
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoryls, their compounds and reactivity: a structure and bonding perspective
Xueying
Guo
 and
Zhenyang
Lin
*
and
Zhenyang
Lin
*
Department of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong. E-mail: chzlin@ust.hk
First published on 1st February 2024
Abstract
Boryls and their compounds are important due to their diverse range of applications in the fields of materials science and catalysis. They are an integral part of boron chemistry, which has attracted tremendous research interest over the past few decades. In this perspective, we provide an in-depth analysis of the reaction chemistry of boryl compounds from a structure and bonding perspective. We discuss the reactivity of boryls in various transition metal complexes and diborane(4) compounds towards different substrate molecules, with a focus on their nucleophilic and electrophilic properties in various reaction processes. Additionally, we briefly discuss the reactivity of boryl radicals. Our analysis sheds new light on the unique properties of boryls and their potential for catalytic applications.
 Xueying Guo | Xueying Guo received her BSc degree from Nankai University in 2017 and obtained her PhD degree in 2021 under the supervision of Professor Zhenyang Lin at The Hong Kong University of Science and Technology, where she focused on theoretical studies of reactivity of TM-boryl complexes and diborane(4) compounds. After completing her doctoral studies, she pursued her postdoctoral research at the same group until May 2022. Following that, she moved to Shanghai Institute of Organic Chemistry to continue her postdoctoral research. In 2024, she will assume a faculty position at Lanzhou University. Her current research focuses on theoretical investigations in main-group chemistry and organometallic chemistry. |
 Zhenyang Lin | Zhenyang Lin received his BSc degree in Chemistry from China University of Geosciences in 1982 and obtained his PhD degree in 1989 under the supervision of Professor Michael P. Mingos at the University of Oxford and carried out his postdoctoral work with Michael B. Hall at Texas A&M University. He joined the Hong Kong University of Science and Technology in 1991 and served as the Department Head of Chemistry from January 2011 to August 2018. He is now a Chair Professor of the university. His research interests include the theoretical aspects of structure, bonding and reactivity of inorganic and organometallic compounds, and homogeneous catalysis. |
1. Introduction
In the vast realm of chemistry, the exploration of novel compounds and their reactivity has been a driving force for many ground-breaking chemistry discoveries. One such area of current intense research is the field of boryls and their compounds, which have gained significant attention due to their unique reactivity.1–5 The study of boryl compounds is expected to pave the way for the development of important applications in catalysis, materials science, and pharmaceuticals.6–12A boryl group refers to a boron centre with two substituents attached to it. The substituents attached to the boron centre can be diverse, including alkyl, aryl, or heteroaryl groups, as well as other functional groups such as halides or alkoxides.13–17Fig. 1 shows the various boryl compounds where the boron centre is typically three-coordinate. Here, we are interested mainly in metal boryl complexes (Fig. 1(b)) and diborane(4) compounds (Fig. 1(c)). The reactivity of organoboron compounds (Fig. 1(a)) is mainly related to synthetic chemistry aspects, such as cross-coupling reactions, and is not the focus of this perspective.18–22
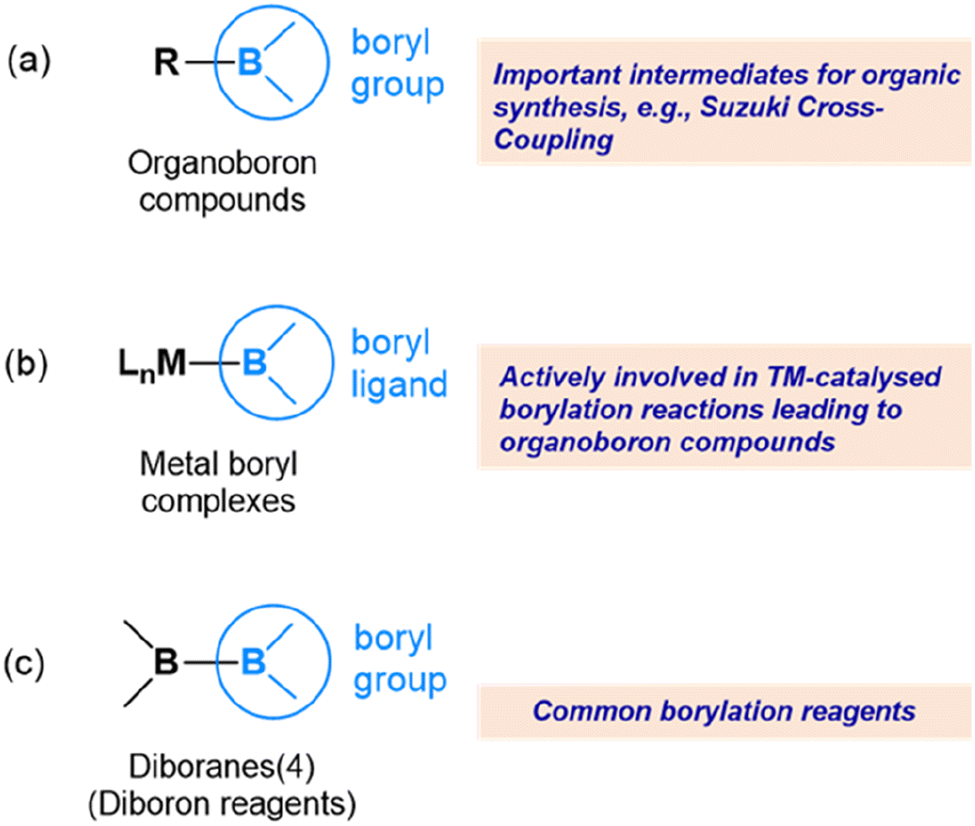 | ||
| Fig. 1 Boryls in various compounds. (a) Organoboron compounds with a boryl group bonded to an organic group. (b) Metal boryl complexes featuring a boryl ligand coordinated to a metal centre. (c) Diborane(4) compounds showcasing two boryl groups bonded via a B–B σ bond. | ||
The reactivity of boryls is a fascinating aspect, characterized by diverse reactivity patterns such as nucleophilic, electrophilic, and radical reactions. Their potential for undergoing various transformations offers tremendous prospects for applications in catalysis. In this article, our focus will be on exploring the versatile reaction chemistry of transition metal boryl complexes and diborane(4) compounds, highlighting their key reactions. It is important to note that this article does not aim to provide a comprehensive review of the topic. Instead, it aims to emphasize crucial points related to the discussed aspects and offer insights into understanding them from a structure and bonding perspective.
2. General aspect
As mentioned above, boryl compounds feature a three-coordinate boron centre. Consequently, the reactivity of boryl compounds is often attributed to the formally “empty” pz orbital on the boron centre (where z is defined along the direction normal to the trigonal plane around the boron centre). This orbital exhibits Lewis acidity and tends to preferentially react with Lewis bases or nucleophiles. This behaviour is particularly pronounced in organoboron compounds (Fig. 1(a)), where the boryl is bonded to an organic group, R.In transition metal boryl complexes and diborane(4) compounds, the boryl boron, being an electropositive element, forms a σ bond with another electropositive element. Consequently, the σ bonding pair of electrons between the boryl group and the electropositive element is likely to play an active role. This leads to various reaction scenarios. Fig. 2 illustrates the three possible reaction scenarios when a boryl group reacts with a substrate molecule. If the σ bonding pair of electrons is associated with the boryl during a reaction, a nucleophilic boryl is formed (Scenario 1). Conversely, if the σ bonding pair of electrons is associated with the other electropositive element, an electrophilic boryl is expected (Scenario 2). Lastly, if the σ bonding pair undergoes homolytic cleavage, radical chemistry may occur (Scenario 3).
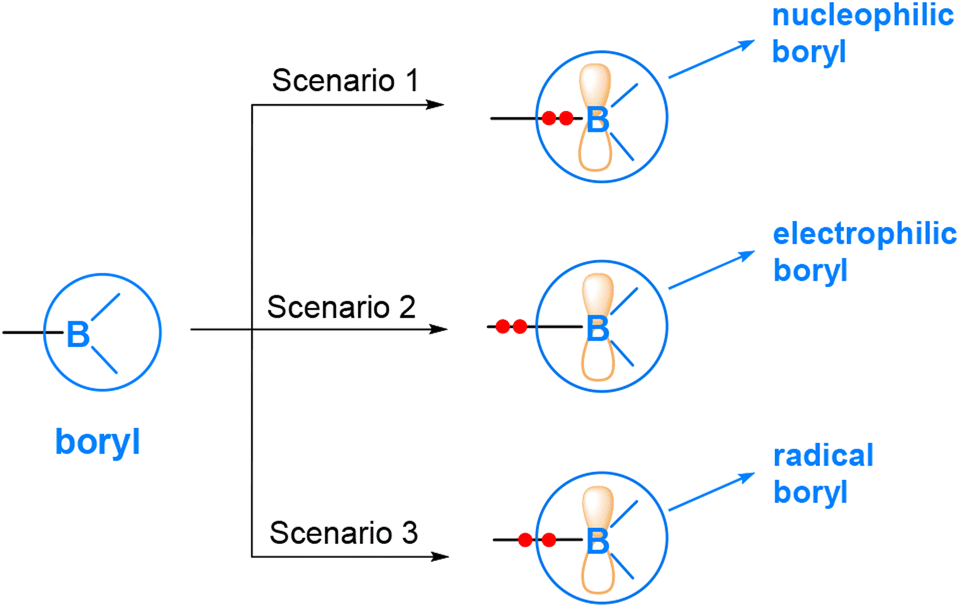 | ||
| Fig. 2 Three possible reaction scenarios when a boryl group reacts with a substrate molecule. | ||
Given the large amount of theoretical work discussed in this perspective, it is necessary to briefly comment on the theoretical methods used in these studies. Density functional theory calculations at various levels have been commonly employed. While the B3LYP functional was often used for both metal and non-metal systems, other functionals such as BP86, ωB97XD and M062X were also popularly used in studies related to reactions of boryls. However, we would like our readers to note that when our focus is on the insights related to the structure and bonding aspects, we can obtain qualitatively similar conclusions regardless of which method is used.
3. Nucleophilic boryls
3.1 Boryl anions
In 2006, Yamashita, Nozaki and their co-workers reported the isolation of the first structurally characterized lithium salt of a boryl anion, LiB(RNCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHNR) (R = 2,6-iPr2C6H3).23 In addition, the magnesium, zinc, calcium, strontium and potassium salts of boryl anions have also been synthesized and structurally characterized.24–26 In these compounds, the boron centre displays highly anionic character.
CHNR) (R = 2,6-iPr2C6H3).23 In addition, the magnesium, zinc, calcium, strontium and potassium salts of boryl anions have also been synthesized and structurally characterized.24–26 In these compounds, the boron centre displays highly anionic character.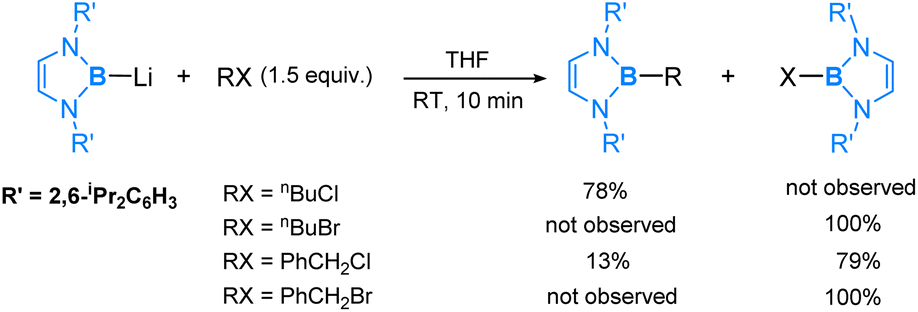 | (1) |
Reactivity studies of the lithium boryl compound show that the boryl anion reacts with a variety of electrophiles such as aldehydes, ketones and carbon dioxide, indicating that the boryl anion is a strong nucleophile.27,28 This is expected in view of the fact that boron is an electropositive element and a lone pair of electrons on the boron centre should be strongly nucleophilic. The reactivity of lithium boryl extends to its reactions with organohalides, as reported in the literature.29 In eqn (1), we present the noteworthy experimental results obtained from the reactions of the lithium boryl with a few representative organohalides (RX). The resulting products, R-boryl, arise from an SN2 substitution wherein the boryl anion replaces the halide, confirming the expected nucleophilic nature of the boryl anion. Interestingly, we also observed halogen-abstraction products, X-boryl, which highlight the reducing capability of the boryl anion, a property not typically associated with carbon nucleophiles. Our DFT studies indicate that these halogen-abstraction products are kinetic products. However, we predict that with prolonged reaction time, the anticipated SN2 substitution products will be eventually formed.30
Fig. 3 illustrates the transition state structures for the two competing pathways. The halogen-abstraction transition state in fact resembles an SN2 transition state occurring at the halogen centre. Thus, an organohalide having a halogen with lower electronegativity and higher capacity for engaging in hypervalent bonding promotes the halogen abstraction pathway. This preference stems from the boryl anion's softer nucleophilic character compared to a carbanion, attributed to the more electropositive boron. Hence, the boryl anion exhibits a greater tendency to attack heavier halide X rather than the organic moiety R in an RX compound.
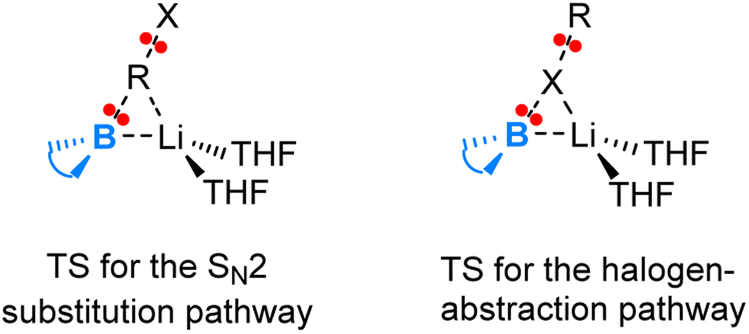 | ||
| Fig. 3 Sketches of the transition state structures for the two competing pathways (SN2 versus halogen-abstraction) in the reaction of lithium boryl with organohalides. | ||
It is also worth noting that McMullin, Hill and co-workers recently reported the synthesis of magnesium(II) diboronate and boryl complexes (Fig. 4), which could deliver a Bpin anion to react with various electrophiles such as organohalides, organic nitriles and carbonyl electrophiles.31–36
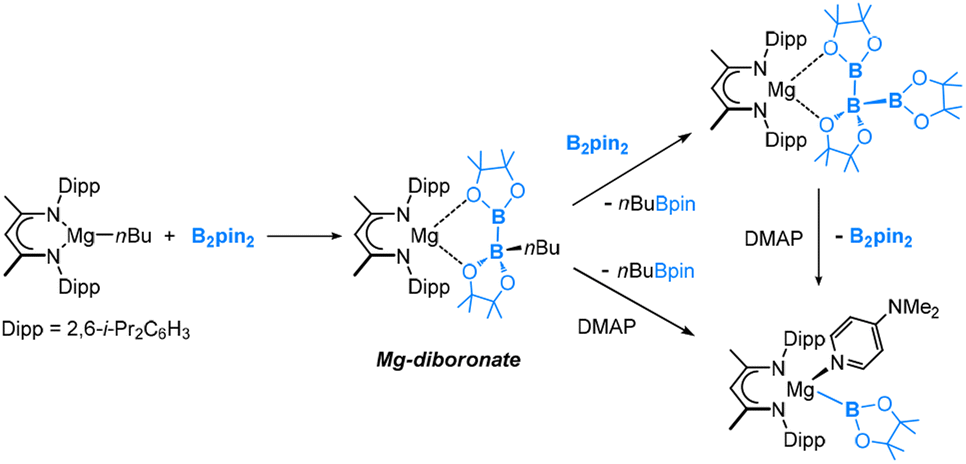 | ||
| Fig. 4 Synthetic routes of magnesium(II) diboronate and boryl complexes. | ||
3.2 Copper(I) boryl complexes
Our interest in the reaction chemistry of boryls dates back to 2006 and 2008 when we conducted investigations into the reaction mechanisms of two closely related reactions. These two reactions employed copper(I) boryl complexes as catalysts and B2pin2 as the borylation reagents, as illustrated in eqn (2) and (3).37–40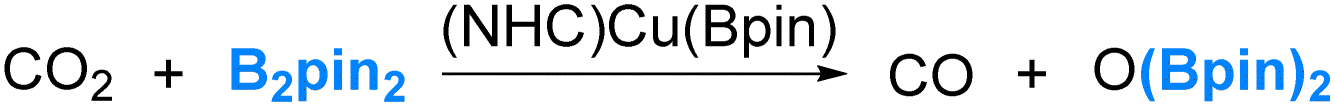 | (2) |
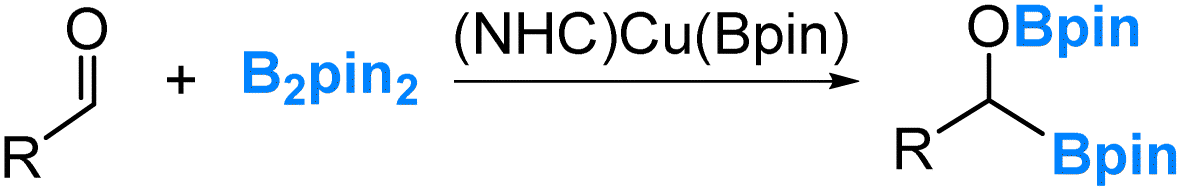 | (3) |
Our studies have led us to establish the nucleophilic nature of the boryl ligands (Scenario 1 in Fig. 2) in these copper(I) boryl complexes,6,37,38,41–43 a property suggested by Miyaura and co-workers as early as 2000.44 Taking the diboration reaction of aldehydes (eqn (2)) as an example, we obtained a mechanism, as depicted in Scheme 1, through our detailed DFT calculations. The mechanism involves aldehyde coordination, insertion and metathesis steps.38
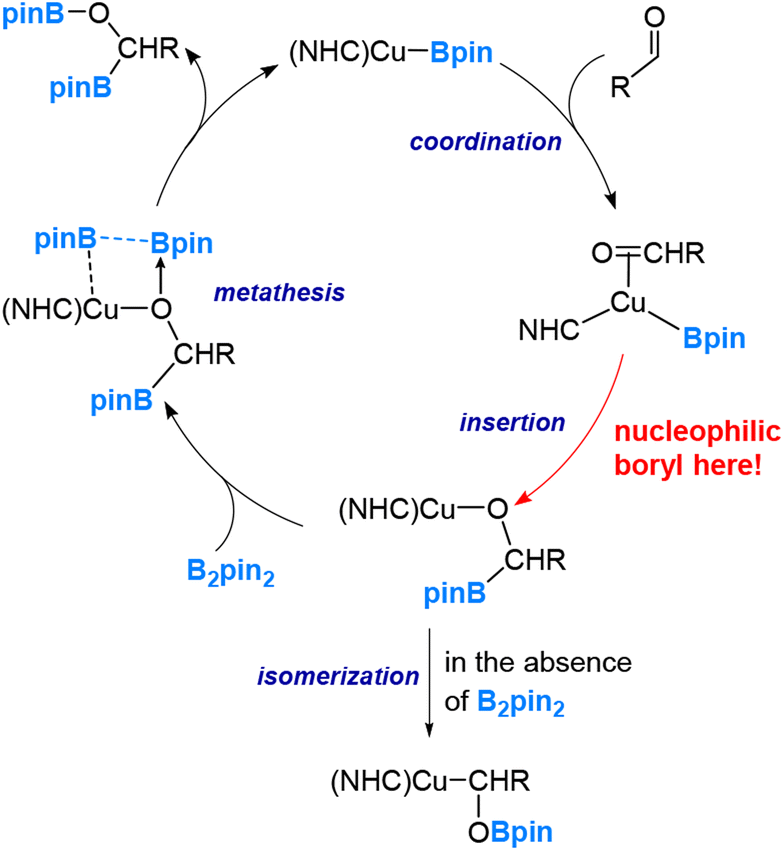 | ||
| Scheme 1 Mechanism of the Cu-boryl-catalyzed diboration reaction of aldehydes. | ||
Notably, the aldehyde insertion was calculated to be the rate-determining step, wherein the boryl ligand acts as a nucleophile attacking the aldehyde carbon centre to form a Cu–O–C–B linkage in the insertion ‘product’. Additionally, our findings indicate that the alternative insertion product, featuring a Cu–C–O–B linkage, observed in the stoichiometric reaction of (NHC)CuBpin with ArCHO in the absence of B2pin2, is a result of isomerization from the favourable insertion “product” the Cu–O–C–B linkage (Scheme 1).
The calculation results demonstrate that the alternative insertion, leading to the formation of the Cu–C–O–B linkage in the insertion ‘product’, presents a substantially higher barrier of 32.1 kcal mol−1, in contrast to the favourable insertion that results in the Cu–O–C–B linkage with a calculated barrier of 11.1 kcal mol−1. Please note that these values were obtained through calculations utilizing formaldehyde, a model aldehyde.38
The pronounced preference of the boryl ligand to attack the carbon centre rather than the oxygen centre of the aldehyde strongly supports the idea of a highly nucleophilic boryl ligand in copper(I) boryl complexes. The nucleophilic properties of boryl ligands in copper-boryl complexes have also been used to explain the experimentally observed regioselectivity in reactions related to substituted olefins42 and α,β-unsaturated carbonyl compounds.43Fig. 5 provides an illustration showing that the Cu–B σ bonding pair of electrons, which occupies the HOMO of the copper(I) boryl complex, is actively involved in the insertion step, resulting in a nucleophilic boryl ligand, while the boron “empty” p orbital primarily serves to stabilize the oxygen lone pairs of electrons. Fig. 6 summarizes how a nucleophilic boryl initiates the reaction with a coordinated substrate, followed by further chemical transformation.
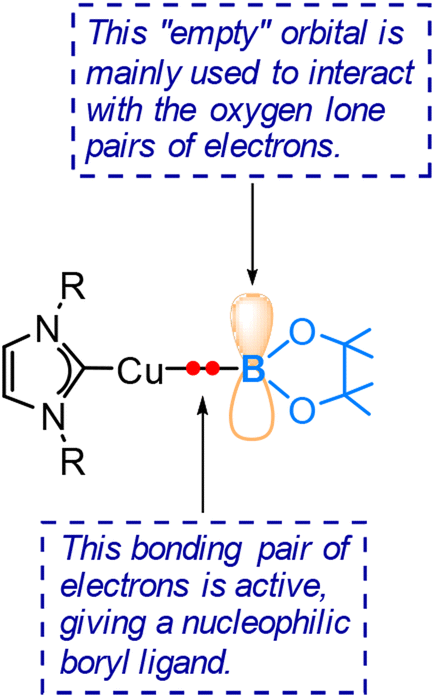 | ||
| Fig. 5 Illustration of the Cu–B σ bonding pair of electrons in boryl ligand nucleophilicity. | ||
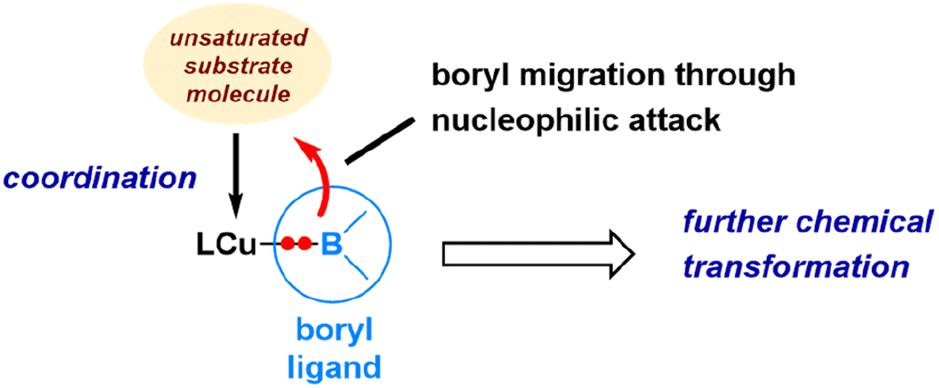 | ||
| Fig. 6 Illustration of how a nucleophilic boryl initiates and is involved in the activation of unsaturated molecules. | ||
4. Electrophilic boryls
4.1 Platinum-catalysed 1,4-diboration of acyclic α,β-unsaturated carbonyl compounds
Electrophilic boryl was first found in Pt-boryl species. In 2012, we carried out a detailed mechanistic study using DFT calculations on the diboration of acyclic α,β-unsaturated carbonyl compounds.45 This reaction was catalyzed by a Pt(0) complex (eqn (4)), as reported by Norman, Marder and co-workers.46 Our investigation revealed that a platinum diboryl species, formed through the oxidative addition of the diboron reagent to the Pt(0) complex, serves as the active species. In this active species, one of the two boryl ligands exhibits electrophilic behaviour by attacking the carbonyl oxygen of a coordinated carbonyl substrate.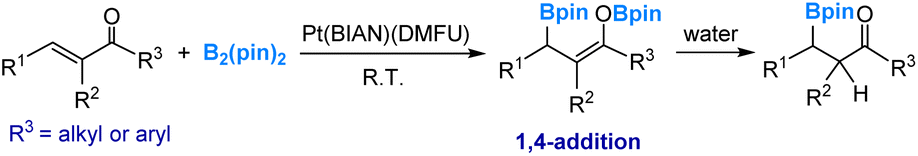 | (4) |
The DFT-derived mechanism, based on the model species MeNCHCHNMe, acrolein and B2(Beg)2, as illustrated in Scheme 2, involves several key steps: oxidative addition of B2(Beg)2, boryl migratory insertion, coordination of acrolein, and subsequent reductive elimination, resulting in the formation of the 1,4-diboration product.
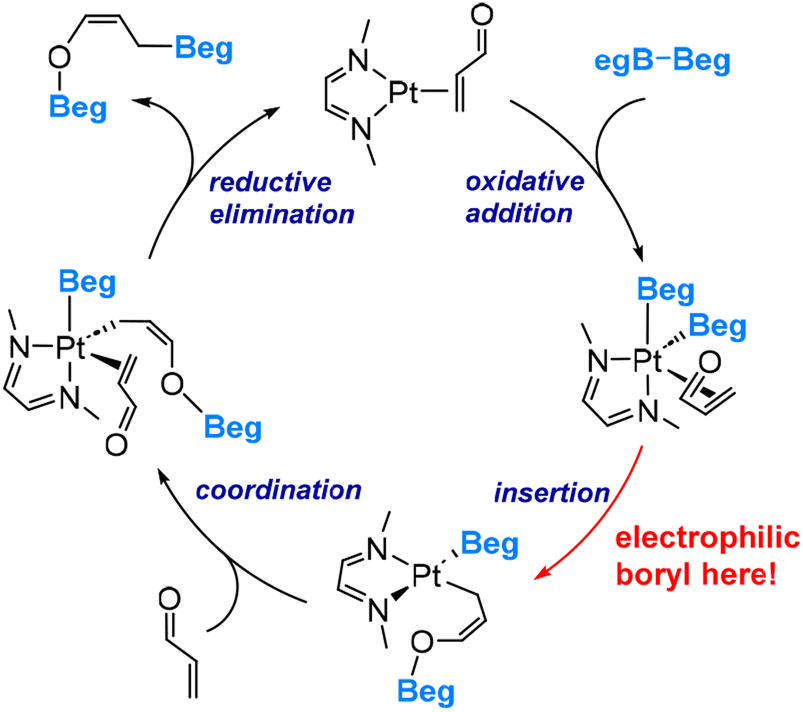 | ||
| Scheme 2 Mechanism of the Pt-boryl-catalyzed diboration reaction of acrolein. | ||
During the boryl migratory insertion, the migrating boryl ligand exhibits electrophilic behaviour by forming a bond with the carbonyl oxygen. The calculated overall insertion barrier is 18.6 kcal mol−1, compared to a substantially higher barrier of 35.7 kcal mol−1 calculated for the migration of the boryl ligand to the terminal carbon of the coordinated acrolein.45 The significant difference in migrating insertion modes highlights the highly electrophilic nature of the boryl ligands in this system (Scenario 2 shown in Fig. 2). It is noteworthy that the polarization of the Pt–boryl bond is primarily directed towards the Pt metal centre, while a Cu–boryl bond, as discussed above, is polarized toward the boron centre. This discrepancy can be attributed to the electronegativity difference between Pt (2.28) and Cu (1.90) versus B (2.04).47 The electronegativity values cited here used the Pauling scale. This observation further demonstrates that a change in the metal centre can switch between the first two scenarios depicted in Fig. 2.
4.2 Rhodium(I)-boryl catalysis
Recently, in collaboration with professor Wanxiang Zhao's research group, we conducted a combined experimental and computational investigation on the rhodium-catalysed deoxygenation and borylation of ketones (eqn (5)).48 Our study involved detailed mechanistic analyses, which provided support for a dual catalytic cycle. In the first cycle, ketones undergo a Rh-catalysed deoxygenation, yielding alkenes through boron enolate intermediates. Subsequently, in the second cycle, the Rh-catalysed dehydrogenative borylation of alkenes occurs, leading to the formation of vinylboronates and diboration products.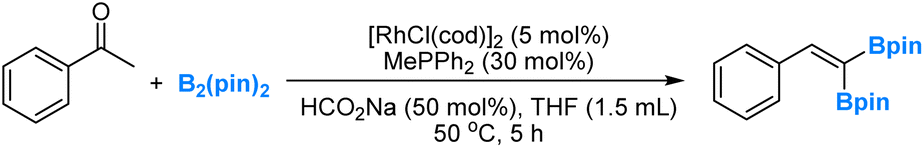 | (5) |
The catalytic cycles involve the active species L3Rh(I)–Bpin (L = phosphine), with the insertion of the ketone into the Rh(I)–Bpin bond as the rate-determining step. Interestingly, the electronegativity of Rh is also 2.28, suggesting that the Rh–Bpin bond is polarized towards the Rh metal centre. This polarization is evident in the rate-determining ketone insertion step (Scheme 3), where Bpin exhibits an electrophilic nature by migrating towards the ketone oxygen. Additionally, our findings indicate a strong back-bonding interaction between the d8 Rh(I) metal centre and the ketone carbon in the insertion transition state.
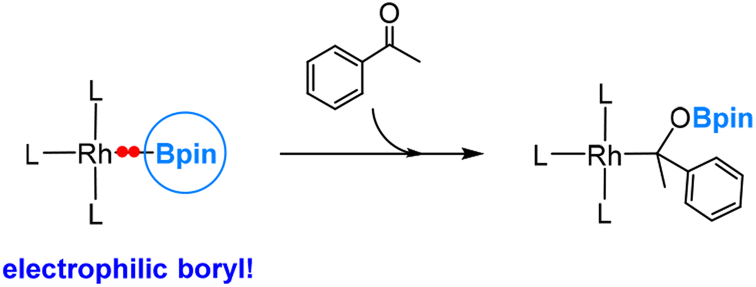 | ||
| Scheme 3 Insertion of a ketone into the Rh(I)–Bpin bond. | ||
4.3 Gold(I)-boryl complexes
In 2021, Yamashita and co-workers reported the synthesis of a gold(I)-diarylboryl complex and its reactions with different electrophiles, including aldehydes and ketones.49 An illustrative example of these reactions is presented in eqn (6). Through DFT calculations, it has been confirmed that the diarylboryl ligand displays an electrophilic nature, indicating a polarization of the Au–B bond towards Au. This observation aligns with our expectations, considering the electronegativity of Au, which is 2.54 on the Pauling scale.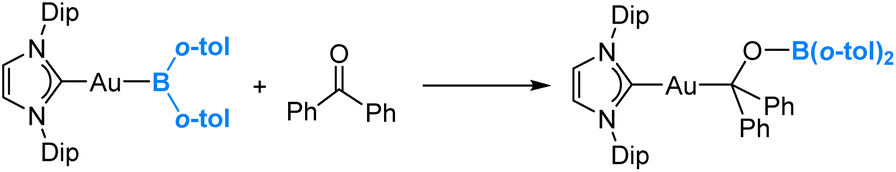 | (6) |
One may argue that the boron centre exhibits high Lewis acidity due to the limited interaction between the empty p orbital on the boron centre of the diarylboryl ligand and the occupied π bonding orbitals of the two aryl substituents. Indeed, our DFT calculations reveal the formation of an intermediate Lewis acid–base adduct, involving the interaction between the ketone oxygen and the boryl boron centre, preceding the insertion of the ketone into the Au(I)–B bond. Further DFT calculations revealed that the two substituents do affect the Lewis acidity of the boron centre in the boryl ligand (vide infra).50
5. Reactivity summary of different transition metal boryl complexes
As indicated in Sections 3 and 4, the electronegativity difference between the metal and boron centre determines the behaviour of the boryl ligands. Based on the discussion above, the following conclusions can be drawn. When the metal centre is more electropositive than boron, e.g., copper, the Cu–B σ-bonding pair of electrons is more polarized towards the boron centre, and the boryl ligand is endowed with nucleophilicity (Scenario 1 shown in Fig. 2), while when the metal centre is much more electronegative than boron, e.g., platinum, rhodium, and gold, the TM–B σ-bond is more polarized towards the TM centre, and the boryl ligand exhibits an electrophilic feature (Scenario 2 shown in Fig. 2). It is also worth mentioning here that, for a transition metal with a non-d10 configuration, the active “valence d electrons” on the metal centre can also play a nucleophilic role, consequently manifesting an electrophilic boryl.Recently, we conducted a computational investigation to explore the reactivity of various transition metal boryl complexes toward benzaldehyde.50 Our approach involved calculating the insertion barriers using DFT calculations. In Fig. 7, we summarize the reactivity patterns revealed through this computational study. Interestingly, we observed that the substituents on the boryl boron also influence the nature of the reactivity. As mentioned earlier, (NHC)CuBpin exhibits a nucleophilic boryl ligand,6 while L3Rh(boryl) (boryl = Bpin and BPh2) and (NHC)AuBPh2 display electrophilic boryl ligands.
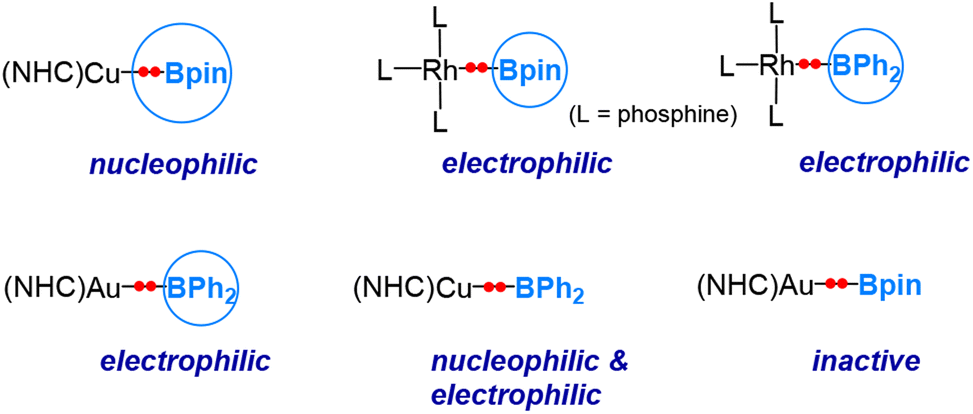 | ||
| Fig. 7 DFT-predicted reactivity of various transition metal boryl complexes towards benzaldehyde. | ||
What surprised us was the discovery that the boryl ligand in (NHC)CuBPh2 demonstrates both nucleophilic and electrophilic characteristics. This suggests that the boron's “empty” p orbital in this particular case plays a pivotal role in conferring the electrophilic nature. Furthermore, (NHC)AuBpin was found to exhibit neither electrophilic nor nucleophilic behaviour, which was unexpected.51
6. Diborane(4) compounds
Containing two 3-coordinated boron centres, diborane(4) (also called diboron(4)) compounds are highly reactive toward small molecule activations.5,52,53 Over the years (2014–2023), by collaborating with the research teams of Yamashita and Braunschweig, we have investigated the mechanism of reactions of various diboranes(4) with a range of unsaturated organic and inorganic molecules, including carbon monoxide, alkynes, nitriles, and azides.54–60 Through our DFT studies, we have elucidated the dual reactivity of diboranes(4), i.e., exhibiting both electrophilic and nucleophilic properties, and how this unique characteristic enables various transformations upon reacting with these molecules.The dual reactivity of diborane(4) is illustrated in Fig. 8. In the initial step, one of the boron centres in diborane(4) functions as a Lewis acidic centre, displaying its electrophilic nature by coordinating with an unsaturated molecule. Then, the remaining uncoordinated boryl moiety exhibits nucleophilic features to migrate as a nucleophile. Subsequently, a wide array of transformations becomes possible, depending upon the specific characteristics of unsaturated molecules involved in the reactions. To illustrate these processes, we will explore a few notable examples that exemplify the variety of transformations leading to distinct products.
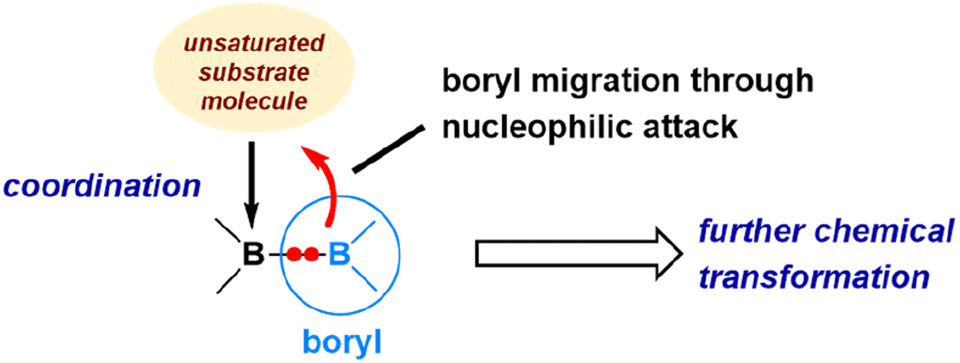 | ||
| Fig. 8 Illustration of the dual reactivity of diborane(4) toward the activation of unsaturated molecules. | ||
6.1 Reaction with CO
Carbon monoxide (CO) is a well-known nucleophile with its lone pair of electrons on the carbon atom. Its reaction with an unsymmetrical diborane(4) compound, pinB-BMes2, was observed to result in a product that involves the cleavage of the B–B bond and one B–C bond within the diborane(4) molecule, as shown in eqn (7).54 Based on our DFT calculations, we have identified the most favourable pathway, as shown in Scheme 4, for the reaction. This pathway entails the initial coordination of carbon monoxide CO to the more electrophilic BMes2 boron centre, followed by a nucleophilic 1,2-Mes migration. Subsequently, a second CO molecule coordinates, and finally, an electrophilic 1,3-Bpin migration occurs, resulting in the formation of the product.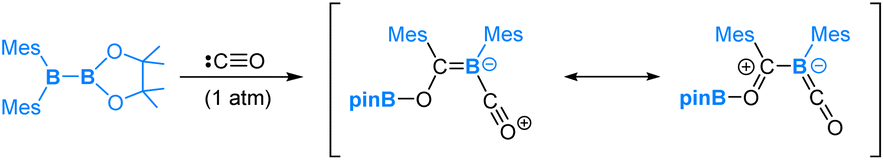 | (7) |
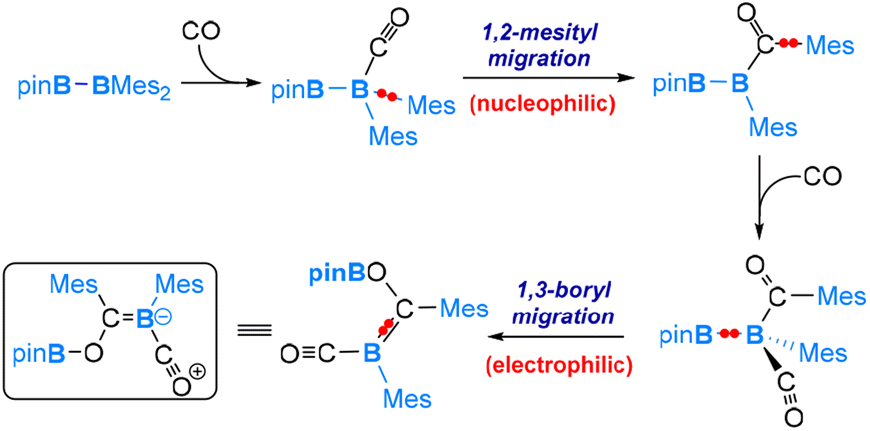 | ||
| Scheme 4 Mechanism of the reaction of pinB-BMes2 with CO. | ||
After the first CO molecule coordination, it is interesting to note that a nucleophilic migration of the Mes moiety takes place instead of the Bpin moiety (Scheme 4). Our DFT results indicate that the nucleophilic Bpin migration is only slightly less favourable. However, it is the subsequent step, characterized by an even higher barrier, that prevents this pathway from being the most favourable.
6.2 Reactions with R–CN and R–NC
Reactions of diborane(4) with organic molecules containing a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N functional group have also been experimentally and theoretically explored. For example, reactions of the unsymmetrical diborane(4) pinB-B(Mes)2 toward isocyanide Xyl-NC55 and the organic nitrile R–CN54,57 have been reported.
N functional group have also been experimentally and theoretically explored. For example, reactions of the unsymmetrical diborane(4) pinB-B(Mes)2 toward isocyanide Xyl-NC55 and the organic nitrile R–CN54,57 have been reported.
In the reaction of pinB-B(Mes)2 with 2,6-dimethylphenyl isocyanide (Xyl-NC), the addition of 1 equiv. of Xyl-NC led to the formation of a spirocyclic oxaboretane product, whereas the addition of 2 equiv. of Xyl-NC afforded an isocyanide-coordinated boraalkene compound, as illustrated in eqn (8).55
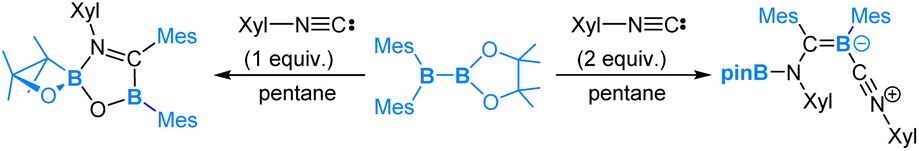 | (8) |
Our DFT calculations have revealed that for the reaction with 1 equivalent of Xyl-NC the mechanism (Scheme 5) begins with Xyl-NC coordinating to the more electrophilic BMes2 centre. This is followed by a nucleophilic 1,2-Bpin migration, then an electrophilic 1,2-Bpin migration. Subsequently, a nucleophilic 1,2-mesityl migration occurs to yield a boraalkene intermediate. Within this boraalkene intermediate, the oxygen lone pair of the pinacol moiety attacks the electrophilic 3-coordinated B-Mes boron centre, leading to the formation of a five-membered ring intermediate, from which an arrangement is followed to neutralize the formal charges and furnish the spirocyclic oxboretane product.
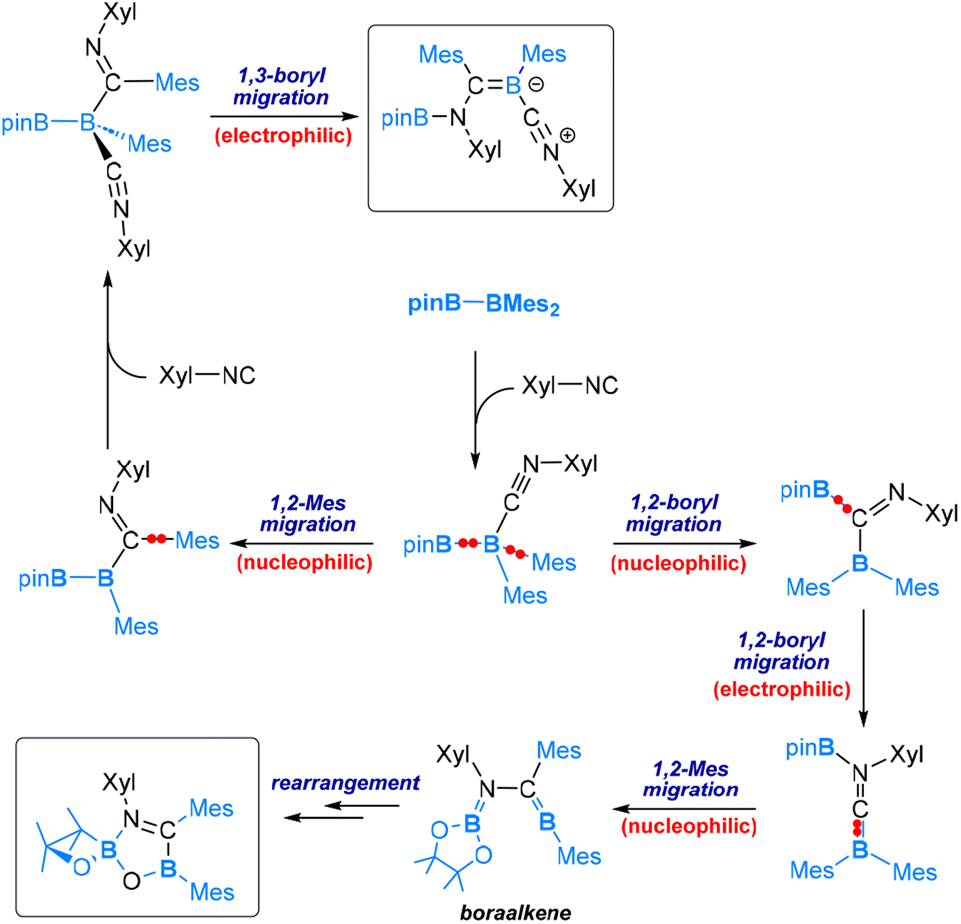 | ||
| Scheme 5 Mechanism of the reaction of pinB-BMes2 with 1 equiv. of Xyl-NC (bottom) and 2 equiv. of Xyl-NC (top). | ||
For the reaction with 2 equivalents of Xyl-NC, the mechanism resembles that of the reaction with CO. Coordination of Xyl-NC to the more electrophilic BMes2 centre takes place as the initial event, followed by a nucleophilic 1,2-mesityl migration, then an electrophilic 1,3-Bpin migration, and finally coordination of the second molecule of Xyl-NC to produce the product (see the top part of Scheme 5).
The mechanisms discussed above for reactions involving one and two equivalents of Xyl-NC differ at the point immediately following the coordination of the first Xyl-NC molecule. Specifically, the mechanism for one equivalent involves nucleophilic 1,2-Bpin migration, while the mechanism for two equivalents involves nucleophilic 1,2-mesityl migration. These results suggest that the nucleophilic 1,2-Bpin and 1,2-mesityl migrations following the coordination of the first Xyl-NC molecule are competitive. In the presence of excess Xyl-NC, the high coordination capability of Xyl-NC enables the occurrence of the 1,2-mesityl migration as well (Scheme 5).
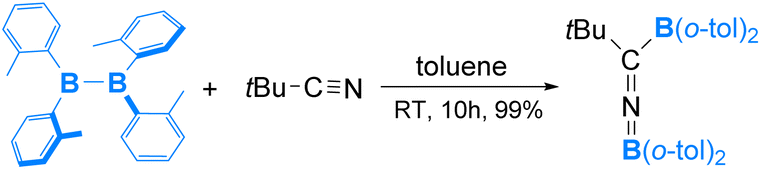 | (9) |
Compared to organic isonitriles (discussed above), organic nitriles exhibit relatively lower reactivity. Therefore, reactions involving organic nitriles require more reactive diboranes(4). Indeed, a highly Lewis acidic diaryldiborane(4) was reported to react with tBuCN in 2019, as shown in eqn (9).57 As expected, the reaction is initiated by coordination of the nitrile nitrogen atom to one electrophilic boron centre of diaryldiborane(4) to form an sp2–sp3 diborane(4) intermediate, from which a nucleophilic 1,3-BAr2 migration occurs and furnishes the final product containing a BNC linkage (Scheme 6).
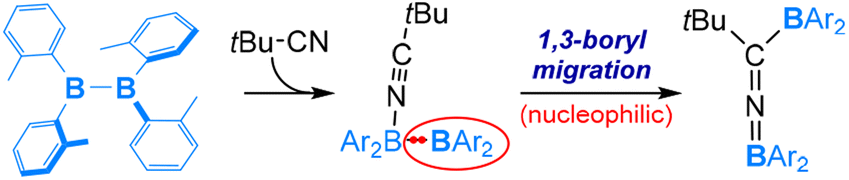 | ||
| Scheme 6 Mechanism of the reaction of B2(o-tol)2 and tBu–NC. | ||
6.3 Reaction with organic azides (RN3)
Reactions of diborane(4) compounds with organic azides have also attracted considerable interest. A variety of reaction modes have also been observed. Here, we give a few examples to illustrate the interesting chemistry discovered.Recently, reactions of tetra(o-tolyl)diborane(4) with organic azides were reported.59 The reaction of tetra(o-tolyl)diborane(4) with 1 equiv. of aryl azides (PhN3 or MesN3) was found to result in an insertion of “aryl nitrene” into the B–B bond to give a diborylamine product (eqn (10)). This reaction shows a nucleophilic 1,2-boryl migration after coordination of the azide via its internal nitrogen, the nucleophilic 1,2-boryl migration is accompanied by a release of an N2 molecule, and thus it can be formally viewed as an SN2-type of reaction at the coordinated N centre.
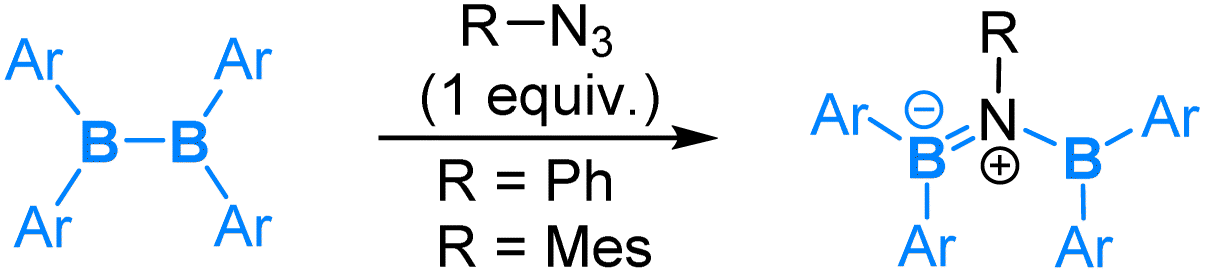 | (10) |
In 2019, by collaborating with the research team of Braunschweig, we theoretically explored the mechanisms for reactions of different substituted diaryl(dihalo)diborane(4) compounds with various organic azides, which resulted in distinct products.58 Here we pick up two examples to show the diverse reactivity of different diaryl(dihalo)diborane(4) compounds toward organic azides.
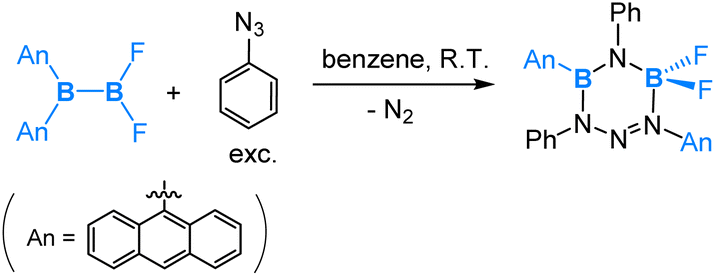 | (11) |
As depicted in eqn (11), the reaction of the unsymmetrical dianthryl(difluoro)diborane(4) with PhN3 yielded a six-membered ring product. DFT-assisted mechanistic studies provided further insight into this transformation as follows (Scheme 7). Initially, the azide molecule coordinates to the more electrophilic BAn2 boron centre, followed by a nucleophilic 1,4-anthryl migration. Subsequently, a cis–trans isomerization takes place, accompanied by the formation of a five-membered ring. In the next step, a second azide molecule coordinates to the Lewis acidic, three-coordinated B–Ar boron centre. This coordination triggers a nucleophilic 1,2-boryl (N-coordinated BF2) migration, accompanied by a release of N2 formally via an SN2-type of reaction at the PhN3 coordinated N centre. This migration and N2 release result in the final formation of the six-membered ring product, as illustrated in Scheme 7.
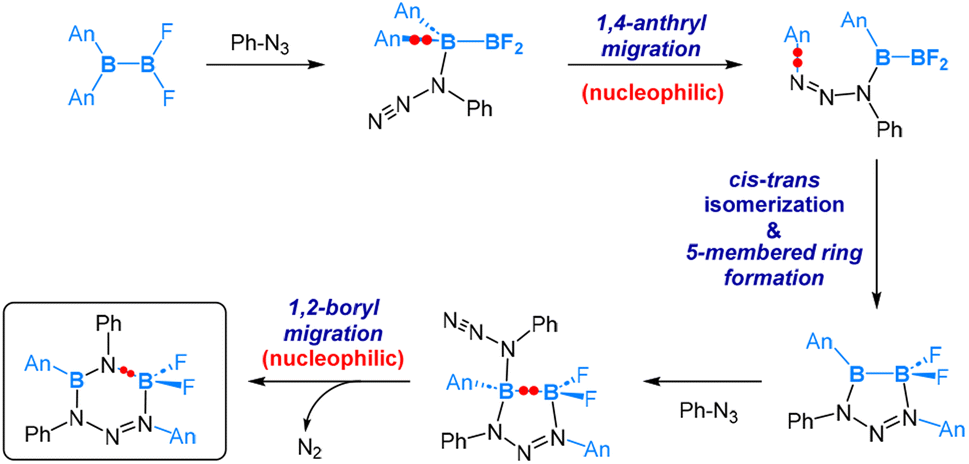 | ||
| Scheme 7 Mechanism of the reaction of B(An)2BF2 and Ph-N3. | ||
Interestingly, in the reaction of a symmetrical dianthryl(dibromo)diborane(4) with PhN3, a [2 + 4] cycloaddition product was obtained as illustrated in eqn (12). DFT calculations revealed that after the coordination of PhN3, [2 + 4] cycloaddition between the N2 moiety and the central aromatic ring of the anthryl group is both kinetically and thermodynamically preferred over the 1,4-anthryl migration. Here we see an interesting situation that the B–B bond is intact.
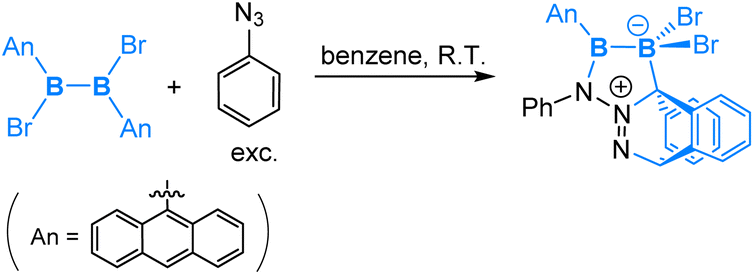 | (12) |
The reaction mechanism described in Scheme 7 reveals that, following the initial coordination of an azide molecule, a nucleophilic 1,4-anthryl migration takes place instead of a nucleophilic boryl migration. Similarly, the mechanism discussed in Scheme 4, concerning the reaction with carbon monoxide, demonstrates that a 1,2-mesityl nucleophilic migration occurs after the initial coordination of CO, rather than a nucleophilic boryl migration. These findings indicate that the nucleophilic boryl migration depicted in Fig. 8 is not always the predominant pathway. It is evident that nucleophilic aryl migrations can effectively compete with nucleophilic boryl migrations. Fig. 9 illustrates these potential migration pathways.
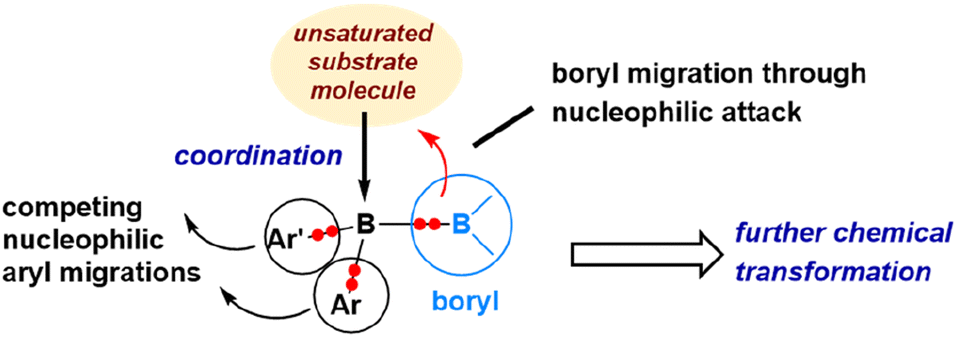 | ||
| Fig. 9 Illustration of the diverse reaction modes of diborane(4) compounds for unsaturated organic substrates. | ||
7. Reactions involving boryl radicals
As Scenario 3 depicted in Fig. 2, a genuine boryl radical species is two-coordinated and highly electron-deficient, with a half-filled sp2-hybridized orbital and an empty pz orbital (Fig. 10). Due to extreme electron-deficiency, two-coordinated boryl radicals are highly reactive and difficult to isolate.61–63 In the 1980s, Roberts and co-workers reported stable boryl radical species coordinated by amines or phosphines.64–68 Later on, N-heterocyclic carbenes (NHC) ligated69–73 and nitrogen heterocycle ligated74–79 boryl radicals were also reported. With Lewis base (LB) coordination, the unpaired electron occupies the “empty” pz orbital of the boron centre, giving a so-called 7-electron (7e) ligated boryl radical species (Fig. 10). When the LB Lewis base contains a π-conjugate system, a ligated boryl radical can be significantly stabilized through π-delocalization. Such 7-electron (7e) boryl radical species are prevalent and widely used in organic synthesis.80–85 In this section, we will briefly discuss the reactivity of Lewis base-stabilized boryl radicals (labelled as 7e boryl radicals) using several examples.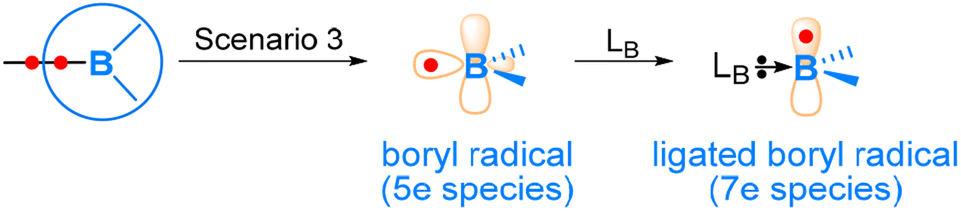 | ||
| Fig. 10 Illustration of the 5e and 7e boryl radical species. | ||
The research group of Wang has reported a series of studies using LB-ligated boryl radicals as catalysts. They applied a spin-centre shift (SCS) strategy, which can also be considered as a radical-relay strategy, to realize numerous borylations and C–X bond activations of unsaturated molecules, including alkenes, alkynes and CO-containing molecules.84,86–94 The SCS strategy shows that LB-ligated boryl radicals prefer to react with an unsaturated bond and coordinate onto the site where the newly formed spin centre can be better stabilized, as exemplified in Scheme 8, which was also confirmed by DFT calculations. After the initial coordination, further transformations, such as hydrogen atom transfer (HAT), halogen atom transfer (XAT), and radical coupling, can occur. Very recently, they also achieved asymmetric radical cycloisomerisation catalysed by chiral (NHC)-ligated boryl radicals by applying the SCS strategy.95
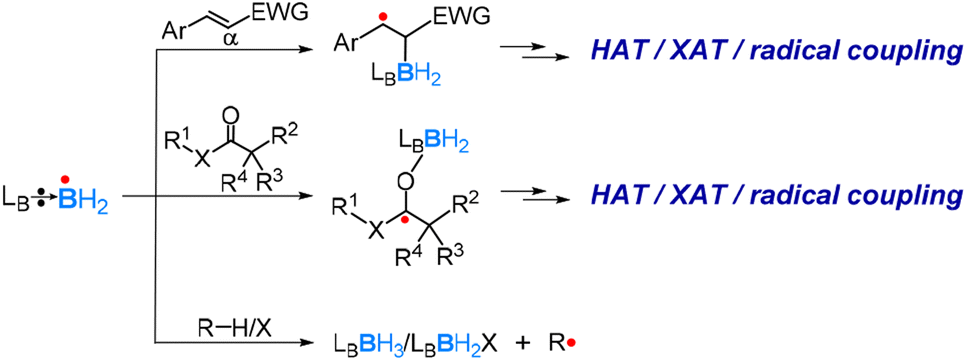 | ||
| Scheme 8 Reactions of a Lewis base ligated boryl radical with unsaturated and saturated molecules. | ||
When reacting with a saturated molecule, the LB-ligated boryl radical can directly abstract an atom from the substrate molecule. For example, Noël and co-workers recently reported a halogen-atom transfer (XAT) by  for C(sp3)–C(sp3) bond formation.96 DFT analysis demonstrated that, in the initial step, the
for C(sp3)–C(sp3) bond formation.96 DFT analysis demonstrated that, in the initial step, the  prefers to undergo iodine atom transfer (IAT) of iodocyclohexane over direct radical addition onto dimethyl maleate. In 2022, Wu, Zhang, and Ma reported a difluoromethylation of unactivated alkenes using freon-22 through an amine-coordinated boryl radical
prefers to undergo iodine atom transfer (IAT) of iodocyclohexane over direct radical addition onto dimethyl maleate. In 2022, Wu, Zhang, and Ma reported a difluoromethylation of unactivated alkenes using freon-22 through an amine-coordinated boryl radical 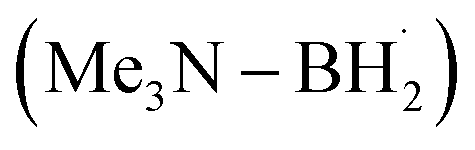 triggered chlorine atom transfer.97 This work revealed that the photo-induced
triggered chlorine atom transfer.97 This work revealed that the photo-induced 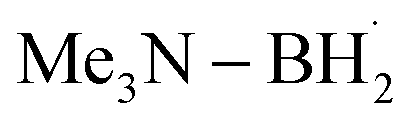 selectively abstracts a chlorine atom from freon-22. Mechanistic investigation confirmed that the ClAT process is both thermodynamically and kinetically more favourable than the HAT process. Direct radical addition onto the alkene substrate was also found to be less likely to proceed.
selectively abstracts a chlorine atom from freon-22. Mechanistic investigation confirmed that the ClAT process is both thermodynamically and kinetically more favourable than the HAT process. Direct radical addition onto the alkene substrate was also found to be less likely to proceed.
8. Summary and outlook
In this perspective, we have provided an in-depth analysis of the reaction chemistry of boryl compounds towards various organic and inorganic substrate molecules, with a focus on their nucleophilic and electrophilic properties from a bonding perspective. Boryl compounds feature a three-coordinate boron centre, with the reactivity often attributed to the formally “empty” p orbital on the boron centre. However, our analysis indicates that a boryl moiety can also be nucleophilic, depending on the element bonded to it. The σ bond to a boryl moiety can be reactive and play a nucleophilic role in a chemical reaction.In transition metal boryl complexes, we found that copper(I) boryl complexes were mainly nucleophilic, while most rhodium(I), platinum(II) and gold(I) boryl complexes were electrophilic. The nucleophilicity is closely related to the polarity of the σ bond to a boryl moiety. Varying the electronic properties of substituents on the boryl ligands can also switch the nature of their reactivity in some cases. While we have discussed the reaction chemistry of copper(I), rhodium(I), platinum(II) and gold(I) boryl complexes in this perspective, a large number of boryl complexes of other transition metals are known, and further systematic studies are needed to fully understand their reactivity.
Diboranes(4) are compounds with two boryl moieties directly σ-bonded. Their reactions with various unsaturated organic and inorganic substrate molecules generally start with coordination of a substrate molecule on the more electrophilic boron centre, where diborane(4) behaves in a Lewis-acidic and electrophilic manner. The subsequent nucleophilic migration of the uncoordinated boryl involving the B–B σ bonding pair of electrons leads to a series of chemical transformations and experimentally observed product(s). However, in reactions with organic azides and carbon monooxide, we have observed circumstances other than the involvement of the B–B σ bonding pair of electrons. Further investigation is needed to understand the factors that affect the preference of one circumstance over the other.
Finally, we briefly discussed reactions involving boryl radicals. Genuine two-coordinate radicals (5e boryl radical species) are scarce. Studies normally focus on base-ligated boryl radicals (7e boryl radicals). Base-ligated boryl radicals usually first react with an unsaturated bond, with the radical being relayed from the boron centre to a carbon centre. When reacting with saturated substrate molecules, atom (preferably heteroatom if any) abstraction occurs, again relaying the radical from the boron centre to a carbon centre. Due to their high reactivity, understanding of their reactions is still in the early stage, and further studies on reactions of boryl radical species is expected to lead to exciting discoveries.
Author contributions
Z. Lin was responsible for designing the structure of this perspective article, while Z. Lin and X. Guo jointly wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Research Grants Council of Hong Kong for financial support (16300021 and 16302222).References
- H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254–5274 CrossRef CAS PubMed.
- H. Braunschweig and D. Rais, Angew. Chem., Int. Ed., 2005, 44, 7826–7828 CrossRef CAS.
- H. Braunschweig and G. R. Whittell, Chem.–Eur. J., 2005, 11, 6128–6133 CrossRef CAS.
- C. Duan and C. Cui, Chem. Soc. Rev., 2024, 53, 361–379 RSC.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed.
- L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987–3995 RSC.
- Z. Lin, Computational Studies in Organometallic Chemistry, Springer International Publishing, 2016, pp. 39–58 Search PubMed.
- D. L. Kays and S. Aldridge, Contemporary Metal Boron Chemistry I: Borylenes, Boryls, Borane σ-Complexes, and Borohydrides, Springer Berlin Heidelberg, 2008, pp. 29–122 Search PubMed.
- H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed.
- J. Cid, H. Gulyás, J. J. Carbó and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC.
- G. J. Irvine, M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685–2722 CrossRef CAS PubMed.
- H. DeFrancesco, J. Dudley and A. Coca, Boron Reagents in Synthesis, American Chemical Society, 2016, vol. 1236, ch. 1, pp. 1–25 Search PubMed.
- U. Kaur, K. Saha, S. Gayen and S. Ghosh, Coord. Chem. Rev., 2021, 446, 214106 CrossRef CAS.
- D. A. Addy, J. I. Bates, D. Vidovic and S. Aldridge, J. Organomet. Chem., 2013, 745–746, 487–493 CrossRef CAS.
- H. Braunschweig, M. Colling, C. Kollann and U. Englert, J. Chem. Soc., Dalton Trans., 2002, 2289–2296 RSC.
- M. L. Buil, M. A. Esteruelas, K. Garcés and E. Oñate, J. Am. Chem. Soc., 2011, 133, 2250–2263 CrossRef CAS PubMed.
- P. M. Rutz, J. Grunenberg and C. Kleeberg, Organometallics, 2022, 41, 3044–3054 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS.
- P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS.
- D. A. Evans, J. L. Katz and T. R. West, Tetrahedron Lett., 1998, 39, 2937–2940 CrossRef CAS.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed.
- A. V. Protchenko, P. Vasko, M. A. Fuentes, J. Hicks, D. Vidovic and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 2064–2068 CrossRef CAS PubMed.
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570–9571 CrossRef CAS PubMed.
- T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606–6610 CrossRef CAS PubMed.
- M. Yamashita, Polar Organometallic Reagents, 2022, pp. 317–335 Search PubMed.
- M. Yamashita and K. Nozaki, Synthesis and Application of Organoboron Compounds, 2015, ch. 1, pp. 1–37 Search PubMed.
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed.
- M. S. Cheung, T. B. Marder and Z. Lin, Organometallics, 2011, 30, 3018–3028 CrossRef CAS.
- A.-F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef PubMed.
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef PubMed.
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef.
- A.-F. Pécharman, N. A. Rajabi, M. S. Hill, C. L. McMullin and M. F. Mahon, Chem. Commun., 2019, 55, 9035–9038 RSC.
- H. T. W. Shere, M. S. Hill, S. E. Neale, M. F. Mahon, C. L. McMullin and A. S. S. Wilson, Z. Anorg. Allg. Chem., 2023, 649, e202200376 CrossRef CAS.
- C. L. McMullin, S. E. Neale and G. L. Young, Eur. J. Inorg. Chem., 2024, 27, e202300393 CrossRef CAS.
- H. Zhao, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2006, 128, 15637–15643 CrossRef CAS PubMed.
- H. Zhao, L. Dang, T. B. Marder and Z. Lin, J. Am. Chem. Soc., 2008, 130, 5586–5594 CrossRef CAS PubMed.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef CAS.
- S. Lin and Z. Lin, Organometallics, 2019, 38, 240–247 CrossRef CAS.
- L. Dang, H. Zhao, Z. Lin and T. B. Marder, Organometallics, 2007, 26, 2824–2832 CrossRef CAS.
- L. Dang, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 4443–4454 CrossRef CAS.
- K. Takahashi, T. Ishiyama and N. Miyaura, Chem. Lett., 2000, 29, 982–983 CrossRef.
- B. Liu, M. Gao, L. Dang, H. Zhao, T. B. Marder and Z. Lin, Organometallics, 2012, 31, 3410–3425 CrossRef CAS.
- Y. G. Lawson, M. J. G. Lesley, N. C. Norman, C. R. Rice and T. B. Marder, Chem. Commun., 1997, 2051–2052 RSC.
- J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, Harper Collins, New York, USA, 4th edn, 1993, p.187 Search PubMed.
- L. Tao, X. Guo, J. Li, R. Li, Z. Lin and W. Zhao, J. Am. Chem. Soc., 2020, 142, 18118–18127 CrossRef CAS PubMed.
- A. Suzuki, X. Guo, Z. Lin and M. Yamashita, Chem. Sci., 2021, 12, 917–928 RSC.
- X. Guo, T. Yang, F. K. Sheong and Z. Lin, ACS Catal., 2021, 11, 5061–5068 CrossRef CAS.
- C. M. Zinser, F. Nahra, L. Falivene, M. Brill, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2019, 55, 6799–6802 RSC.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- H.-J. Himmel, Eur. J. Inorg. Chem., 2018, 2018, 2139–2154 CrossRef CAS.
- H. Asakawa, K. H. Lee, Z. Lin and M. Yamashita, Nat. Commun., 2014, 5, 4245 CrossRef CAS.
- Y. Katsuma, H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, Organometallics, 2016, 35, 2563–2566 CrossRef CAS.
- Y. Katsuma, N. Tsukahara, L. Wu, Z. Lin and M. Yamashita, Angew. Chem., Int. Ed., 2018, 57, 6109–6114 CrossRef CAS.
- Y. Katsuma, L. Wu, Z. Lin, S. Akiyama and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 317–321 CrossRef CAS PubMed.
- D. Prieschl, G. Bélanger-Chabot, X. Guo, M. Dietz, M. Müller, I. Krummenacher, Z. Lin and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 1065–1076 CrossRef CAS PubMed.
- M. Yamamoto, W. C. Chan, Z. Lin and M. Yamashita, Chemistry, 2023, 29, e202302027 CrossRef CAS PubMed.
- C. Kojima, K.-H. Lee, Z. Lin and M. Yamashita, J. Am. Chem. Soc., 2016, 138, 6662–6669 CrossRef CAS PubMed.
- D. Lu, C. Wu and P. Li, Org. Lett., 2014, 16, 1486–1489 CrossRef CAS PubMed.
- C. Wu, X. Hou, Y. Zheng, P. Li and D. Lu, J. Org. Chem., 2017, 82, 2898–2905 CrossRef CAS PubMed.
- P. R. Rablen and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4648–4653 CrossRef CAS.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- J. A. Baban and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1983, 1224–1226 RSC.
- J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1988, 1195–1200 RSC.
- V. Paul and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1988, 1895–1901 RSC.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS.
- S.-H. Ueng, M. M. Brahmi, É. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS PubMed.
- S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS.
- T. Matsumoto and F. P. Gabbaï, Organometallics, 2009, 28, 4252–4253 CrossRef CAS.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed.
- J. C. Walton, Angew. Chem., Int. Ed., 2009, 48, 1726–1728 CrossRef CAS PubMed.
- J. Lalevée, N. Blanchard, A.-C. Chany, M.-A. Tehfe, X. Allonas and J.-P. Fouassier, J. Phys. Org. Chem., 2009, 22, 986–993 CrossRef.
- J. Lalevée, N. Blanchard, M.-A. Tehfe, A.-C. Chany and J.-P. Fouassier, Chem.–Eur. J., 2010, 16, 12920–12927 CrossRef PubMed.
- G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985–5989 CrossRef CAS PubMed.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS PubMed.
- L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC.
- F. Barth, F. Achrainer, A. M. Pütz and H. Zipse, Chem.–Eur. J., 2017, 23, 13455–13464 CrossRef CAS PubMed.
- T. Taniguchi, Chem. Soc. Rev., 2021, 50, 8995–9021 RSC.
- J. H. Kim, T. Constantin, M. Simonetti, J. Llaveria, N. S. Sheikh and D. Leonori, Nature, 2021, 595, 677–683 CrossRef CAS PubMed.
- T. Y. Peng, F. L. Zhang and Y. F. Wang, Acc. Chem. Res., 2023, 56, 169–186 CrossRef CAS.
- T. Y. Peng, Z. Y. Xu, F. L. Zhang, B. Li, W. P. Xu, Y. Fu and Y. F. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201329 CrossRef CAS PubMed.
- J. Qi, F.-L. Zhang, J.-K. Jin, Q. Zhao, B. Li, L.-X. Liu and Y.-F. Wang, Angew. Chem., Int. Ed., 2020, 59, 12876–12884 CrossRef CAS PubMed.
- J.-K. Jin, H.-M. Xia, F.-L. Zhang and Y.-F. Wang, Chin. J. Org. Chem., 2020, 40, 2185–2194 CrossRef CAS.
- T. Y. Peng, F. L. Zhang and Y. F. Wang, Acc. Chem. Res., 2023, 56, 169–186 CrossRef CAS.
- T. T. Simur, F. W. Dagnaw, Y.-J. Yu, F.-L. Zhang and Y.-F. Wang, Chin. J. Chem., 2022, 40, 577–581 CrossRef CAS.
- T.-Y. Peng, Z.-Y. Xu, F.-L. Zhang, B. Li, W.-P. Xu, Y. Fu and Y.-F. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201329 CrossRef CAS PubMed.
- Q. Zhao, B. Li, X. Zhou, Z. Wang, F.-L. Zhang, Y. Li, X. Zhou, Y. Fu and Y.-F. Wang, J. Am. Chem. Soc., 2022, 144, 15275–15285 CrossRef CAS PubMed.
- F.-L. Zhang, B. Li, K. N. Houk and Y.-F. Wang, JACS Au, 2022, 2, 1032–1042 CrossRef CAS.
- T. Ye, F.-L. Zhang, H.-M. Xia, X. Zhou, Z.-X. Yu and Y.-F. Wang, Nat. Commun., 2022, 13, 426 CrossRef CAS PubMed.
- Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS.
- S.-C. Ren, F.-L. Zhang, A.-Q. Xu, Y. Yang, M. Zheng, X. Zhou, Y. Fu and Y.-F. Wang, Nat. Commun., 2019, 10, 1934 CrossRef PubMed.
- A.-Q. Xu, F.-L. Zhang, T. Ye, Z.-X. Yu and Y.-F. Wang, CCS Chem., 2019, 1, 504–512 CrossRef CAS.
- C.-L. Wang, J. Wang, J.-K. Jin, B. Li, Y. L. Phang, F.-L. Zhang, T. Ye, H.-M. Xia, L.-W. Hui, J.-H. Su, Y. Fu and Y.-F. Wang, Science, 2023, 382, 1056–1065 CrossRef CAS PubMed.
- T. Wan, L. Capaldo, D. Ravelli, W. Vitullo, F. J. de Zwart, B. de Bruin and T. Noël, J. Am. Chem. Soc., 2023, 145, 991–999 CrossRef CAS PubMed.
- Z.-Q. Zhang, Y.-Q. Sang, C.-Q. Wang, P. Dai, X.-S. Xue, J. L. Piper, Z.-H. Peng, J.-A. Ma, F.-G. Zhang and J. Wu, J. Am. Chem. Soc., 2022, 144, 14288–14296 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |