
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic enantioselective synthesis of Csp2–N atropisomers via formal Csp2–O bond amination†
Chenxiao
Qian‡
ab,
Jing
Huang‡
b,
Tingting
Huang‡
a,
Lijuan
Song
 c,
Jianwei
Sun
*b and
Pengfei
Li
*a
c,
Jianwei
Sun
*b and
Pengfei
Li
*a
aDepartment of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, College of Science, Southern University of Science and Technology Guangming Advanced Research Institute, Southern University of Science and Technology (SUSTech), Shenzhen 518055, China. E-mail: lipf@sustech.edu.cn; flyli1980@gmail.com
bDepartment of Chemistry and the Hong Kong Branch of Chinese National Engineering Research Centre for Tissue Restoration & Reconstruction, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: sunjw@ust.hk
cSchool of Science, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, China
First published on 9th February 2024
Abstract
Compared with well-developed construction of Csp2–Csp2 atropisomers, the synthesis of Csp2–N atropisomers remains in its infancy, which is recognized as both appealing and challenging. Herein, we achieved the first organocatalyzed asymmetric synthesis of Csp2–N atropisomers by formal Csp2–O amination. With the aid of a suitable acid, 3-alkynyl-3-hydroxyisoindolinones reacted smoothly with 1-methylnaphthalen-2-ols to afford a wide range of atropisomers by selective formation of the Csp2–N axis. Particularly, both the kinetic (Z)-products and the thermodynamic (E)-products could be selectively formed. Furthermore, the rarely used combination of two chiral Brønsted acid catalysts achieved excellent enantiocontrol, which is intriguing and unusual in organocatalysis. Based on control experiments and DFT calculations, a cascade dehydration/addition/rearrangement process was proposed. More importantly, this work provided a new plat-form for direct atroposelective construction of the chiral Csp2–N axis.
Introduction
Atropisomers, the most widely recognized class of compounds featuring axial chirality, consist of conformers with restricted rotation around a single bond that enable their isolation in a stable form. In recent decades, significant advancements have been made in the construction of atropisomeric scaffolds, particularly with respect to highly developed Csp2–Csp2 axis biaryls1 and styrenes.2 In stark contrast, despite the widespread occurrence of C–N axis in bioactive compounds3 (Fig. 1A) and chiral ligands4 (Fig. 1B), the catalytic asymmetric synthesis of these atropisomeric scaffolds is seldom reported due to their inherently less stable configuration and lower rotational barrier.5 Traditional strategies for the construction of such enantioenriched frameworks rely on the functional group conversion of the existing Csp2–N axis,6 such as N-functionalizations,7 desymmetrizations,8 asymmetric cyclizations.9 Although central-to-axial chirality conversion can induce chiral C–N axes, it is not a one-step synthesis.10 In this context, catalytic enantioselective construction of the Csp2–N axis is the most direct strategy, but this is also a highly challenging task that has been limitedly explored.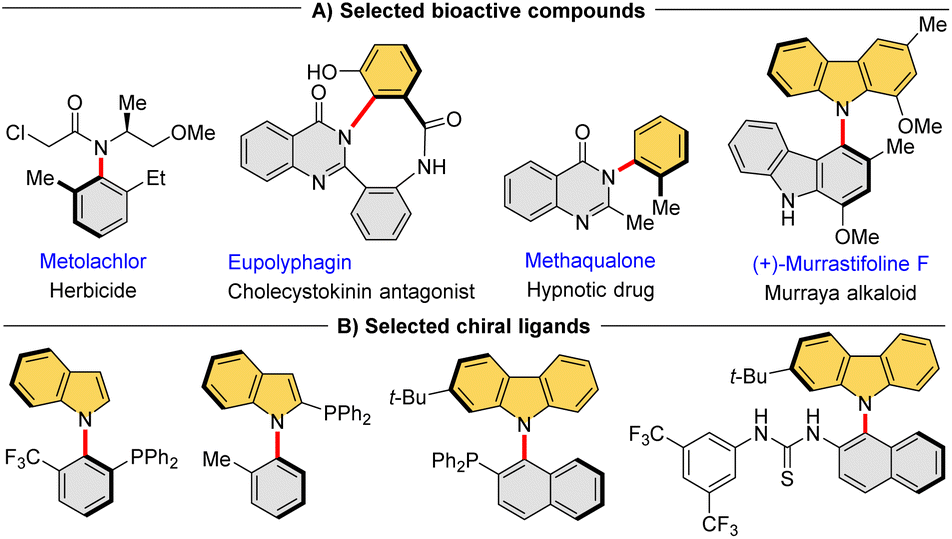 | ||
| Fig. 1 Selected C–N atropisomeric compounds. | ||
Currently, direct atroposelective construction of chiral Csp2–N axis mainly relies on two strategies: (i) organocatalytic asymmetric amination reactions, including electrophilic amination of electron-rich naphthalenes with azodicarboxylates/quinone diimines11 and nucleophilic amination of azonaphthalenes with electron-rich amines4c (Scheme 1A); (ii) metal-catalyzed asymmetric coupling reactions of secondary amines with aryl halides12 or diazo compounds13 (Scheme 1B). In both scenarios, limited substrate scope and harsh reaction conditions constitute the major issues. Particularly, Lin et al. gracefully disclosed a chiral phosphoric acid (CPA)-catalyzed atroposelective cascade reaction among 2,3-diketoesters, aromatic amines and 1,3-cyclohexanediones for the construction of axially chiral N-arylindoles (Scheme 1C).14 The reaction, however, still necessitated the utilization of dichloromethane at a temperature of 65 °C. Consequently, new efficient methods under mild conditions for the direct atroposelective construction of the Csp2–N bond remain in high demand.
