
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn adsorbate biased dynamic 3D porous framework for inverse CO2 sieving over C2H2†
Nivedita
Sikdar‡§
 ,
Subhajit
Laha‡
,
Rohan
Jena
,
Anupam
Dey
,
Faruk Ahamed
Rahimi
and
Tapas Kumar
Maji
*
,
Subhajit
Laha‡
,
Rohan
Jena
,
Anupam
Dey
,
Faruk Ahamed
Rahimi
and
Tapas Kumar
Maji
*
Molecular Materials Laboratory, Chemistry and Physics of Materials Unit, School of Advanced Materials (SAMat), International Centre for Materials Science (ICMS), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore, 560064, India. E-mail: tmaji@jncasr.ac.in; Fax: +91-80-2208-2766; Tel: +91-80-2208-2826
First published on 17th April 2024
Abstract
Separating carbon dioxide (CO2) from acetylene (C2H2) is one of the most critical and complex industrial separations due to similarities in physicochemical properties and molecular dimensions. Herein, we report a novel Ni-based three-dimensional framework {[Ni4(μ3-OH)2(μ2-OH2)2(1,4-ndc)3](3H2O)}n (1,4-ndc = 1,4-naphthalenedicarboxylate) with a one-dimensional pore channel (3.05 × 3.57 Å2), that perfectly matches with the molecular size of CO2 and C2H2. The dehydrated framework shows structural transformation, decorated with an unsaturated Ni(II) centre and pendant oxygen atoms. The dynamic nature of the framework is evident by displaying a multistep gate opening type CO2 adsorption at 195, 273, and 298 K, but not for C2H2. The real time breakthrough gas separation experiments reveal a rarely attempted inverse CO2 selectivity over C2H2, attributed to open metal sites with a perfect pore aperture. This is supported by crystallographic analysis, in situ spectroscopic inspection, and selectivity approximations. In situ DRIFTS measurements and DFT-based theoretical calculations confirm CO2 binding sites are coordinatively unsaturated Ni(II) and carboxylate oxygen atoms, and highlight the influence of multiple adsorption sites.
Introduction
C2H2 holds crucial significance as a monomeric organic feedstock in both chemical and polymer industries, along with extensive use as a fuel in welding apparatus.1 The production of ultra-pure C2H2 is essential for industrial chemical manufacturing, but the presence of impurities such as CO2 and CH4 in reactor products from hydrocarbon cracking poses a significant challenge.2–4 Purifying C2H2 and separating it from persistent CO2 impurities is regarded as one of the most formidable tasks due to the similarities in molecular dimensions (C2H2: 3.32 × 3.34 × 5.7 Å3 and CO2: 3.18 × 3.33 × 5.36 Å3).5 The current purification technology necessitates operation under cryogenic temperatures and high pressures, adding complexity to the separation process due to the closely matched boiling points (C2H2, 189.3 K; CO2, 194.7 K).4,6 Therefore, the aforementioned conventional methods for gas separation, such as solvent extraction or cryogenic distillation, are expensive, inefficient, and highly energy-consuming. In this context, adsorptive separation through porous materials is attracting significant interest as it is more environmentally friendly, cost-effective, and involves lower energy expenses, making it a next-generation separation technology.Benefiting from precise control over pore accessibility and the pore environment, metal–organic frameworks (MOFs) demonstrate significant potential in the field of gas storage and separation.7–12 Regrettably, the majority of reported MOFs exhibit a very similar binding affinity for both C2H2 and CO2, with only a few designed specifically for the exclusive separation of C2H2 from CO2.13,14 In those few successful reports, pores are typically engineered to favor the preferential adsorption of C2H2 over CO2. This is achieved by employing a hydrophobic pore environment, leveraging acid–base interactions with the binding site, and incorporating linkers with highly polarizable π-electrons.15–19 Nevertheless, in each case, the blowdown step necessitates an extra step involving a deep vacuum to achieve the recovery of ultrapure C2H2 due to its strong binding to the pore.20 Hence, achieving inverse selectivity, where CO2 adsorption is favored over C2H2, could eliminate the need for the co-current blowdown step, making the overall separation process more straightforward and energy-efficient.
In this regard, Yang et al. were the first to report the preferential adsorption of CO2 over C2H2 by incorporating both pendant carboxyl and pyridyl groups in the channels.21 Recently, Kitagawa and coworkers showcased the selective adsorption of CO2 based on a flexible PCP (porous coordination polymer) driven by C–H⋯π and π–π interactions. However, it also delimits a significant C2H2 uptake of 86.2 mL g−1 STP at 195 K.22 Other approaches involve post-synthetically modifying the pore surface with ions, immobilizing methyl groups to enhance van der Waals interactions, and introducing pendant polar OH groups, among others.23–25 However, a trend of both CO2 and C2H2 adsorption was observed. The structural flexibility and dynamics of MOFs can potentially enhance shape matching and provide specific binding towards particular gas molecules.26–34 In this study, our strategy for preferential CO2 sieving involves introducing an open metal site while maintaining the specific geometry of the pore. In comparison to functional group interactions (from linkers) with CO2, as discussed earlier, the open metal site interaction with CO2 is expected to be stronger. This could potentially enable the sieving out of target CO2 molecules by minimizing the co-adsorption of counter gas (C2H2) molecules.
In this work, we report the synthesis and structural characterization of a 3D metal–organic framework {[Ni4(μ3-OH)2(μ2-OH2)2(1,4-ndc)3](3H2O)}n (1), embodied by 1,4-naphthalenedicarboxylate linkers (1,4-ndc). The resulting framework features an accessible 1D pore channel with high polarity and an aperture size of approximately 3.3 Å, facilitating preferential CO2 binding within the undulating 1-D channels. Consecutive temperature-dependent powder X-ray diffraction (PXRD) studies, conducted immediately after obtaining the vapor adsorption isotherm, collectively offer a more comprehensive understanding of the structural dynamics of the 3D framework (Scheme 1). The desolvated framework 1 (1′) exhibits a gate-opening type multistep adsorption profile for CO2 at 195, 273, and 298 K but not for C2H2, demonstrating selective adsorption of CO2 over C2H2 at ambient temperature (298 K). The real time breakthrough gas separation measurements are performed to elucidate the experimental performance of CO2vs. C2H2 and CO2vs. CH4 separation under ambient conditions. Additional investigations were carried out using in situ DRIFTS measurements and DFT calculations to gain a deeper understanding of the CO2 binding sites on the pore surface.
 | ||
| Scheme 1 Schematic representation of dynamicity in a Ni-MOF and its preferential CO2 sieving over C2H2. | ||
Results and discussion
Crystal structure and framework stability of {[Ni4(μ3-OH)2(μ2-OH2)2(1,4-ndc)3](3H2O)}n (1)
The reaction of the 1,4-H2ndc ligand with Ni(NO3)2 in water under basic pH resulted in the formation of dark green block-shaped crystals of 1. Single-crystal X-ray diffraction analysis indicates that 1 crystallized in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit consists of four Ni(II) centers, three 1,4-ndc linkers, two μ3-OH groups, two μ2-OH2 molecules, and three guest water molecules (Fig. 1a). The 3D framework adopts a square grid structure, formed through the crosslinking of fused Ni(II) octahedral chains with 1,4-ndc linkers, as depicted in Fig. 1b and c. This structure consists of four independent Ni(II) atoms assembled into two parallel Ni–O–Ni chains. The parallel chains in the structure consist of Ni1 and Ni2 connected with a μ3-OH group, μ2-OH2, and μ-O from carboxylate ligands. On the other hand, Ni3 and Ni4 are connected with carboxylate groups through syn–syn bridging (Fig. 1a–c). Consequently, in the parallel chains, each Ni2 and Ni3 are associated with three different carboxylate oxygen atoms, whereas Ni1 and Ni4 are attached to one μ3-OH, one μ2-OH2, two μ-O, and two carboxylates from four different linkers, forming distorted NiO6 octahedra to create four alternate triangular arrays. The intrachain Ni1–Ni2 separation distances are 3.306 Å (syn–syn bridging) and 3.774 Å (μ2-OH2 bridging), while for Ni3–Ni4, they are 3.274 Å (syn–syn bridging) and 3.805 Å (μ2-OH2 bridging). The Ni–O distances with 1,4-ndc oxygen atoms and μ3-OH are all comparable and fall in the range of 1.974(14)–2.210(16) Å. The O–Ni–O cisoid angles range from 79.50(6)° to 102.92(6)°, and the transoid angles vary from 166.79(7)° to 176.45(6)°, indicating the degree of deviation from the ideal octahedron (Tables S1–S3†). The dicarboxylate group of the 1,4-ndc linkers diverges from the chain and connects to the Ni(II) centers of the adjacent chains, forming a 3D framework with 1-D channels along the a-axis (Fig. 1b) with guest water molecules (Fig. 1d). Excluding the guest molecules, 1 has undulating channels (9.8% void space) along the a-axis with a channel size of 3.05 × 3.57 Å2. The removal of the water molecules would provide the structure with the unsaturated Ni(II) sites, which can capture CO2 molecules having a quadrupole moment.
space group. The asymmetric unit consists of four Ni(II) centers, three 1,4-ndc linkers, two μ3-OH groups, two μ2-OH2 molecules, and three guest water molecules (Fig. 1a). The 3D framework adopts a square grid structure, formed through the crosslinking of fused Ni(II) octahedral chains with 1,4-ndc linkers, as depicted in Fig. 1b and c. This structure consists of four independent Ni(II) atoms assembled into two parallel Ni–O–Ni chains. The parallel chains in the structure consist of Ni1 and Ni2 connected with a μ3-OH group, μ2-OH2, and μ-O from carboxylate ligands. On the other hand, Ni3 and Ni4 are connected with carboxylate groups through syn–syn bridging (Fig. 1a–c). Consequently, in the parallel chains, each Ni2 and Ni3 are associated with three different carboxylate oxygen atoms, whereas Ni1 and Ni4 are attached to one μ3-OH, one μ2-OH2, two μ-O, and two carboxylates from four different linkers, forming distorted NiO6 octahedra to create four alternate triangular arrays. The intrachain Ni1–Ni2 separation distances are 3.306 Å (syn–syn bridging) and 3.774 Å (μ2-OH2 bridging), while for Ni3–Ni4, they are 3.274 Å (syn–syn bridging) and 3.805 Å (μ2-OH2 bridging). The Ni–O distances with 1,4-ndc oxygen atoms and μ3-OH are all comparable and fall in the range of 1.974(14)–2.210(16) Å. The O–Ni–O cisoid angles range from 79.50(6)° to 102.92(6)°, and the transoid angles vary from 166.79(7)° to 176.45(6)°, indicating the degree of deviation from the ideal octahedron (Tables S1–S3†). The dicarboxylate group of the 1,4-ndc linkers diverges from the chain and connects to the Ni(II) centers of the adjacent chains, forming a 3D framework with 1-D channels along the a-axis (Fig. 1b) with guest water molecules (Fig. 1d). Excluding the guest molecules, 1 has undulating channels (9.8% void space) along the a-axis with a channel size of 3.05 × 3.57 Å2. The removal of the water molecules would provide the structure with the unsaturated Ni(II) sites, which can capture CO2 molecules having a quadrupole moment.
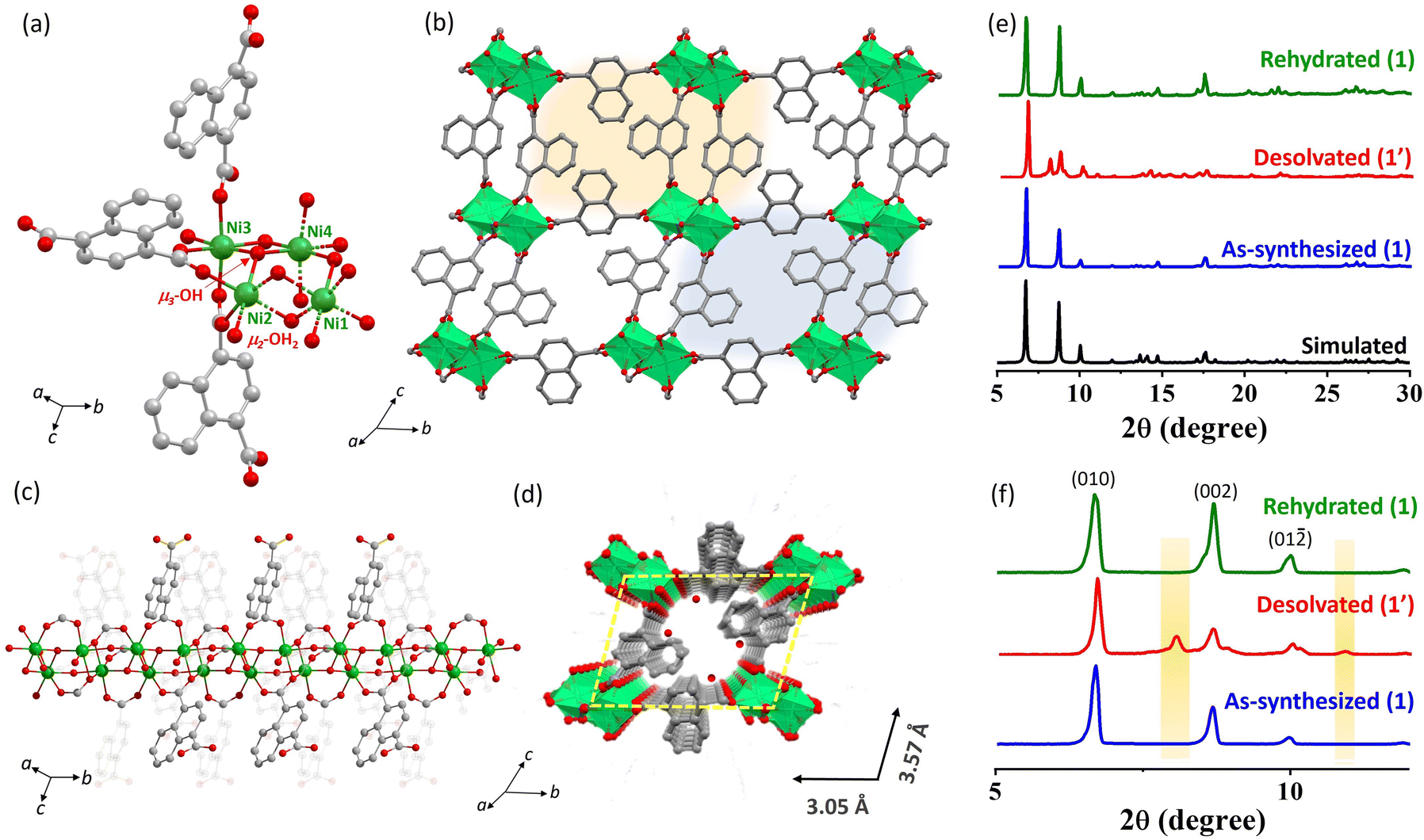 | ||
| Fig. 1 (a) Asymmetric unit of 1 executing different Ni(II) environments. (b) Three-dimensional view along the a direction. (c) View of the bilayer {Ni2-(μ-OH)(μ-OH2)} chains supported by 1,4-ndc showing the bridging water and hydroxyl groups. (d) Pore view of 1 along the a direction. (e) Structural transformation of the as-synthesized → desolvated → rehydrated MOF as evident from PXRD analysis. (f) A close-up view of (e) reveals the differences in Bragg's reflection between the different phases. | ||
The PXRD pattern of the as-synthesized 1 showed high crystallinity, and good correspondence with the simulated pattern indicates its purity in the bulk phase (Fig. 1e and f) and this was further supported by thermogravimetric analysis (TGA) (Fig. S1†). TGA of 1 revealed the loss of three water molecules at 127 °C with a weight loss of 5.14%, consistent with the calculated value (5.7%) and this phase is tenable up to 225 °C. At 225–280 °C, further weight loss (expt 3.22%, cald 3.8%) was accomplished due to the release of two bridging water molecules and the dehydrated framework is stable up to 300 °C (Fig. S1†). After complete dehydration, 1 transformed to an orthorhombic 1′ phase [cell parameters of a = 20.449 Å, b = 9.794 Å, c = 8.913 Å, α = β = γ = 90° and V = 1785.23 Å3] with approximately 6.7% reduction in cell volume, suggesting overall structural contraction to a denser phase (Fig. 1e, f and Table S4†).30 Further rehydration of 1′ (exposed to water vapor for 48 h) revives the as-synthesized phase as realized from the resemblances of PXRD patterns and indexing of the powder pattern [a = 7.081 Å, b = 13.802 Å, c = 20.864 Å, α = 98.486°, β = 96.655°, γ = 101.691° and V = 1952.52 Å3] (Fig. 1e, f and Table S4†). The stability test of framework 1 in various solvents was conducted, including hot water (80 °C), dimethylformamide (DMF), methanol (CH3OH), and chloroform (CHCl3). The results, depicted in Fig. S3,† indicate structural stability under these conditions. However, the MOF exhibited structural degradation under acidic (1 M HCl) and basic conditions (1 M KOH).
Gas adsorption study
The gas adsorption properties were investigated with N2 at 77 K and CO2 at 195 K (Fig. 2a). There is no significant N2 adsorption observed, indicating the non-porous nature of the dehydrated framework. In contrast, 1′ exhibited a gate-opening, multistep CO2 adsorption profile at 195 K, with a total uptake of 61 cm3 g−1 (equivalent to 11.9 wt% and 2.47 molecules per formula unit). The gated CO2 uptake can be attributed to the smaller kinetic diameter of CO2 (3.3 Å) compared to N2 (3.64 Å) and the quadrupolar nature of CO2, which facilitates interactions with the pore surface decorated with unsaturated Ni(II) sites and oxygen atoms from the bridging –OH and carboxyl groups.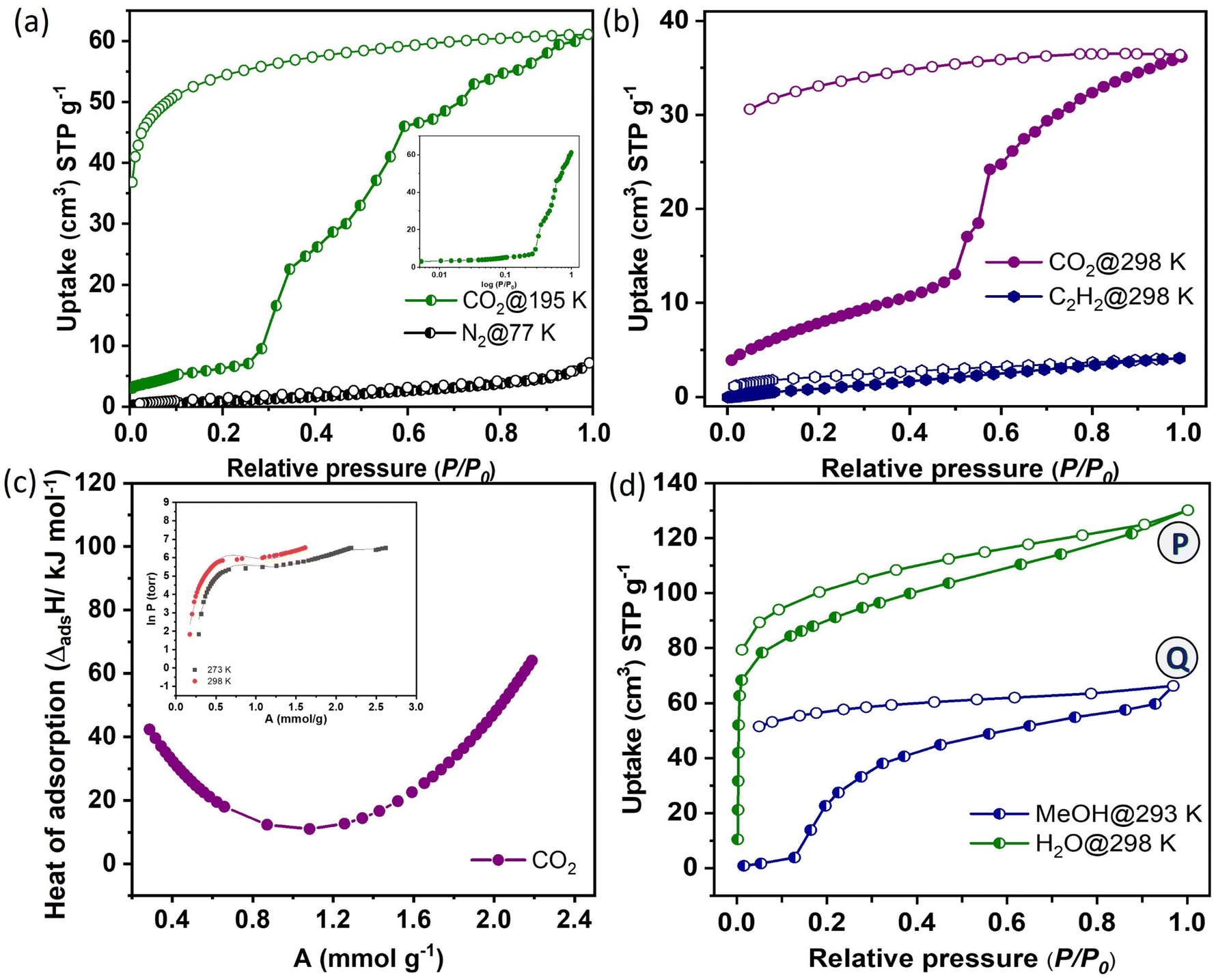 | ||
| Fig. 2 (a) N2 and CO2 adsorption isotherms of 1′ at 77 and 195 K (inset; log curve for a clear understanding of the close-to-open phase transition). (b) Adsorption isotherms of CO2 and C2H2 at 298 K. (c) Isosteric heat of adsorption of CO2 for 1′ at different loading concentrations with virial fitting in the inset. (d) Vapor adsorption isotherms of H2O (298 K) and MeOH (293 K). In the adsorption isotherms, at points P and Q, the adsorbed samples were subjected to PXRD measurement which revealed a similar pattern to the as-synthesized framework after adsorbing water and methanol molecules as shown in Fig. S7.† | ||
The adsorption isotherm showed negligible CO2 uptake up to P/P0 ∼ 0.25, followed by an abrupt increase in uptake and then saturating at P/P0 = 0.99. The non-coincidence of desorption graph with respect to the adsorption isotherm with a broad hysteresis suggested strong confinement of CO2 molecules inside the pore. CO2 adsorption isotherms were also measured at 273 and 298 K (Fig. S4a†). The total uptake capacities are 49 cm3 g−1 (9.8 wt% and 2.02 molecules per formula unit, at 273 K) and 37 cm3 g−1 (7.8 wt% and 1.62 molecules per formula unit, at 298 K). The threshold gate opening pressure (P/P0) shifted to 0.3 to 0.5 at 273 and 298 K, respectively (Fig. S4b†). The gate opening pressure increases with rising measurement temperatures. The isosteric heat of adsorption (Qst) for CO2 is calculated using the adsorption isotherms at 273 and 298 K, employing the Clausius–Clapeyron equation.35–38 This calculation reveals a high value of 42.35 kJ mol−1 at zero loading, indicating a strong adsorbate–adsorbent interaction (Fig. 2c). The value sharply increases at the high loading pressure of CO2, suggesting structural expansion from a non-porous to an open porous structure. This transition from a non-porous to a porous state is further supported by high-pressure CO2 adsorption isotherms. At pressures exceeding 1.0 bar, the adsorption profile displays a sharp increase resulting in a total adsorption capacity of 57 cm3 g−1 at 273 K and 56 cm3 g−1 at 298 K, finally reaching the same amount. This observation strongly confirms the CO2 adsorption capabilities in the open phase. The dynamic pore chemistry and geometry, combined with the appropriate pore aperture of 3.05 × 3.57 Å2, motivated us to assess the adsorption capabilities of 1′ for various small, lightweight hydrocarbons (CH4, C2H2, C2H4, C2H6) and H2 under ambient conditions (Fig. 2b, S5a and Table S6†). However, the adsorption of C2H4 (4.16 Å) and C2H6 (4.44 Å) was excluded due to the inconvenience of size between the pore aperture and their kinetic diameters (Fig. 2b and c). Given that C2H2 possesses a rod-shaped anisotropic linear geometry similar to CO2, with acidic hydrogen at both ends, it was anticipated that 1′ would also serve as a suitable adsorbent for C2H2. Surprisingly, 1′ showed minimal adsorption of C2H2 (4.1 cm3 g−1), which is notably lower than the CO2 uptake observed under similar conditions. This suggested that the interactions of C2H2 molecules with the framework may not be sufficiently strong to trigger the gate-opening mechanism of 1′. Specific interactions, including Lewis acid–base interactions, electrostatic interactions, and hydrogen bonding, between CO2 molecules and the pore surface, adorned with unsaturated metal sites and various functional groups from the organic linker, are recognized as pivotal factors in achieving selective molecular sieving.39 The adsorption kinetics of CO2 was further assessed by fitting them into the Linear Driving Force (LDF) model. The rate constant value at the 2nd point (P ∼ 4 kPa) is k = 2.21 × 10−2 s−1 (Fig. S6†). It is worth mentioning that N2, H2 and CH4 molecules have either zero or lower quadrupole moment compared to CO2 (Table S6†). C2H4 and C2H6 exhibit larger kinetic diameters compared to CO2, and they also possess relatively lower quadrupole moments.40 C2H2 and CO2 have similar kinetic diameters and quadrupole moments.41 The large quadrupole moment of CO2 can interact effectively with the dehydrated framework decorated with the unsaturated Ni(II) sites and oxygen atoms from carboxylate groups which help to diffuse the CO2 molecule inside the pore.22,42 This process results in the transformation from a non-porous to a porous state with gated sorption behaviour. In the case of C2H2, due to its inverse polarizability, it is expected to bind laterally, which hinders its entry into the pore due to its larger size (5.7 Å) along the z-direction.22 Therefore, for CO2, it exhibits a discriminatory gate-opening sorption isotherm, which is further stabilized by strong host–guest interactions.
To better understand the nature of the pore surface, solvent vapour (water; H2O (298 K) and methanol; CH3OH (293 K)) adsorption has been carried out with activated 1 (1′). Notably, the H2O vapor (with a kinetic diameter of 2.64 Å) adsorption isotherm for 1′ displayed a rapid and steep uptake in the low-humidity range, reaching a saturation uptake of 128 cm3 g−1, which is equivalent to 5.1 molecules per formula unit and similar to the H2O molecules (bridging and guest) present in the as-synthesized framework (Fig. 2d). This steep uptake in a relatively low-pressure region suggests a strong interaction between H2O molecules and the pore surface, which is decorated with unsaturated Ni(II) centers, as well as oxygen atoms from the bridging –OH and –OCO groups from the 1,4-ndc linker. As anticipated, the pore exhibited less affinity for CH3OH molecules, exhibiting a distinct gate-opening-type profile due to their larger size (3.62 Å) and lower polarity compared to H2O (2.64 Å).
This observation is further supported by the βE0 values obtained from the Dubinin–Radushkevich (D–R) equation43,44 which reveal the adsorbate–adsorbent interactions. The values are found to be 7.8 and 2.5 kJ mol−1 for H2O and CH3OH, respectively. To gain deeper insights into the transition in porosity upon guest loading, we conducted PXRD measurements on samples collected at two points, P and Q, during H2O and CH3OH vapor adsorption, respectively (Fig. S7†). The PXRD patterns at these points closely matched the diffraction patterns of the as-synthesized structure (1), indicating the expansion of the framework from the activated shrunk phase (1′) back to its original state (1, open-pore phase) to accommodate guest molecules. The adsorption isotherm and the observed structural transformation undeniably support the notion that the hydrophilic pore surface is biased for polar molecules, such as H2O and CO2.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) with CO2 on 1′
To understand the mode of interactions, we have performed in situ DRIFTS with CO2 over 1′. This experiment has been performed with an evenly coated activated sample on the glass disc placed in a removable vacuum tight sample compartment (evaluable/purgeable) against an MCT (mercury–cadmium–telluride) detector in an FT-IR (Vertex 70B) spectrometer. The sample compartment, made of zinc selenide (ZnSe), is resistant to high temperature and pressure. In this case, the CO2 concentration in the chamber has been regulated through an MFC (mass flow controller). At first, the IR peaks attributed exclusively to molecular CO2 were identified with increased dosing of only CO2 (99.995% dry) in the chamber, in the absence of sample (Fig. S8–S10†). Further, the activated MOF was recorded in the chamber without CO2 flow (Fig. S9†). To isolate the spectrum attributed exclusively to the CO2⋯MOF interaction, the spectra from previous experiments were utilized as background references. For signals related to CO2⋯MOF interaction, the CO2 flow in the chamber was gradually increased under similar conditions. The emerging peaks became prominent with higher CO2 dosing which is indicative of CO2 interactions with the MOF. As depicted in Fig. 3, with a gradual increase in CO2 flow, prominent IR features emerged when comparing the blue background (BG) spectrum to the other spectra in the dataset. As illustrated in Fig. 3 and S9,† the ν3 mode of CO2 antisymmetric stretching was observed in a narrow spectral interval of 2383–2300 cm−1 (Table S7†).45–49 Therefore, the appearance of strong bands when the sample is subjected to CO2 pressure can be directly correlated with the interaction of CO2 molecules with the available sites in the framework. Two strong asymmetric vibrations were observed at 2352 and 2331 cm−1 (with a difference of ∼20 cm−1), when only CO2 was passed without the MOF. When CO2 is flowed over activated 1′, four peaks are prominently observed with different intensities suggesting different modes of CO2 interaction with the functional sites. The vibration frequencies observed are 2383 and 2362 cm−1 (with a difference of 20 cm−1) and 2340 and 2312 cm−1 (with a difference of 28 cm−1). According to the literature, all these vibrational frequencies can be correlated with the ν3(CO2) asymmetric modes of O–C–O interacting with open metal site NiII (Fig. 3).45,50 The relative shifting of these peaks may originate from varying strengths of interactions between CO2 and open metal sites (Niδ+⋯Oδ−–C–O type interaction). Likewise, the two weak bands at 1645 and 1455 cm−1 may be attributed to carboxylate stretching frequencies.51,52 Notably, activated 1′, in the absence of CO2, showed carboxylate stretching frequencies of 1576 and 1369 cm−1, indicating changes in the chemical environment of the 1,4-ndc-carboxylate group when dozed with CO2 pressure (Fig. S9†). Several other bands were also observed in the range between 3734 and 3600 cm−1, and their interpretation is complex. We believe that the presence of multiple peaks, such as those at 3734, 3723, and 3695 cm−1, can be attributed to both OH vibrational frequencies and CO2 overtones (ν1 + ν2).53–55 The graph clearly illustrates that the peak at 3723 cm−1 increases with the rising CO2 flow in comparison to other peaks. Hence, in accordance with the available literature, this phenomenon can be attributed to either (1) CO2 overtones or (2) the presence of bicarbonate (OOC⋯OHδ−–Mδ+) in that specific region.45,51,52,56 The in situ IR study confirmed that CO2 adsorption occurs within the pores of 1′, facilitated primarily by NiII⋯Oδ−CO interaction with possible additional interaction sites, which is evident in the gated multistep CO2 adsorption profile at 298 K.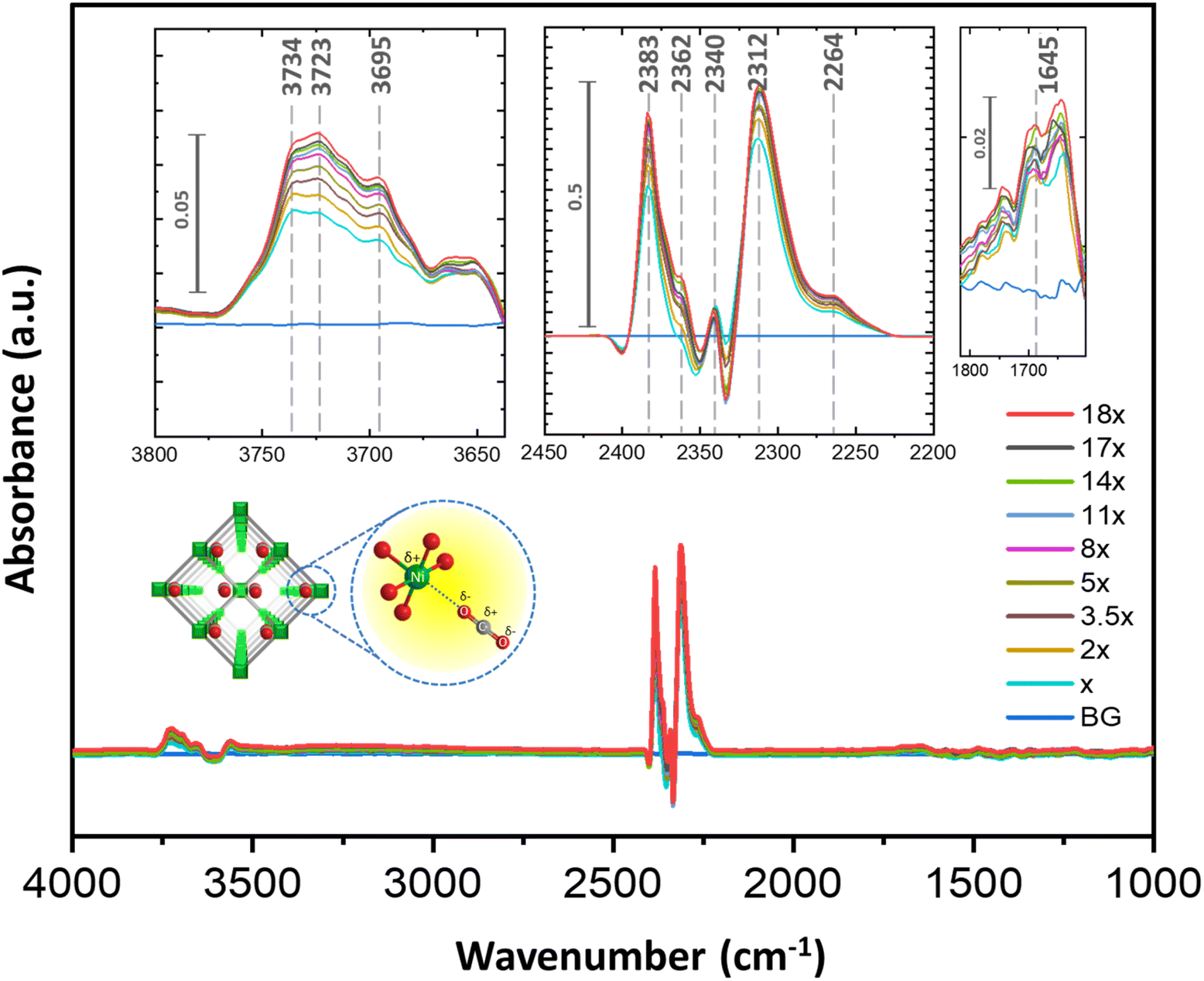 | ||
| Fig. 3 In situ DRIFTS of CO2 adsorption in 1′ studied under ambient conditions. The X is the flow rate of CO2 dosing inside the chamber to develop increased concentration inside the sample chamber. X = 10 L per hour. The schematic of CO2–MOF interaction as anticipated from the adsorption, PXRD and DRIFTS study. | ||
Theoretical calculations
We conducted density functional theory (DFT) calculations to investigate the interactions between the dehydrated framework and gas (CO2 and C2H2) molecules. A fragmented model consisting of four Ni(II) centers (Ni1–Ni4) without a bridging water molecule was considered for DFT calculations (Table S10†). In the fragmented model Ni1 and Ni2 are the unsaturated metal sites, which would serve as the potential adsorption sites for CO2 and C2H2.50 Accordingly, CO2 and C2H2 adsorbed models were designed for DFT calculations (Fig. 4 and Tables S8 and S9†). The results revealed that the CO2 molecule could be bound to the Ni(II) centers (Ni1 and Ni2) through its O atom in an end-on fashion. The strong binding of the CO2 molecule with Ni1 and Ni2 could be characterized by effective bond distances of 2.73 Å (O1–Ni1) and 2.84 Å (O1–Ni2), as depicted in Fig. 4a.57 Additionally, weak interactions between the electron deficient carbon atom of CO2 and two adjacent carboxylate oxygen atoms of the framework (2.96 and 2.87 Å) were also observed resulting in a slight deviation of CO2 bond angle to 177.8°, which showed overall strong binding and stabilization of CO2 molecules within the framework (Fig. 4a).50,58 In contrast to this, C2H2 barely has any interaction with any Ni(II) or oxo-sites in the framework.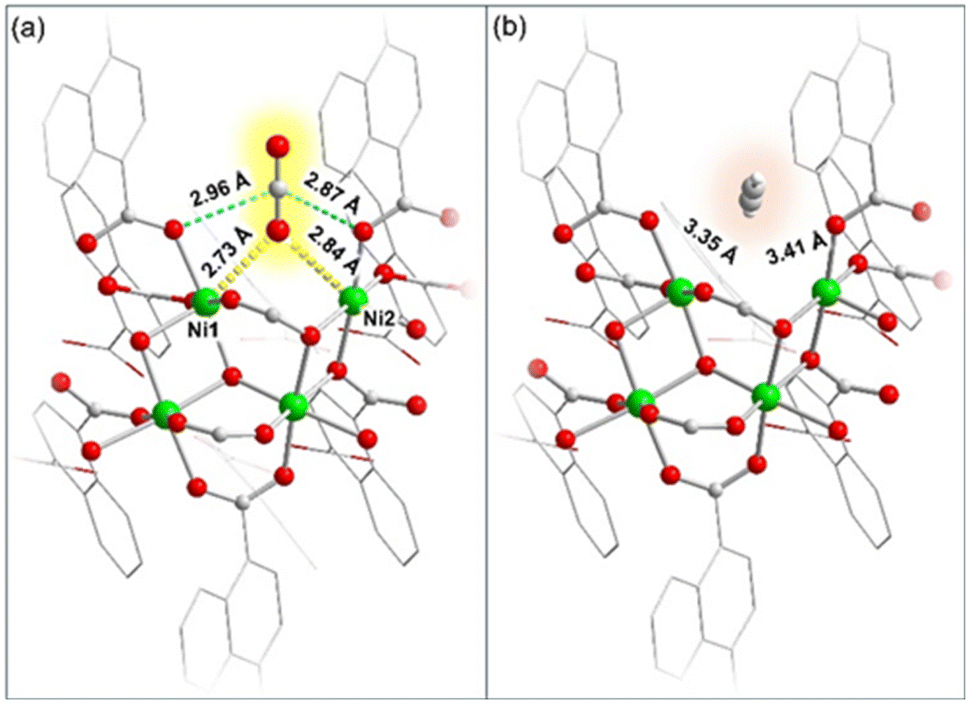 | ||
| Fig. 4 Density functional theory (DFT) results showing (a) interactions between CO2 and unsaturated Ni(II) sites along with carboxylate oxygen atoms and (b) no such interactions between C2H2 and the framework. | ||
C2H2 could be located in a spatially isolated floating position, with a distance of ∼3.4 Å from Ni(II) sites (Fig. 4b and Table S9†). Moreover, the binding energy of the CO2 and C2H2 molecules with the MOF was calculated to quantify the respective binding affinities. As anticipated, CO2 exhibited a higher binding energy of −42.5 kJ mol−1, significantly higher than that of C2H2 (ΔE = −5.8 kJ mol−1), consistent with the earlier observations.38 Therefore, the theoretical findings strongly support the experimentally observed selective uptake of CO2 compared to C2H2. The NiII⋯OCO interaction observed in DRIFTS experiments aligns well with the predictions of DFT calculations, confirming a strong NiII⋯OCO interaction.
Adsorptive separation based on dynamic breakthrough measurement
In the following step, to project the selectivity of 1′ in the real time separation of industrially crucial CO2/H2, CO2/N2, CO2/CH4, and CO2/C2H2 gas mixtures, a calculation was conducted using the ideal adsorbed solution theory (IAST) after fitting isotherms to the dual-site Langmuir–Freundlich equation at ambient temperature (298 K) (Fig. S11†). As depicted in Fig. 5, the adsorption selectivity for gas mixtures of CO2/CH4 and CO2/C2H2 at 110 kPa and 298 K was calculated to be 1.33 and 1.5, respectively. This value is comparable to other reported values, and the uptake ratio is reasonably higher (∼9), comparable to a few other literature reports at saturated vapor pressure.15,21,22,59,60 The real-time breakthrough separation was performed by passing similar binary compositions through a packed column bed of 1′ with a 2.8–2.2 mL min−1 flow rate (Fig. S12†). The breakthrough separation reveals that 1′ can achieve satisfactory separation of a CO2/C2H2 (0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5; v/v) mixture, where C2H2 was first eluted and quickly approached purity without any detectable CO2 (Fig. 6, S12†).
:0.5; v/v) mixture, where C2H2 was first eluted and quickly approached purity without any detectable CO2 (Fig. 6, S12†).
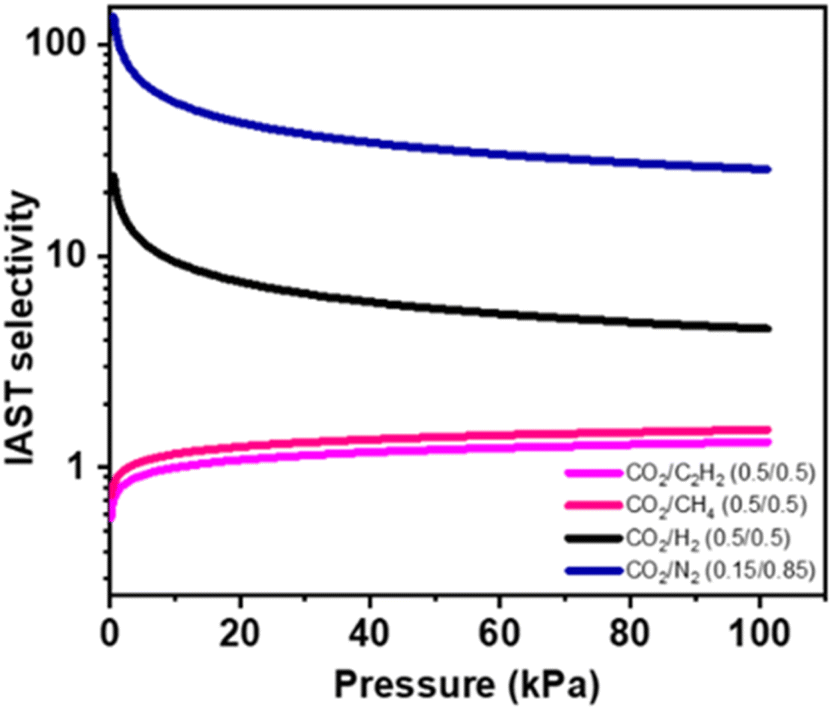 | ||
| Fig. 5 IAST selectivity approximation studied from a 298 K adsorption isotherm by fitting it in a dual-site Langmuir–Freundlich (DSLF) equation. | ||
 | ||
| Fig. 6 (a) The bi-cyclic dynamic breakthrough separation of equimolar C2H2/CO2 for 1′ and (b) respective concentration of C2H2 in the outlet (plotting cycle 2 with 0 s start time) considering the flow rate through the bed of 2.8–2.2 mL min−1. | ||
For CO2/C2H2 separation, CO2 was retained in the column bed sufficiently longer compared to C2H2 (300 s after the initiation of feed gas dosing) until the material reached its saturation uptake (Fig. 6 and Table S13†). Upon saturation, CO2 broke through, and corresponding outlet gas mixtures quickly reached equimolar proportions. Thereafter, by stopping feed mixtures and flowing a purge of carrier gas (He here) through the packed column the retained amount of CO2 could be removed with high purity by making the column regenerated for subsequent separations. Most of the reported frameworks are selective towards C2H2 and as a result polymer-grade C2H2 can be only eluted in the downstream desorption phase of fixed bed adsorptive operation. Unlike the usual technique, fine matching of the dynamic pore of 1′ allows CO2 sieving exclusively as pure C2H2 can be produced during the adsorption phase, leading to the separation becoming easier and more cost-effective. The breakthrough separation experiments of other gas mixtures such as CO2/H2 (0.5:0.5, v/v), CO2/N2 (0.85:0.15, v/v) and CO2/CH4 (0.5:0.5, v/v) were also performed where H2, N2 and CH4 were first eluted and quickly approached high purity without any detectable CO2 (Fig. S12†).
Conclusions
In conclusion, a contemporary three-dimensional Ni-based framework has been synthesized, characterized and studied for separating industrially important gas mixtures with similar size and physical properties. The structural flexibility and pore environment have been thoroughly investigated by several diffraction and adsorption tools (H2O and MeOH). The framework exhibits multistep gated adsorption exclusively towards CO2 due to likeliness in size, polarizability and alignment in the pore governed by unsaturated metal and carboxylate(O)⋯CO2 interactions, further confirmed by in situ DRIFTS analysis and theoretical investigation. Such discriminatory sorption of CO2 makes this framework highly selective from its industrial spin-off components in particular C2H2 and CH4. The practical separation feasibility of this framework is further tested under continuous flowing conditions by using CO2/C2H2, CO2/CH4, CO2/N2 and CO2/H2 gas mixtures at ambient temperature. Therefore, these results will provide a new strategy for designing and utilizing a dynamic porous framework for biased CO2 capture and an inverse separation from the most important industrial analogue C2H2.Data availability
All associated data are in the ESI† or deposited with CCDC (2310563).Author contributions
N. S., S. L. and R. J. have performed synthesis and basic characterizations. N. S. and R. J. have analysed the single crystal structure, while N. S. has executed advanced characterizations such as sorption dependent PXRD and adsorption isotherms both in vapor and gaseous phases. S. L. has carried out hydrocarbon adsorption isotherms, selectivity study, breakthrough analysis and co-written the manuscript with N. S. S. L. and A. D. have organized the in situ DRIFTS instrumental assembly and performed spectroscopic measurements under varied conditions. F. A. R. has performed theoretical calculation. T. K. M. has supervised the overall work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the authors thankfully acknowledge SERB, Dept. of Science and Technology (DST), Council of Scientific and Industrial Research (CSIR), Govt. of India for financial support (project no. SPR/2021/000592, CRG/2019/005951). The SAMat, ICMS, SSL research facility and Sheikh Saqr senior fellowship (T. K. M.) are also gratefully acknowledged.Notes and references
- P. J. Stang and F. Diederich, Modern acetylene chemistry, John Wiley & Sons, 2008 Search PubMed.
- J.-W. Zhang, M.-C. Hu, S.-N. Li, Y.-C. Jiang, P. Qu and Q.-G. Zhai, Chem. Commun., 2018, 54, 2012–2015 RSC.
- Y. Ye, Z. Ma, R.-B. Lin, R. Krishna, W. Zhou, Q. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2019, 141, 4130–4136 CrossRef CAS PubMed.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS PubMed.
- C. R. Reid and K. M. Thomas, Langmuir, 1999, 15, 3206–3218 CrossRef CAS.
- M. Bühl and G. Wipff, ChemPhysChem, 2011, 12, 3095–3105 CrossRef PubMed.
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273–274, 139–164 CrossRef CAS.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- A. Hazra, A. Jain, M. S. Deenadayalan, S. A. Adalikwu and T. K. Maji, Inorg. Chem., 2020, 59, 9055–9064 CrossRef CAS PubMed.
- A. Chakraborty, S. Roy, M. Eswaramoorthy and T. K. Maji, J. Mater. Chem. A, 2017, 5, 8423–8430 RSC.
- A. Hazra, S. Jana, S. Bonakala, S. Balasubramanian and T. K. Maji, Chem. Commun., 2017, 53, 4907–4910 RSC.
- R. Haldar, N. Sikdar and T. K. Maji, Mater. Today, 2015, 18, 97–116 CrossRef CAS.
- J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2009, 131, 5516–5521 CrossRef CAS PubMed.
- K.-J. Chen, H. S. Scott, D. G. Madden, T. Pham, A. Kumar, A. Bajpai, M. Lusi, K. A. Forrest, B. Space, J. J. Perry and M. J. Zaworotko, Chem, 2016, 1, 753–765 CAS.
- R.-B. Lin, L. Li, H. Wu, H. Arman, B. Li, R.-G. Lin, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 8022–8028 CrossRef CAS PubMed.
- A. Luna-Triguero, J. M. Vicent-Luna, R. M. Madero-Castro, P. Gómez-Álvarez and S. Calero, ACS Appl. Mater. Interfaces, 2019, 11, 31499–31507 CrossRef CAS PubMed.
- W. Fan, S. Yuan, W. Wang, L. Feng, X. Liu, X. Zhang, X. Wang, Z. Kang, F. Dai, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 8728–8737 CrossRef PubMed.
- H.-G. Hao, Y.-F. Zhao, D.-M. Chen, J.-M. Yu, K. Tan, S. Ma, Y. Chabal, Z.-M. Zhang, J.-M. Dou, Z.-H. Xiao, G. Day, H.-C. Zhou and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16067–16071 CrossRef CAS PubMed.
- J. Wang, Y. Zhang, Y. Su, X. Liu, P. Zhang, R.-B. Lin, S. Chen, Q. Deng, Z. Zeng, S. Deng and B. Chen, Nat. Commun., 2022, 13, 200 CrossRef CAS PubMed.
- R. Krishna, ACS Omega, 2020, 5, 16987–17004 CrossRef CAS PubMed.
- W. Yang, A. J. Davies, X. Lin, M. Suyetin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, J. E. Parker, C. C. Tang, M. W. George, P. Hubberstey, S. Kitagawa, H. Sakamoto, E. Bichoutskaia, N. R. Champness, S. Yang and M. Schröder, Chem. Sci., 2012, 3, 2993–2999 RSC.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- S.-Q. Yang, R. Krishna, H. Chen, L. Li, L. Zhou, Y.-F. An, F.-Y. Zhang, Q. Zhang, Y.-H. Zhang, W. Li, T.-L. Hu and X.-H. Bu, J. Am. Chem. Soc., 2023, 145, 13901–13911 CrossRef CAS PubMed.
- Y. Shi, Y. Xie, H. Cui, Y. Ye, H. Wu, W. Zhou, H. Arman, R.-B. Lin and B. Chen, Adv. Mater., 2021, 33, 2105880 CrossRef CAS PubMed.
- Y. Xie, H. Cui, H. Wu, R.-B. Lin, W. Zhou and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 9604–9609 CrossRef CAS PubMed.
- S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- T. K. Maji, R. Matsuda and S. Kitagawa, Nat. Mater., 2007, 6, 142–148 CrossRef CAS PubMed.
- J. D. Evans, V. Bon, I. Senkovska, H.-C. Lee and S. Kaskel, Nat. Commun., 2020, 11, 2690 CrossRef CAS PubMed.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- T. K. Maji, K. Uemura, H.-C. Chang, R. Matsuda and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 3269–3272 CrossRef CAS PubMed.
- S. Bhattacharyya and T. K. Maji, Coord. Chem. Rev., 2022, 469, 214645 CrossRef CAS.
- N. Sikdar, S. Bonakala, R. Haldar, S. Balasubramanian and T. K. Maji, Chem.–Eur. J., 2016, 22, 6059–6070 CrossRef CAS PubMed.
- N. Sikdar, A. Hazra and T. K. Maji, Inorg. Chem., 2014, 53, 5993–6002 CrossRef CAS PubMed.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- L. W. Bingel and K. S. Walton, Langmuir, 2023, 39, 4475–4482 CrossRef CAS PubMed.
- M.-H. Yu, B. Space, D. Franz, W. Zhou, C. He, L. Li, R. Krishna, Z. Chang, W. Li, T.-L. Hu and X.-H. Bu, J. Am. Chem. Soc., 2019, 141, 17703–17712 CrossRef CAS PubMed.
- S. Laha, N. Dwarkanath, A. Sharma, D. Rambabu, S. Balasubramanian and T. K. Maji, Chem. Sci., 2022, 13, 7172–7180 RSC.
- M. Chen, S. Chen, W. Chen, B. E. G. Lucier, Y. Zhang, A. Zheng and Y. Huang, Chem. Mater., 2018, 30, 3613–3617 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. M. Junquera-Hernández, J. Sánchez-Marín and D. Maynau, Chem. Phys. Lett., 2002, 359, 343–348 CrossRef.
- K. A. Moltved and K. P. Kepp, J. Phys. Chem. C, 2019, 123, 18432–18444 CrossRef CAS.
- N. D. Hutson and R. T. Yang, Adsorption, 1997, 3, 189–195 CrossRef CAS.
- T. K. Maji, G. Mostafa, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2005, 127, 17152–17153 CrossRef CAS PubMed.
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Chem. Rev., 2021, 121, 1286–1424 CrossRef CAS PubMed.
- M. Mihaylov, K. Chakarova, S. Andonova, N. Drenchev, E. Ivanova, A. Sabetghadam, B. Seoane, J. Gascon, F. Kapteijn and K. Hadjiivanov, J. Phys. Chem. C, 2016, 120, 23584–23595 CrossRef CAS.
- S. Andonova, S. S. Akbari, F. Karadaş, I. Spassova, D. Paneva and K. Hadjiivanov, J. CO2 Util., 2021, 50, 101593 CrossRef CAS.
- N. R. Walker, R. S. Walters, G. A. Grieves and M. A. Duncan, J. Chem. Phys., 2004, 121, 10498–10507 CrossRef CAS PubMed.
- W. Chen, Z.-R. Li, D. Wu, F.-L. Gu, X.-Y. Hao, B.-Q. Wang, R.-J. Li and C.-C. Sun, J. Chem. Phys., 2004, 121, 10489–10494 CrossRef CAS PubMed.
- P. D. C. Dietzel, R. E. Johnsen, H. Fjellvåg, S. Bordiga, E. Groppo, S. Chavan and R. Blom, Chem. Commun., 2008, 41, 5125–5127 RSC.
- W. Zhou, Z. Ma, S. Guo, M. Wang, J. Wang, M. Xia, L. Jia, B. Hou, D. Li and Y. Zhao, Appl. Surf. Sci., 2018, 427, 867–873 CrossRef CAS.
- L. Proaño, E. Tello, M. A. Arellano-Trevino, S. Wang, R. J. Farrauto and M. Cobo, Appl. Surf. Sci., 2019, 479, 25–30 CrossRef.
- S. Eckle, H.-G. Anfang and R. J. Behm, J. Phys. Chem. C, 2011, 115, 1361–1367 CrossRef CAS.
- G. Garbarino, D. Bellotti, E. Finocchio, L. Magistri and G. Busca, Catal. Today, 2016, 277, 21–28 CrossRef CAS.
- Q. Guan, X. Cheng, R. Li and W. Li, J. Catal., 2013, 299, 1–9 CrossRef CAS.
- J. Baltrusaitis, J. H. Jensen and V. H. Grassian, J. Phys. Chem. B, 2006, 110, 12005–12016 CrossRef CAS PubMed.
- S. A. FitzGerald, J. M. Schloss, C. J. Pierce, B. Thompson, J. L. C. Rowsell, K. Yu and J. R. Schmidt, J. Phys. Chem. C, 2015, 119, 5293–5300 CrossRef CAS.
- K. Jayaramulu, S. K. Reddy, A. Hazra, S. Balasubramanian and T. K. Maji, Inorg. Chem., 2012, 51, 7103–7111 CrossRef CAS PubMed.
- J. Lee, C. Y. Chuah, J. Kim, Y. Kim, N. Ko, Y. Seo, K. Kim, T. H. Bae and E. Lee, Angew. Chem., Int. Ed., 2018, 57, 7869–7873 CrossRef CAS PubMed.
- Y. Du, Y. Chen, Y. Wang, C. He, J. Yang, L. Li and J. Li, Sep. Purif. Technol., 2021, 256, 117749 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic details, TGA, adsorption isotherms, and DRIFTS data. CCDC 2310563. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06611h |
| ‡ These authors contributed equally. |
| § Present address: Department of Chemistry, School of Science, Gandhi Institute of Technology and Management (GITAM), Hyderabad 502329, India. |
| This journal is © The Royal Society of Chemistry 2024 |