
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous–crystalline RuTi nanosheets enhancing OH species adsorption for efficient hydrogen oxidation catalysis†
Licheng
Wei
a,
Nan
Fang
a,
Fei
Xue
a,
Shangheng
Liu
a,
Wei-Hsiang
Huang
d,
Chih-Wen
Pao
d,
Zhiwei
Hu
e,
Yong
Xu
 *c,
Hongbo
Geng
*b and
Xiaoqing
Huang
*a
*c,
Hongbo
Geng
*b and
Xiaoqing
Huang
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: hxq006@xmu.edu.cn
bSchool of Materials Engineering, Changshu Institute of Technology, Changshu, 215500, China
ci-Lab Suzhou Institute of Nano-Tech and Nano-Bionics (SINANO), Chinese Academy of Sciences (CAS), 398 Ruoshui Road, Suzhou, 215123, China
dNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 30076, Taiwan
eMax Planck Institute for Chemical Physics of Solids, Nothnitzer Strasse 40, Dresden 01187, Germany
First published on 30th January 2024
Abstract
Anion exchange membrane fuel cells are a potentially cost-effective energy conversion technology, however, the electrocatalyst for the anodic hydrogen oxidation reaction (HOR) suffers from sluggish kinetics under alkaline conditions. Herein, we report that Ru-based nanosheets with amorphous–crystalline heterointerfaces of Ru and Ti-doped RuO2 (a/c-Ru/Ti-RuO2) can serve as a highly efficient HOR catalyst with a mass activity of 4.16 A mgRu−1, which is 19.8-fold higher than that of commercial Pt/C. Detailed characterization studies show that abundant amorphous–crystalline heterointerfaces of a/c-Ru/Ti-RuO2 nanosheets provide oxygen vacancies and unsaturated coordination bonds for balancing adsorption of hydrogen and hydroxyl species on Ru active sites to elevate HOR activity. Moreover, Ti doping can facilitate CO oxidation, leading to enhanced strength to CO poisoning. This work provides a strategy for enhancing alkaline HOR performance over Ru-based catalysts with heteroatom and heterointerface dual-engineering, which will attract immediate interest in chemistry, materials science and beyond.
Introduction
Anion exchange membrane fuel cells (AEMFCs) have been regarded as one of the most effective technologies for converting hydrogen fuel to alleviate the current energy crisis and environmental concerns. The mild alkaline conditions of AEMFCs can accommodate more cell components and more durable stability than proton-exchange membrane fuel cells (PEMFCs).1–4 However, the kinetics of the hydrogen oxidation reaction (HOR) in AEMFCs is two orders of magnitude lower than that in PEMFCs. Under these circumstances, one needs to increase the contents of platinum-group-metals (PGMs) to reach the desired HOR performance, which leads to an increase in cost.5–7 Moreover, industrial hydrogen produced from the conventional steam reforming process inevitably contains trace amounts of CO, which can poison PGMs during catalysis. Therefore, improving alkaline kinetics, strengthening resistance to CO poisoning, and reducing the loading amounts of PGMs during the HOR are of great importance yet formidably challenging.Owing to its similar hydrogen affinity to Pt, Ru has been widely recognized as a promising candidate for the HOR. In principle, ideal HOR electrocatalysts should be capable of having appropriate adsorption strengths to H, OH, and CO.8–10 Optimizing interfaces/surfaces of catalysts to regulate the surface electron structure and reaction energy barrier is the key point for balancing intermediates adsorption.11–15 Over the past few decades, substantial efforts have been devoted to constructing surface and interfacial synergy for regulating hydrogen binding energy (HBE) and hydroxyl binding energy (OHBE) and thus modulating HOR performance.12,16,17 Despite great progress in catalyst design for the HOR, high-efficiency HOR electrocatalysis is limited by the sluggish alkaline kinetics and unsatisfactory long-term stability of catalysts. It is of great importance to design efficient electrocatalysts for alkaline HOR yet it is formidably challenging.
Inspired by the significance of amorphous materials with abundant unsaturated sites and defects in catalytic performance,18–21 here, we synthesize Ru-based nanosheets with amorphous–crystalline heterointerfaces of Ru and Ti-doped RuO2 (a/c-Ru/Ti-RuO2) as a highly active and durable catalyst for the HOR. Detailed characterization studies show that a/c-Ru/Ti-RuO2 nanosheets with abundant amorphous–crystalline heterointerfaces provide oxygen vacancies and unsaturated coordination bonds for balancing adsorption of hydrogen and hydroxyl species on Ru active sites to elevate HOR activity. Besides, doping Ti significantly weakened CO adsorption for improving CO tolerance of the a/c-Ru/Ti-RuO2 catalyst. Impressively, a/c-Ru/Ti-RuO2 nanosheets exhibit a current density and mass activity of 2.48 mA cm−2 and 4.16 A mgRu−1, respectively, at an overpotential of 50 mV, as well as much superior stability and resistance to CO poisoning to commercial Pt/C.