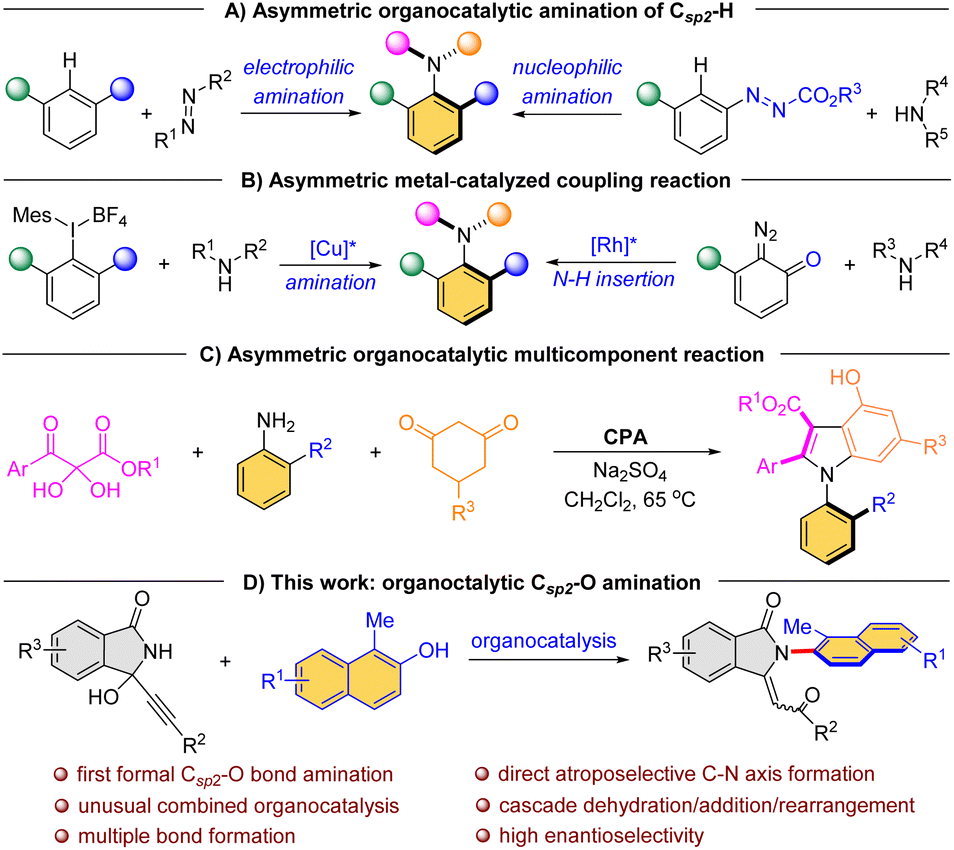 | ||
| Scheme 1 Direct construction of Csp2–N atropisomers. | ||
It is known to be challenging to break a Csp2–O bond attached to electron-rich aromatic rings, as a result of the high C–O bond dissociation energy. Generally, transition metal catalysts, including Ni- and Pd-catalysts, are required for such C–O bond activation.15 It is also known that aryl C–O bonds can be converted to C–N bonds with transition-metal-catalyzed catalysis, but no success has been achieved for direct construction of a chiral Csp2–N axis by Csp2–O bond activation.16 Moreover, transition-metal-free amination often necessitates harsh conditions, such as high temperature and strong alkali.17 Based on our previous research on indolinone derivatives18 and in continuation of our efforts on the organocatalytic reactions of functionalized alcohols,19 herein we discovered an intriguing dehydration/addition/rearrangement reaction between 1-substituted naphthols and indolinone derivatives leading to the formation of unusual Csp2–N atropisomeric scaffolds (Scheme 1D). More importantly, the catalytic asymmetric variant of this process has also been achieved by the combination of two chiral Brønsted acid catalysts.20
Results and discussion
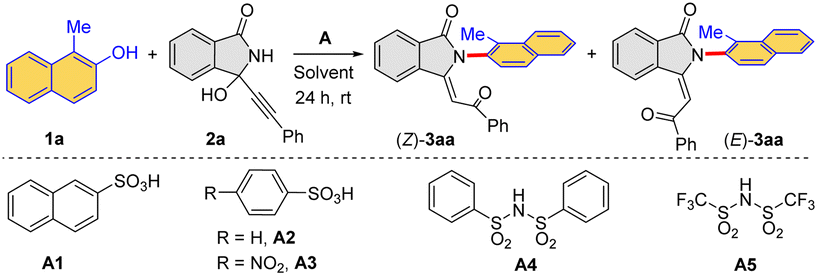
We employed 1-methylnaphthalen-2-ol 1a (ref. 21) and 3-alkynyl-3-hydroxyisoindolinone 2a as the model substrates for this study. Initially, different Brønsted acids were chosen as catalysts in view of their proven performance (Table 1). In the presence of different sulfonic acids (A1–A3) and sufonimides (A4–A5) (Table 1, entries 1–5), two isomeric products (Z)-3aa and (E)-3aa were observed, which contained a newly formed Csp2–N axis. Furthermore, it was also found that compound (Z)-3aa was the kinetic product, and the thermodynamic product (E)-3aa could be obtained from (Z)-3aa under the stronger Brønsted acidic conditions (details in mechanism studies). After careful screening of Brønsted acids, A1 was identified as the optimal catalyst to obtain the product (Z)-3aa with a yield of 63% (Table 1, entry 1). Alternatively, A3 could enable the formation of (E)-3aa with an improved yield of 83% (Table 1, entry 3). The further solvent evaluation revealed that CH2Cl2 was the optimal choice (Table 1, entries 6–8). Importantly, by adjusting substrate concentration, an improved yield of product (Z)-3aa was achieved (Table 1, entries 9–11).|
|
||||
|---|---|---|---|---|
| Entrya | A | Solvent | Yield [%]b | |
| a Unless noted, under Ar atmosphere, a mixture of 1a (0.05 mmol), 2a (0.10 mmol) and A (10 mol%) in the solvent (0.5 mL) was stirred at room temperature (rt) for 24 h. b Determined by 1H NMR with CH2Br2 as internal standard. | ||||
| 1 | A1 | CH2Cl2 | (Z)-3aa, 63 | (E)-3aa, 15 |
| 2 | A2 | CH2Cl2 | (Z)-3aa, 10 | (E)-3aa, 60 |
| 3 | A3 | CH2Cl2 | (Z)-3aa, 14 | (E)-3aa, 83 |
| 4 | A4 | CH2Cl2 | (Z)-3aa, <5 | (E)-3aa, <5 |
| 5 | A5 | CH2Cl2 | (Z)-3aa, 10 | (E)-3aa, 76 |
| 6 | A1 | MeCN | (Z)-3aa, <5 | (E)-3aa, <5 |
