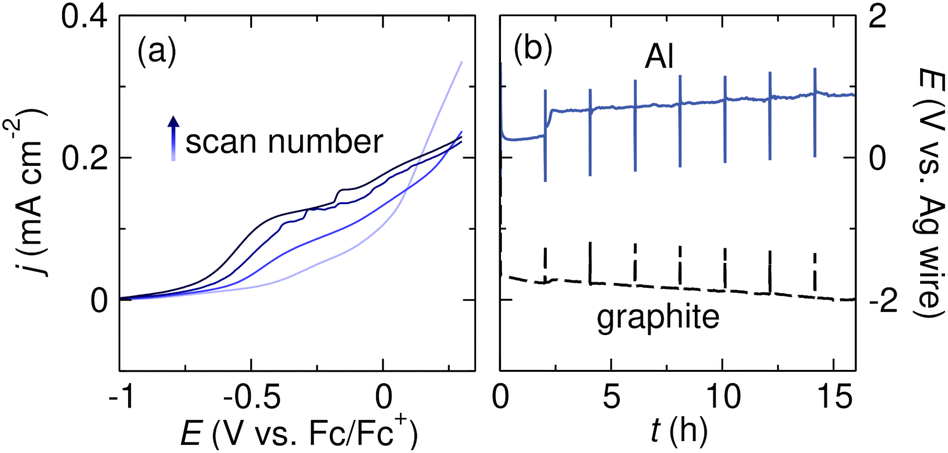 | ||
| Fig. 5 (a) Linear sweep voltammograms of an Al sacrificial anode in THF with 0.05 M TBABF4 + 0.05 M TBABr supporting electrolyte. The voltammograms were collected at 5 mV s−1 scan rate with 85% iR compensation. (b) Voltage profiles of Al and graphite electrodes during galvanostatic Al stripping in THF with 0.25 M TBABF4 + 0.25 M TBABr supporting electrolyte. The Al stripping experiment was conducted with tBuBr as a sacrificial reductant. Adapted from ref. 91 with permission from the Royal Society of Chemistry. | ||
Similar strategies can be applied in systems with Mg sacrificial anodes. Addition of Br− decreased the thickness of the passivation layer formed on a Mg anode and formed an ionically conducting surface film that permitted Mg stripping while limiting chemical reactions between the Mg anode and the electrolyte (see Section 4).97 In a Mg battery system, addition of MgCl2 to a solution of Mg(TFSI)2 in dimethoxyethane (DME) resulted in significant improvements in Mg oxidation compared to the solution without MgCl2, likely due to destabilization of the surface oxide film.79 MgCl2 was later shown to suppress the passivation of Mg metal by PF6− ions.85 Li et al. showed that, as an alternative to Cl-containing salts, small amounts of I2 could be added to an electrolyte consisting of Mg(TFSI)2 and DME to form an ionically conductive MgI2 surface film that mitigated the Mg oxidation overpotential.105
If no satisfactory means of suppressing the formation of an insulating surface film on Mg or Al can be found, less oxophilic anodes such as Zn may be more applicable to the system at hand. Zn anodes are chemically compatible with a wide range of solvents and supporting electrolytes and are less likely to exhibit high stripping overpotentials due to passivation via inherent reactivity.106–108
4. Passivation by products formed during anodic stripping
Insulating surface films can also form due to processes that occur during the electrolysis in addition to or in lieu of the films that form immediately upon contact with the electrolyte, as described in the previous section. During stripping, fresh metal surface is exposed to the electrolyte and the corresponding metal cation is generated, ideally in solution. The fresh metal surface can react with the electrolyte components differently than the original metal surface because the surface layers described in the previous section can be anodically destroyed. The newly exposed metal can react with the electrolyte to form a new surface film composed of decomposition products from the supporting electrolyte, solvent, or organic substrate.109,110 Further, though stripping produces metal cations that ideally dissolve into the electrolyte, the metal cations can react at the anode/electrolyte interface to form insulating deposits.4.1 Examples of passivation by insulating salt nucleation
Passivation of the sacrificial anode during electrolysis has been observed in several synthetic reactions. Here we define passivation as the evolution of a high impedance surface film that shuts down electrochemistry at that electrode. In many cases, this passivation is linked to the use of strongly reducing anodes like Mg or Al.First we focus on examples using Mg anodes. While attempting to scale up reactions involving electrochemical cross-electrophile coupling of alkyl halides, Zhang et al. initially observed high cell voltage accompanied by visible formation of a thick passivating film during the first few hours of electrolysis.98 Lu et al. observed similar passivation of the Mg sacrificial anode in an electrochemically driven three-component cross-electrophile coupling reaction; though conversion of the starting material exceeded 95%, the desired product was obtained in low yields due to the growth of a thick passivating film at the anode and accompanying high cell voltage.111 Anode passivation can also be influenced by seemingly unrelated components. In the electroreduction of epoxides, Huang et al. employed tripyrrolidinophosphoric acid triamide (TPPA) as a cosolvent to improve the solubility of the LiCl supporting electrolyte in THF and to prevent cathodic reduction of the sacrificial metal cations. When the electroreduction was run in the absence of TPPA, the Mg sacrificial anode was coated in insulating salts, which passivated the anode and caused extreme cell voltages that stopped the reaction early.112
Mg is not the only sacrificial anode that can be passivated during electrolysis. In a Ni-catalyzed cross-electrophile coupling reaction, Perkins et al. observed significant precipitation of metal halide salts when either a Mg or Al sacrificial anode was used. Incomplete conversion of the starting material was observed when an Al anode was used, and no reaction was observed when using a Mg anode.113 Walker et al. observed high cell voltage and passivation of a Zn sacrificial anode during electrolysis in an electrochemical Ni-catalyzed Mizoroki–Heck reaction. Due to the low yields and incomplete conversion of starting material, the authors posit that the Ni catalyst is incompatible with the high cell voltage that arises from the anodic passivation.55
Several of the passivation reactions described above can be attributed to salt nucleation at the anode surface. As metal cations are stripped into solution, they coordinate to anions in solution and form a surface layer of insoluble salts that prevents further metal stripping. Salt deposition is common with multivalent cations like Mg2+ and Al3+ since multivalent cations are difficult to solvate due to their high charge density.114,115 A rough or uneven anode surface can exacerbate nucleation of these salts at the anode/electrolyte interface.97,116 The surface roughness may arise from either insufficient electrode polishing or corrosion/pitting reactions with other species in solution, such as organic halides or halide complexes.97,116,117 When an anodic potential is applied, the rough electrode surface generates an uneven electric field, which leads to non-uniform metal stripping and makes the surface even rougher (Fig. 6). The roughness increases the surface area of the electrode, leaving more space for salt nucleation. Further, the nanoscale heterogeneities can change the contact angle between a salt crystal and the electrode, which can reduce the energetic barrier for nucleation.118 An insulating salt layer thus forms and will grow over the anode surface during the reaction. While anode corrosion will need to be managed through reaction design, surface roughness can be mitigated by employing appropriate electrode polishing procedures (vide supra).
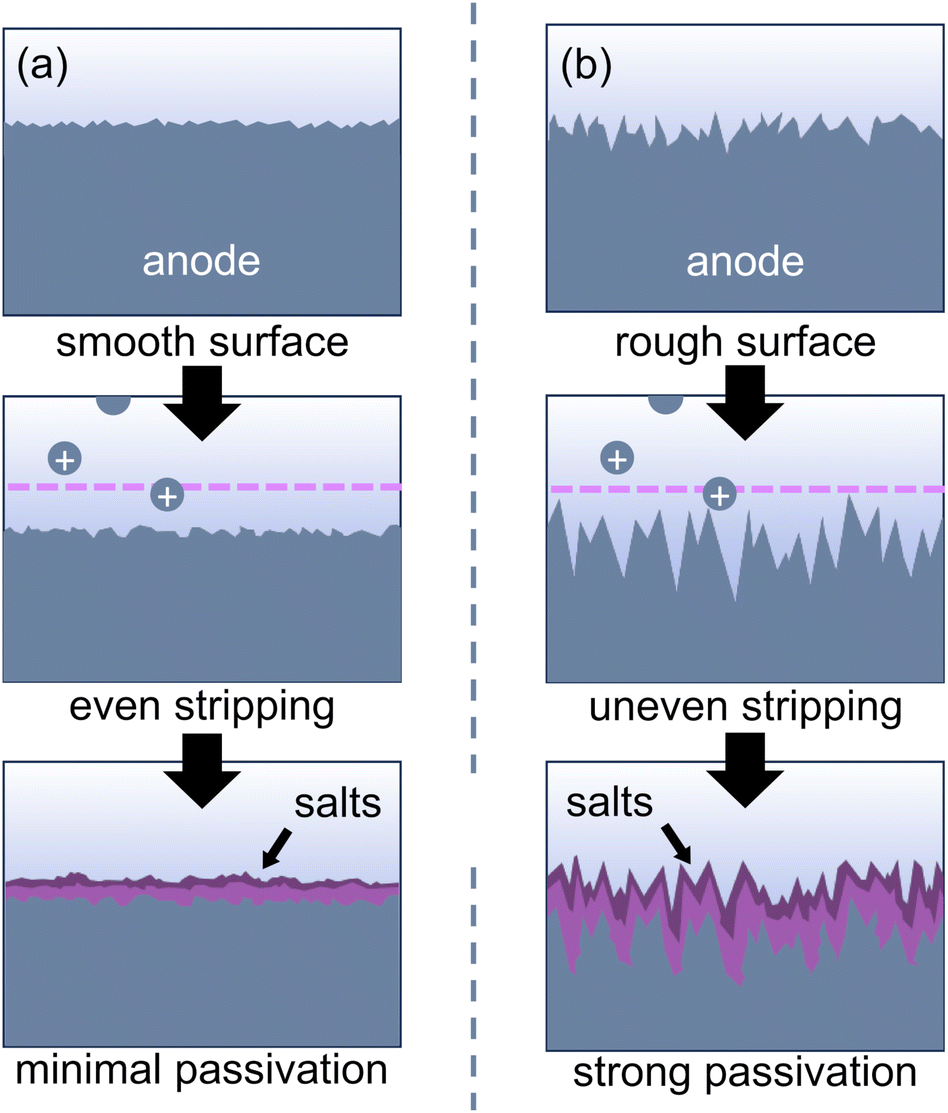 | ||
| Fig. 6 (a) An even electric field leads to smooth metal stripping, while (b) an uneven electric field caused by surface heterogeneities leads to nucleation of metal salts as the anode is oxidized. | ||
4.2 Symptoms of passivation by products formed during anodic stripping
Anode passivation by insulating salts or by corrosion during the reaction is usually readily apparent. Extreme anode fouling is common in such reactions and marked by the growth of a thick, visible surface coating. The coatings typically look black in color due to their rough nature and can appear as crystalline deposits, a tacky surface layer, or a conformal film. In a constant current experiment, the cell voltage typically increases over the first few minutes to hours of the electrolysis as the insulating surface blocks cation transport. Depending on the nature of the surface reactions, the anode fouling may result in low product yields, incomplete conversion of starting materials, and/or extreme cell voltages that reach the compliance limits of the instrument before the reaction is complete.4.3 Diagnosing the problem
The appearance of a thick coating on the anode is usually enough to suggest that high cell voltages are related to processes happening at the anode. The growth of this film is often unrelated to the cathodic reaction and can thus unnecessarily limit the range of applicable conditions for the reductive reaction of interest. To verify that the anodic surface coating is insulating and prevents Mg stripping (and that the high cell voltage is not caused by some concurrent cathodic process), the voltage of both electrodes can be monitored during the electrolysis. This experiment requires the use of a reference electrode.92–95 Large fluctuations or a sharp increase in anode voltage suggest that anode passivation may be limiting conversion.97,98,112A sharp increase in anode voltage is usually related to a mechanically stable surface coating. However, not all surface coatings are tightly bound to the anode, and mechanical stirring or inadvertent scraping of the anode during the reaction can dislodge salt deposits at the anode surface, exposing fresh metal. Fig. 7a shows an example of a fluctuating anodic voltage profile that is characteristic of salts dislodging from the Mg anode. As the surface deposits grow, the anode voltage increases. When part of the insulating salt coating is dislodged from the anode surface by contact with the stir bar, for instance, fresh Mg metal is exposed, and Mg stripping can again occur at lower overpotentials. The anode potential then decreases, and the Mg anode is oxidized as in the beginning of the reaction. However, any salt remaining on the anode contributes to surface roughening, which exacerbates further salt nucleation when the Mg is oxidized. The salt layer regrows over the exposed Mg, and the anode voltage increases once again. This process repeats until the end of the reaction.97
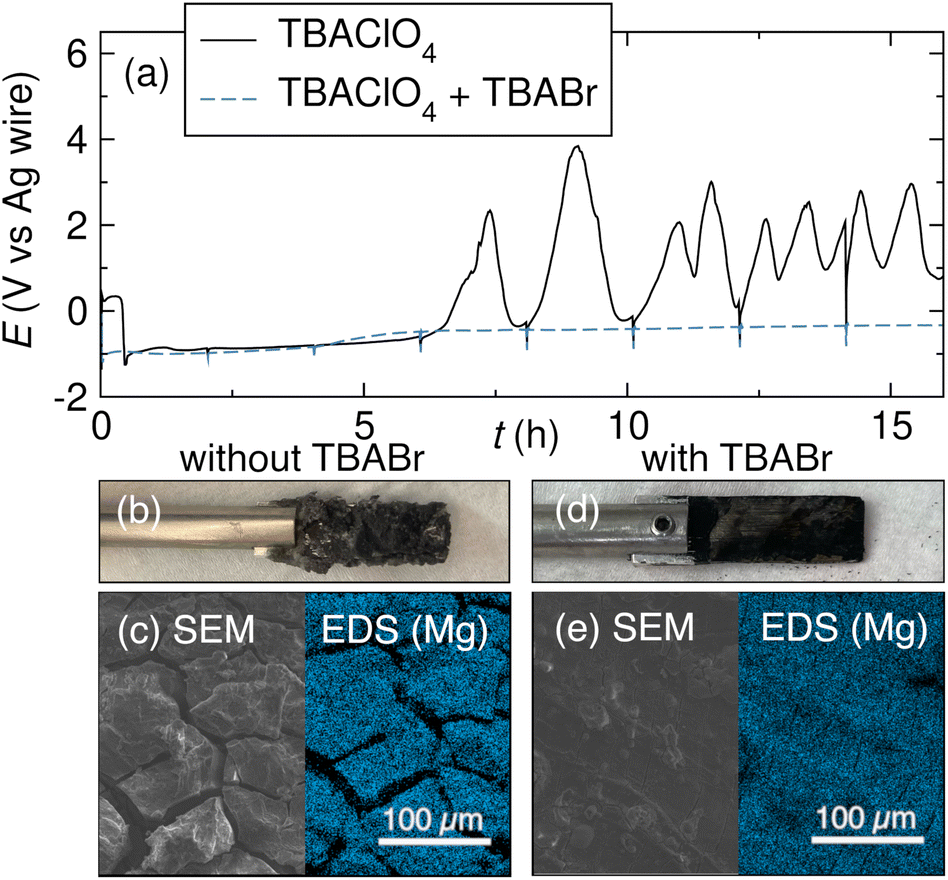 | ||
| Fig. 7 (a) Voltage profiles of a Mg anode during Mg oxidation in the presence of 0.5 M tBuBr using either 0.5 M TBAClO4 or 0.25 M TBAClO4 + 0.25 M TBABr supporting electrolyte in THF. (b) Photo of the Mg anode with insulating salt nucleation after electrolysis with the TBAClO4 electrolyte. (c) SEM/EDS images of the Mg anode after removal of the insulating salt coating. (d) Photo and (e) SEM/EDS images of the Mg anode with minimal salt buildup after electrolysis with the TBAClO4 + TBABr electrolyte. Reprinted with permission from ref. 97 licensed under CC BY-NC-ND 4.0. Copyright (2023) The Authors. | ||
If anode passivation is determined to be the cause of a failed electrolysis reaction, the next step is to determine the reaction that causes passivation. Characterization of the surface film by XRD, XPS, SEM, EDS, X-ray fluorescence, or infrared or Raman spectroscopy can indicate the nature of species present at the anode surface. XPS gives the most detailed chemical information about the surface film and has been used to characterize surface reactions at sacrificial anodes previously,97,98 but depending on the nature of the reaction, other techniques may also provide sufficient information to understand the surface reaction.
4.4 Troubleshooting the problem
Once the nature of the surface reaction has been determined, several strategies can be used to troubleshoot the issue. If the surface reaction is due to passivation by salt nucleation, proper polishing procedures as described above can minimize the initial sites available for salt nucleation. Nucleation sites can form from corrosion reactions even if the anode is well polished, though, so chemical strategies for removing the salt layer may be required. Coordinating solvents can be added to improve the solubility of metal salts. Zhang et al. added DME as a cosolvent to dissolve Mg(ClO4)2 and MgBr2 which deposited at the Mg surface during cross-electrophile coupling of alkyl halides.98 Lu et al. employed the same strategy in a three-component cross-electrophile coupling reaction.111 Both Mg(ClO4)2 and MgBr2 are poorly soluble in THF, which was used as the reaction medium, but linear ethers and diethers chelate more strongly to the Mg2+ ions and solubilize Mg salts.119,120 As described above, TPPA plays a similar role in supporting Mg sacrificial anode oxidation in THF; a passivating salt layer forms on the anode in the absence of TPPA but does not prevent Mg oxidation when TPPA is present.112 Peters et al. observed that TPPA could prevent passivation of a galvanized steel wire electrode following Mg deposition during an electrochemical Birch reaction.57 The cosolvent/additive strategy is not limited to reactions in THF: during an electrocatalytic sulfination of aryl halides, Lou et al. demonstrated that passivation of a Zn sacrificial anode by a solution of SO2 in acetonitrile could be mitigated by running the reaction in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile:dimethylacetamide.121 In pure dimethylacetamide, the black passivating deposits that formed on the electrode peeled off, suggesting that dimethylacetamide solvates the deposits better than acetonitrile.
:1 acetonitrile:dimethylacetamide.121 In pure dimethylacetamide, the black passivating deposits that formed on the electrode peeled off, suggesting that dimethylacetamide solvates the deposits better than acetonitrile.
Reactions between the anode and a substrate can be mitigated by including halide salts as additives or co-supporting electrolytes, similarly to managing anode passivation by reactions prior to electrolysis. Halide-containing salts can react at the metal surface to form an ionically conductive surface film, preventing nucleation of insulating species during the reaction. Zhang et al. demonstrated the utility of this strategy with a Mg electrode during the reduction of organohalides. During the reduction of tBuBr using a supporting electrolyte of 0.5 M TBAClO4 in THF, the anode voltage fluctuates as described above (Fig. 7a), and a thick surface film consisting of Mg(ClO4)2 and MgBr2 forms at the anode. Fig. 7 shows the anode after electrolysis (b) and after removal of the salt layer (c); the rough and insulating surface coating prevents effective Mg oxidation, and cracks in the Mg surface provide ample sites for additional salt nucleation. By contrast, when a supporting electrolyte consisting of 0.25 M TBAClO4 and 0.25 M TBABr is used, the anodic voltage remains stable for the full reaction (Fig. 7a) and the Mg surface is smooth and crack-free (Fig. 7d and e). The Br-containing supporting electrolyte is thought to form an ionically conductive surface film on the Mg anode that prevents contact between the corrosive organohalide and the anode.97 This passivation-preventing behavior has also been observed with a LiBr co-supporting electrolyte in Mg battery chemistry.122
5. Reduction of anodically generated cations at the cathode
Once effective metal stripping has been enabled, metal cations will be introduced to the electrolyte throughout the electrolysis reaction. When using an undivided cell, it is possible to reduce the newly generated metal cations at the cathode. Competitive reduction of the metal cations and the substrate leads to low faradaic efficiencies for electrochemical reactions. In some cases, the metal deposits can passivate the cathode, preventing further reduction of the substrate. Both situations can produce low product yields. Product distribution can also change if the kinetics of the reduction reaction at the plated metal are different from those of the original cathode material; sometimes this leads to new reactivity.3 If reduction of the cation is significantly easier than reduction of the substrate, metal deposits at the cathode can build up and eventually reach the anode, leading to extremely hazardous short circuits.123 In all cases, the plated metal is thus no longer innocent in the reaction, even if the anode metal and the cations in solution do not otherwise interact with components of the reductive reaction. Fig. 8 shows the possible outcomes of competitive reduction of the organic substrate and anodically generated cations.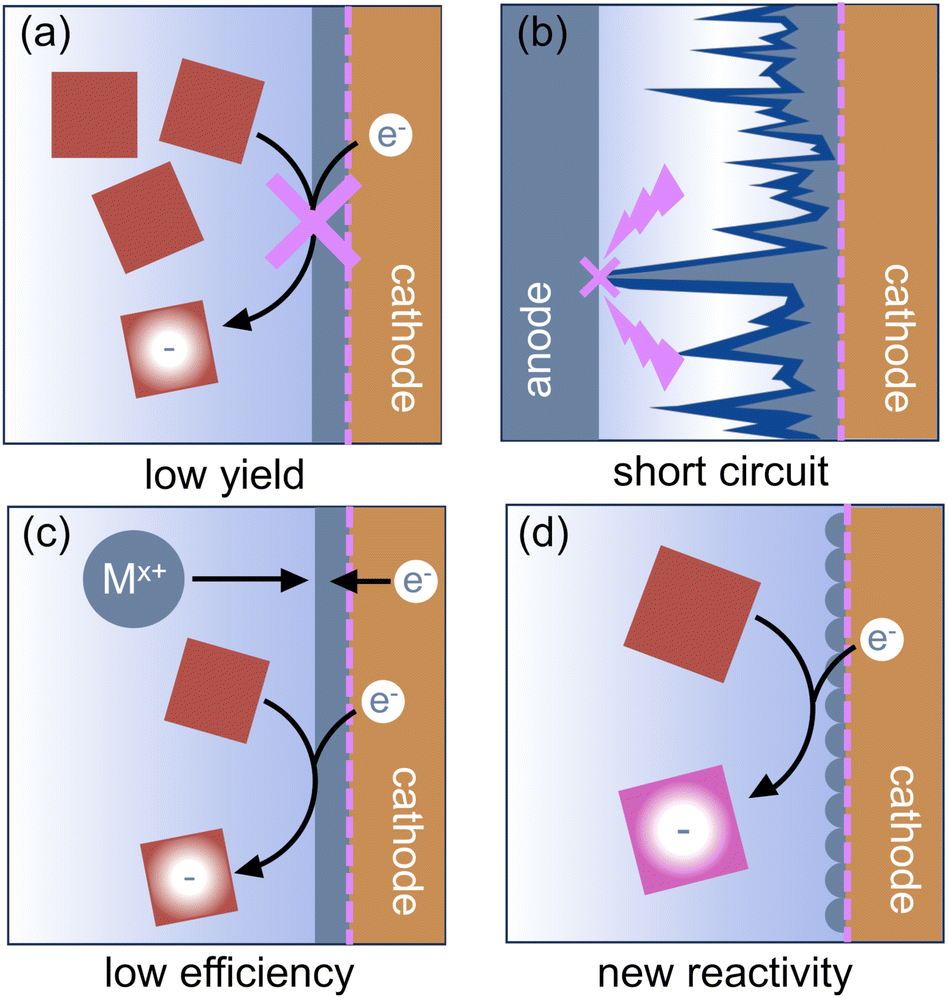 | ||
| Fig. 8 Reduction of anodically generated cations can outcompete reduction of the organic substrate at the cathode, leading to (a) low product yields, (b) hazardous short circuits, and (c) low faradaic efficiency. (d) In certain cases, metal deposition from the anode can lead to new or better controlled reactivity at the cathode. | ||
Special attention must be given to systems in which cross-plating of the metal cations is likely to occur. In a constant current electrolysis experiment, the applied current and reaction time are often chosen assuming that 100% of the electrons passed will go towards conversion of the reactants, i.e. the faradaic efficiency approaches 100%. However, when metal cations are reduced at the cathode at the same time as the substrate, some of the electrons passed at the cathode will go toward metal plating rather than substrate reduction, and product yields will be correspondingly lower. While competitive reduction of cations from the sacrificial anode is often an undesired side reaction that must be mitigated to improve reaction efficiency, reduction of the cations is not necessarily chemically incompatible with reduction of the substrate, and employing strategies to prevent metal plating can lead to higher product yields under otherwise similar conditions. Thus, care must be taken not to mistake low faradaic efficiency for bad or incompatible reaction conditions, even though both result in lower-than-expected yields of the product.