Results and discussion
a/c-Ru/Ti-RuO2 was synthesized by mixing ruthenium acetylacetonate and titanium acetylacetonate with a salt template of sodium chloride, which experienced a thermal treatment at 250 °C (Fig. 1a). It was found that a/c-Ru/Ti-RuO2 exhibited smooth nanosheets with a molar ratio of Ru/Ti of ∼3 (Fig. 1b and S1†). The thickness was measured to be ∼4.4 nm based on the atomic force microscopy (AFM) result (Fig. 1c). The TEM image and elemental mapping reveal that O, Ru and Ti are uniformly distributed in a/c-Ru/Ti-RuO2 nanosheets (Fig. 1d and e). HRTEM images show that a/c-Ru/Ti-RuO2 nanosheets consist of crystalline and amorphous structures with interlaced heterointerfaces (Fig. 1f and S2†). According to fast Fourier-transform (FFT) and inverse fast Fourier-transform (IFFT) images transformed HRTEM of a/c-Ru/Ti-RuO2 (Fig. 1f and g), the measured lattice distances of 0.227 nm and 0.205 nm correspond to the (111) plane of RuO2 and (101) plane of Ru, respectively. The diffusion ring without diffraction spots on the FFT image transformed F2 region confirmed the co-existence of amorphous and crystalline phases on a/c-Ru/Ti-RuO2 (Fig. 1h), which was further validated by XRD. Based on the XRD and HRTEM images (Fig. S3 and S4†), the crystal structure and morphology of a/c-Ru/RuO2 is similar to those of a/c-Ru/Ti-RuO2. The lattice distances of 0.205 nm, 0.214 nm, and 0.222 nm in the HRTEM image of a/c-Ru/RuO2 can be indexed as the (101) and (002) planes of metallic Ru and (110) plane of RuO2, respectively. Note that the lattice distance of RuO2 (0.227 nm) in the HRTEM image of a/c-Ru/Ti-RuO2 is larger than that in a/c-Ru/RuO2 (0.222 nm), which can be ascribed to the lattice expansion of RuO2 after Ti doping. Moreover, crystalline Ti doped Ru nanosheets and Ru nanosheets, which were named c-Ti-RuO2 and c-RuO2, respectively, were prepared by increasing synthetic temperature to 300 °C (see the ESI† for details). In the XRD of c-Ti-RuO2 nanosheets, the peaks at 28.0°, 35.3°, and 54.4° are indexed to the (110), (101), and (211) planes of RuO2, respectively (Fig. 1h and S5,† PDF: 70-2662). Comparing with c-RuO2, the peak of the (110) plane in the XRD pattern of c-Ti-RuO2 negatively shifts by 0.26°, implying that doping of Ti onto RuO2 induces the formation of lattice expansion.22 Despite the similar morphology, agglomerated nanoparticles appear on c-Ti-RuO2 nanosheets due to the increase in temperature during synthesis (Fig. S6†). Moreover, the HRTEM image and FFT and IFFT images of c-Ti-RuO2 further indicate that c-Ti-RuO2 is mainly composed of interlaced crystalline heterointerfaces. The lattice distances of 0.324 nm, 0.257 nm and 0.322 nm in the HRTEM image of c-Ti-RuO2 are ascribed to the (110), (101) and (110) planes of RuO2, respectively (Fig. 1i and j). Note that the lattice distance of the (110) plane for c-Ti-RuO2 is larger than that of RuO2 (0.317 nm), further confirming lattice expansion after Ti doping, which is consistent with the XRD results.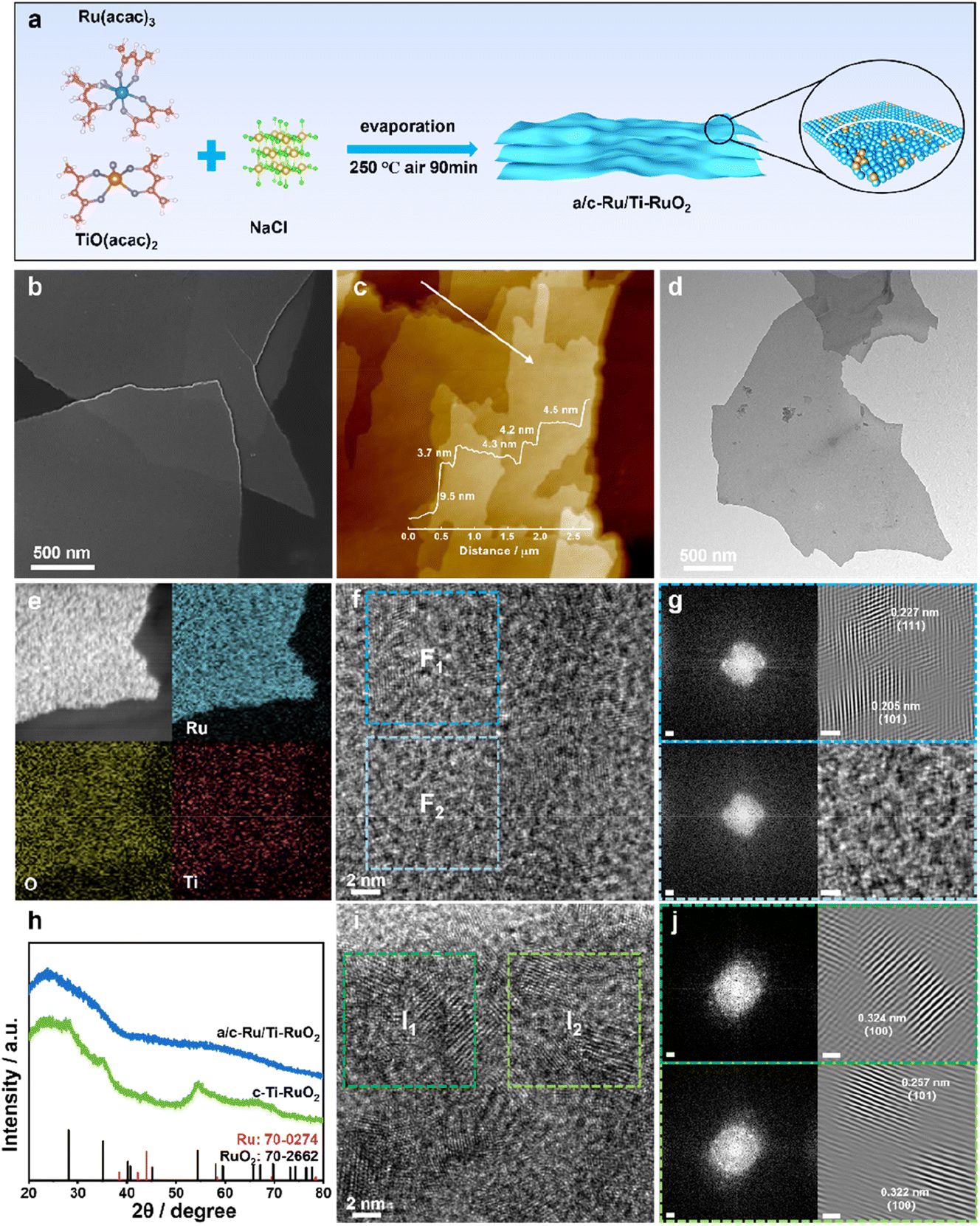 | ||
| Fig. 1 (a) Schematic illustration for the synthesis of a/c-Ru/Ti-RuO2 nanosheets. (b) SEM, (c) AFM, (d) TEM, (e) elemental mapping, and (f) HRTEM image of a/c-Ru/Ti-RuO2. (g) FFT and IFFT images transformed from (f). (h) XRD patterns of a/c-Ru/Ti-RuO2 and c-Ti-RuO2. (i) HRTEM image of c-Ti-RuO2. (j) FFT and IFFT images transformed from (i). The scale bars in FFT and IFFT images are 1/2 nm and 1 nm, respectively. | ||
To investigate the effects of Ti doping on a/c-Ru/Ti-RuO2 with amorphous–crystalline heterointerfaces, electron paramagnetic resonance (EPR) spectroscopy was performed.23 As shown in Fig. 2a and S7,† it was found that Ti doping and the amorphous–crystalline heterointerface both contribute to the formation of oxygen vacancies. Moreover, the surface electronic properties of a/c-Ru/Ti-RuO2 and c-Ti-RuO2 were measured by XPS (Fig. S8†). It was noted that Ru consists of Ru0 and Ru4+ in a/c-Ru/Ti-RuO2 and c-Ti-RuO2 (Fig. 2b). Compared to c-Ti-RuO2, the position of Ru4+ in the XPS spectrum of a/c-Ru/Ti-RuO2 negatively shifted by 0.12 eV, which was attributed to the amorphous structure of metallic Ru0.24,25 A similar negative shift was observed for the Ti 2p peak (Fig. 2c) in the XPS spectra of a/c-Ru/Ti-RuO2, which could be attributed to the formation of oxygen vacancies and the amorphous–crystalline heterointerface.23,26,27 In O 1s XPS spectra, the peaks at 529.6 eV, 530.6 eV and 532.5 eV were ascribed to Ru–O/Ti–O, surface bridge oxide of RuO2 (Ru–OBRI) and OH species on coordinatively unsaturated Ru sites (Ru–OCUS), respectively (Fig. 2d).28 Moreover, the larger amounts of Ru–OBRI in a/c-Ru/Ti-RuO2 compared with c-Ti-RuO2 confirmed more oxygen vacancy formation in a/c-Ru/Ti-RuO2 (Table S1†), which was in line with the EPR results.
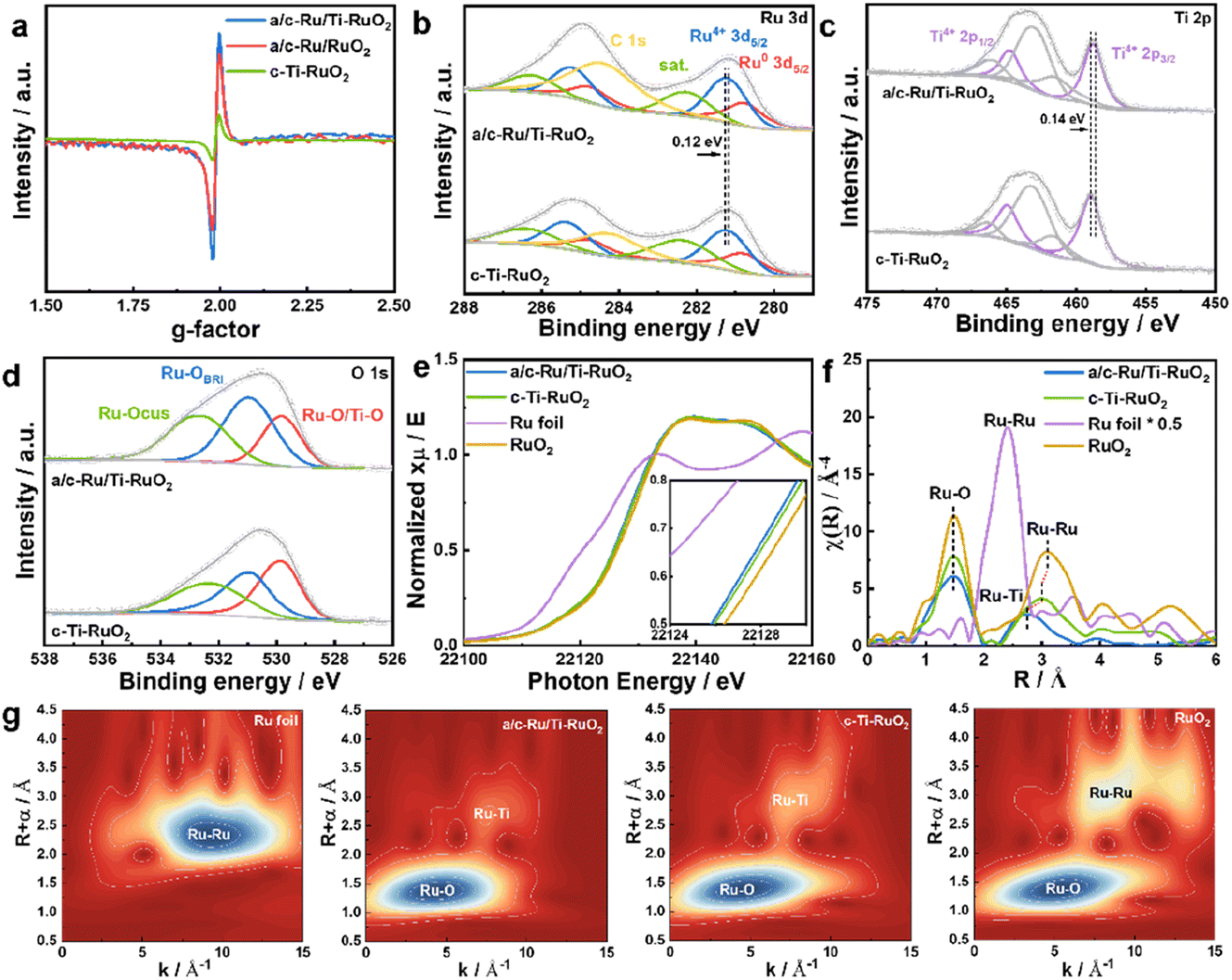 | ||
| Fig. 2 (a) EPR spectra of a/c-Ru/RuO2, a/c-Ru/Ti-RuO2 and c-Ti-RuO2. (b–d) Ru 3d, Ti 2p and O 1s spectra of a/c-Ru/Ti-RuO2 and c-Ti-RuO2. (e) Normalized XANES spectra of Ru foil, a/c-Ru/Ti-RuO2, c-Ti-RuO2, and RuO2 at the Ru-K edge. (f) The Fourier-transform (FT) k3-weighted χ(k) function of the EXAFS spectra of Ru foil, a/c-Ru/Ti-RuO2, c-Ti-RuO2, and RuO2 at the Ru-K edge. (g) Wavelet transformation for the k3-weighted EXAFS of the Ru-K edge for Ru foil, a/c-Ru/Ti-RuO2, c-Ti-RuO2, and RuO2. | ||
Furthermore, X-ray absorption near-edge structure spectroscopy (XANES) was performed to investigate the electronic structures of a/c-Ru/Ti-RuO2 and c-Ti-RuO2. Compared to RuO2, the smaller energy shift of the Ru-K edge position suggested that the valence of Ru in a/c-Ru/Ti-RuO2 and c-Ti-RuO2 is slightly lower than that in RuO2 (Fig. 2e). Compared to c-Ti-RuO2, the Ru-K edge of a/c-Ru/Ti-RuO2 slightly shifted to lower energy, suggesting that the amorphous structure of metallic Ru and the formation of oxygen vacancies in a/c-Ru/Ti-RuO2 influenced the electronic properties of Ru. In the corresponding Fourier-transformed extended X-ray absorption fine structure spectroscopy (EXAFS) spectra, the peaks at 1.47 Å and 3.10 Å in Ru–K EXAFS spectra were ascribed to the first Ru–O and second Ru–Ru coordination of RuO2, respectively, while the peak at 2.39 Å represented the Ru–Ru coordination of Ru foil (Fig. 2f). Compared to c-Ti-RuO2, the lower peak intensity of Ru–O coordination in the EXAFS spectrum of a/c-Ru/Ti-RuO2 compared with that of RuO2 implied unsaturated Ru–O coordination due to the formation of oxygen vacancies in a/c-Ru/Ti-RuO2.29–31 Furthermore, the feature at 3.01 Å was ascribed to Ru–Ti coordination in c-Ti-RuO2 (Fig. 2f). Compared to c-Ti-RuO2 (∼3.01 Å), the peak of Ru–Ti coordination in the EXAFS spectrum of a/c-Ru/Ti-RuO2 shifted to shorter distance and obviously weakened, indicating the higher disorder degree of a/c-Ru/Ti-RuO2 (Fig. 2f), which was further confirmed by the wavelet transformation (WT) contour plots (Fig. 2g). In addition, the absence of Ru–Ru coordination in the EXAFS spectrum of a/c-Ru/Ti-RuO2 may be caused by the disorder structure and small proportion of metallic Ru.29
HOR performance was performed using a typical three-electrode system on a rotating disk electrode (RDE) with H2-saturated alkaline electrolyte (0.1 M KOH). All electrochemical performance of catalysts was tested after activating for 100 cycles at a scan rate of 0.5 V s−1 at potential from −0.2 to 0.4 V vs. RHE. Screening experiments suggested that a/c-Ru/Ti-RuO2 with a Ru/Ti ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was the optimal HOR catalyst (Fig. S9†). Furthermore, the HOR polarization curve of a/c-Ru/Ti-RuO2 tested N2-saturated 0.1 M KOH solution (Fig. S10†) showed that current density is close to zero, indicating the occurrence of the HOR in H2-saturated 0.1 M KOH solution. As shown in Fig. 3a, a/c-Ru/Ti-RuO2 exhibited a current density of 2.48 mA cm−2 at 50 mV, which was higher than that of c-Ti-RuO2 (1.54 mA cm−2) and a/c-Ru/RuO2 (2.00 mA cm−2), indicating that the amorphous–crystalline heterointerface and Ti doping synergistically improved alkaline HOR activity. Moreover, the HOR polarization curves of catalysts were collected at different rotating rates from 400 to 2500 rpm to analyse the electron transport process (Fig. 3b and S11a–c†). The fitting slopes in Koutecky–Levich plots of a/c-Ru/Ti-RuO2, a/c-Ru/RuO2, c-Ti-RuO2, and Pt/C (inset of Fig. 3b and S11d†) were 13.83, 13.49, 14.86, and 14.79 cm2 mA−1 rpm1/2, respectively, showing limiting current densities of 2.89, 2.96, 2.69, and 2.70 mA cm−2 at 1600 rpm, being consistent with the theoretical value.32 The kinetic currents (ik) were calculated by Butler–Volmer fitting. a/c-Ru/Ti-RuO2 displayed higher kinetic current than the other catalysts at all potentials, suggesting its fastest HOR kinetics (Fig. 3c). The exchange current (i0) of a/c-Ru/Ti-RuO2, which was calculated via the Butler–Volmer equation with nonlinear fitting, was 1.49 mA, which was 3.6, 1.9, and 5.3 times compared with that of c-Ti-RuO2 (0.42 mA), a/c-Ru/RuO2 (0.77 mA), and commercial Pt/C (0.28 mA). To quantitatively compare HOR activity, the mass activity and specific activity of these catalysts were normalized to the mass of Ru or Pt and electrochemically surface area (ECSA, marked as jk,m and j0,s). The loading mass of Ru was measured by thermogravimetry (TG) and energy dispersive spectroscopy (EDS) (Fig. S1, S6 and S12†). As shown in Fig. 3d, the jk,m of a/c-Ru/Ti-RuO2 was 4.16 A mgRu−1, which was 9.2, 7.7, and 19.8 times higher than that of c-Ti-RuO2 (0.41 A mgRu−1), a/c-Ru/RuO2 (0.48 A mgRu−1), and commercial Pt/C (0.20 A mgPt−1), respectively. Note that the mass activity of a/c-Ru/Ti-RuO2 has surpassed that of most reported catalysts (Fig. S13 and Table S2†). Moreover, ECSAs were obtained by Cu underpotential deposition (CuUPD) stripping (Fig. S14†). Then, ECSA normalized specific activities of a/c-Ru/Ti-RuO2, c-Ti-RuO2, a/c-Ru/RuO2 and commercial Pt/C were 1.65, 0.59, 0.68, and 0.51 mA cm−2, respectively. The highest mass activity and specific activity of a/c-Ru/Ti-RuO2 indicated the best kinetic and intrinsic activities of a/c-Ru/Ti-RuO2. Furthermore, the stability of catalysts was tested in H2-saturated 0.1 M KOH electrolyte by chronoamperometry technology at 0.1 V vs. RHE. a/c-Ru/Ti-RuO2 could maintain 80% of the initial current after stability testing for 12000 s, which was much longer than that for a/c-Ru/RuO2 (2900 s), c-Ti-RuO2 (1560 s), and Pt/C (940 s) (Fig. 3e). Specifically, the morphology of a/c-Ru/Ti-RuO2 was well maintained with only negligible Ru dissolution after durability testing (Fig. S15†).
:1 was the optimal HOR catalyst (Fig. S9†). Furthermore, the HOR polarization curve of a/c-Ru/Ti-RuO2 tested N2-saturated 0.1 M KOH solution (Fig. S10†) showed that current density is close to zero, indicating the occurrence of the HOR in H2-saturated 0.1 M KOH solution. As shown in Fig. 3a, a/c-Ru/Ti-RuO2 exhibited a current density of 2.48 mA cm−2 at 50 mV, which was higher than that of c-Ti-RuO2 (1.54 mA cm−2) and a/c-Ru/RuO2 (2.00 mA cm−2), indicating that the amorphous–crystalline heterointerface and Ti doping synergistically improved alkaline HOR activity. Moreover, the HOR polarization curves of catalysts were collected at different rotating rates from 400 to 2500 rpm to analyse the electron transport process (Fig. 3b and S11a–c†). The fitting slopes in Koutecky–Levich plots of a/c-Ru/Ti-RuO2, a/c-Ru/RuO2, c-Ti-RuO2, and Pt/C (inset of Fig. 3b and S11d†) were 13.83, 13.49, 14.86, and 14.79 cm2 mA−1 rpm1/2, respectively, showing limiting current densities of 2.89, 2.96, 2.69, and 2.70 mA cm−2 at 1600 rpm, being consistent with the theoretical value.32 The kinetic currents (ik) were calculated by Butler–Volmer fitting. a/c-Ru/Ti-RuO2 displayed higher kinetic current than the other catalysts at all potentials, suggesting its fastest HOR kinetics (Fig. 3c). The exchange current (i0) of a/c-Ru/Ti-RuO2, which was calculated via the Butler–Volmer equation with nonlinear fitting, was 1.49 mA, which was 3.6, 1.9, and 5.3 times compared with that of c-Ti-RuO2 (0.42 mA), a/c-Ru/RuO2 (0.77 mA), and commercial Pt/C (0.28 mA). To quantitatively compare HOR activity, the mass activity and specific activity of these catalysts were normalized to the mass of Ru or Pt and electrochemically surface area (ECSA, marked as jk,m and j0,s). The loading mass of Ru was measured by thermogravimetry (TG) and energy dispersive spectroscopy (EDS) (Fig. S1, S6 and S12†). As shown in Fig. 3d, the jk,m of a/c-Ru/Ti-RuO2 was 4.16 A mgRu−1, which was 9.2, 7.7, and 19.8 times higher than that of c-Ti-RuO2 (0.41 A mgRu−1), a/c-Ru/RuO2 (0.48 A mgRu−1), and commercial Pt/C (0.20 A mgPt−1), respectively. Note that the mass activity of a/c-Ru/Ti-RuO2 has surpassed that of most reported catalysts (Fig. S13 and Table S2†). Moreover, ECSAs were obtained by Cu underpotential deposition (CuUPD) stripping (Fig. S14†). Then, ECSA normalized specific activities of a/c-Ru/Ti-RuO2, c-Ti-RuO2, a/c-Ru/RuO2 and commercial Pt/C were 1.65, 0.59, 0.68, and 0.51 mA cm−2, respectively. The highest mass activity and specific activity of a/c-Ru/Ti-RuO2 indicated the best kinetic and intrinsic activities of a/c-Ru/Ti-RuO2. Furthermore, the stability of catalysts was tested in H2-saturated 0.1 M KOH electrolyte by chronoamperometry technology at 0.1 V vs. RHE. a/c-Ru/Ti-RuO2 could maintain 80% of the initial current after stability testing for 12000 s, which was much longer than that for a/c-Ru/RuO2 (2900 s), c-Ti-RuO2 (1560 s), and Pt/C (940 s) (Fig. 3e). Specifically, the morphology of a/c-Ru/Ti-RuO2 was well maintained with only negligible Ru dissolution after durability testing (Fig. S15†).
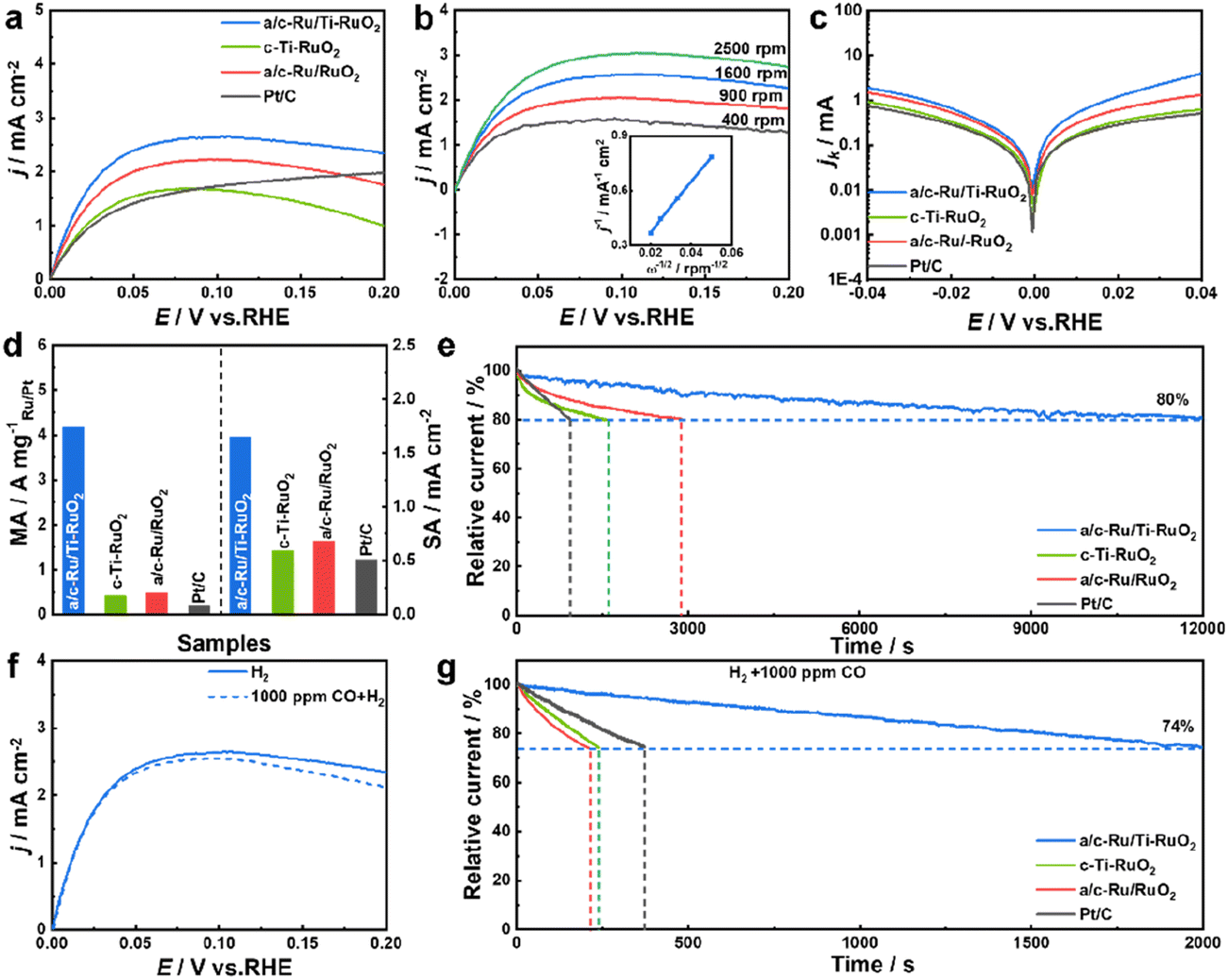 | ||
| Fig. 3 (a) HOR polarization curves of a/c-Ru/Ti-RuO2, c-Ti-RuO2, a/c-Ru/RuO2 and commercial Pt/C. (b) HOR polarization curves of a/c-Ru/Ti-RuO2 at different rotating speeds (inset: Koutecky–Levich plots of the catalyst at 0.1 V vs. RHE). (c) Tafel plots and (d) kinetic current density and exchange current density for various catalysts. (e) Chronoamperometric responses at an overpotential of 0.1 V vs. RHE of catalysts in H2-saturated 0.1 M KOH. (f) HOR polarization curves of a/c-Ru/Ti-RuO2 in H2-saturated and 1000 CO ppm + H2-saturated 0.1 M KOH electrolyte. (g) Chronoamperometric responses at an overpotential of 0.1 V vs. RHE of catalysts in 1000 CO ppm + H2-saturated 0.1 M KOH electrolyte. | ||
In addition, the resistance to CO poisoning of a/c-Ru/Ti-RuO2 was tested by introducing 1000 ppm CO (Fig. 3f). A slight decline in the HOR polarization curve of a/c-Ru/Ti-RuO2 was observed in 1000 ppm CO + H2-saturated 0.1 M KOH electrolyte, which was much smaller than that of c-Ti-RuO2, a/c-Ru/RuO2, and commercial Pt/C (Fig. S16†), indicating that Ti doping and the heterointerface efficiently block CO species from poisoning active sites. Besides, the superior resistance of a/c-Ru/Ti-RuO2 to CO poisoning was further evaluated by chronoamperometry at 0.1 V vs. RHE. As shown in Fig. 3g, the currents of c-Ti-RuO2, a/c-Ru/RuO2, and commercial Pt/C sharply decreased to 74% of the initial current after being tested for 240 s, 210 s, and 370 s, respectively, while 74% of the initial current was maintained after 2000 s for a/c-Ru/Ti-RuO2, indicating the superior CO tolerance of a/c-Ru/Ti-RuO2 to other references.
To deeply understand the enhanced HOR performance over a/c-Ru/Ti-RuO2, CV, in situ FTIR, H/D isotope experiment and CO stripping experiment were performed. The larger area of the CV curve of a/c-Ru/Ti-RuO2 than that of c-Ti-RuO2 and a/c-Ru/RuO2 implied that more active sites were exposed on a/c-Ru/Ti-RuO2 (Fig. S17†). The intense peak, appearing at ∼0.1 V vs. RHE in the CV curve after activating for 100 cycles, corresponded to the adsorption of hydrogen and exchange reaction of hydrogen to adsorbed hydroxyl, which contributed to exposure of active sites of a/c-Ru/Ti-RuO2 after activation (Fig. 4a).7 Note that the morphology and structure of a/c-Ru/Ti-RuO2 were largely preserved after the present activation (Fig. S18a–d†). In XPS spectra, the positive shift of the Ru 3d peak for a/c-Ru/Ti-RuO2 after activation was attributed to the adsorption of OHad species in alkaline electrolyte (Fig. S18e†),33–35 which was further confirmed by the positive shifts of Ru–OCUS and Ru–OBRI peaks in the XPS spectrum of activated a/c-Ru/Ti-RuO2 (Fig. 4b).36–38 The new peak at 535.3 eV was ascribed to the O–F bond due to the Nafion binder. Analysis of XANES and EXAFS spectra of the fresh and activated a/c-Ru/Ti-RuO2 further confirmed that the present activation could significantly enhance OHad adsorption, due to lower coordination of Ru–Ru after activation than fresh a/c-Ru/Ti-RuO2 (Fig. 4c and S18f†). Furthermore, based on comparison CV curves of catalysts after activating for 100 cycles, the larger oxidation peak area of a/c-Ru/Ti-RuO2 than that of c-Ti-RuO2, a/c-Ru/RuO2, and commercial Pt/C suggested stronger interaction between Ru and hydrogen/hydroxyl species, while the lower potential in the CV peak further indicated its weak binding ability with adsorbed hydrogen (Fig. 4a, d and S19†). This is due to the optimized electronic structure of Ru active sites by oxygen vacancies and unsaturated coordination of a/c-Ru/Ti-RuO2 with amorphous–crystallines interface. Moreover, an isotope experiment was performed to further evaluate the enhanced OHad adsorption.8,39 As shown in Fig. S20,† the decreased current density ratio of jKOH/jKOD with potential implies that the replacement of OH− with OD− reduces the HOR activity of catalysts, which is attributed to the different pD/pH values of KOD and KOH.40,41 Note that the ratio of jKOH/jKOD for a/c-Ru/Ti-RuO2 is much smaller than that of commercial Pt/C, indicating its strong adsorption ability towards both OH− and OD− on a/c-Ru/Ti-RuO2, as a result of significantly enhanced HOR performance of a/c-Ru/Ti-RuO2 (Fig. 4e).42
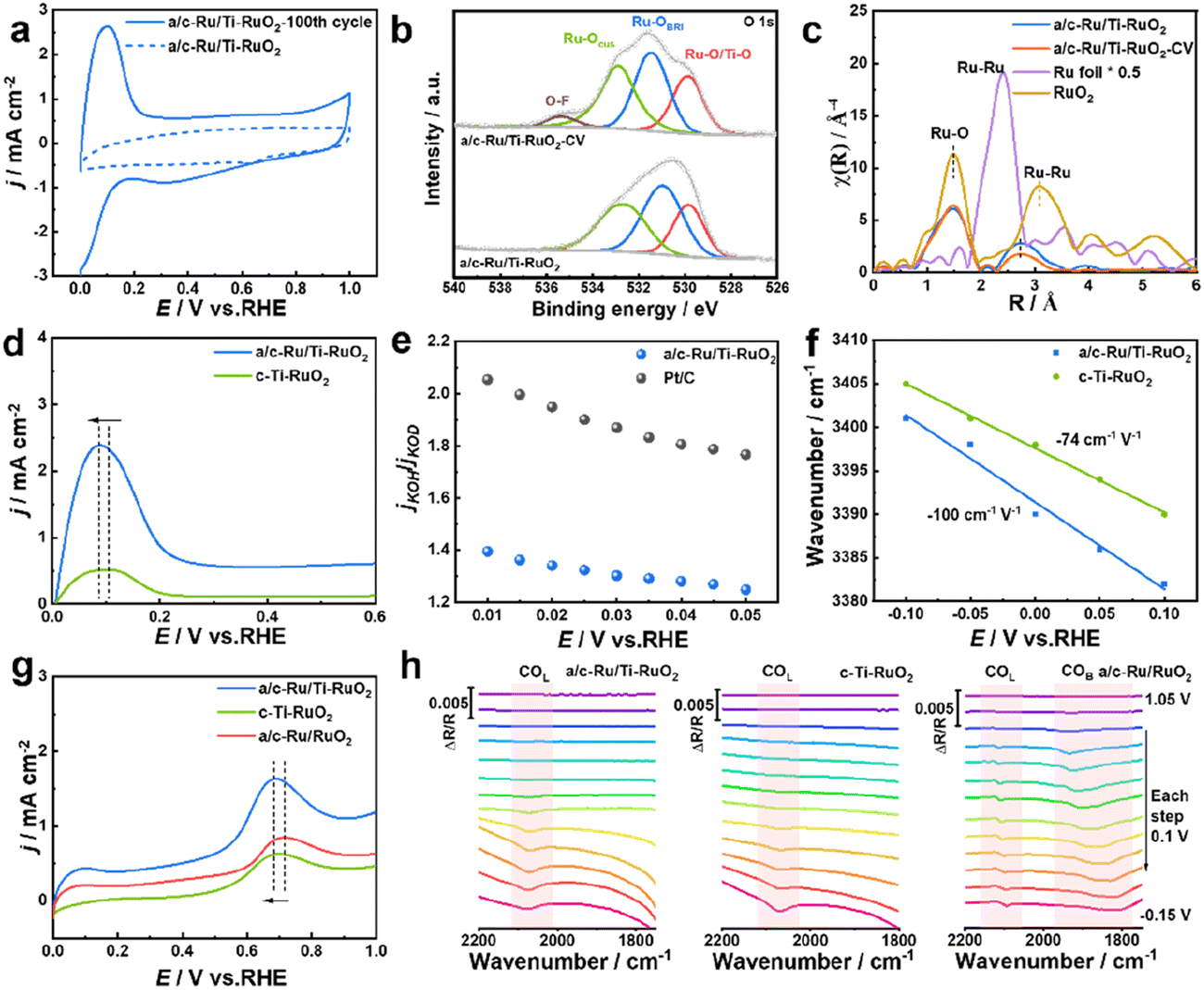 | ||
| Fig. 4 (a) CV curves collected in 0.1 M KOH during 0–1 V vs. RHE of a/c-Ru/Ti-RuO2 before and after activating for 100 cycles. (b) O 1s spectra and (c) Ru K-edge Fourier-transform (FT) k3-weighted χ(k) function of the EXAFS spectra of a/c-Ru/Ti-RuO2 before and after CV activation. (d) CV peaks of a/c-Ru/Ti-RuO2 and c-Ti-RuO2 in 0.1 M KOH. (e) The ratio of current density in 0.1 M KOH and KOD electrolyte for a/c-Ru/Ti-RuO2 and commercial Pt/C. (f) In situ FTIR wavenumber change of O–H stretching vibration for a/c-Ru/Ti-RuO2 and c-Ti-RuO2. (g) CO-stripping voltammetry of a/c-Ru/Ti-RuO2, c-Ti-RuO2, and a/c-Ru/RuO2. (h) In situ FTIR spectra of a/c-Ru/Ti-RuO2, c-Ti-RuO2, and a/c-Ru/RuO2 for CO oxidation recorded during stepping the potential from −0.15 to 1.05 V vs. RHE in 0.1 M KOH electrolyte. | ||
Besides, in situ Fourier transform infrared spectroscopy (FTIR) was performed in H2-saturated 0.1 M KOH electrolyte to evaluate the HBE and OHBE for a/c-Ru/Ti-RuO2. As shown in Fig. S21,† a vibrational band is observed at 2064 cm−1 for a/c-RuTi, which is ascribed to the Ru–H bond.7 Note that the Ru–H vibration of a/c-Ru/Ti-RuO2 negatively shifts to a lower wavenumber compared with that of c-Ti-RuO2 (2071 cm−1), suggesting that the binding strength of H on a/c-Ru/Ti-RuO2 is weaker than that on c-Ti-RuO2.43,44 The band at ∼3400 cm−1 in in situ FTIR spectra was ascribed to O–H stretching vibration of interfacial water,7,45,46 where a larger Stark tuning rate for a/c-Ru/Ti-RuO2 (−100 cm−1 V−1) than that of c-Ti-RuO2 (−74 cm−1 V−1) indicates that the heterointerface of a/c-Ru/Ti-RuO2 is beneficial to enhance interaction with OHad species (Fig. 4f).45 Additionally, a CO-stripping experiment was performed to reveal the enhanced resistance to CO poisoning of a/c-Ru/Ti-RuO2. As shown in Fig. 4g and S22,† the peak potential of CO stripping follows the trend as a/c-Ru/Ti-RuO2 (0.689 V vs. RHE) < c-Ti-RuO2 (0.702 V vs. RHE) < a/c-Ru/RuO2 (0.718 V vs. RHE), suggesting that Ti doping efficiently enhances OH species affinity of a/c-Ru/RuO2 for facilitating adsorbed CO oxidation. In the in situ FTIR spectra collected from −0.15 to 1.05 V vs. RHE, the adsorption peaks at ∼2094 cm−1 and ∼1850 cm−1 could be assigned to CO linear and bridged adsorption, respectively (Fig. 4h).47–50 Compared to a/c-Ru/RuO2, the absence of the peak of CO bridged adsorption in FTIR spectra of a/c-Ru/Ti-RuO2 and c-Ti-RuO2 suggests that Ti-doping can weaken CO adsorption.47 Besides, the peak intensity of CO linear adsorption for a/c-Ru/Ti-RuO2 and c-Ti-RuO2 decreases much faster than that of a/c-Ru/RuO2 with the potentials, further confirming the weakened CO adsorption on a/c-Ru/Ti-RuO2 and c-Ti-RuO2.51,52 In addition, the peak of CO adsorption completely disappears at 0.65 V vs. RHE in the in situ FTIR spectrum of a/c-Ru/Ti-RuO2, while the obvious peaks of CO adsorption in the FTIR spectra of a/c-Ru/RuO2 at the same potential demonstrate that the strong synergy at the heterointerface can significantly weaken the binding strength of CO on a/c-Ru/Ti-RuO2 (Fig. S23†). Based on the above results, the amorphous–crystalline heterointerface is beneficial for formation of oxygen vacancies, which can weaken binding energy of Ru–H and promote OHad adsorption, as a result of improved HOR performance.
Conclusions
In summary, we have demonstrated that a/c-Ru/Ti-RuO2 nanosheets can serve as an efficient catalyst for alkaline HOR. The results from XAFS, the isotopic experiment, and in situ FTIR suggest that the amorphous–crystalline heterointerface of a/c-Ru/Ti-RuO2 provides abundant oxygen vacancies, which can weaken binding energy of Ru–H and strengthen OHad adsorption, leading to a significant enhancement in alkaline HOR activity. Impressively, the mass activity and specific activity of a/c-Ru/Ti-RuO2 reach 4.16 A mgRu−1 and 1.65 mA cm−2, which are 19.8 times and 2.2 times higher than that of commercial Pt/C, respectively. Moreover, a/c-Ru/Ti-RuO2 with amorphous–crystalline heterointerfaces displays fast CO oxidation efficiently enhancing its CO tolerance. This work provides a strategy for enhancing alkaline HOR performance over Ru-based catalysts, which will attract immediate interest in chemistry, materials science and beyond.Data availability
The data supporting the findings of this study, including chemicals, sample preparation, instrumentation, supplementary tests, and corresponding supplementary discussions, are available in ESI.†Author contributions
Licheng Wei: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, visualization. Nan Fang, Fei Xue, Shangheng Liu, Wei-Hsiang Huang, Chih-Wen Pao, Zhiwei Hu: formal analysis, investigation. Yong Xu: writing – review & editing, supervision. Hongbo Geng: writing – review & editing, supervision, funding acquisition. Xiaoqing Huang: conceptualization, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2020YFB1505802), the Ministry of Science and Technology of China (2017YFA0208200), the National Natural Science Foundation of China (22025108, U21A20327, and 22121001), the start-up support from Xiamen University, the Natural Science Foundation of the Jiangsu Higher Education Institutions (22KJA430009), the Science and Technology Development Plan of Suzhou (ZXL2022176), and the Guangdong Provincial Natural Science Fund for Distinguished Young Scholars (2021B1515020081). We acknowledge support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials.Notes and references
- X. Wang, X. Li, D. Kong, L. Zhao, Y. Cui, Y. Wang, T. Cai, Q. Xue, Z. Yan and W. Xing, Nano Energy, 2022, 104, 107877 CrossRef CAS.
- L. Su, D. Gong, Y. Jin, D. Wu and W. Luo, J. Energy Chem., 2022, 66, 107–122 CrossRef CAS.
- W. Moschkowitsch, O. Lori and L. Elbaz, ACS Catal., 2022, 12, 1082–1089 CrossRef CAS.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187–200 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2015, 8, 177–181 RSC.
- Y. Xue, L. Shi, X. Liu, J. Fang, X. Wang, B. P. Setzler, W. Zhu, Y. Yan and Z. Zhuang, Nat. Commun., 2020, 11, 5651 CrossRef CAS PubMed.
- B. Zhang, B. Zhang, G. Zhao, J. Wang, D. Liu, Y. Chen, L. Xia, M. Gao, Y. Liu, W. Sun and H. Pan, Nat. Commun., 2022, 13, 5894 CrossRef CAS PubMed.
- L. Su, Y. Zhao, Y. Jin, Z. Liu, H. Cui and W. Luo, Adv. Funct. Mater., 2022, 32, 2113047 CrossRef CAS.
- T. Wang, L. Y. Li, L. N. Chen, T. Sheng, L. Chen, Y. C. Wang, P. Zhang, Y. H. Hong, J. Ye, W. F. Lin, Q. Zhang, P. Zhang, G. Fu, N. Tian, S. G. Sun and Z. Y. Zhou, J. Am. Chem. Soc., 2022, 144, 9292–9301 CrossRef CAS PubMed.
- L. Zhang, H. Jang, H. Liu, M. G. Kim, D. Yang, S. Liu, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2021, 60, 18821–18829 CrossRef CAS PubMed.
- F. Yang, X. Bao, P. Li, X. Wang, G. Cheng, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2019, 58, 14179–14183 CrossRef CAS PubMed.
- L. Su, Y. Jin, D. Gong, X. Ge, W. Zhang, X. Fan and W. Luo, Angew. Chem., Int. Ed., 2023, 62, e202215585 CrossRef CAS PubMed.
- W. Wang, S. Guo, I. Lee, K. Ahmed, J. Zhong, Z. Favors, F. Zaera, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4452 CrossRef PubMed.
- J. Xu and X. Kong, Small Methods, 2022, 6, e2101432 CrossRef PubMed.
- M. Kundu, R. Mishra, T. Bhowmika and S. Barman, J. Mater. Chem. A, 2018, 6, 23531–23541 RSC.
- Y. Yang, X. Sun, G. Han, X. Liu, X. Zhang, Y. Sun, M. Zhang, Z. Cao and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 10644–10649 CrossRef CAS PubMed.
- Y. Zhou, W. Hao, X. Zhao, J. Zhou, H. Yu, B. Lin, Z. Liu, S. J. Pennycook, S. Li and H. J. Fan, Adv. Mater., 2022, 34, e2100537 CrossRef PubMed.
- Y. Zhang, J. Fang, L. Zhang, D. Wei, W. Zhu and Z. Zhuang, Chem. Commun., 2022, 58, 4488–4491 RSC.
- Z. Ma, C. Chen, X. Cui, L. Zeng, L. Wang, W. Jiang and J. Shi, ACS Appl. Mater. Interfaces, 2021, 13, 44224–44233 CrossRef CAS PubMed.
- J. Kang, G. Liu, Q. Hu, Y. Huang, L.-M. Liu, L. Dong, G. Teobaldi and L. Guo, J. Am. Chem. Soc., 2023, 145, 25143–25149 CrossRef CAS PubMed.
- J. Zhang, G. Ren, D. Li, Q. Kong, Z. Hu, Y. Xu, S. Wang, L. Wang, M. Cao and X. Huang, Sci. Bull., 2022, 67, 2103–2111 CrossRef CAS PubMed.
- J. Kang, X. Qiu, Q. Hu, J. Zhong, X. Gao, R. Huang, C. Wan, L. M. Liu, X. Duan and L. Guo, Nat. Catal., 2021, 4, 1050–1058 CrossRef CAS.
- A. Foelske, O. Barbieri, M. Hahn and R. Kötz, Electrochem. Solid-State Lett., 2006, 9, 268–272 CrossRef.
- S. Liu, L. Dai, Y. Qu, Y. Qiu, J. Fan, X. Li, Q. Zhangc and X. Guo, Mater. Chem. Front., 2021, 5, 6648–6658 RSC.
- G. Feng, M. Hu, S. Yuan, J. Nan and H. Zeng, Nanomaterials, 2021, 11, 2801 CrossRef CAS PubMed.
- J. Xu, W. Dong, C. Song, Y. Tang, W. Zhao, Z. Hong and F. Huang, J. Mater. Chem. A, 2016, 4, 15698–15704 RSC.
- J. P. Zheng, P. J. Cygan and T. R. Jow, J. Electrochem. Soc., 1995, 142, 2699–2703 CrossRef CAS.
- G. Wu, X. Zheng, P. Cui, H. Jiang, X. Wang, Y. Qu, W. Chen, Y. L. J. Ge, Y. Yao, R. Sun, Y. Wu, L. Gu, X. Hong and Y. Li, Nat. Commun., 2019, 10, 4855 CrossRef PubMed.
- D. McKeown, P. Hagans, L. L. Carette, A. Russell, K. Swider and D. Rolison, J. Phys. Chem. B, 1999, 103, 4825–4832 CrossRef CAS.
- Q. Pei, J. Yu, G. Qiu, K. Tan, J. Wen, Y. Yu, J. Wang, J. Guo, J. Guo, L. Rao, T. He and P. Chen, Appl. Catal., B, 2023, 336, 122947 CrossRef CAS.
- C. Zhan, Y. Xu, L. Bu, H. Zhu, Y. Feng, T. Yang, Y. Zhang, Z. Yang, B. Huang, Q. Shao and X. Huang, Nat. Commun., 2021, 12, 6261 CrossRef CAS PubMed.
- Q. Yao, B. Huang, N. Zhang, M. Sun, Q. Shao and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 13983–13988 CrossRef CAS PubMed.
- J. Wang, Y. Ji, R. Yin, Y. Li, Q. Shao and X. Huang, J. Mater. Chem. A, 2019, 7, 6411–6416 RSC.
- Y. Dong, Q. Sun, C. Zhan, J. Zhang, H. Yang, T. Cheng, Y. Xu, Z. Hu, C. W. Pao, H. Geng and X. Huang, Adv. Funct. Mater., 2022, 33, 2210328 CrossRef.
- R. R. Rao, M. J. Kolb, J. Hwang, A. F. Pedersen, A. Mehta, H. You, K. A. Stoerzinger, Z. Feng, H. Zhou, H. Bluhm, L. Giordano, I. E. L. Stephens and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 17802–17811 CrossRef CAS.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- T. F. Hsieh, C. C. Chuang, W. J. Chen, J. H. Huang, W. T. Chen and C. M. Shu, Carbon, 2012, 50, 1740–1747 CrossRef CAS.
- E. Liu, L. Jiao, J. Li, T. Stracensky, Q. Sun, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2020, 13, 3064–3074 RSC.
- E. Liu, L. Jiao, J. Li, T. Stracensky, Q. Sun, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2020, 13, 3064–3074 RSC.
- L. Rebollar, S. Intikhab, J. D. Snyder and M. H. Tang, J. Phys. Chem. Lett., 2020, 11, 2308–2313 CrossRef CAS PubMed.
- B. Zhang, B. Zhang, G. Zhao, J. Wang, D. Liu, Y. Chen, L. Xia, M. Gao, Y. Liu, W. Sun and H. Pan, Nat. Commun., 2022, 13, 5894 CrossRef CAS PubMed.
- S. Zhu, X. Qin, Y. Yao and M. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef PubMed.
- J. Kubota and K. Aika, J. Chem. Soc., Chem. Commun., 1992, 8, 661–662 RSC.
- Z. Zhang, X. Tian, B. Zhang, L. Huang, F. Zhu, X. Qu, L. Liu, S. Liu, Y. Jiang and S. Sun, Nano Energy, 2017, 34, 224–232 CrossRef CAS.
- S. Zhu, X. Qin, Y. Yao and M. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef PubMed.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220–1230 CrossRef CAS.
- C. Fang, X. Jiang, J. Hu, J. Song, N. Sun, D. Zhang and L. Kuai, ACS Appl. Mater. Interfaces, 2021, 13, 5079–5087 CrossRef CAS PubMed.
- M. J. S. Farias, W. Cheuquepán, A. A. Tanaka and J. M. Feliu, ACS Catal., 2017, 7, 3434–3445 CrossRef CAS.
- C. Elmasides, D. I. Kondarides, S. G. Neophytides and X. E. Verykios, J. Catal., 2001, 198, 195–207 CrossRef CAS.
- Z. Dong, Y. Nan, T. Tang, X.-Z. Liu, J. Fu, H.-R. Pan, Z. Jiang, L. Ding, X. Cheng, L.-R. Zheng, J. Zhang, X. Chang, B. Xu and J.-S. Hu, ACS Catal., 2023, 13, 7822–7830 CrossRef CAS.
- T. Komatsu and Y. Fukui, Appl. Catal., A, 2005, 279, 173–180 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06705j |
| This journal is © The Royal Society of Chemistry 2024 |