| 7 | A1 | EtOAc | (Z)-3aa, 46 | (E)-3aa, <5 |
| 8 | A1 | THF | (Z)-3aa, 22 | (E)-3aa, <5 |
| 9 | A1 | CH2Cl2 (1.0 mL) | (Z)-3aa, 82 | (E)-3aa, <5 |
| 10 | A1 | CH2Cl2 (1.2 mL) | (Z)-3aa, 90 | (E)-3aa, <5 |
| 11 | A1 | CH2Cl2 (1.5 mL) | (Z)-3aa, 84 | (E)-3aa, <5 |
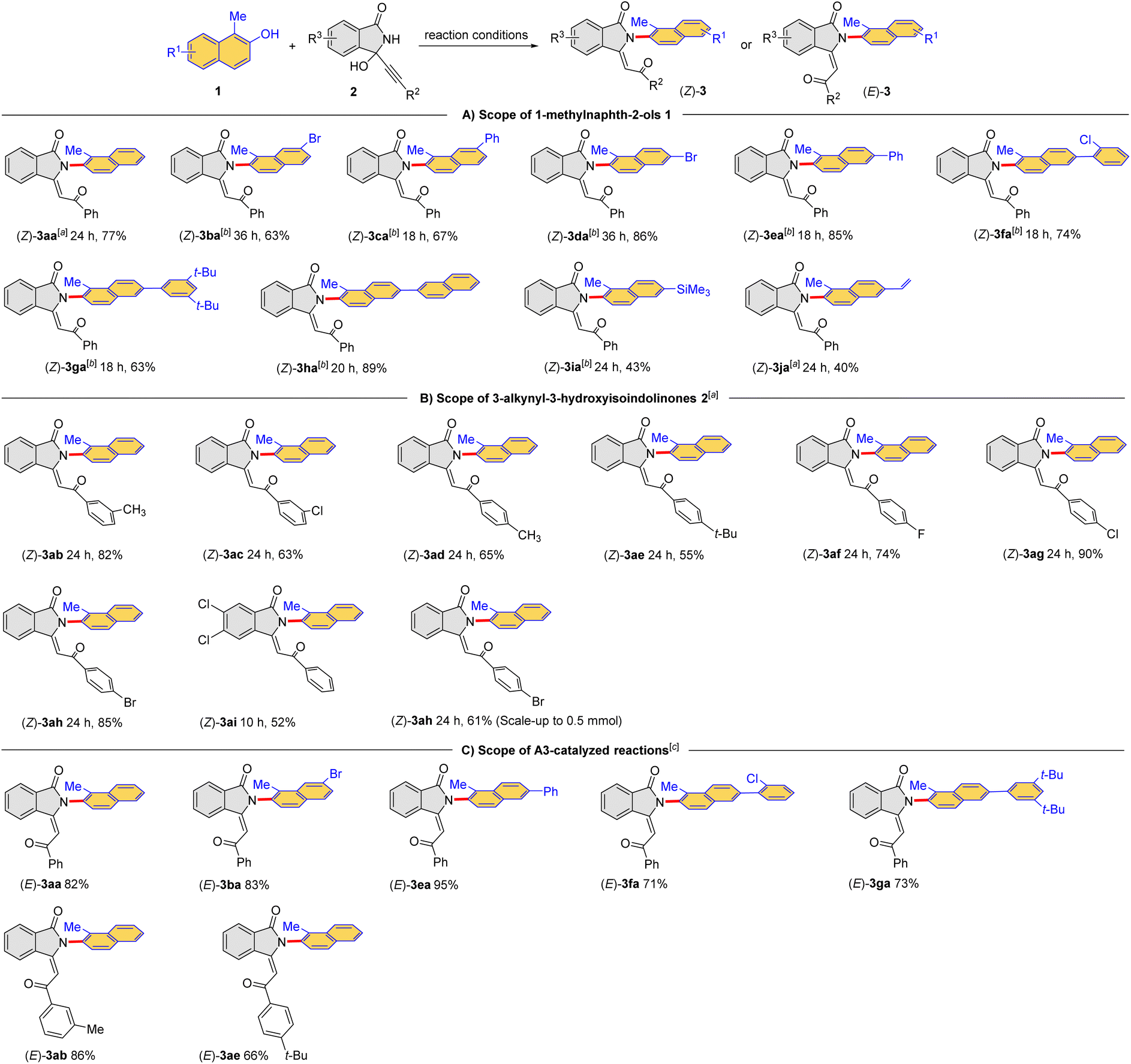
Under the optimized conditions, the scope of the cascade dehydration/addition/rearrangement reaction between 1-methylnaphthalen-2-ols 1 and 3-alkynyl-3-hydroxyisoindolinones 2 was examined at a relatively larger scale. Because the target products (Z)-3 could be converted to the thermodynamic products (E)-3, the time of each reaction was monitored by thin-layer chromatography (TLC). In some of these reactions, the amount of A1 was increased to 20 mol% to shorten the reaction time to reduce isomerization. As shown in Table 2A, a range of naphthols 1a–j reacted smoothly with 3-alkynyl-3-hydroxyisoindolinone 2a to provide the corresponding (Z)-2-(naphthalen-2-yl)-isoindolin-1-ones (Z)-3aa–ja in moderate yields. These encouraging results indicated that the protocol could be applied to naphthols bearing different substituents, including trimethylsilyl (SiMe3, Table 2, (Z)-3ia) and terminal olefin (Table 2, (Z)-3ja). In addition, the A1-catalyzed reaction also showed excellent compatibility with 3-alkynyl-3-hydroxyisoindolinones bearing different substituents in the benzene ring, affording the desired products (Z)-3ab–ah in moderate to high yields (Table 2B). However, the methyl group in the 1-position of naphthol seemed to be essential. Replacing it by other substituents (H, Et) would lead to a complex mixture instead of desired product. The reaction of 1,3-dimethylnaphthalen-2-ol also resulted in the complex mixture. The configuration of (Z)-3aa was unambiguously confirmed by X-ray crystallography (CCDC 2294518). The configurations of other products were assigned by analogy.
| a A mixture of 1 (0.20 mmol), 2 (0.40 mmol) and A1 (10 mol%) in CH2Cl2 (4.8 mL) was stirred at room temperature (rt) under Ar atmosphere for noted time. b A mixture of 1 (0.20 mmol), 2 (0.40 mmol) and A1 (20 mol%) in CH2Cl2 (6.0 mL) was stirred at 30 °C under Ar atmosphere for the noted time. c A mixture of 1 (0.20 mmol), 2 (0.40 mmol) and A3 (10 mol%) in CH2Cl2 (2.4 mL) was stirred at rt under Ar atmosphere for 24 h. |
|---|
|
The generality for the formation of the thermodynamic products (E)-3 was also examined (Table 2C). Notably, the presented products were obtained in high yields after further optimizations. Exceptionally, some thermodynamic products were either formed in low efficiency or hard to isolated from the reaction mixture. The configuration of (E)-3aa was confirmed by X-ray crystallography (CCDC 2294530). Taken together, a divergent cascade reaction between 1-methylnaphthalen-2-ols and 3-alkynyl-3-hydroxyisoindolinones was achieved for the direct construction of Csp2–N bond from Csp2–O bond to furnish the kinetic products (Z)-3 and the thermodynamic products (E)-3, respectively.