5.1 Examples of competitive reduction
The competitive reduction of metal cations makes certain anodes, particularly those metals with mild reduction potentials, inapplicable in electrochemical reactions that require strongly reducing potentials at the cathode.124 Zinc cations are particularly prone to cross-plating in undivided cells (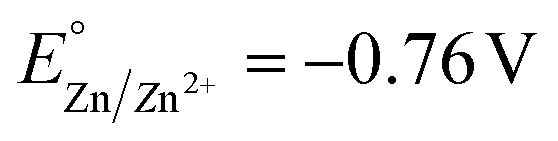 vs. SHE, compared to
vs. SHE, compared to 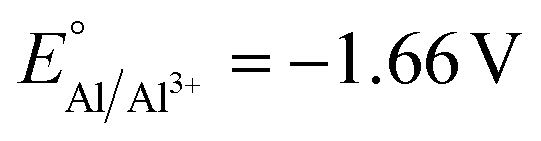 and
and 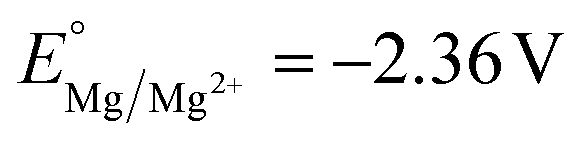 ), and the reduction of the metal cations competes with the reductive reaction of interest. Yuan et al. noted that the competitive reduction of Zn cations and CO2 or styrene substrates at a Ni cathode leads to low yields for styrene carboxylation. Such low yields were not observed when the sacrificial anode was switched to Al or Mg, which are thermodynamically more difficult to reduce.125 Zhang et al. reported that reduction of Zn2+ generated at a Zn sacrificial anode competes with reduction of 4-tert-butylstyrene in an electrochemical carbofunctionalization reaction with alkyl bromides; a Mg sacrificial anode did not result in similar challenges.126 In a Ni-catalyzed electrochemical cyclization of alkynyl aryl iodides, Déjardin et al. observed incomplete reduction of the substrate by the Ni catalyst, likely because reduction of anodically generated Zn2+ competes with reduction of the Ni catalyst. Switching to an Al sacrificial anode significantly increased product yields.127
), and the reduction of the metal cations competes with the reductive reaction of interest. Yuan et al. noted that the competitive reduction of Zn cations and CO2 or styrene substrates at a Ni cathode leads to low yields for styrene carboxylation. Such low yields were not observed when the sacrificial anode was switched to Al or Mg, which are thermodynamically more difficult to reduce.125 Zhang et al. reported that reduction of Zn2+ generated at a Zn sacrificial anode competes with reduction of 4-tert-butylstyrene in an electrochemical carbofunctionalization reaction with alkyl bromides; a Mg sacrificial anode did not result in similar challenges.126 In a Ni-catalyzed electrochemical cyclization of alkynyl aryl iodides, Déjardin et al. observed incomplete reduction of the substrate by the Ni catalyst, likely because reduction of anodically generated Zn2+ competes with reduction of the Ni catalyst. Switching to an Al sacrificial anode significantly increased product yields.127
While the mild reduction potential of Zn makes it particularly prone to competitive reduction and cross-plating, other anodically generated cations can also be reduced at the cathode under strongly reducing potentials. Lu et al. observed cathodic deposition of Mg metal from the sacrificial anode at the end of an electrolysis reaction involving electroreductive disilylation of alkenes. In dry, nonacidic solutions, the Mg plating does not pose any experimental hazards, but in wet solvent or in the presence of acid, the plated Mg is combustible in air and the reaction workup must be handled with caution.64 Durandetti et al. observed cross-plating of Fe from a Fe sacrificial anode during a electroassisted zinc-free Reformatsky-type reaction catalyzed by Fe species generated in situ. Fe deposition did not interfere with the reaction and high product yields could be obtained, but excess charge was passed to account for the low faradaic efficiency.128 In an electroreductive coupling of organic halides in ethanol or methanol, reduced yields were attributed to cathodic reduction of anodically generated Fe(II).129 Ishifune et al. observed competitive reduction of several anodically generated cations during the electroreduction of p-methoxyisopropylbenzene.130
5.2 Symptoms of competitive reduction
Competitive reduction of anodically generated cations is most often evidenced by lower-than-expected product yields and low faradaic efficiency, especially in constant current electrolysis. Depending on the possibility and nature of substrate reduction (chemical or electrochemical) at the plated metal, competitive reduction may result in low substrate conversion, full conversion but an unexpected product distribution, or degradation of the substrate and/or products as described in previous sections. Cathode fouling may also be observed, though whether or not the metal deposition is visible by eye will depend on the size of the deposits and extent of deposition.5.3 Diagnosing the problem
Depending on the extent of metal deposition, microscopy techniques may be required to confirm that competitive cation reduction is occurring. SEM and EDS can easily identify micron-sized metal deposits and spatially map the elemental composition of the surface, which will indicate whether the reduced cations cover the entire cathode surface or whether the original cathode material is still accessible.131 Once cross-plating is confirmed, electrochemical techniques can be used to determine if the cross-plating limits yields by any mechanism other than competition for electrons. Cyclic voltammetry (CV) experiments will indicate the reduction potentials of individual components in the system, including the substrate(s) and anodically generated cations. A reference electrode or non-interfering reference couple is required to accurately determine the relative reduction potentials of each component. Conducting CV experiments before electrolysis can hint at whether competitive reduction will occur during electrolysis — if the metal cation is reduced at a potential that is similar to or more positive than the substrate or catalyst to be reduced during electrolysis, competitive reduction is likely to occur.5.4 Troubleshooting the problem
Competitive reduction can be prevented most easily by switching to a sacrificial anode with a more negative reduction potential. However, in many cases, using a more reducing metal introduces additional complications as described in the previous three sections. If an anode with a mild reduction potential must be used to prevent substrate degradation or anodic passivation, a divided cell can physically separate the anode and cathode, preventing cross-plating and short circuits.123If the reduction potential of the anodically generated cations is more negative than that of the substrate but competitive reduction still occurs during constant current electrolysis, then reduction of the cation is kinetically faster than reduction of the substrate. In this situation, instead of performing constant current electrolysis, electrolysis can be conducted at a constant potential which is more negative than the reduction potential of the substrate but less negative than that of the anodically generated cation. Constant potential electrolysis can thus improve selectivity for substrate reduction over cation reduction.15 Conducting electrolysis at a constant potential prevents reduction reactions that are less thermodynamically accessible (i.e., the reduction of species with more negative reduction potentials) but longer electrolysis times may be required to fully reduce the substrate. In a constant potential reductive electrolysis, the anode/counter electrode should only be used as the reference electrode (i.e. in a two-electrode cell) in cases where no electrode passivation or undesired reactivity is observed. Otherwise, the potential will drift at the counter/reference electrode, which will cause the working electrode to experience more oxidizing conditions than initially expected.
As in the case of side reactions between the anode and the substrate, it is important to acknowledge that cross-plating of anodically generated cations can be beneficial in some reactions. In certain cases, plating at the cathode generates active sites for important chemical and electrochemical transformations (Fig. 8d). For example, Huang et al. found that Zn particles deposited at the cathode from sacrificial anode cations mediate the allylation of aldehydes in aqueous ammonia solutions by forming nucleophilic allylzinc species at the electrode surface.131 Similarly, reduction of anodically generated Zn2+ can form nanostructured Zn at the cathode, which can mediate the electroreduction of epoxides to alkenes and of imines and alkyl halides to amines.132,133 Reduction of cations from the sacrificial anode can also pin the cathodic potential at such a level as to avoid over-reducing the substrate to undesired products. In a constant current experiment, the electrode potentials are controlled by the potentials of the fastest oxidation and reduction events. Deposition of an easily reducible metal can thus prevent extreme reducing potentials that would degrade products at the cathode, albeit while reducing faradaic efficiency. Gosmini et al. observed that reducing anodically generated Zn at the cathode kept the cathode potential high enough to prevent direct reduction of an aryl halide species and instead promoted the desired cross-coupling of aryl halides with 2-halopyridine.134 Similarly, in the electrochemical carboxylation of benzal diacetates, Senboku et al. found that reduction of anodically generated Zn2+ at the cathode prevented extreme cathodic potentials that would lead to over-reduction of the desired metal acetate product.135 Cross-plated cations generated at the sacrificial anode can play a role in the reductive synthetic reaction; this synergy can be considered a form of paired electrolysis.
6. Summary and future outlook
Although the oxidation of a sacrificial metal anode is assumed to be a straightforward and simple counter reaction, processes involving the anode are integral to the overall reaction and can dictate the success or failure of an electrosynthetic transformation. To develop robust electrochemically-driven reactions, it is beneficial if the chemistry at the anode is understood and optimized alongside the cathodic reaction. The chemical and electrochemical effects of the sacrificial anode processes can affect every aspect of the synthetic reaction, from the faradaic efficiency to the scalability to the stability of the substrate and reactants. Sacrificial anode chemistry should not be ignored in reaction optimization.When selecting a sacrificial anode for an electrosynthetic reaction, it becomes clear that the anode's interactions with every component of the system must be taken into account. While sacrificial anode performance is strongly reaction-dependent, broad guidelines can be developed to direct initial screening experiments. In general, Zn or Fe anodes can be used to avoid chemically reducing easily reducible substrates or decomposing the solvent, while Mg or Al anodes can be employed to prevent cross-plating at the cathode in undivided cells when strongly reducing potentials are required to drive the cathodic reaction.
A major roadblock to the development of effective half reactions using sacrificial anodes is the relative lack of data on the anodes. It is not sufficient to report only the best-performing sacrificial anode in an optimization table, even if a reaction fails exclusively because of an anodic process and not because of anything related to the reductive reaction of interest. At minimum, several of the most common sacrificial anodes should be screened, and a description of the behavior of each anode under a given set of reaction conditions should be reported in the ESI. In particular, visible fouling of either electrode and changes in the product distribution that are directly tied to the anode chemistry should be highlighted. Only by reporting both successes and failures with different sacrificial anodes can the process of optimizing the anode chemistry be streamlined in future reactions.
Though visual observations are useful to begin troubleshooting issues at the anode, more detailed analytical data are necessary to develop a fundamental understanding of the anodic processes and how they influence the cathodic reaction. Though some instruments have been optimized to enable inexpensive and rapid screening of synthetic conditions, they are often insufficient for analytical characterization of electrochemical processes at either electrode, especially when screening multiple sacrificial anodes. Control over potential or current across multiple cells with reference electrodes and the ability to analyze the electrochemical data are required for exploring and understanding sacrificial anode processes in more detail.
To date, most of the analysis of sacrificial anode behavior has been conducted for reactions run in THF as the solvent. Because the choice of solvent is often dictated by the requirements of the synthetic reaction, the chosen sacrificial anode should not react with the solvent, or such reactions should be mitigated through electrolyte design. Moving forward, more attention should be paid to anode processes in other commonly used solvents, such as acetonitrile and N,N-dimethylformamide. The nature of the surface film formed at the anode surface, including continuous oxidation of the anode, will depend on the nature of solvent decomposition at a given metal. The higher donor number and coordinating ability of these solvents may affect the solubility of the sacrificial cations and their corresponding salts, potentially affecting the presence or extent of surface passivation by insulating products during electrolysis. New electrolyte design strategies may be required to enable effective metal oxidation in solvents other than THF. Furthermore, detailed investigations of the behavior of common sacrificial anodes such as Zn and Fe in various solvents and electrolyte systems should be undertaken to better understand the impacts of anode chemistry on such reactions.
In addition to electrolyte design strategies, altering the chemistry of the anode itself may enable new reactivity. Ionically conductive but electronically insulating surface coatings, similar to SEIs on battery anodes, could permit efficient metal stripping while minimizing chemical or electrochemical degradation of electrolyte components at the anode surface.115 Such surface layers can be generated in situ from reactions between the metal and the electrolyte or ex situ through solid state synthesis or reaction in a different solution than the one used for reductive electrosynthesis. Native SEIs, i.e. those formed in contact with electrolyte, and artificial SEIs on metals commonly used as sacrificial anodes (Mg, Zn, Al) are being studied in the battery literature; this research can provide a starting point for the development of surface coatings for use in electrosynthesis.
Alternatively, new nonmetallic anodes can be designed to mitigate or prevent the common failure mechanisms detailed here. Materials that undergo oxidative deintercalation mechanisms, for instance, exhibit a wide range of chemical potentials, allowing the potential at the anode to be tuned. Further, the deintercalation reactions can generate a variety of different cations in solution depending on the application. Our group is currently working toward these goals.
Many strategies for improving sacrificial anode performance involve introducing new species or new tunable parameters, adding to the already long list of variables that must be optimized in each reaction. In the future, machine learning may assist in predicting the performance of a particular sacrificial anode under a given set of reaction conditions.136 Such predictive technology presents an attractive complement to traditional synthetic screening experiments. However, to effectively train a model to predict sacrificial anode behavior, both successful and failed reactions must be reported in the literature. A balanced training set requires high-quality data on the anode chemistry for each reaction conducted in the optimization of the sacrificial anode, even if the reaction results in low product yields.
Although sacrificial anodes will continue to be necessary for optimizing individual half reactions, a long-term goal of electrosynthesis is to move towards a paired electrolysis setup to maximize atom economy by taking advantage of desirable reactions at both electrodes. The processes at each electrode can be related if targeting a convergent electrosynthesis procedure, but the two half reactions are not required to contribute to the same overall transformation as long as neither reaction is deleterious to the other.21 Before incorporating a reductive reaction into a paired electrolysis setup, detailed mechanistic studies must first be undertaken to ensure that the sacrificial anode and its byproducts are not required for the reductive reaction to proceed. Divided cell studies which separate the sacrificial anode cations from the reductive reaction are particularly instructive in these cases.
Ultimately, a deep understanding of sacrificial anode behavior in electrosynthetic conditions and the development of experimental guidelines for sacrificial anode choice will require cross-disciplinary collaborations between synthetic chemists, electrochemists, and surface scientists. A thorough understanding of sacrificial anode chemistry will streamline reaction optimization and expand the chemical space compatible with electrosynthetic reactions. We hope that by addressing the major pitfalls described herein, sacrificial anodes will not limit the development of new and interesting reductive electrochemistry.
Author contributions
S. D. W., W. Z., and K. A. S. conceptualized the perspective. S. D. W., W. Z., and W. G. performed the literature review. S. D. W. wrote the manuscript. W. G. and S. L. provided critical insights into synthetic reactions. K. A. S. provided guidance on content and organization. All authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation Center for Synthetic Organic Electrochemistry (CHE-2002158). K. A. S. acknowledges support from the David and Lucile Packard Foundation, Alfred P. Sloan Foundation, and Camille and Henry Dreyfus Foundation. The authors thank Nicholas V. Dulock for graphic design assistance.References
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, Organic Electrosynthesis: From Academia to Industry, React. Chem. Eng., 2020, 5, 977–990 RSC.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis?, Chem, 2020, 6, 2484–2496 CAS.
- H.-Y. Zhou, H.-T. Tang and W.-M. He, The Future of Organic Electrochemistry Current Transfer, Chin. J. Catal., 2023, 46, 4–10 CrossRef CAS.
- D. Pletcher, Organic Electrosynthesis – A Road to Greater Application. A Mini Review, Electrochem. Commun., 2018, 88, 1–4 CrossRef CAS.
- D. Pollok and S. R. Waldvogel, Electro-Organic Synthesis – a 21 St Century Technique, Chem. Sci., 2020, 11, 12386–12400 RSC.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Electrifying Organic Synthesis, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- A. Shatskiy, H. Lundberg and M. D. Kärkäs, Organic Electrosynthesis: Applications in Complex Molecule Synthesis, ChemElectroChem, 2019, 6, 4067–4092 CrossRef CAS.
- X.-F. Liu, K. Zhang, L. Tao, X.-B. Lu and W.-Z. Zhang, Recent Advances in Electrochemical Carboxylation Reactions Using Carbon Dioxide, Green Chem. Eng., 2022, 3, 125–137 CrossRef.
- B. Huang, Z. Sun and G. Sun, Recent Progress in Cathodic Reduction-Enabled Organic Electrosynthesis: Trends, Challenges, and Opportunities, eScience, 2022, 2, 243–277 CrossRef.
- Electrochemistry in Organic Synthesis, ed. L. Ackermann, Georg Thieme Verlag KG, Stuttgart, 1st edn, 2022, p. b000000126 Search PubMed.
- B. A. Frontana-Uribe, R. Daniel Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Organic Electrosynthesis: A Promising Green Methodology in Organic Chemistry, Green Chem., 2010, 12, 2099–2119 RSC.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an Enabling Technology for Organic Synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, Organic Electrochemistry: Molecular Syntheses with Potential, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS.
- M. Rafiee, M. N. Mayer, B. T. Punchihewa and M. R. Mumau, Constant Potential and Constant Current Electrolysis: An Introduction and Comparison of Different Techniques for Organic Electrosynthesis, J. Org. Chem., 2021, 86, 15866–15874 CrossRef CAS PubMed.
- E. C. R. McKenzie, S. Hosseini, A. G. C. Petro, K. K. Rudman, B. H. R. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Versatile Tools for Understanding Electrosynthetic Mechanisms, Chem. Rev., 2022, 122, 3292–3335 CrossRef CAS.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, A Synthetic Chemist's Guide to Electroanalytical Tools for Studying Reaction Mechanisms, Chem. Sci., 2019, 10, 6404–6422 RSC.
- C. Costentin and J.-M. Savéant, Concepts and Tools for Mechanism and Selectivity Analysis in Synthetic Organic Electrochemistry, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11147–11152 CrossRef CAS PubMed.
- W. Li, T. Nonaka and T.-C. Chou, Paired Electrosynthesis of Organic Compounds, Electrochemistry, 1999, 67, 4–10 CrossRef CAS.
- L. Muchez, D. E. De Vos and M. Kim, Sacrificial Anode-Free Electrosynthesis of α-Hydroxy Acids via Electrocatalytic Coupling of Carbon Dioxide to Aromatic Alcohols, ACS Sustainable Chem. Eng., 2019, 7, 15860–15864 CrossRef CAS.
- T. Wu and K. D. Moeller, Organic Electrochemistry: Expanding the Scope of Paired Reactions, Angew. Chem., Int. Ed., 2021, 60, 12883–12890 CrossRef CAS PubMed.
- A. Claraz and G. Masson, Recent Advances in C(Sp3)–C(Sp3) and C(Sp3)–C(Sp2) Bond Formation through Cathodic Reactions: Reductive and Convergent Paired Electrolyses, ACS Org. Inorg. Au, 2022, 2, 126–147 CrossRef CAS PubMed.
- F. Marken, A. J. Cresswell and S. D. Bull, Recent Advances in Paired Electrosynthesis, Chem. Rec., 2021, 21, 2585–2600 CrossRef CAS PubMed.
- Y. Li, L. Wen and W. Guo, A Guide to Organic Electroreduction Using Sacrificial Anodes, Chem. Soc. Rev., 2023, 52, 1168–1188 RSC.
- G. Silvestri, S. Gambino, G. Filardo, M. Tiitta, M. Sjostrom, S. Wold, R. Berglind and B. Karlsson, Use of Sacrificial Anodes in Synthetic Electrochemistry. Processes Involving Carbon Dioxide, Acta Chem. Scand., 1991, 45, 987–992 CrossRef CAS.
- J. Chaussard, J.-C. Folest, J.-Y. Nedelec, J. Perichon, S. Sibille and M. Troupel, Use of Sacrificial Anodes in Electrochemical Functionalization of Organic Halides, Synthesis, 1990, 1990, 369–381 CrossRef.
- D. M. Heard and A. J. J. Lennox, Electrode Materials in Modern Organic Electrochemistry, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS.
- M. Izzi, M. C. Sportelli, N. Ditaranto, R. A. Picca, M. Innocenti, L. Sabbatini and N. Cioffi, Pros and Cons of Sacrificial Anode Electrolysis for the Preparation of Transition Metal Colloids: A Review, ChemElectroChem, 2020, 7, 386–394 CrossRef CAS.
- A. Rodríguez and J. A. García-Vázquez, The Use of Sacrificial Anodes for the Electrochemical Synthesis of Metallic Complexes, Coord. Chem. Rev., 2015, 303, 42–85 CrossRef.
- D. J. Charboneau, N. Hazari, H. Huang, M. R. Uehling and S. L. Zultanski, Homogeneous Organic Electron Donors in Nickel-Catalyzed Reductive Transformations, J. Org. Chem., 2022, 87, 7589–7609 CrossRef CAS PubMed.
- C. J. Burrows, J. B. Harper, W. Sander and D. J. Tantillo, Solvation Effects in Organic Chemistry, J. Org. Chem., 2022, 87, 1599–1601 CrossRef CAS PubMed.
- A. M. Couper, D. Pletcher and F. C. Walsh, Electrode Materials for Electrosynthesis, Chem. Rev., 1990, 90, 837–865 CrossRef CAS.
- F. Aymard, J.-Y. Nédélec and J. Périchon, An Efficient Inexpensive Electrochemical Preparation of Ruppert's Reagent, Tetrahedron Lett., 1994, 35, 8623–8624 CrossRef CAS.
- D. Franco, A. Riahi, F. Hénin, J. Muzart and E. Duñach, Electrochemical Reduction of a Racemic Allyl β-Keto Ester Catalyzed by Nickel Complexes: Asymmetric Induction, Eur. J. Org Chem., 2002, 2002, 2257–2259 CrossRef.
- J. Y. Nedelec, H. A. H. Mouloud, J. C. Folest and J. Perichon, Electrochemical Cross-Coupling of Alkyl Halides in the Presence of a Sacrificial Anode, J. Org. Chem., 1988, 53, 4720–4724 CrossRef CAS.
- T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, One-Pot Electrochemical Nickel-Catalyzed Decarboxylative Sp2–Sp3 Cross-Coupling, Org. Lett., 2019, 21, 816–820 CrossRef CAS.
- S. B. Beil, D. Pollok and S. R. Waldvogel, Reproducibility in Electroorganic Synthesis—Myths and Misunderstandings, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS.
- M. Klein and S. R. Waldvogel, Counter Electrode Reactions—Important Stumbling Blocks on the Way to a Working Electro-organic Synthesis, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS.
- M. C. Leech and K. Lam, A Practical Guide to Electrosynthesis, Nat. Rev. Chem, 2022, 6, 275–286 CrossRef.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, A Survival Guide for the “Electro-curious”, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Making Electrochemistry Easily Accessible to the Synthetic Chemist, Green Chem., 2020, 22, 3358–3375 RSC.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- D. Seyferth, The Grignard Reagents, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- Hydroboration and Organic Synthesis: 9-Borabicyclo[3.3.1]Nonane (9-BBN), ed. R. S. Dhillon, Springer, Berlin, Heidelberg, 2007, pp. 271–281 Search PubMed.
- S. E. Koon, C. E. Oyler, J. H. M. Hill and W. J. Bowyer, Visualization of the Areal Distribution of the Reactivity of Magnesium Surfaces in the Formation of Grignard Reagents, J. Org. Chem., 1993, 58, 3225–3226 CrossRef CAS.
- C. E. Teerlinck and W. J. Bowyer, Reactivity of Magnesium Surfaces during the Formation of Grignard Reagents, J. Org. Chem., 1996, 61, 1059–1064 CrossRef CAS.
- S. Condon, C. Zou and J.-Y. Nédélec, Electrochemical Alkyl Transfer Reaction from Trialkylboranes to Polyhalo Compounds, J. Organomet. Chem., 2006, 691, 3245–3250 CrossRef CAS.
- S. Durandetti, S. Sibille and J. Perichon, Electrochemical Allylation of Carbonyl Compounds Using Nickel Catalyst and Zinc(II) Species, J. Org. Chem., 1989, 54, 2198–2204 CrossRef CAS.
- T. Ohno, M. Sakai, Y. Ishino, T. Shibata, H. Maekawa and I. Nishiguchi, Mg-Promoted Regio- and Stereoselective C-Acylation of Aromatic α,β-Unsaturated Carbonyl Compounds, Org. Lett., 2001, 3, 3439–3442 CrossRef CAS.
- Y. Yamamoto, H. Maekawa, S. Goda and I. Nishiguchi, Novel One-Pot Vicinal Double C-Acylation of Styrenes and Methacrylates by Electroreduction, Org. Lett., 2003, 5, 2755–2758 CrossRef CAS PubMed.
- Y. Kita, H. Maekawa, Y. Yamasaki and I. Nishiguchi, Highly Selective and Facile Synthesis of Dihydro- and Tetrahydropyridine Dicarboxylic Acid Derivatives Using Electroreduction as a Key Step1, Tetrahedron, 2001, 57, 2095–2102 CrossRef CAS.
- X. Wu, C. N. Gannett, J. Liu, R. Zeng, L. F. T. Novaes, H. Wang, H. D. Abruña and S. Lin, Intercepting Hydrogen Evolution with Hydrogen-Atom Transfer: Electron-Initiated Hydrofunctionalization of Alkenes, J. Am. Chem. Soc., 2022, 144, 17783–17791 CrossRef CAS PubMed.
- M. C. C. Areias, L. W. Bieber, M. Navarro and F. B. Diniz, Metal-Free Electrochemical Reformatsky Reaction in Water: Further Evidence for a Radical Mechanism, J. Electroanal. Chem., 2003, 558, 125–130 CrossRef CAS.
- C. Saboureau, M. Troupel and J. Perichon, Organic Electrosynthesis with a Sacrificial Anode. Chemical Reductive Degradation of the Solvent N,N-dimethyl Formamide, J. Appl. Electrochem., 1990, 20, 97–101 CrossRef CAS.
- B. R. Walker and C. S. Sevov, An Electrochemically Promoted, Nickel-Catalyzed Mizoroki–Heck Reaction, ACS Catal., 2019, 9, 7197–7203 CrossRef CAS.
- Y.-L. Lai and J.-M. Huang, Palladium-Catalyzed Electrochemical Allylic Alkylation between Alkyl and Allylic Halides in Aqueous Solution, Org. Lett., 2017, 19, 2022–2025 CrossRef CAS PubMed.
- B. K. Peters, et al., Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery Chemistry, Science, 2019, 363, 838–845 CrossRef CAS.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Electrochemically Mediated Atom Transfer Radical Polymerization of n-Butyl Acrylate on Non-Platinum Cathodes, Polym. Chem., 2016, 7, 5357–5365 RSC.
- J. Luo, M. Chavez, C. Durante, A. Gennaro, A. A. Isse and M. Fantin, Improvement of Electrochemically Mediated Atom Transfer Radical Polymerization: Use of Aluminum as a Sacrificial Anode in Water, Electrochim. Acta, 2022, 432, 141183 CrossRef CAS.
- U. S. Yousef, Electroinitiated Cationic Polymerization of P-Chloromethyl Styrene Using Different Sacrificial Anode Materials in Nitromethane, Eur. Polym. J., 1998, 34, 637–643 CrossRef CAS.
- L. Sun and M. Mellah, Efficient Electrosynthesis of SmCl2, SmBr2, and Sm(OTf)2 from a “Sacrificial” Samarium Anode: Effect of nBu4NPF6 on the Reactivity, Organometallics, 2014, 33, 4625–4628 CrossRef CAS.
- L. Sun, K. Sahloul and M. Mellah, Use of Electrochemistry to Provide Efficient SmI2 Catalytic System for Coupling Reactions, ACS Catal., 2013, 3, 2568–2573 CrossRef CAS.
- K. Sahloul, L. Sun, A. Requet, Y. Chahine and M. Mellah, A Samarium “Soluble” Anode: A New Source of SmI2 Reagent for Electrosynthetic Application, Chem.–Eur. J., 2012, 18, 11205–11209 CrossRef CAS PubMed.
- L. Lu, J. C. Siu, Y. Lai and S. Lin, An Electroreductive Approach to Radical Silylation via the Activation of Strong Si–Cl Bond, J. Am. Chem. Soc., 2020, 142, 21272–21278 CrossRef CAS.
- S. Manabe, C. M. Wong and C. S. Sevov, Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine, J. Am. Chem. Soc., 2020, 142, 3024–3031 CrossRef CAS PubMed.
- C. Gosmini, J. Y. Nédélec and J. Périchon, Electrochemical Cross-Coupling between Functionalized Aryl Halides and 2-Chloropyrimidine or 2-Chloropyrazine Catalyzed by Nickel 2,2′-Bipyridine Complex, Tetrahedron Lett., 2000, 41, 201–203 CrossRef CAS.
- S. Oudeyer, E. Léonel, J. P. Paugam and J.-Y. Nédélec, Epoxide Formation by Indirect Electroreductive Coupling between Aldehydes or Ketones and Activated Gem-Dichloro Compounds, Synthesis, 2004, 389–400 CAS.
- R. T. Ribeiro, I. L. de Mattos, S. Sengmany, R. Barhdadi, E. Léonel, C. Cachet-Vivier and M. Navarro, Iron Role in the Electrochemical Cyclopropanation Reaction of Activated Olefins and Halogenated Compounds, Electrochim. Acta, 2011, 56, 7352–7360 CrossRef CAS.
- G. Silvestri, S. Gambino, G. Filardo, G. Greco and A. Gulotta, Electrochemical Carboxylation of Benzal Chloride, Tetrahedron Lett., 1984, 25, 4307–4308 CrossRef CAS.
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. D. Vos, Electrocarboxylation: Towards Sustainable and Efficient Synthesis of Valuable Carboxylic Acids, Beilstein J. Org. Chem., 2014, 10, 2484–2500 CrossRef.
- M. Wang, C. Zhang, C. Ci, H. Jiang, P. H. Dixneuf and M. Zhang, Room Temperature Construction of Vicinal Amino Alcohols via Electroreductive Cross-Coupling of N-Heteroarenes and Carbonyls, J. Am. Chem. Soc., 2023, 145, 10967–10973 CrossRef CAS.
- G. Hilt and K. I. Smolko, Electrochemical Regeneration of Low-Valent Indium(I) Species as Catalysts for C-C Bond Formations, Angew. Chem., Int. Ed., 2001, 40, 3399–3402 CrossRef CAS.
- N. W. J. Ang and L. Ackermann, Electroreductive Nickel-Catalyzed Thiolation: Efficient Cross-Electrophile Coupling for C-S Formation, Chem.–Eur. J., 2021, 27, 4883–4887 CrossRef CAS PubMed.
- T. Leisegang, F. Meutzner, M. Zschornak, W. Münchgesang, R. Schmid, T. Nestler, R. A. Eremin, A. A. Kabanov, V. A. Blatov and D. C. Meyer, The Aluminum-Ion Battery: A Sustainable and Seminal Concept?, Front. Chem., 2019, 7, 268 CrossRef CAS.
- S. S. Kim and K. A. See, Activating Magnesium Electrolytes through Chemical Generation of Free Chloride and Removal of Trace Water, ACS Appl. Mater. Interfaces, 2021, 13, 671–680 CrossRef CAS.
- J. Ghijsen, H. Namba, P. A. Thiry, J. J. Pireaux and P. Caudano, Adsorption of Oxygen on the Magnesium (0001) Surface Studied by XPS, Appl. Surf. Sci., 1981, 8, 397–411 CrossRef CAS.
- P. A. Thiry, J. Ghijsen, R. Sporken, J. J. Pireaux, R. L. Johnson and R. Caudano, Incipient Oxidation of Magnesium: A High-Resolution Electron-Energy-Loss and Photoemission Study, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 3620–3631 CrossRef CAS PubMed.
- S. He, J. Luo and T. Leo Liu, MgCl 2/AlCl 3 Electrolytes for Reversible Mg Deposition/Stripping: Electrochemical Conditioning or Not?, J. Mater. Chem. A, 2017, 5, 12718–12722 RSC.
- I. Shterenberg, M. Salama, H. D. Yoo, Y. Gofer, J.-B. Park, Y.-K. Sun and D. Aurbach, Evaluation of (CF3SO2)2N- (TFSI) Based Electrolyte Solutions for Mg Batteries, J. Electrochem. Soc., 2015, 162, A7118 CrossRef CAS.
- Z. W. B. Iton and K. A. See, Multivalent Ion Conduction in Inorganic Solids, Chem. Mater., 2022, 34, 881–898 CrossRef CAS.
- H. J. T. Ellingham, Reducibility of Oxides and Sulphides in Metallurgical Processes, J. Soc. Chem. Ind., 1944, 63, 125–160 CrossRef CAS.
- J. G. Connell, B. Genorio, P. P. Lopes, D. Strmcnik, V. R. Stamenkovic and N. M. Markovic, Tuning the Reversibility of Mg Anodes via Controlled Surface Passivation by H2O/Cl– in Organic Electrolytes, Chem. Mater., 2016, 28, 8268–8277 CrossRef CAS.
- J. H. Nordlien, S. Ono, N. Masuko and K. Nisancioglu, A TEM Investigation of Naturally Formed Oxide Films on Pure Magnesium, Corros. Sci., 1997, 39, 1397–1414 CrossRef CAS.
- T. J. DeLano and S. E. Reisman, Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis, ACS Catal., 2019, 9, 6751–6754 CrossRef CAS PubMed.
- I. Shterenberg, M. Salama, Y. Gofer and D. Aurbach, Hexafluorophosphate-Based Solutions for Mg Batteries and the Importance of Chlorides, Langmuir, 2017, 33, 9472–9478 CrossRef CAS.
- S. J. Harwood, M. D. Palkowitz, C. N. Gannett, P. Perez, Z. Yao, L. Sun, H. D. Abruña, S. L. Anderson and P. S. Baran, Modular Terpene Synthesis Enabled by Mild Electrochemical Couplings, Science, 2022, 375, 745–752 Search PubMed.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, On the Electrochemical Behavior of Magnesium Electrodes in Polar Aprotic Electrolyte Solutions, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef CAS.
- H. Shuai, J. Xu and K. Huang, Progress in Retrospect of Electrolytes for Secondary Magnesium Batteries, Coord. Chem. Rev., 2020, 422, 213478 CrossRef CAS.
- H. D. Yoo, S.-D. Han, I. L. Bolotin, G. M. Nolis, R. D. Bayliss, A. K. Burrell, J. T. Vaughey and J. Cabana, Degradation Mechanisms of Magnesium Metal Anodes in Electrolytes Based on (CF3SO2)2N– at High Current Densities, Langmuir, 2017, 33, 9398–9406 CrossRef CAS PubMed.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Anode-Electrolyte Interfaces in Secondary Magnesium Batteries, Joule, 2019, 3, 27–52 CrossRef CAS.
- W. Zhang, W. Guan, Y. Wang, S. Lin and K. A. See, Enabling Al Sacrificial Anodes in Tetrahydrofuran Electrolytes for Reductive Electrosynthesis, Chem. Sci., 2023, 14, 13108–13118 RSC.
- R. Dugas, J. D. Forero-Saboya and A. Ponrouch, Methods and Protocols for Reliable Electrochemical Testing in Post-Li Batteries (Na, K, Mg, and Ca), Chem. Mater., 2019, 31, 8613–8628 CrossRef CAS PubMed.
- C. A. Paddon and R. G. Compton, A Reference Electrode for Electrochemical and Cryoelectrochemical Use in Tetrahydrofuran Solvent, Electroanalysis, 2005, 17, 1919–1923 CrossRef CAS.
- F. M. Schuett, S. J. Zeller, M. J. Eckl, F. M. Matzik, M.-K. Heubach, T. Geng, J. M. Hermann, M. Uhl, L. A. Kibler, A. K. Engstfeld and T. Jacob, Versatile 3D-Printed Micro-Reference Electrodes for Aqueous and Non-Aqueous Solutions, Angew. Chem., 2021, 133, 22965–22972 CrossRef.
- M. Ciobanu, J. P. Wilburn and D. A. Lowy, Miniaturized Reference Electrodes. II. Use in Corrosive, Biological, and Organic Media, Electroanalysis, 2004, 16, 1351–1358 CrossRef CAS.
- S. Wang, J. Zhang, O. Gharbi, V. Vivier, M. Gao and M. E. Orazem, Electrochemical Impedance Spectroscopy, Nat. Rev. Methods Primers, 2021, 1, 1–21 CrossRef.
- W. Zhang, C. Gu, Y. Wang, S. D. Ware, L. Lu, S. Lin, Y. Qi and K. A. See, Improving the Mg Sacrificial Anode in Tetrahydrofuran for Synthetic Electrochemistry by Tailoring Electrolyte Composition, JACS Au, 2023, 3, 2280–2290 CrossRef CAS.
- W. Zhang, L. Lu, W. Zhang, Y. Wang, S. D. Ware, J. Mondragon, J. Rein, N. Strotman, D. Lehnherr, K. A. See and S. Lin, Electrochemically Driven Cross-Electrophile Coupling of Alkyl Halides, Nature, 2022, 604, 292–297 CrossRef CAS PubMed.
- E. McCafferty, Sequence of Steps in the Pitting of Aluminum by Chloride Ions, Corros. Sci., 2003, 45, 1421–1438 CrossRef CAS.
- N. R. Levy, M. Auinat and Y. Ein-Eli, Tetra-Butyl Ammonium Fluoride – An Advanced Activator of Aluminum Surfaces in Organic Electrolytes for Aluminum-Air Batteries, Energy Storage Mater., 2018, 15, 465–474 CrossRef.
- S. S. A. Rehim, H. H. Hassan and M. A. Amin, Corrosion and Corrosion Inhibition of Al and Some Alloys in Sulphate Solutions Containing Halide Ions Investigated by an Impedance Technique, Appl. Surf. Sci., 2002, 187, 279–290 CrossRef CAS.
- A. A. Mazhar, W. A. Badawy and M. M. Abou-Romia, Impedance Studies of Corrosion Resistance of Aluminium in Chloride Media, Surf. Coat. Technol., 1986, 29, 335–345 CrossRef CAS.
- W. Li, T. Cochell and A. Manthiram, Activation of Aluminum as an Effective Reducing Agent by Pitting Corrosion for Wet-chemical Synthesis, Sci. Rep., 2013, 3, 1229 CrossRef.
- P. M. Natishan and W. E. O'Grady, Chloride Ion Interactions with Oxide-Covered Aluminum Leading to Pitting Corrosion: A Review, J. Electrochem. Soc., 2014, 161, C421 CrossRef CAS.
- X. Li, T. Gao, F. Han, Z. Ma, X. Fan, S. Hou, N. Eidson, W. Li and C. Wang, Reducing Mg Anode Overpotential via Ion Conductive Surface Layer Formation by Iodine Additive, Adv. Energy Mater., 2018, 8, 1701728 CrossRef.
- S.-D. Han, N. N. Rajput, X. Qu, B. Pan, M. He, M. S. Ferrandon, C. Liao, K. A. Persson and A. K. Burrell, Origin of Electrochemical, Structural, and Transport Properties in Nonaqueous Zinc Electrolytes, ACS Appl. Mater. Interfaces, 2016, 8, 3021–3031 CrossRef CAS PubMed.
- M. S. Chae, J. W. Heo, H. H. Kwak, H. Lee and S.-T. Hong, Organic Electrolyte-Based Rechargeable Zinc-Ion Batteries Using Potassium Nickel Hexacyanoferrate as a Cathode Material, J. Power Sources, 2017, 337, 204–211 CrossRef CAS.
- Y. Cheng, L. Luo, L. Zhong, J. Chen, B. Li, W. Wang, S. X. Mao, C. Wang, V. L. Sprenkle, G. Li and J. Liu, Highly Reversible Zinc-Ion Intercalation into Chevrel Phase Mo6S8 Nanocubes and Applications for Advanced Zinc-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 13673–13677 CrossRef CAS PubMed.
- D. W. Shoesmith, S. Sunder, M. G. Bailey, G. J. Wallace and F. W. Stanchell, Anodic Oxidation of Copper in Alkaline Solutions: Part IV. Nature of the Passivating Film, J. Electroanal. Chem. Interfacial Electrochem., 1983, 143, 153–165 CrossRef CAS.
- T. Vorauer, J. Schöggl, S. G. Sanadhya, M. Poluektov, W. D. Widanage, L. Figiel, S. Schädler, B. Tordoff, B. Fuchsbichler, S. Koller and R. Brunner, Impact of Solid-Electrolyte Interphase Reformation on Capacity Loss in Silicon-Based Lithium-Ion Batteries, Commun. Mater., 2023, 4, 1–12 CrossRef.
- L. Lu, Y. Wang, W. Zhang, W. Zhang, K. A. See and S. Lin, Three-Component Cross-Electrophile Coupling: Regioselective Electrochemical Dialkylation of Alkenes, J. Am. Chem. Soc., 2023, 145, 22298–22304 CrossRef CAS PubMed.
- C. Huang, W. Ma, X. Zheng, M. Xu, X. Qi and Q. Lu, Epoxide Electroreduction, J. Am. Chem. Soc., 2022, 144, 1389–1395 CrossRef CAS.
- R. J. Perkins, D. J. Pedro and E. C. Hansen, Electrochemical Nickel Catalysis for Sp2-Sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides, Org. Lett., 2017, 19, 3755–3758 CrossRef CAS.
- K. S. Han, N. T. Hahn, K. R. Zavadil, N. R. Jaegers, Y. Chen, J. Z. Hu, V. Murugesan and K. T. Mueller, Factors Influencing Preferential Anion Interactions during Solvation of Multivalent Cations in Ethereal Solvents, J. Phys. Chem. C, 2021, 125, 6005–6012 CrossRef CAS.
- H. Zhang, L. Qiao, H. Kühnle, E. Figgemeier, M. Armand and G. Gebresilassie Eshetu, From Lithium to Emerging Mono- and Multivalent-Cation-Based Rechargeable Batteries: Non-Aqueous Organic Electrolyte and Interphase Perspectives, Energy Environ. Sci., 2023, 16, 11–52 RSC.
- X. Liu, A. Du, Z. Guo, C. Wang, X. Zhou, J. Zhao, F. Sun, S. Dong and G. Cui, Uneven Stripping Behavior, an Unheeded Killer of Mg Anodes, Adv. Mater., 2022, 34, 2201886 CrossRef CAS PubMed.
- P. He and J. Huang, Detrimental Effects of Surface Imperfections and Unpolished Edges on the Cycling Stability of a Zinc Foil Anode, ACS Energy Lett., 2021, 6, 1990–1995 CrossRef CAS.
- Q. Zeng and S. Xu, Thermodynamics and Characteristics of Heterogeneous Nucleation on Fractal Surfaces, J. Phys. Chem. C, 2015, 119, 27426–27433 CrossRef CAS.
- Y. Shao, T. Liu, G. Li, M. Gu, Z. Nie, M. Engelhard, J. Xiao, D. Lv, C. Wang, J.-G. Zhang and J. Liu, Coordination Chemistry in Magnesium Battery Electrolytes: How Ligands Affect Their Performance, Sci. Rep., 2013, 3, 3130 CrossRef PubMed.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, Theoretical Analysis on De-Solvation of Lithium, Sodium, and Magnesium Cations to Organic Electrolyte Solvents, J. Electrochem. Soc., 2013, 160, A2160 CrossRef CAS.
- T. S.-B. Lou, Y. Kawamata, T. Ewing, G. A. Correa-Otero, M. R. Collins and P. S. Baran, Scalable, Chemoselective Nickel Electrocatalytic Sulfinylation of Aryl Halides with SO2, Angew. Chem., Int. Ed., 2022, 61, e202208080 CrossRef CAS PubMed.
- R. Li, Y. Li, R. Zhang, M. He, Y. Ma, H. Huo, P. Zuo and G. Yin, Voltage Hysteresis of Magnesium Anode: Taking Magnesium-Sulfur Battery as an Example, Electrochim. Acta, 2021, 369, 137685 CrossRef CAS.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, Reductive Electrophotocatalysis: Merging Electricity and Light To Achieve Extreme Reduction Potentials, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- W. Zhang, W. Guan, J. I. Martinez Alvarado, L. F. T. Novaes and S. Lin, Deep Electroreductive Chemistry: Harnessing Carbon- and Silicon-Based Reactive Intermediates in Organic Synthesis, ACS Catal., 2023, 13, 8038–8048 CrossRef CAS.
- G.-Q. Yuan, H.-F. Jiang, C. Lin and S.-J. Liao, Efficient Electrochemical Synthesis of 2-Arylsuccinic Acids from CO2 and Aryl-Substituted Alkenes with Nickel as the Cathode, Electrochim. Acta, 2008, 53, 2170–2176 CrossRef CAS.
- W. Zhang and S. Lin, Electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed.
- C. Déjardin, A. Renou, J. Maddaluno and M. Durandetti, Nickel-Catalyzed Electrochemical Cyclization of Alkynyl Aryl Iodide and the Domino Reaction with Aldehydes, J. Org. Chem., 2021, 86, 8882–8890 CrossRef PubMed.
- M. Durandetti, C. Meignein and J. Périchon, Iron-Mediated Electrochemical Reaction of α-Chloroesters with Carbonyl Compounds, Org. Lett., 2003, 5, 317–320 CrossRef CAS PubMed.
- V. Courtois, R. Barhdadi, M. Troupel and J. Périchon, Electroreductive Coupling of Organic Halides in Alcoholic Solvents. An Example: The Electrosynthesis of Biaryls Catalysed by Nickel-2,2′ Bipyridine Complexes, Tetrahedron, 1997, 53, 11569–11576 CrossRef CAS.
- M. Ishifune, H. Yamashita, Y. Kera, N. Yamashita, K. Hirata, H. Murase and S. Kashimura, Electroreduction of Aromatics Using Magnesium Electrodes in Aprotic Solvents Containing Alcoholic Proton Donors, Electrochim. Acta, 2003, 48, 2405–2409 CrossRef CAS.
- J.-M. Huang and Y. Dong, Zn-Mediated Electrochemical Allylation of Aldehydes in Aqueous Ammonia, Chem. Commun., 2009, 3943–3945 RSC.
- J.-M. Huang, Z.-Q. Lin and D.-S. Chen, Electrochemically Supported Deoxygenation of Epoxides into Alkenes in Aqueous Solution, Org. Lett., 2012, 14, 22–25 CrossRef CAS PubMed.
- J.-M. Huang, X.-X. Wang and Y. Dong, Electrochemical Allylation Reactions of Simple Imines in Aqueous Solution Mediated by Nanoscale Zinc Architectures, Angew. Chem., Int. Ed., 2011, 50, 924–927 CrossRef CAS PubMed.
- C. Gosmini, S. Lasry, J.-Y. Nedelec and J. Perichon, Electrochemical Cross-Coupling between 2-Halopyridines and Aryl or Heteroaryl Halides Catalysed by Nickel-2,2′-Bipyridine Complexes, Tetrahedron, 1998, 54, 1289–1298 CrossRef CAS.
- H. Senboku, K. Sakai, A. Fukui, Y. Sato and Y. Yamauchi, Efficient Synthesis of Mandel Acetates by Electrochemical Carboxylation of Benzal Diacetates, ChemElectroChem, 2019, 6, 4158–4164 CrossRef CAS.
- M. Dörr, M. M. Hielscher, J. Proppe and S. R. Waldvogel, Electrosynthetic Screening and Modern Optimization Strategies for Electrosynthesis of Highly Value-added Products, ChemElectroChem, 2021, 8, 2621–2629 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |