
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObtaining giant Rashba–Dresselhaus spin splitting in two-dimensional chiral metal–organic frameworks†
Shanshan
Liu
a,
Ke
Xu
*c,
Xingxing
Li
 *ab,
Qunxiang
Li
ab and
Jinlong
Yang
ab
*ab,
Qunxiang
Li
ab and
Jinlong
Yang
ab
aKey Laboratory of Precision and Intelligent Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: lixx@ustc.edu.cn
bHefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China
cHubei Key Laboratory of Low Dimensional Optoelectronic Materials and Devices, School of Physics and Electronic Engineering, Hubei University of Arts and Science, Xiangyang, 441053, China. E-mail: xuke@hbuas.edu.cn
First published on 5th April 2024
Abstract
Two-dimensional (2D) nonmagnetic semiconductors with large Rashba–Dresselhaus (R–D) spin splitting at valence or conduction bands are attractive for magnetic-field-free spintronic applications. However, so far, the number of 2D R–D inorganic semiconductors has been quite limited, and the factors that determine R–D spin splitting as well as rational design of giant spin splitting, remain unclear. For this purpose, by exploiting 2D chiral metal–organic frameworks (CMOFs) as a platform, we theoretically develop a three-step screening method to obtain a series of candidate 2D R–D semiconductors with valence band spin splitting up to 97.2 meV and corresponding R–D coupling constants up to 1.37 eV Å. Interestingly, the valence band spin texture is reversible by flipping the chirality of CMOFs. Furthermore, five keys for obtaining giant R–D spin splitting in 2D CMOFs are successfully identified: (i) chirality, (ii) large spin–orbit coupling, (iii) narrow band gap, (iv) valence and conduction bands having the same symmetry at the Γ point, and (v) strong ligand field.
Introduction
One of the primary goals in spintronics is to utilize the spin degree of freedom of electrons for data storage and information transmission.1,2 One promising approach is using the so-called Rashba–Dresselhaus (R–D) spin–orbit coupling (SOC) effect, which relies on the spin–orbit interaction of carriers in an inversion-asymmetric environment.3–11 Due to such a SOC effect, the spin and momentum are entangled together, forming a spin texture in the momentum space. It holds significant importance for generating, manipulating, and detecting spin currents.12 The most notable feature of the R–D effect is its capacity to manipulate spin exclusively through electric fields, without the need for external magnetic fields or magnetic exchange interactions, enabling potential applications in spin field-effect transistors (SFETs).13,14 Despite the great potential of the R–D effect, achieving giant spin splitting in 2D materials is limited to a few inorganic semiconductors.15–29 Moreover, the underlying factors influencing spin splitting and the general method to achieve giant spin splitting remain to be explored.Considering the limited structural diversity and tunability of inorganic materials, we turn our attention to 2D metal–organic frameworks (MOFs).30 Compared with inorganic materials, the properties of 2D MOFs can be easily designed by adjusting the metal nodes, ligand linkers, or connection modes between them. For the occurrence of R–D spin splitting, one of the basic requirements is to break the spatial inversion symmetry. Here, the combined term, R–D spin splitting, refers to either Rashba or Dresselhaus spin splitting. In this aspect, 2D chiral metal–organic frameworks (CMOFs) with the absence of inversion and mirror symmetries, provide a natural platform.31–35 Constructed from chiral molecules and metals, 2D CMOFs serve as an important subclass of the MOF family and have received extensive attention in recent years,36–42 especially in asymmetric catalysis43 and enantioselective44,45 applications. However, whether and how notable R–D spin splitting can be achieved in 2D CMOFs and the correlation between chirality and the R–D effect are still unclear.
To solve the above issues, we choose octahedrally coordinated metals (Sr–Sn and Ba–Pb) and different substituted bipyridine ligands with axial chirality to form a family of 2D CMOFs. Through a three-step screening process based on first-principles calculations, more than 20 2D CMOFs with obvious R–D spin splitting at the valence band (VB) are obtained. Among them, five structures possess R–D energies (ΔE) greater than 80 meV at the Γ point in the VB and R–D coupling constants (α) exceeding 1.20 eV Å, which are comparable to those reported for 2D inorganic materials.15,16 In particular, the α of the 2D CMOF structure OsH2(bipyridine_CN) reaches 1.37 eV Å, with the ΔE being 97.2 meV. Furthermore, we find that the VB spin texture is reversible via chirality inversion of 2D CMOFs. Finally, through systematical analysis of the above CMOFs, five main factors affecting the spin splitting are revealed: in addition to (i) chirality and (ii) strength of SOC, the following three factors also play a crucial role, that is, (iii) the size of the band gap, (iv) the symmetry of the VB and conduction band (CB) at the Γ point, and (v) ligand field strength.
Results and discussion
2D CMOF screening workflow
To generate the R–D spin splitting, two basic conditions need to be satisfied: the systems (i) break the spatial inversion symmetry3,4 and (ii) have large spin–orbit coupling.46 In addition, it is also generally accepted that the system is not magnetic. To fulfill these conditions, we utilize two inorganic ligands (L1) coordinated with heavy metal atoms (M) as nodes and axially chiral ligands47–49 (L2) as linkers to form 2D CMOFs in a tetragonal lattice (Fig. 1a). In this structure, the two L1 ligands are axially connected to M, while the L2 ligands form four coordination bonds with M equatorially. Such an arrangement results in the metal center with an octahedral coordination configuration (Fig. 1a). For metal atoms (M), those from the fifth period (Sr to Sn) and sixth period (Ba to Pb) are selected. For the axial inorganic ligands (L1), the options are –I, –Br, –Cl, –F, –CN, and –H. For equatorial organic ligands (L2), 4,4′-bipyridine derivatives with axial chirality are chosen, and the axial chirality of the L2 ligand varies by using different substituent groups for X and Y (Fig. 1a).50,51 If the Y group is –H, the L2 ligand is represented as bipyridine_X, and if the Y group is changed, it is represented as bipyridine_X_Y. The 2D CMOFs are denoted by ML1(bipyridine_X_Y). By adjusting the three degrees of freedom (M, L1, and L2), the 2D CMOFs with giant R–D energy (ΔE) are screened out through the following three steps (Fig. 1b):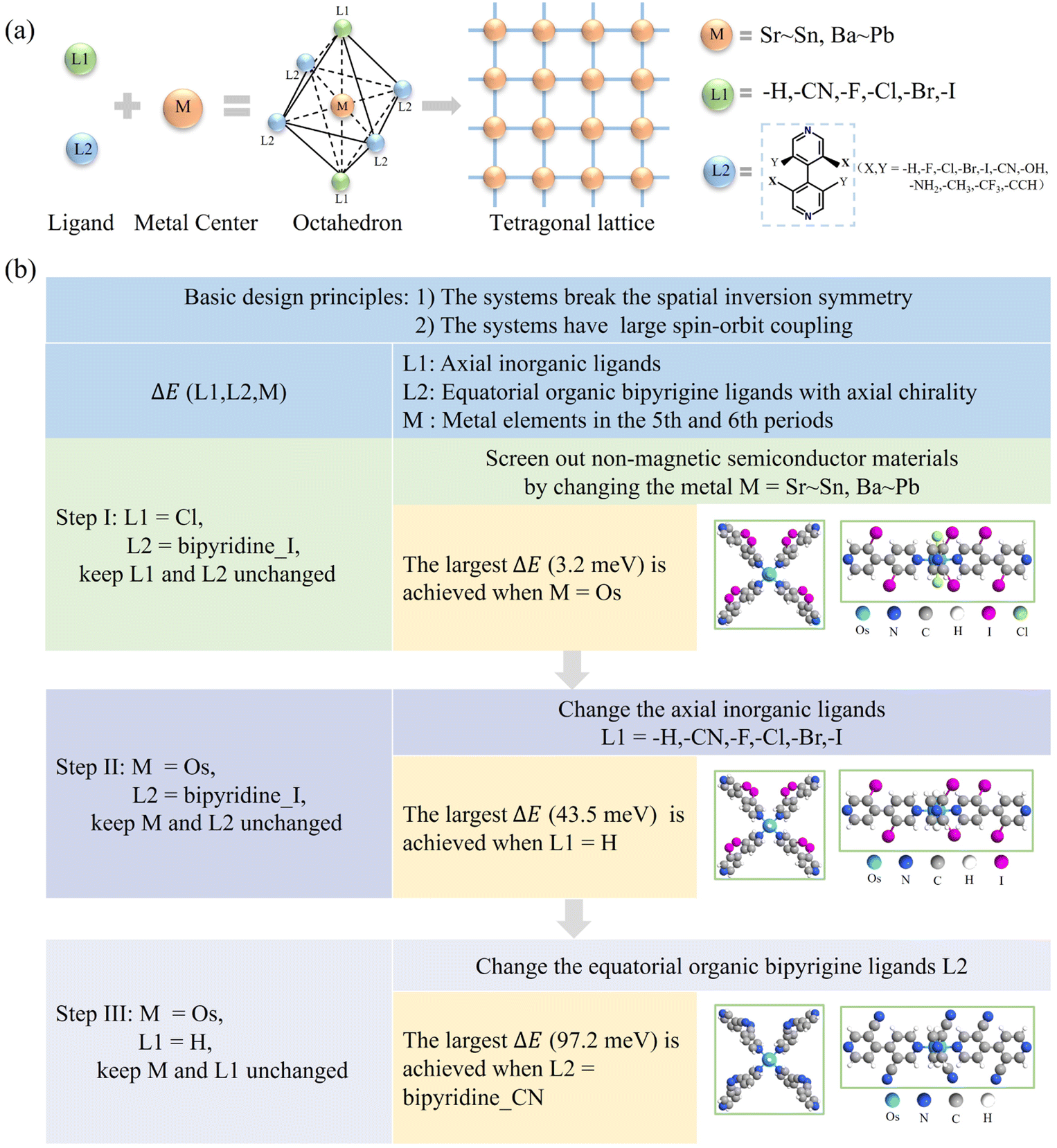 | ||
| Fig. 1 (a) Structural composition diagram of 2D chiral metal–organic frameworks (CMOFs) in a tetragonal lattice. (b) The proposed three-step screening procedure to obtain 2D Rashba–Dresselhaus (R–D) semiconductors with large R–D spin splitting in CMOFs. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) [4,4′-bipyridine]-3,3′-diiodo). Note that such an axially chiral ligand can be easily synthesized in experiments.52 By changing the metal center M, we obtain 23 CMOFs with the C2 space group and screen out 10 non-magnetic semiconductors. When M is Sr, Ru, Cd, Os, Pt, or Pb, spin splitting occurs in either the valence band or the conduction band (see Table S1† for more details). Among them, the splitting for Os-based CMOFs is significantly larger than that of other metal-based CMOFs, i.e., the ΔE of OsCl2(bipyridine_I) is 3.2 meV.
[4,4′-bipyridine]-3,3′-diiodo). Note that such an axially chiral ligand can be easily synthesized in experiments.52 By changing the metal center M, we obtain 23 CMOFs with the C2 space group and screen out 10 non-magnetic semiconductors. When M is Sr, Ru, Cd, Os, Pt, or Pb, spin splitting occurs in either the valence band or the conduction band (see Table S1† for more details). Among them, the splitting for Os-based CMOFs is significantly larger than that of other metal-based CMOFs, i.e., the ΔE of OsCl2(bipyridine_I) is 3.2 meV.
Obviously, the combination of optimal L1 (–H) and L2 (bipyridine_CN) ligands helps largely improve the ΔE for Os-based CMOFs from 3.2 to 97.2 meV. Such enhancement is expected to be also applicable to other metal-based CMOFs. For instance, when the metal center is 4d transition metal Ru, the ΔE of RuH2(bipyridine_CN) is enhanced to be 14.8 meV, representing a 28-fold increase compared to that of RuCl2(bipyridine_I) (see Table S4†).
Dynamic and thermal stability analysis
Taking the outstanding OsH2(bipyridine_CN) as an example, the structural stability is examined by phonon spectroscopy and ab initio molecular dynamics (AIMD) simulations. As shown in Fig. S1,† no obvious imaginary frequency is observed from the phonon spectrum, indicating that OsH2(bipyridine_CN) is dynamically stable. Although some extremely small imaginary frequencies (less than 0.78 cm−1) exist near the Γ point, they are considered to be unphysical and caused by size and boundary effects,53 and a similar phenomenon has also been observed in other reported MOFs.54–56 In addition, during 9 ps AIMD simulation at 600 K (Fig. S2†), the total energy fluctuates near its equilibrium value without any sudden drops, and the lattice structure remains intact without undergoing any reconstruction, confirming the good thermal stability of the structure.Electronic structure and chirality
Fig. 2b and c show the band structures of the R-chiral OsH2(bipyridine_CN) calculated using PBE and PBE + SOC, respectively. When the SOC is not considered, OsH2(bipyridine_CN) is a direct band gap semiconductor (0.84 eV) with the valence band maximum (VBM) and conduction band minimum (CBM) located at the M point. All bands are spin degenerate. After including the SOC effect, the band gap is reduced to 0.60 eV. Moreover, the spin degeneracy is lifted, and its VBM and CBM are changed to locate between Γ and M points (near the Γ point) (Fig. 2c). In particular, the valence band presents a typical Dresselhaus spin splitting. The spin texture in the k-space for the split upper and lower branches is plotted in Fig. 2d, respectively. Near the Γ point, the in-plane spin texture of the upper branch exhibits anti-vortex geometry, which indicates a typical Dresselhaus-type SOC.57 Interestingly, the spin texture of the lower branch shows a reverse anti-vortex geometry. The PBE + SOC band structure is further confirmed by using HSE06 + SOC calculations (Fig. S3†), which qualitatively give the same results.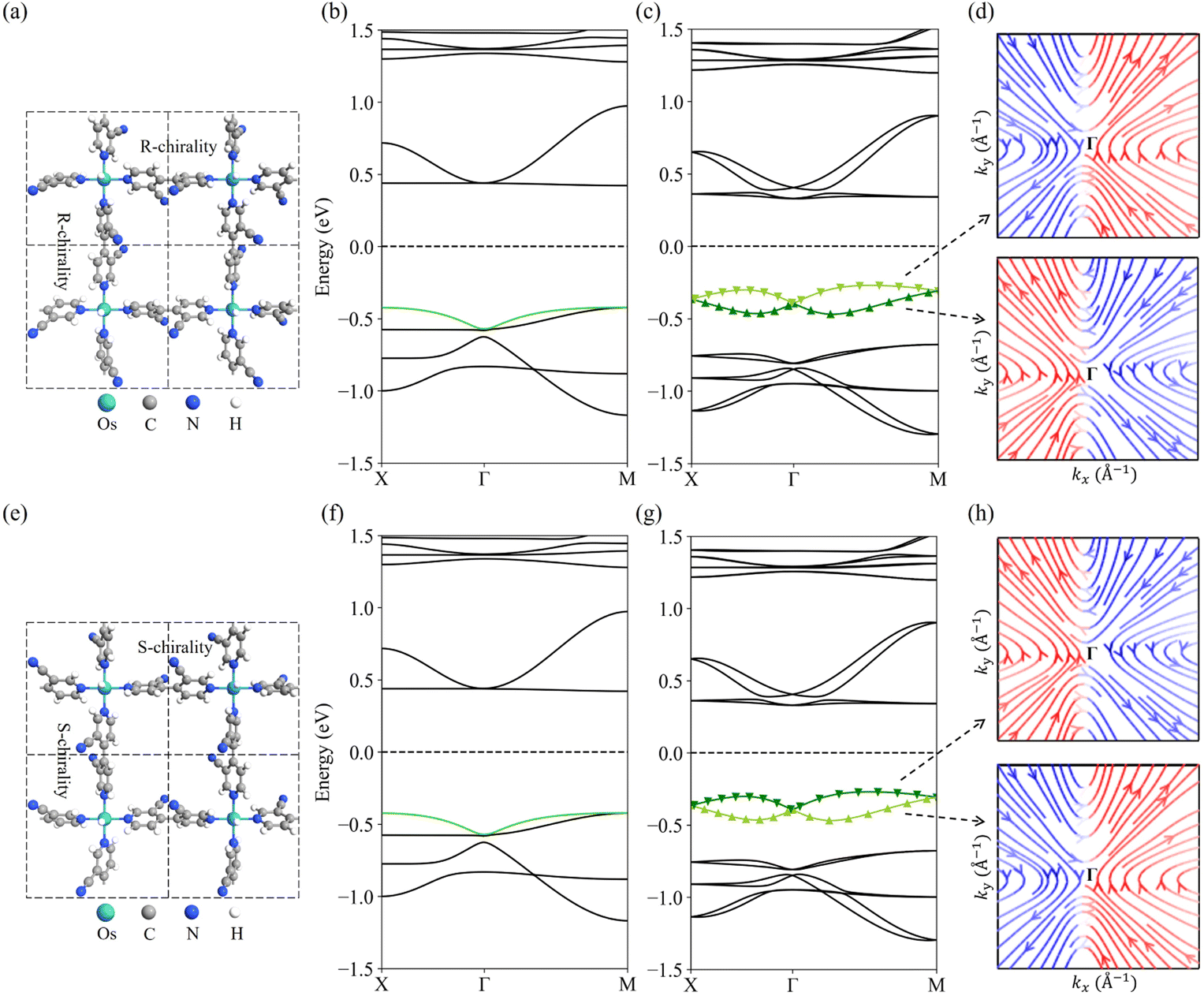 | ||
| Fig. 2 (a) Structure of optimized R-chiral OsH2(bipyridine_CN). Band structures calculated with (b) PBE and (c) PBE + SOC for R-chiral OsH2(bipyridine_CN), and (d) its spin texture of upper and lower Dresselhaus bands in the valence band (VB). (e), (f), (g) and (h) are those for S-chiral OsH2(bipyridine_CN). The tangent vectors of the streamlines (directions indicated by the arrows) in the spin texture represent the spin vector {sx,sy} in the (kx, ky) plane, and the red (blue) color represents the positive (negative) spin component sy. The Fermi levels are set to zero. | ||
Fig. 2f and g show the band structures of the S-chiral OsH2(bipyridine_CN) calculated with the PBE and PBE + SOC, respectively. The band gap of S-chirality is the same as that of R-chirality. However, the valence band spin texture is totally reversed compared to R-chirality (Fig. 2h). This indicates that the chirality inversion is a potential route to control the spin splitting. Experimentally, the chirality inversion could be realized using temperature, pressure, solvent, light, etc.58–61 Here, a two-step transition path from R-chiral to S-chiral OsH2(bipyridine_CN) is simulated (Fig. S4†), where the energy barrier is determined to be 0.7 eV. Such a big energy barrier allows the two enantiomers to coexist independently at room temperature. The space group of the metastable intermediate state is Cm, with the coexistence of R- and S-chiral bipyridine_CN ligands. Interestingly, the structure possesses a vortex Rashba-type spin texture, with the ΔE being 109 meV.
The effect of the band gap
To reveal the factors that affect the magnitude of spin splitting in 2D CMOFs, we plot ΔE versus band gap in Fig. 3a (see Tables S1–S3†). As shown in the upper-left corner of Fig. 3a, the systems OsH2(bipyridine_CN), OsH2(bipyridine_CCH), OsH2(bipyridine_H), OsH2(bipyridine_F) and OsH2(bipyridine_OH) exhibit narrow band gaps and large R–D energies. The general trend is that the smaller the band gap, the bigger the spin splitting. However, there are several exceptions (marked by triangles), indicating that the band gap is not the only factor affecting spin splitting. For example, OsH2(bipyridine_H) (0.52 eV) has a smaller band gap than OsH2(bipyridine_CN) (0.6 eV), but its ΔE (88.5 meV) is smaller than that of OsH2(bipyridine_CN) (97.2 meV). To explain this anomaly, factors other than the band gap need to be considered. Bahramy et al.62 suggest that in the presence of a strong SOC, two states with the same symmetry can lead to a large spin splitting. In the next section, the effect of symmetry on the spin splitting will be investigated.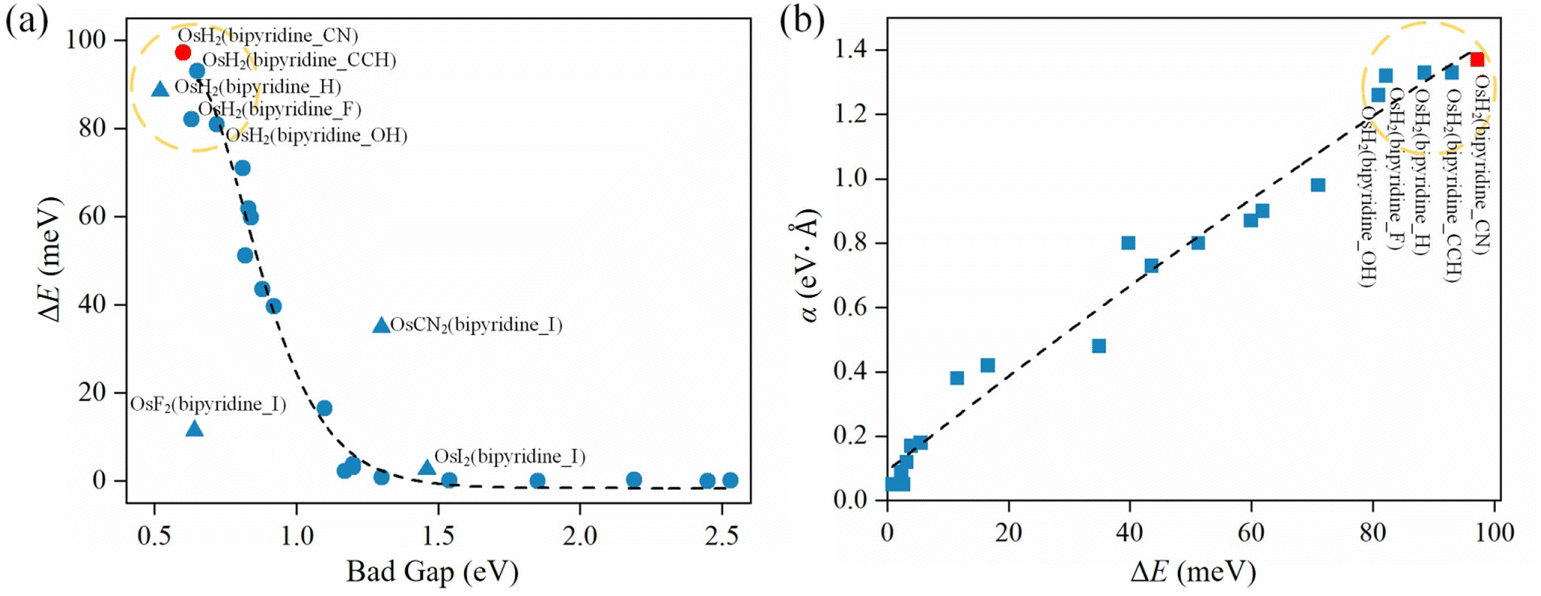 | ||
| Fig. 3 (a) The relationship between the band gap and ΔE. (b) The relationship between ΔE and α. | ||
Fig. 3b shows the relationship between the ΔE and the α (see Tables S2 and S3† for more details). The R–D coupling constant is calculated using α = 2ΔE/k0, where k0 is the momentum-offset from the Γ point and ΔE is the R–D energy of spin splitting. In SFETs, a big α helps shorten the channel length,63 which is beneficial for maintaining spin coherence and improving integration of the spintronic devices. It is clear that the α is almost proportional to ΔE. When the ΔE is the largest (97.2 meV), the corresponding α reaches 1.37 eV Å. Therefore, to obtain a significant α, a large ΔE is needed.
The effect of symmetry
The symmetry characteristics of the VB and CB at the Γ point are determined by first-principles calculations combined with group-theory analysis. In the absence of SOC, for OsH2(bipyridine_CN) with the C2 space group, all the bands at the Γ point are transformed according to the single-group representations of C2. An important consideration is the presence of crystal field splitting (CFS), which affects the electronic structure of the system.64 The projected band structure of OsH2(bipyridine_CN) in Fig. 4a implies that the CB can be simply regarded as contributed by the p-orbital, while the VB is contributed by the d-orbital. The character table of the C2 point group is shown in Table S5:† for the p-type CB, {pz} → A and {px, py} → B and for the d-type VB, {dxy, dz2} → A and {dyz, dxz} → B. As shown in Fig. 4b, at the Γ point, the ordering of CFS for the VB and CB follows the sequence B → A and A → B, respectively, with increasing energy. When SOC is introduced, the previously defined single-group representation is transformed into a double-group representation. In the C2 double point group, the reduction is A → Γ3 ⊕ Γ4 and B → Γ3 ⊕ Γ4 (Γ3 ⊕ Γ4 is simplified as Γ3,4). Fig. 4b shows that the VB and CB have the same symmetry Γ3,4 at the Γ point. Furthermore, the SOC introduces a splitting between the j = 3/2 and j = 1/2 states of the p-level, and the j = 5/2 and j = 3/2 states of the d-level (a detailed discussion is in Note S1 of the ESI†).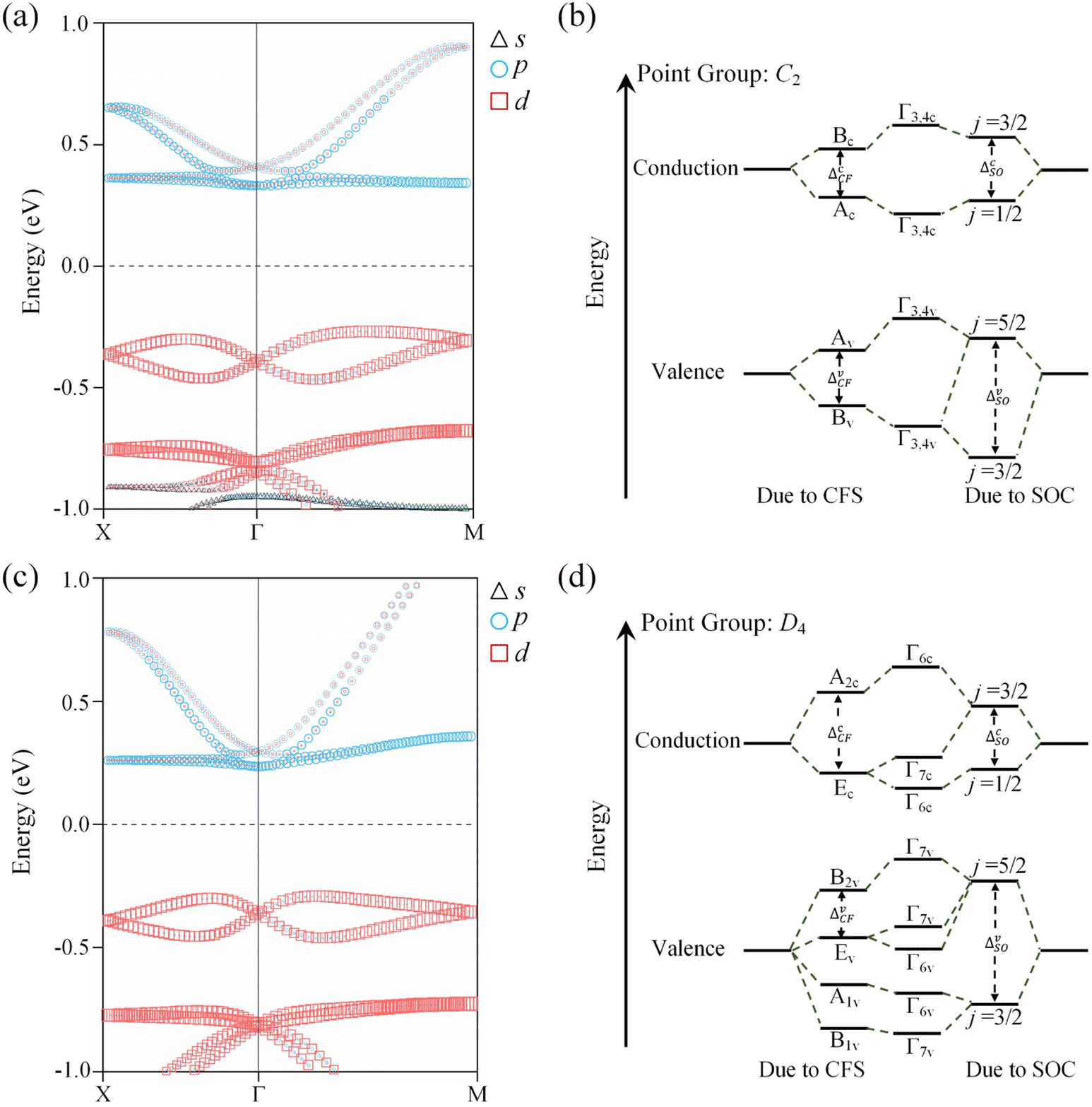 | ||
| Fig. 4 Projected band structure of (a) OsH2(bipyridine_CN) and (c) OsH2(bipyridine_H). Illustration of the band splitting of (b) OsH2(bipyridine_CN) and (d) OsH2(bipyridine_H) due to a combination of crystal field splitting (CFS) and spin–orbit coupling (SOC), where ΔCF denotes the crystal field splitting energy and ΔSO denotes the atomic SOC energy. | ||
For OsH2(bipyridine_H), it belongs to the P422 space group and the Γ point has D4 symmetry. Without SOC, all the bands at Γ are thus transformed according to the single-group representations of D4. In Fig. 4c, the projected band structure of OsH2(bipyridine_H) shows the dominant contributions of the d-orbital at the VB and the p-orbital at the CB. From the character table of the D4 point group (Table S6†), it can be seen for the p-type CB that {px, py} → E and {pz} → A2 and for the d-type VB, {dxy} → B2, {dxz, dyz} → E, {dz2} → A1 and {dx2−y2} → B1. Fig. 4d gives the diagrammatic representation of band splitting caused by CFS and SOC. Without SOC, CFS for the VB (CB) at the Γ point follows the sequence E → B2 (E → A2), in an increasing order of energy. When SOC is incorporated, the previously defined single-group representation is transformed into a double-group representation. In the D4 double point group, the reduction is A2 → Γ6, E → Γ6 ⊕ Γ7, B2 → Γ7, A1 → Γ6 and B1 → Γ7. In other words, B1 and B2 transform to Γ7 and A1 and A2 transform to Γ6, whereas E splits into two two-fold bands: Γ6 and Γ7. Obviously, the VB and CB at the Γ point exhibit different symmetries. Since two states with the same symmetry are beneficial for enhancing spin splitting,62 OsH2(bipyridine_CN), which has the same symmetry at the Γ point in the VB and CB, possesses a larger ΔE than OsH2(bipyridine_H).
The effect of ligand field strength
According to ligand field theory,65 the ligand field strength can be described by the spectrochemical series: –I < –Br < –Cl < –F < –CN.66,67 During the screening process, we find that the ligand field strength also has a notable influence on the spin splitting (see Table S2†). The ligand field strength indirectly affects the ΔE by varying the spin–orbit coupling. A stronger ligand field can increase the proportion of metal atoms in the VB, thereby inducing a more efficient SOC.68 In Table S7†, the proportion of Os metal atoms in the VB increases with the increase of ligand field strength, which results in an enhancement in ΔE. According to the ligand field strength, the anomalies of Os(CN)2(bipyridine_I), OsF2(bipyridine_I) and OsI2(bipyridine_I) in Fig. 3a can be well explained. For OsF2(bipyridine_I) and Os(CN)2(bipyridine_I), their VB and CB have the same symmetry at the Γ point, and the band gap of the former (0.64 eV) is smaller than that of the latter (1.30 eV). Without considering the influence of ligand field strength, the ΔE of OsF2(bipyridine_I) is expected to be larger than that of Os(CN)2(bipyridine_I). However, the stronger ligand field strength of –CN endows Os(CN)2(bipyridine_I) (34.9 meV) with a larger ΔE than OsF2(bipyridine_I) (11.5 meV). Additionally, we have tested other strong field ligands including NCS−, NO2−, PH3, and CO. The calculation results show that the energy of spin splitting increases as the ligand field strength increases (see Table S8†). Another factor worth noting is the SOC strength of the ligand itself. For OsI2(bipyridine_I) and OsBr2(bipyridine_I), though the ligand field strength of –I is weaker than that of –Br, the ligand –I is heavier than –Br, resulting in a much stronger SOC of OsI2(bipyridine_I). Consequently, OsI2(bipyridine_I) has a larger ΔE than OsBr2(bipyridine_I) (see Table S2†).Finally, based on the above discussions, five conditions required for obtaining giant spin splitting in 2D CMOFs are identified: (i) the presence of chiral ligands, which breaks the inversion symmetry, (ii) the existence of heavy atoms, which leads to a significant spin–orbit coupling, (iii) a narrow band gap, (iv) the same symmetry of the VB and CB at the Γ point, and (v) the presence of strong field ligands.69,70
An important factor for designing spin field-effect transistors (SFETs) based on R–D semiconductors is the channel length L = πℏ2/(2m*α),63 over which the spin precesses by π (180°). To improve the integration, a short channel length is desired, which requires a large α. Taking the best OsH2(bipyridine_CN) as an example, the calculated carrier effective mass m* and coupling constant α of the valence band are 1.96 me and 1.37 eV Å, respectively. According to the formula L = πℏ2/(2m*α), we find that to achieve a spin precession of 180°, the required channel length (L) is 0.45 nm. This value is challenging to attain with previous 2D semiconductors and is ideal for miniaturized nano-spintronic devices. Moreover, a possible synthesis route for OsH2(bipyridine_CN) is proposed (more details can be found in Note S2 of the ESI†).
Conclusions
In summary, based on first-principles calculations, we report an unprecedented class of 2D CMOFs with significant R–D spin splitting. Among them, OsH2(bipyridine_CN) demonstrates an R–D energy ΔE of 97.2 meV and a coupling constant A of 1.37 eV Å. Additionally, the underlying factors that control the R–D spin splitting have been revealed, which can lead to the future development of 2D R–D semiconductors with giant spin splitting.Computational methods
Rashba spin–orbit coupling
The Rashba spin–orbit coupling term is induced by the structure inversion asymmetry. The effective Rashba Hamiltonian can be represented as:71 | (1) |
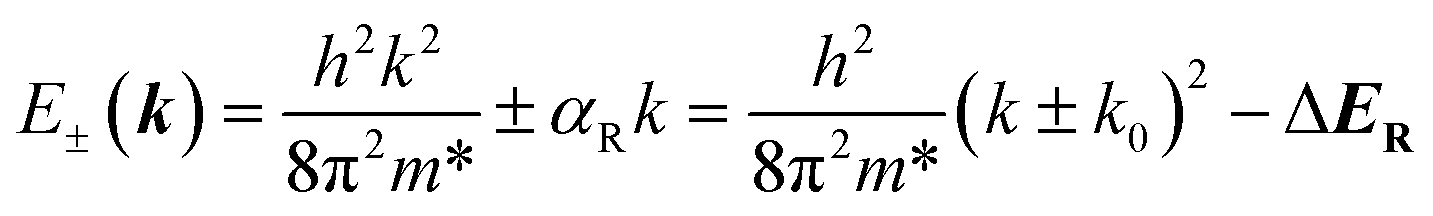 | (2) |
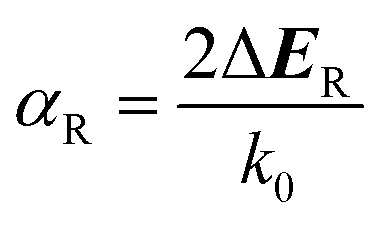 | (3) |
Dresselhaus spin–orbit coupling
The Dresselhaus spin–orbit coupling term arises from bulk inversion asymmetry. The 2D Dresselhaus Hamiltonian can be represented as:72| HD(k) = αD(kxσx − kyσy) | (4) |
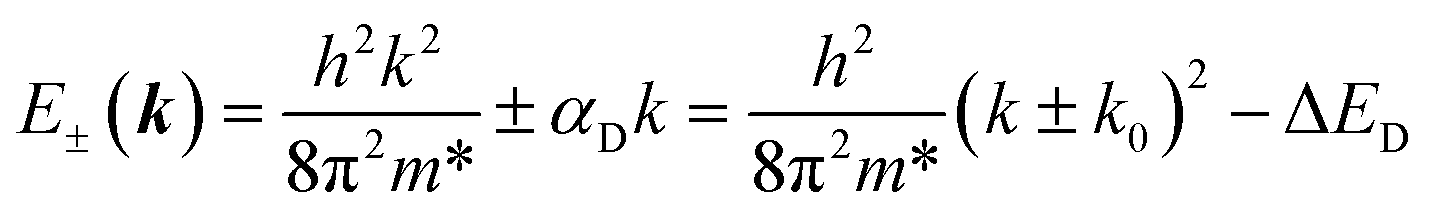 | (5) |
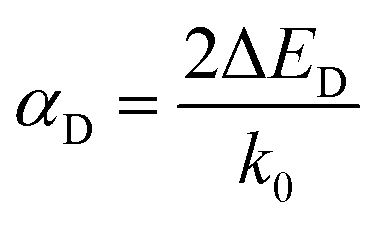 | (6) |
For calculating the αR/D, we find k0 and ΔER/D through the parabolic fitting of the energy band where spin splitting occurs and calculate the αR/D through eqn (3) and (6). ΔER and ΔED are indistinguishable solely from the band structure (see Fig. S5a and b†) but can be distinguished through the spin texture. In general, the Rashba and Dresselhaus spin textures exhibit vortex and antivortex geometries in the kx–ky plane (see Fig. S5c and d†).
The electronic structures of 2D CMOFs are calculated based on the density functional theory (DFT) method within the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA)73 implemented in the Vienna ab initio simulation package (VASP).74 A projector augmented wave (PAW) potential and a plane-wave cut-off energy of 500 eV are used. The vacuum space along the z direction is 15 Å. The first Brillouin zone is sampled with a Monkhorst–Pack grid of 3 × 3 × 1. Both the lattice constant and the positions of all atoms are relaxed until the force is less than 0.01 eV Å−1. The criterion for the total energy is set as 1 × 10−6 eV. In our calculations of all the materials, each transition metal atom is given a notable initial magnetic moment to find its optimal value. By checking the converged local magnetic moments, the magnetic properties of the material are identified, that is, if the local magnetic moment of each atom is zero, the material is determined to be non-magnetic, otherwise, it is magnetic. The HSE06 functional (ref. 75) is used to ensure the accuracy of the PBE results, and then all materials are screened using the PBE to save the computation time. The phonon spectrum is simulated by using the finite displacement method as implemented in the Phonopy package interfaced with the VASP.76 A 3 × 3 × 1 supercell with a Monkhorst–Pack k-point mesh of 1 × 1 × 1 is adopted. The thermal stability is assessed according to AIMD simulations at 600 K by using a 2 × 2 × 1 supercell. The band symmetry is calculated using the Quantum Espresso package.77
Author contributions
X. Li conceived the idea for this study and designed the experiments. S. Liu performed the calculations. K. Xu helped with the analysis and interpretation of the data. S. Liu wrote the manuscript and all authors assisted with editing, analysis, and interpretation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22288201, 22273092, 22322304, and 22373095), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450101), the Innovation Program for Quantum Science and Technology (2021ZD0303306), and USTC Tang Scholar.References
- X. L. Yan, X. Su, J. Chen, C. Jin and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202305408 CrossRef CAS PubMed.
- S. H. Yang, R. Naaman, Y. Paltiel and S. S. P. Parkin, Int. Rev. Phys., 2021, 3, 328–343 Search PubMed.
- E. I. Rashba, Sov. Phys. Solid State, 1960, 2, 1109–1122 Search PubMed.
- G. Dresselhaus, Phys. Rev., 1955, 100, 580–586 CrossRef CAS.
- Y. Wang, C. C. Xiao, M. G. Chen, C. Q. Hu, J. D. Zou, C. Wu, J. Z. Jiang, S. Y. A. Yang, Y. H. Lu and W. Ji, Mater. Horiz., 2018, 5, 521–528 RSC.
- H. Q. Ai, X. K. Ma, X. F. Shao, W. F. Li and M. W. Zhao, Phys. Rev. Mater., 2019, 3, 054407 CrossRef CAS.
- M. U. Farooq, L. D. Xian and L. Huang, Phys. Rev. B, 2022, 105, 245405 CrossRef CAS.
- F. Nagasawa, A. A. Reynoso, J. P. Baltanas, D. Frustaglia, H. Saarikoski and J. Nitta, Phys. Rev. B, 2018, 98, 245301 CrossRef CAS.
- C. E. Whittaker, T. Dowling, A. V. Nalitov, A. V. Yulin, B. Royall, E. Clarke, M. S. Skolnick, I. A. Shelykh and D. N. Krizhanovskii, Nat. Photonics, 2021, 15, 193–196 CrossRef CAS.
- A. Stroppa, D. Di Sante, P. Barone, M. Bokdam, G. Kresse, C. Franchini, M. H. Whangbo and S. Picozzi, Nat. Commun., 2014, 5, 5900 CrossRef CAS PubMed.
- L. Meier, G. Salis, I. Shorubalko, E. Gini, S. Schön and K. Ensslin, Nat. Phys., 2007, 3, 650–654 Search PubMed.
- A. Soumyanarayanan, N. Reyren, A. Fert and C. Panagopoulos, Nature, 2016, 539, 509–517 CrossRef CAS PubMed.
- J. Ingla-Aynes, F. Herling, J. Fabian, L. E. Hueso and F. Casanova, Phys. Rev. Lett., 2021, 127, 047202 CrossRef CAS PubMed.
- H. C. Koo, S. B. Kim, H. Kim, T. E. Park, J. W. Choi, K. W. Kim, G. Go, J. H. Oh, D. K. Lee, E. S. Park, I. S. Hong and K. J. Lee, Adv. Mater., 2020, 32, 2002117 CrossRef CAS PubMed.
- K. Wu, J. J. Chen, H. H. Ma, L. Y. Wan, W. Hu and J. L. Yang, Nano Lett., 2021, 21, 740–746 CrossRef CAS PubMed.
- J. J. Che, K. Wu, W. Hu and J. L. Yang, J. Am. Chem. Soc., 2022, 144, 20035–20046 CrossRef PubMed.
- M. K. Jana, R. Y. Song, H. L. Liu, D. R. Khanal, S. M. Janke, R. D. Zhao, C. Liu, Z. V. Vardeny, V. Blum and D. B. Mitzi, Nat. Commun., 2020, 11, 4699 CrossRef CAS PubMed.
- X. M. Fu, C. Jia, L. Sheng, Q. X. Li, J. L. Yang and X. X. Li, J. Phys. Chem. Lett., 2023, 14, 11292–11297 CrossRef CAS PubMed.
- S. Singh and A. H. Romero, Phys. Rev. B, 2017, 95, 165444 CrossRef.
- T. Hu, F. H. Jia, G. D. Zhao, J. Y. Wu, A. Stroppa and W. Ren, Phys. Rev. B, 2018, 97, 235404 CrossRef CAS.
- X. Y. Chin, D. Cortecchia, J. Yin, A. Bruno and C. Soci, Nat. Commun., 2015, 6, 7383 CrossRef CAS PubMed.
- A. B. Maghirang, R. A. B. Villaos, M. N. R. Perez, L. Y. Feng, Z. Q. Huang, C. H. Hsu and F. C. Chuang, ACS Appl. Electron. Mater., 2022, 4, 5308–5316 CrossRef.
- J. F. Khoury, J. G. He, J. E. Pfluger, I. Hadar, M. Balasubramanian, C. C. Stoumpos, R. Zu, V. Gopalan, C. Wolverton and M. G. Kanatzidis, Chem. Sci., 2020, 11, 870–878 RSC.
- A. B. Maghirang, G. Macam, A. L. Sufyan, Z. Q. Huang, C. H. Hsu and F. C. Chuang, Chin. J. Phys., 2022, 77, 2346–2354 CrossRef CAS.
- P. A. L. Sino, L. Y. Feng, R. A. B. Villaos, H. N. Cruzado, Z. Q. Huang, C. H. Hsu and F. C. Chuang, Nanoscale Adv., 2021, 3, 6608–6616 RSC.
- B. Maurer, C. Vorwerk and C. Draxl, Phys. Rev. B, 2022, 105, 8 Search PubMed.
- M. K. Jana, R. Y. Song, Y. Xie, R. D. Zhao, P. C. Sercel, V. Blum and D. B. Mitzi, Nat. Commun., 2021, 12, 4982 CrossRef CAS PubMed.
- M. Kepenekian, R. Robles, C. Katan, D. Sapori, L. Pedesseau and J. Even, ACS Nano, 2015, 9, 11557–11567 CrossRef CAS PubMed.
- M. T. Pham, E. Amerling, T. A. Ngo, H. M. Luong, K. Hansen, H. T. Pham, T. N. Vu, H. Tran, L. Whittaker-Brooks and T. D. Nguyen, Adv. Opt. Mater., 2022, 10, 2101232 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- L. D. Barron, Chem. Soc. Rev., 1986, 15, 189–223 RSC.
- S. Weigelt, C. Busse, L. Petersen, E. Rauls, B. Hammer, K. V. Gothelf, F. Besenbacher and T. R. Linderoth, Nat. Mater., 2006, 5, 112–117 CrossRef CAS PubMed.
- J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- M. H. Liu, L. Zhang and T. Y. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- Y. Yang, J. Liang, F. Pan, Z. Wang, J. Q. Zhang, K. Amin, J. Fang, W. J. Zou, Y. L. Chen, X. H. Shi and Z. X. Wei, Nat. Commun., 2018, 9, 3808 CrossRef PubMed.
- W. Gong, Z. J. Chen, J. Q. Dong, Y. Liu and Y. Cui, Chem. Rev., 2022, 122, 9078–9144 CrossRef CAS PubMed.
- Q. S. Cheng, Q. Ma, H. B. Pei, H. Liang, X. J. Zhang, X. N. Jin, N. J. Liu, R. B. Guo and Z. L. Mo, Coord. Chem. Rev., 2023, 484, 215120 CrossRef CAS.
- Y. H. Liu, L. M. Liu, X. Chen, Y. Liu, Y. Han and Y. Cui, J. Am. Chem. Soc., 2021, 143, 3509–3518 CrossRef CAS PubMed.
- J. Guo, Y. Zhang, Y. F. Zhu, C. Long, M. T. Zhao, M. He, X. F. Zhang, J. W. Lv, B. Han and Z. Y. Tang, Angew. Chem., Int. Ed., 2018, 57, 6873–6877 CrossRef CAS PubMed.
- Z. Sharifzadeh, K. Berijani and A. Morsali, Coord. Chem. Rev., 2021, 445, 214083 CrossRef CAS.
- T. H. Zhao, J. L. Han, Y. H. Shi, J. Zhou and P. F. Duan, Adv. Mater., 2021, 33, 2101797 CrossRef CAS PubMed.
- L. A. Hall, D. M. D'Alessandro and G. Lakhwani, Chem. Soc. Rev., 2023, 52, 3567–3590 RSC.
- H. Zhang, L. L. Lou, K. Yu and S. X. Liu, Small, 2021, 17, 2005686 CrossRef CAS PubMed.
- Z. Z. Weng, H. Xu, W. Zhang, G. L. Zhuang, L. S. Long, X. J. Kong and L. S. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 37402–37411 Search PubMed.
- J. P. Zhao, J. Luo, Z. H. Lin, X. Chen, G. H. Ning, J. Z. Liu and D. Li, Inorg. Chem. Front., 2022, 9, 4907–4912 RSC.
- M. Nagano, A. Kodama, T. Shishidou and T. Oguchi, J. Phys.: Condens. Matter, 2009, 21, 064239 CrossRef PubMed.
- S. Y. Yan, W. Xia, S. Y. Li, Q. L. Song, S. H. Xiang and B. Tan, J. Am. Chem. Soc., 2020, 142, 7322–7327 CrossRef CAS PubMed.
- D. Zhang and Q. R. Wang, Coord. Chem. Rev., 2015, 286, 1–16 CrossRef CAS.
- M. McCarthy and P. J. Guiry, Tetrahedron, 2001, 57, 3809–3844 CrossRef CAS.
- A. Rang, M. Engeser, N. M. Maier, M. Nieger, W. Lindner and C. A. Schalley, Chem.–Eur. J., 2008, 14, 3855–3859 CrossRef CAS PubMed.
- V. Mamane, E. Aubert, P. Peluso and S. Cossu, J. Org. Chem., 2012, 77, 2579–2583 CrossRef CAS PubMed.
- J. Richard, J. Joseph, C. Wang, A. Ciesielski, J. Weiss, P. Samori, V. Mamane and J. A. Wytko, J. Org. Chem., 2021, 86, 3356–3366 CrossRef CAS PubMed.
- J. L. Lu, G. Chen, W. Luo, J. Iñiguez, L. Bellaiche and H. J. Xiang, Phys. Rev. Lett., 2019, 122, 227601 CrossRef CAS PubMed.
- X. Y. Li, Q. B. Liu, Y. S. Tang, W. Li, N. Ding, Z. Liu, H. H. Fu, S. Dong, X. X. Li and J. L. Yang, J. Am. Chem. Soc., 2023, 145, 7869–7878 CrossRef CAS PubMed.
- H. F. Lv, X. Y. Li, D. X. Wu, Y. Liu, X. X. Li, X. J. Wu and J. L. Yang, Nano Lett., 2022, 22, 1573–1579 CrossRef CAS PubMed.
- C. Tang, L. Zhang, S. Sanvito and A. J. Du, J. Am. Chem. Soc., 2023, 145, 2485–2491 CrossRef CAS PubMed.
- C. M. Acosta, L. D. Yuan, G. M. Dalpian and A. Zunger, Phys. Rev. B, 2021, 104, 104408 CrossRef.
- J. B. Yao, W. H. Wu, W. T. Liang, Y. J. Feng, D. Y. Zhou, J. J. Chruma, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, Angew. Chem., Int. Ed., 2017, 56, 6869–6873 CrossRef CAS PubMed.
- C. Xiao, W. H. Wu, W. T. Liang, D. Y. Zhou, K. Kanagaraj, G. Cheng, D. Su, Z. H. Zhong, J. J. Chruma and C. Yang, Angew. Chem., Int. Ed., 2020, 59, 8094–8098 CrossRef CAS PubMed.
- L. B. Wang, L. Yin, W. Zhang, X. L. Zhu and M. Fujiki, J. Am. Chem. Soc., 2017, 139, 13218–13226 CrossRef CAS PubMed.
- H. K. Bisoyi and Q. Li, Angew. Chem., Int. Ed., 2016, 55, 2994–3010 CrossRef CAS PubMed.
- M. S. Bahramy, R. Arita and N. Nagaosa, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 041202 CrossRef.
- S. Gupta and B. I. Yakobson, J. Am. Chem. Soc., 2021, 143, 3503–3508 CrossRef CAS PubMed.
- F. A. Cotton, Chemical applications of group theory, John Wiley & Sons, 1991 Search PubMed.
- J. Griffith and L. Orgel, Q. Rev., Chem. Soc., 1957, 11, 381–393 RSC.
- J. Glerup, O. Monsted and C. E. Schaffer, Inorg. Chem., 1976, 15, 1399–1407 CrossRef CAS.
- L. Carlton, Magn. Reson. Chem., 2004, 42, 760–768 CrossRef CAS PubMed.
- Z. R. Hao, K. Zhang, K. Chen, P. Wang, Z. Y. Lu, W. G. Zhu and Y. Liu, Dalton Trans., 2020, 49, 8722–8733 RSC.
- N. Kunkel, H. Kohlmann, A. Sayede and M. Springborg, Inorg. Chem., 2011, 50, 5873–5875 CrossRef CAS PubMed.
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305–3366 CrossRef CAS PubMed.
- T. Etienne, E. Mosconi and F. De Angelis, J. Phys. Chem. Lett., 2016, 7, 1638–1645 CrossRef CAS PubMed.
- M. Studer, M. P. Walser, S. Baer, H. Rusterholz, S. Schon, D. Schuh, W. Wegscheider, K. Ensslin and G. Salis, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 235320 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Solid State, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Group theory analysis, a possible route for the experimental synthesis, phonon spectrum and AIMD simulations for OsH2(bipyridine_CN), HSE06 + SOC band structure for OsH2(bipyridine_CN), the energy barrier for the transition from R-chiral to S-chiral OsH2(bipyridine_CN) and the structure and Rashba spin splitting for the metastable intermediate state, R–D band splitting and spin texture schematic diagram, all 2D CMOFs generated during three-step screening, character table for C2 and D4 point groups, proportion of heavy elements in the valence band, and effects of strong field ligands on R–D spin splitting. See DOI: https://doi.org/10.1039/D3SC06636C |
| This journal is © The Royal Society of Chemistry 2024 |