The successful construction of the Csp2–N axis inspired us to further develop an enantioselective variant of this process. Thus, the reaction between 1a and 2a was studied for the initial evaluation of some chiral phosphoric acids (Table 3, entries 1–2) and chiral phosphoramides (Table 3, entries 3–7) as potential catalysts. Indeed, chiral phosphoramide C1 was found to be a promising catalyst, affording the desired (R, Z)-3aa in 64% yield with 65% ee (Table 3, entry 3). While a single catalyst could not eventually improve the enantioselectivity to an excellent level, we were curious about the use of combined Brønstred acid systems. The combination of a chiral phosphoric acid and a chiral phosphoramide was then surveyed (Table 3, entries 8–10). Pleasingly, when phosphoramide C1 was used together with chiral phosphoric acid B2, the desired (R, Z)-3aa was formed with 90% ee (Table 3, entry 10). Notably, control experiments indicated that the use of any other combination or any single acid could not reach such a high level of enantiocontrol. Other solvents could not improve the reaction outcome (Table 3, entries 11–13). Notably, the loading of B2 and C1 could be further reduced to 8.0 mol% without compromising the efficiency (Table 3, entry 14). No product was obtained when the reaction was carried out at 0 °C (Table 3, entry 16). It is worth noting that substrate 2a has a low solubility at 0 °C. Therefore, a programmed cooling protocol was used to smoothly afford the target product (R, Z)-3aa with 93% ee (Table 3, entry 17).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | CPA | Solvent | Temp [oC] | Yield [%]b | ee [%]c |
| a Unless noted, under Ar atmosphere, a mixture of 1a (0.05 mmol), 2a (0.10 mmol) and CPA (10 mol%) in the solvent (2.0 mL) was stirred at room temperature (rt) for 24 h. b Determined by 1H NMR with CH2Br2 as internal standard. c Determined by chiral-HPLC analysis. d C1 (10 mol%), B1 (10 mol%). e C2 (10 mol%), B2 (10 mol%). f C1 (10 mol%), B2 (10 mol%). g C1 (8 mol%), B2 (8 mol%). h C1 (5 mol%), B2 (5 mol%). i The mixture was stirred at rt under Ar atmosphere for 5 min (2a was totally dissolved), then it was cooled to 0 °C for 6 days. | |||||
| 1 | B1 | CH2Cl2 | rt | Trace | — |
| 2 | B2 | CH2Cl2 | rt | Trace | — |
| 3 | C1 | CH2Cl2 | rt | 64 | 65 |
| 4 | C2 | CH2Cl2 | rt | 20 | 46 |
| 5 | C3 | CH2Cl2 | rt | 44 | 53 |
| 6 | C4 | CH2Cl2 | rt | Trace | — |
| 7 | C5 | CH2Cl2 | rt | 18 | 7 |
| 8d | C1/B1 | CH2Cl2 | rt | 40 | 61 |
| 9e | C2/B2 | CH2Cl2 | rt | 34 | 90 |
| 10f | C1/B2 | CH2Cl2 | rt | 44 | 90 |
| 11f | C1/B2 | MeCN | rt | Trace | — |
| 12f | C1/B2 | EtOAc | rt | Trace | — |
| 13f | C1/B2 | THF | rt | Trace | — |
| 14g | C1/B2 | CH2Cl2 | rt | 44 | 90 |
| 15h | C1/B2 | CH2Cl2 | rt | 32 | 88 |
| 16g | C1/B2 | CH2Cl2 | 0 | Trace | — |
| 17g,i | C1/B2 | CH2Cl2 | rt to 0 | 56 | 93 |
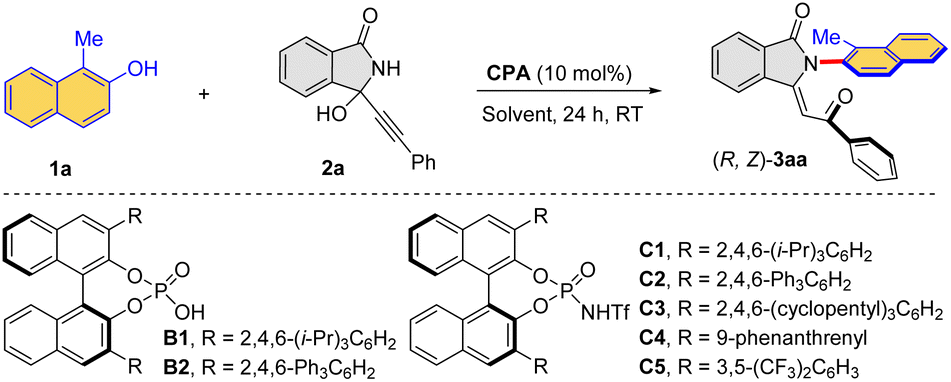
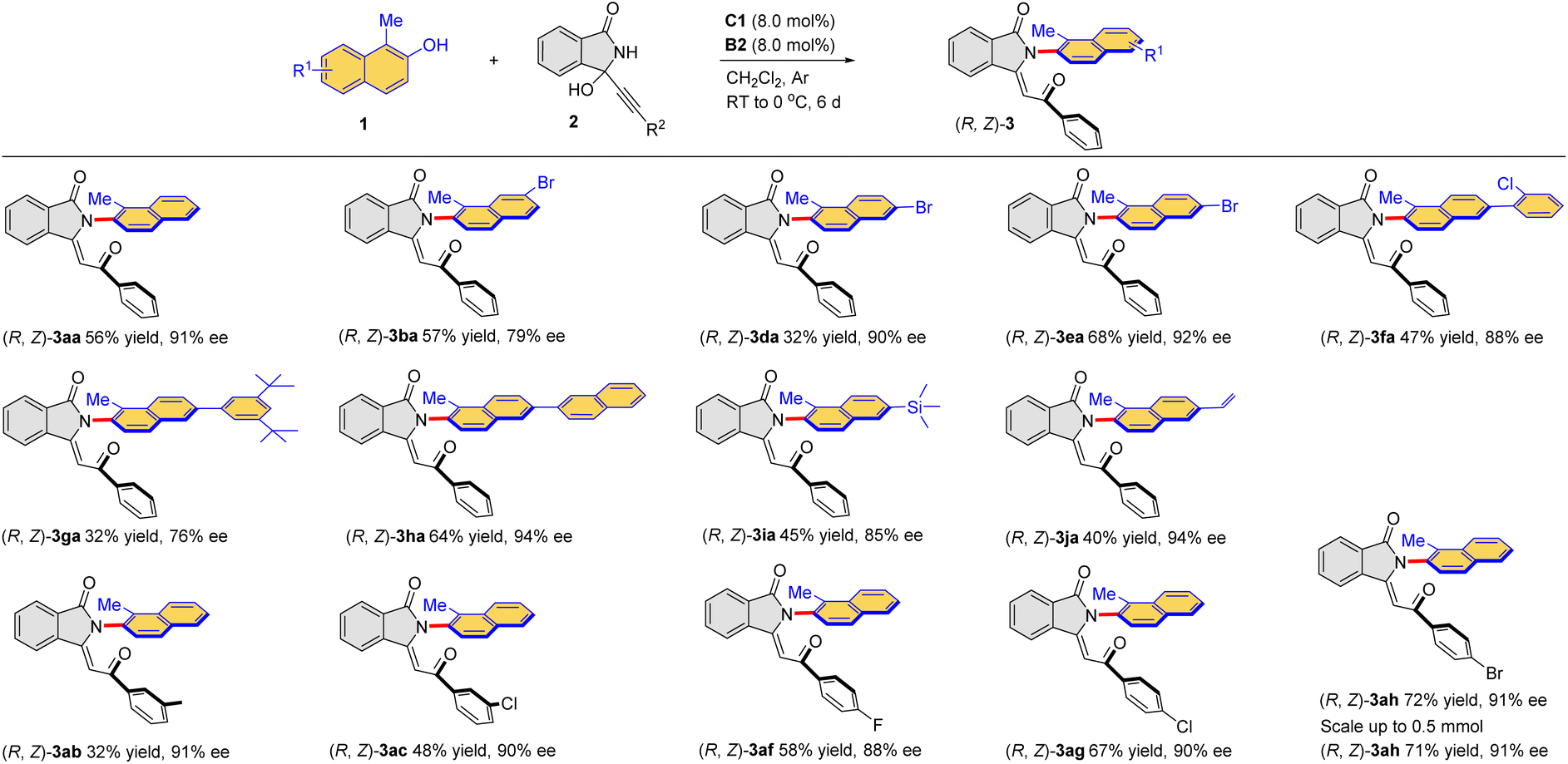
Under the established conditions, the scope of this combined catalytic system was examined for this intriguing process for the direct formation of atroposelective Csp2–N axis (Table 4). The kinetic products (R, Z)-3 were exclusively obtained under the optimized conditions. More specifically, a range of naphthols 1a–j reacted smoothly with 3-alkynyl-3-hydroxyisoindolinone 2a to provide the corresponding enantioenriched products (R, Z)-3aa–ja in moderate yields. The halogen (Br, Cl) could be introduced into the aromatic ring of naphthol, affording (R, Z)-3ba–fa in 32–68% yields with 79–92% ee. The reaction of naphthol with bulky substituents (3,5-(t-Bu)2C6H3, 2-naphthyl) could also proceed smoothy, furnishing the corresponding products (R, Z)-3ga in 32% yield with 76% ee and (R, Z)-3ha in 64% yield with 94% ee, respectively. Particularly, trimethylsilyl and the terminal olefin unit were also compatible, and (R, Z)-3ia was obtained in 45% yield with 85% ee and (R, Z)-3ja in 40% yield with 94% ee. Moreover, a series of 3-alkynyl-3-hydroxyisoindolinones with different substituents (3-MeC6H4, 3-ClC6H4, 4-FC6H4, 4-ClC6H4, 4-BrC6H4) were also reactive to afford the desired products in generally moderate yields with high enantioselectivities. The reactions could be run at a relatively large scale (0.5 mmol), which afforded the desired products with comparable results as the small-scale reactions. The configuration of (R, Z)-3aa was confirmed by X-ray crystallography (CCDC 2294540).
| a Under Ar atmosphere, a mixture of 1 (0.20 mmol), 2 (0.40 mmol), C1 (8 mol%) and B2 (8 mol%) in CH2Cl2 (8.0 mL) was stirred at room temperature (rt) for 5 min, then it was cooled to 0 °C for 6 h. Products (R, Z)-3 were obtained in isolated yields. The ee was determined by chiral-HPLC analysis. |
|---|
|
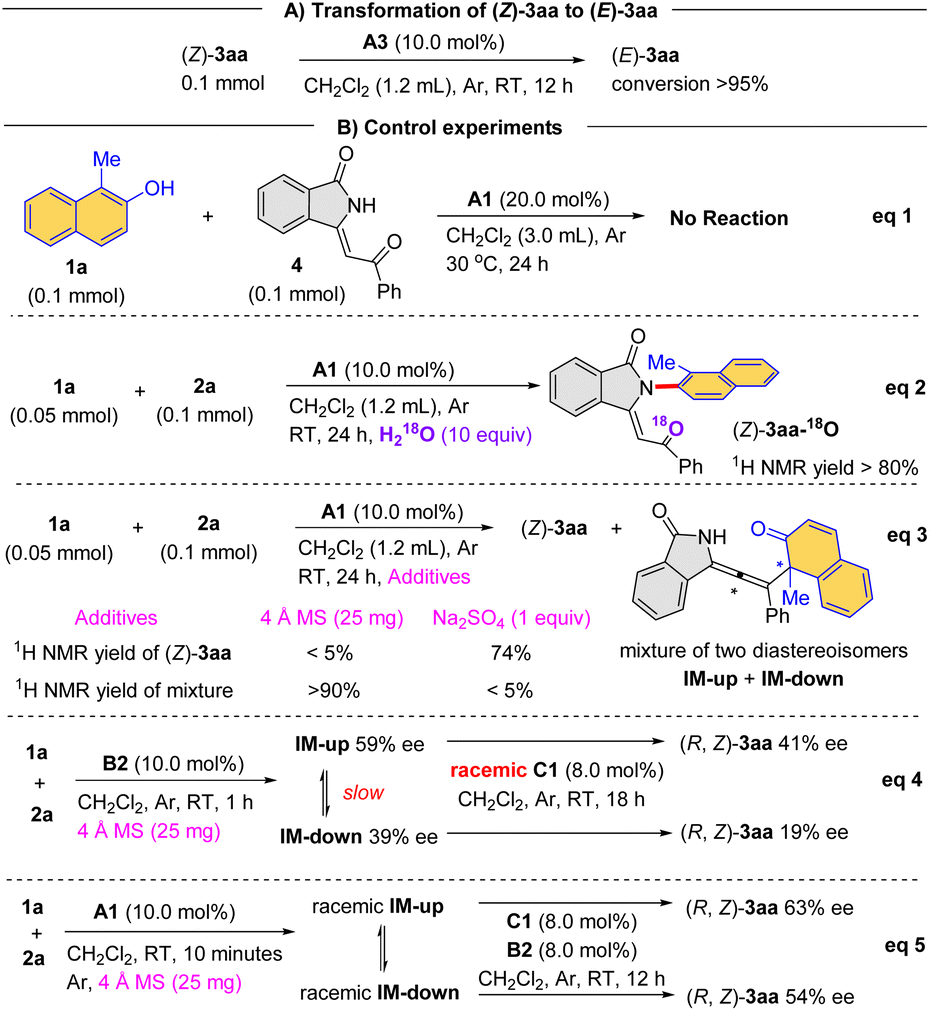
To gain more information about the reaction mechanism, we have performed a series of control experiments. First of all, product (Z)-3aa could be converted to (E)-3aa in the presence of catalyst A3, which confirmed that the former is the kinetic product and the latter is the thermodynamic product (Scheme 2A).
 | ||
| Scheme 2 Further investigations. | ||
Furthermore, no reaction took place between naphthalen-2-ol 1a and enone 4 from 3-alkynyl-3-hydroxyisoindolinone 2a under the standard conditions, suggesting that enone 4 was not a viable intermediate (Scheme 2B, eqn (1)). When H218O was added to the A1-mediated reaction, the 18O atom was incorporated into the product (Z)-3aa-18O (Scheme 2B, eqn (2)). Importantly, with 4 Å molecular sieves as additive, this process provided an allene as product, which was a mixture of diastereomers IM-up (weak polarity) and IM-down (strong polarity). The structure of this allene product was confirmed un-ambiguously by X-ray crystallography (CCDC 2294537, Scheme 2B, eqn (3)). Interestingly, the diastereomeric ratio (IM-up and IM-down) changed over time, likely via reverse reaction. Furthermore, with B2 as catalyst, the allene diastereomers could be isolated as enantioenriched form (59% ee for IM-up, 39% ee for IM-down), both of which could lead to enantioenriched product (R, Z)-3aa in the presence of the racemic catalyst C1 (Scheme 2B, eqn (4)), suggesting that the conversion from allene to product could be stereospecific. Moreover, the racemic allene diastereomers were also generated in the presence of catalyst A1 combined with 4 Å molecular sieves. Notably, treated with the combined catalysts, chiral C1 and B2, enantioenriched (R, Z)-3aa could be still obtained (Scheme 2B, eqn (5)). We reasoned that, even though racemic, the allene intermediate could reverse to their precursors in the presence of the chiral acid to generate enantioenriched allene, which resulted in the enantioenrichment in the final product. In this case, the rate-determining step is likely at a later stage (after allene formation), which would allow reversible steps before the addition of water to allene.
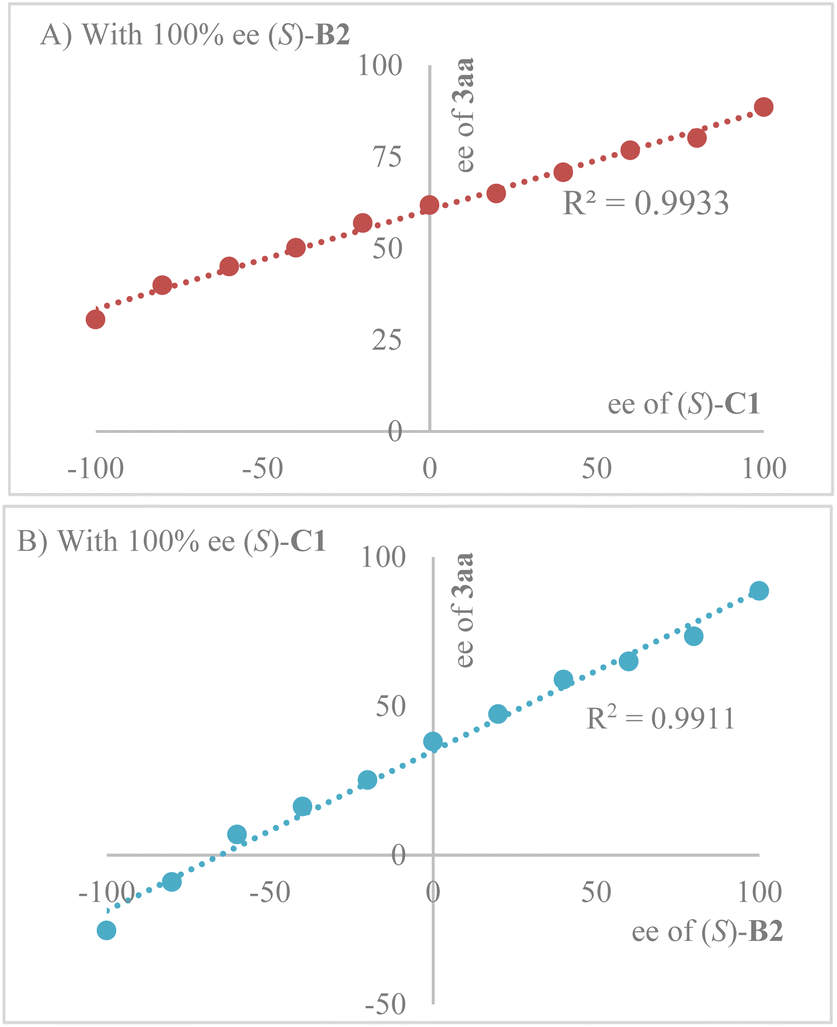
Interestingly, enantiopurity of product (R, Z)-3aa has a linear correlation to both the enantiopurity of C1 (Scheme 3A) and B2 (Scheme 3B), suggesting that both of them are contributing to the key enantiodetermining transition state. It is worth noting that such a scenario with two different chiral Brønsted acids contributing to enantiocontrol is extremely uncommon, to the best of our knowledge.
 | ||
| Scheme 3 Non-linear effect. | ||
To understand the mechanism, density functional theory (DFT) calculations were performed on the racemic reaction. The results indicated that this intriguing process comprises of dehydration, allene formation, water addition, cyclization, and ring-opening (Fig. 2). It starts substrate coordination to naphthalene-2-sulfonic acid (A1) by hydrogen bonding followed by elimination of water to form intermediate INT3 with an energy barrier of 16.7 kcal mol−1. Next, a nucleophilic attack from the naphthol to the resulting imine INT4 occurs together with synergistic proton transfer, leading to the allene intermediate INT5, with an energy barrier 12.4 kcal mol−1. In the presence of water, the electron-rich allene motif can be further activated by acid and trigger a water addition to form INT9. Next, an intramolecular cyclization takes place by the nucleophilic attack of the N-atom to the carbonyl group to form a C–N bond. Further dehydration forms iminium INT13. Finally, a facile ring-opening process driven by the aromatization furnishes the final product. It is worth noting that the water elimination from INT11 is the rate-determining step, which is consistent with the control experiments showing that the allene formation might be likely reversible.
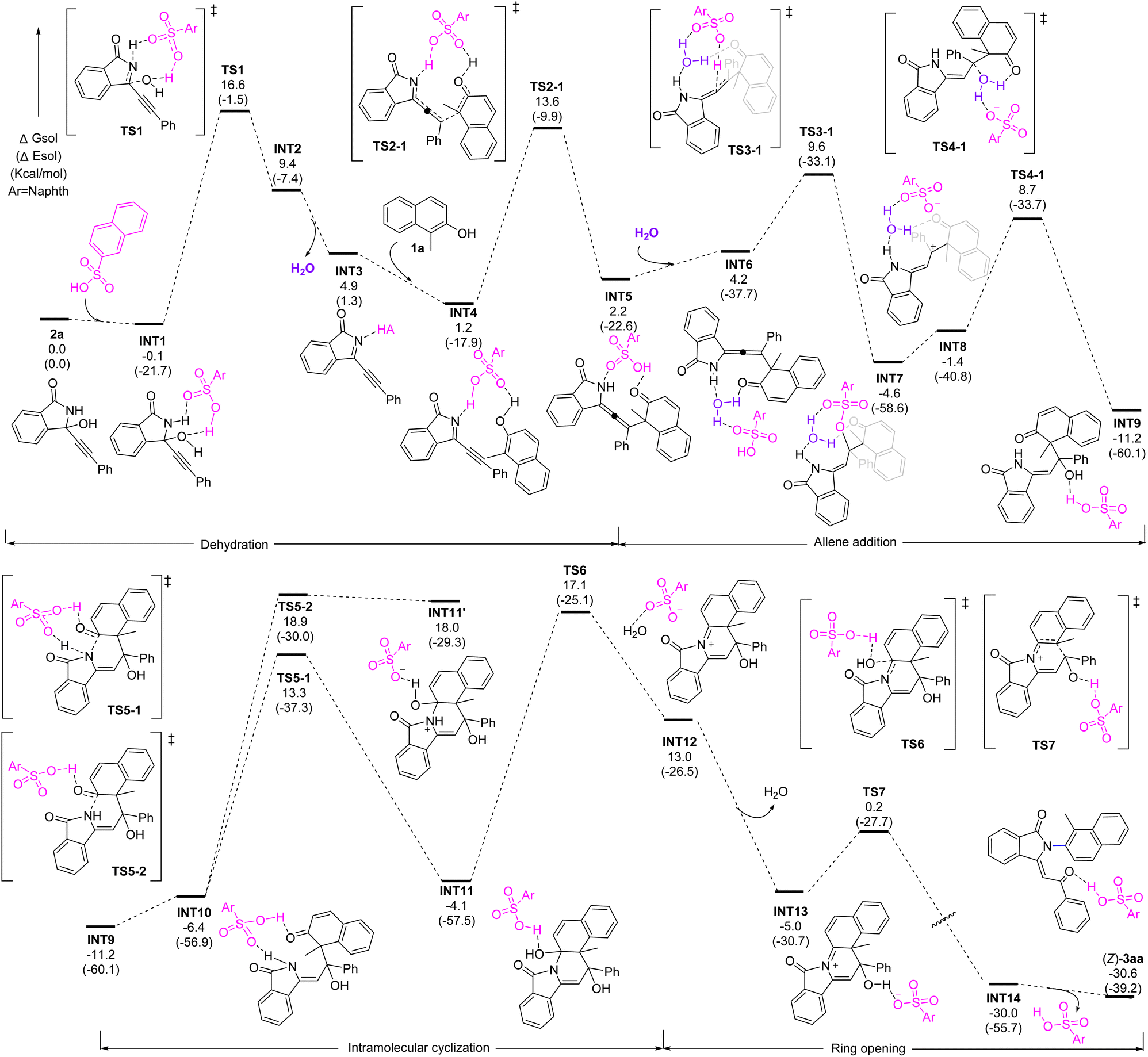 | ||
| Fig. 2 DFT study. Energetic profiles of the reaction at the M06-2X-D3/6-311G(d,p)-SMD(DCM)//M06-2X-D3/6-31G(d) level of theory and the energy values are in given kcal mol−1. | ||
Conclusions
In summary, we have successfully developed the first organocatalyzed asymmetric synthesis of Csp2–N atropisomers by formal Csp2–O amination. Different from known methods for the access to Csp2–N atropisomers by functionalization of existing C–N bonds, our process involved construction of the key C–N bond with concomitant asymmetric control. By adjusting the strength of the acid catalyst and reaction time, both the kinetic product (Z)-isomers and the thermodynamic product (E)-isomers could be selectively formed. More importantly, the rarely used combination of two chiral Brønsted acid catalysts proved critical to the excellent enantiocontrol. Control experiments confirmed that both of them are required for the high enantioselectivity and both are involved in the enantiodetermining transition state. However, more accurate information about the enantiodetermining transition state is unknown at this point. DFT calculations also provided important information to the general reaction pathway, which involves allene as the key intermediate. Additional studies on this intriguing process are under-way in our laboratories.Data availability
Experimental procedures, spectral data and DFT data can be found in the ESI.†Author contributions
Pengfei Li, Jianwei Sun and Chenxiao Qian wrote the manuscript. Pengfei Li and Jianwei Sun supervised the project. Pengfei Li conceived the project. Chenxiao Qian and Tingting Huang performed condition optimization, scope study and control experiments. Jing Huang and Lijuan Song performed DFT studies. All the authors proofread and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Shenzhen Innovation of Science and Technology Commission (20200925151614002), Guangdong Innovative Program (2019BT02Y335), and Guangdong Provincial Key Laboratory of Catalysis (2020B121201002). The authors acknowledge the assistance of SUSTech Core Research Facilities, Xiaoyong Chang (X-ray), and Yang Yu (HRMS).Notes and references
- Selected reviews, see: (a) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed; (b) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (c) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (d) M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898–12919 CrossRef CAS; (e) H.-H. Zhang and F. Shi, Acc. Chem. Res., 2022, 55, 2562–2580 CrossRef CAS PubMed; (f) J. K. Cheng, S.-H. Xiang and B. Tan, Acc. Chem. Res., 2022, 55, 2920–2937 CrossRef CAS PubMed.
- Selected reports, see: (a) S.-C. Zheng, S. Wu, Q. Zhou, L. W. Chung, L. Ye and B. Tan, Nat. Commun., 2017, 8, 15238–15245 CrossRef PubMed; (b) Y.-B. Wang, P. Yu, Z.-P. Zhou, J. Zhang, J. Wang, S.-H. Luo, Q.-S. Gu, K. N. Houk and B. Tan, Nat. Catal., 2019, 2, 504–513 CrossRef CAS; (c) L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497–511 CrossRef CAS; (d) C. Ma, F.-T. Sheng, H.-Q. Wang, S. Deng, Y.-C. Zhang, Y. Jiao, W. Tan and F. Shi, J. Am. Chem. Soc., 2020, 142, 15686–15696 CrossRef CAS PubMed.
- (a) G. Chesters, G. V. Simsiman, J. Levy, B. J. Alhajjar, R. N. Fathulla and J. M. Harkin, Rev. Environ. Contam. Toxicol., 1989, 110, 1–74 CAS; (b) H.-L. Jiang, X.-H. Luo, X.-Z. Wang, J.-L. Yang, X.-J. Yao, P. Crews, F. A. Valeriote and Q.-X. Wu, Fitoterapia, 2012, 83, 1275–1280 CrossRef CAS PubMed; (c) Y.-B. Wang, S.-C. Zheng, Y.-M. Hu and B. Tan, Nat. Commun., 2017, 8, 15489–15497 CrossRef PubMed.
- (a) T. Mino, S. Komatsu, K. Wakui, H. Yamada, H. Saotome, M. Sakamoto and T. Fujita, Tetrahedron: Asymmetry, 2010, 21, 711–718 CrossRef CAS; (b) T. Mino, M. Ishikawa, K. Nishikawa, K. Wakui and M. Sakamoto, Tetrahedron: Asymmetry, 2023, 24, 499–504 CrossRef; (c) W. Xia, Q.-J. An, X.-H. Xiang, S. Li, Y.-B. Wang and B. Tan, Angew. Chem., Int. Ed., 2020, 59, 6775–6779 CrossRef CAS PubMed.
- Selected reviews, see: (a) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem. – Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed; (b) O. Kitagawa, Acc. Chem. Res., 2021, 54, 719–730 CrossRef CAS PubMed; (c) G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855–1893 CrossRef CAS; (d) J. Feng, C.-J. Lu and R.-R. Liu, Acc. Chem. Res., 2023, 56, 2537–2554 CrossRef CAS PubMed.
- Selected reviews, see: (a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (b) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed; (c) Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117–136 CrossRef; (d) S. Choppin and J. Wencel-Delord, Acc. Chem. Res., 2023, 56, 189–202 CrossRef CAS PubMed.
- Selected reports, see: (a) O. Kitagawa, M. Kohriyama and T. Taguchi, J. Org. Chem., 2002, 67, 8682–8684 CrossRef CAS PubMed; (b) J. Terauchi and D. P. Curran, Tetrahedron: Asymmetry, 2003, 14, 587–592 CrossRef CAS; (c) O. Kitagawa, M. Takahashi, M. Yoshikawa and T. Taguchi, J. Am. Chem. Soc., 2005, 127, 3676–3677 CrossRef CAS PubMed; (d) O. Kitagawa, M. Yoshikawa, H. Tanabe, T. Morita, M. Takahashi, Y. Dobashi and T. Taguchi, J. Am. Chem. Soc., 2006, 128, 12923–12931 CrossRef CAS PubMed; (e) S. Shirakawa, K. Liu and K. Maruoka, J. Am. Chem. Soc., 2012, 134, 916–919 CrossRef CAS PubMed; (f) S.-L. Li, C. Yang, Q. Wu, H.-L. Zheng, X. Li and J.-P. Cheng, J. Am. Chem. Soc., 2018, 140, 12836–12843 CrossRef CAS PubMed; (g) G.-H. Yang, H. Zheng, X. Li and J.-P. Cheng, ACS Catal., 2020, 10, 2324–2333 CrossRef CAS.
- Selected reports, see: (a) D. P. Curran, H. Qi, S. J. Geib and N. C. DeMello, J. Am. Chem. Soc., 1994, 116, 3131–3132 CrossRef CAS; (b) W.-L. Duan, Y. Imazaki, R. Shintani and T. Hayashi, Tetrahedron, 2007, 63, 8529–8536 CrossRef CAS; (c) J.-W. Zhang, J.-H. Xu, D.-J. Cheng, C. Shi, X.-Y. Liu and B. Tan, Nat. Commun., 2016, 7, 10677–10686 CrossRef PubMed; (d) L.-L. Zhang, J.-W. Zhang, S.-H. Xiang, Z. Guo and B. Tan, Org. Lett., 2018, 20, 6022–6026 CrossRef CAS PubMed; (e) C. Parida, S. K. Dave, K. Das and S. C. Pan, Adv. Synth. Catal., 2023, 365, 1185–1190 CrossRef CAS.
- Selected reports, see: (a) K. Tanaka, K. Takeishi and K. Noguchi, J. Am. Chem. Soc., 2006, 128, 4586–4587 CrossRef CAS PubMed; (b) H. Liu, W. Feng, C. W. Kee, D. Leow, W.-T. Loh and C.-H. Tan, Adv. Synth. Catal., 2010, 352, 3373–3379 CrossRef CAS; (c) N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem. – Eur. J., 2010, 16, 6752–6755 CrossRef CAS PubMed; (d) T. Li, C. Mou, P. Qi, X. Peng, S. Jiang, G. Hao, W. Xue, S. Yang, L. Hao, Y. R. Chi and Z. Jin, Angew. Chem., Int. Ed., 2021, 60, 9362–9367 CrossRef CAS PubMed; (e) Q.-J. An, W. Xia, W.-Y. Ding, H.-H. Liu, S.-H. Xiang, Y.-B. Wang, G. Zhong and B. Tan, Angew. Chem., Int. Ed., 2021, 60, 24888–24893 CrossRef CAS PubMed.
- (a) C. Sun, X. Qi, X.-L. Min, X.-D. Bai, P. Liu and Y. He, Chem. Sci., 2020, 11, 10119–10126 RSC; (b) V. Corti, M. K. Thøgersen, V. J. Enemærke, N. M. Rezayee, C. L. Barløse and K. A. Jørgensen, Chem. – Eur. J., 2022, 28, e202202395 CrossRef CAS PubMed.
- (a) S. Brandes, M. Bella, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 1147–1151 CrossRef CAS PubMed; (b) S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard and K. A. Jørgensen, Chem. – Eur. J., 2006, 12, 6039–6052 CrossRef CAS PubMed; (c) H.-Y. Bai, F.-X. Tan, T.-Q. Liu, G.-D. Zhu, J.-M. Tian, T.-M. Ding, Z.-M. Chen and S.-Y. Zhang, Nat. Commun., 2019, 10, 3063–3071 CrossRef PubMed; (d) J. Qin, T. Zhou, T.-P. Zhou, L. Tang, H. Zuo, H. Yu, G. Wu, Y. Wu, R.-Z. Liao and F. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202205159 CrossRef CAS PubMed.
- (a) J. Rae, J. Frey, S. Jerhaoui, S. Choppin, J. Wencel-Delord and F. Colobert, ACS Catal., 2018, 8, 2805–2809 CrossRef CAS; (b) J. Frey, A. Malekafzali, I. Delso, S. Choppin, F. Colobert and J. Wencel-Delord, Angew. Chem., Int. Ed., 2020, 59, 8844–8848 CrossRef CAS PubMed.
- (a) Q. Ren, T. Cao, C. He, M. Yang, H. Liu and L. Wang, ACS Catal., 2021, 11, 6135–6140 CrossRef CAS; (b) C. Niu, Y. Zhou, Q. Chen, Y. Zhu, S. Tang, Z.-X. Yu and J. Sun, Org. Lett., 2022, 24, 7428–7433 CrossRef CAS PubMed.
- L. Wang, J. Zhong and X. Lin, Angew. Chem., Int. Ed., 2019, 58, 15824–15828 CrossRef CAS PubMed.
- S.-Q. Zhang and X. Hong, Acc. Chem. Res., 2021, 54, 2158–2171 CrossRef CAS PubMed.
- Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed.
- X. Chang, Q. Zhang and C. Guo, Org. Lett., 2019, 21, 4915–4918 CrossRef CAS PubMed.
- (a) C. Qian, M. Liu, J. Sun and P. Li, Org. Chem. Front., 2022, 9, 1234–1240 RSC; (b) C. Qian, T. Huang, J. Sun and P. Li, Org. Lett., 2022, 24, 6472–6476 CrossRef CAS PubMed; (c) Y. Xia, M. Liu, C. Qian, P. Li, M. Dong and W. Li, Org. Chem. Front., 2023, 10, 30–34 RSC.
- (a) S. Liu, K. L. Chan, Z. Lin and J. Sun, J. Am. Chem. Soc., 2023, 145, 12802–12811 CrossRef CAS PubMed; (b) M. Liu, B. Shen, C. Liu, P. Yu and P. Li, J. Am. Chem. Soc., 2023, 145, 14562–14569 CrossRef CAS PubMed.
- For pioneering examples of CPA catalysis, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed; (c) D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (e) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed; (f) T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS PubMed; (g) J. Kikuchi and M. Terada, Chem. – Eur. J., 2021, 27, 10215–10225 CrossRef CAS PubMed.
- A. B. Woldegiorgis, Z. Han and X.-F. Lin, Org. Lett., 2021, 23, 6606–6611 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06707f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |