
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the photocatalytic upcycling of polystyrene to benzoic acid: a combined computational-experimental approach for acridinium catalyst design†
Albert
Ong‡
 a,
Zi Cheng
Wong‡
b,
Kang Le Osmund
Chin
c,
Wei Wei
Loh
a,
Ming Hui
Chua
*c,
Shi Jun
Ang
*bc and
Jason Y. C.
Lim
*ad
a,
Zi Cheng
Wong‡
b,
Kang Le Osmund
Chin
c,
Wei Wei
Loh
a,
Ming Hui
Chua
*c,
Shi Jun
Ang
*bc and
Jason Y. C.
Lim
*ad
aInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis #08-03, Singapore 138634, Republic of Singapore. E-mail: jason_lim@imre.a-star.edu.sg
bInstitute of High Performance Computing (IHPC), Agency for Science, Technology and Research (A*STAR), 1 Fusionopolis Way, Connexis, #16-16, Singapore 138632, Republic of Singapore. E-mail: ang_shi_jun@ihpc.a-star.edu.sg
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore. E-mail: chua_ming_hui@isce2.a-star.edu.sg
dDepartment of Materials Science and Engineering, National University of Singapore (NUS), 9 Engineering Drive 1, Singapore 117576, Republic of Singapore
First published on 18th December 2023
Abstract
Converting polystyrene into value-added oxygenated aromatic compounds is an attractive end-of-life upcycling strategy. However, identification of appropriate catalysts often involves laborious and time-consuming empirical screening. Herein, after demonstrating the feasibility of using acridinium salts for upcycling polystyrene into benzoic acid by photoredox catalysis for the first time, we applied low-cost descriptor-based combinatorial in silico screening to predict the photocatalytic performance of a family of potential candidates. Through this approach, we identified a non-intuitive fluorinated acridinium catalyst that outperforms other candidates for converting polystyrene to benzoic acid in useful yields at low catalyst loadings (≤5 mol%). In addition, this catalyst also proved effective with real-life polystyrene waste containing dyes and additives. Our study underscores the potential of computer-aided catalyst design for valorizing polymeric waste into essential chemical feedstock for a more sustainable future.
1. Introduction
Polystyrene (PS), one of the most produced thermoplastics worldwide,1 is indispensable in the packaging and construction industries.2 However, it has become one of the most prevalent pollutants in the environment3 due to its chemical inertness and resulting slow rates of natural degradation. Because of the low existing global collection and recycling rates,4 the existing life-cycle of PS is largely linear, with expanded PS constituting a large fraction of landfill waste.5 Instead of disposal, upcycling end-of-life plastics into financially more-valuable chemicals and materials is of great interest,6–10 with PS being especially valued as a possible source of industrially-relevant aromatics.11,12 Amongst them, benzoic acid (BA) is of particular importance, with annual demand exceeding 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons13 across multiple industrial applications, such as food preservatives, antimicrobial agents and chemical precursors. However, PS oxidative degradation is hindered by the chemical inertness of its saturated C–C backbone, which traditionally necessitates harsh oxidation conditions involving high temperatures and pressures.14–16
000 metric tons13 across multiple industrial applications, such as food preservatives, antimicrobial agents and chemical precursors. However, PS oxidative degradation is hindered by the chemical inertness of its saturated C–C backbone, which traditionally necessitates harsh oxidation conditions involving high temperatures and pressures.14–16
Photoredox catalysis offers unique opportunities for PS degradation to occur under mild conditions (e.g. near-ambient temperature) to produce oxygenated aromatics.17–20 This stems from their ability to facilitate hydrogen atom transfer (HAT) from the PS backbone, generating reactive radicals that elicit backbone oxidation and chain scission reactions. Although several inorganic catalysts have been reported to produce oxygenated aromatics from PS,18,21 the use of metal-free low molecular weight organo-photoredox catalysts22 has thus far been largely under-exploited. In this respect, we hypothesized that acridinium salts, which are able to access highly-oxidizing excited states upon visible light photo-activation,23 can serve as effective catalysts for PS oxidative degradation. Furthermore, low-energy visible light photoexcitation of acridinium salts can also enable better control of PS backbone degradation to achieve improved product selectivity. Acridinium salts are amongst the most structurally-diverse families of organo-photoredox catalysts available, offering ample opportunities to modulate their photophysical properties (e.g. excited state redox potential) and catalytic efficacy by diversification of the scaffold substituents.24 However, this makes a systematic empirical exploration of the vast catalyst design space for PS degradation impractically laborious, with multi-stepped de novo synthesis of each acridinium candidate required. To overcome this bottleneck, we present a combined experimental and computational catalyst screening approach,25 allowing us to identify a high-performing fluorinated acridinium catalyst candidate able to produce BA with yield surpassing other structural analogues synthesized.
2. Results and discussion
As PS degradation using acridinium salts is hitherto unexplored, we commenced the study with the model Ph-Acr-Ph catalyst (nomenclature: [9-substituent]-Acr-[N-substituent]) to identify the essential reaction conditions for BA production from commercial PS resins (Table 1). When irradiated with a 390 nm LED light source for 24 h under an O2 atmosphere (entry 1), Ph-Acr-Ph (10 mol% w.r.t. PS monomers) and 10 mol% HCl (aq) afforded BA in 33% yield as the dominant aromatic product, with some formic acid detected (Fig. S4†). Trace aromatic molecules such as benzaldehyde were also detected by ESI-MS (Fig. S5 and S23†), but were present in insufficient quantities for 1H NMR detection. In addition, gel permeation chromatography (GPC) analysis of the crude product mixture revealed that short-chain oligomers with a bimodal distribution (Fig. S6†) were also formed during the reaction.|
|
|||||
|---|---|---|---|---|---|
| S/N | Ph-Acr-Ph loadinga (mol%) | Acid additiveb (mol% loading) | Deviation from above conditions | BA yieldc (%) | M n (kDa) |
| a Although no external heat source was used, the reaction temperature was maintained at (∼35 ± 1) °C throughout reaction duration due to heating caused by the LED light source. b w.r.t. PS monomers present. c Determined by quantitative 1H-NMR using 1,3,5-trimethoxybenzene as internal standard (Section S4). d Determined via GPC analysis with THF as the mobile phase against monodisperse polystyrene standards. Mn of PS starting material = 42.7 kDa by GPC analysis. e Molecular weight of PS oligomers are lower than the accurate GPC mass range. f Bimodal GPC product distribution observed. | |||||
| 1 | 10 | HCl (10) | — | 33 | <0.30e,f |
| 2 | — | HCl (10) | — | 6 | 1.71 |
| 3 | 10 | HCl (10) | Argon atmosphere | 0 | 6.90 |
| 4 | 10 | HCl (10) | Reaction in the dark | 0 | 42.3 |
| 5 | 10 | — | — | 20 | <0.30e,f |
| 6 | 5 | HCl (200) | — | 46 | <0.30e,f |
| 7 | 5 | HCl (500) | — | 50 | <0.30e,f |
| 8 | 5 | HBr (200) | — | 7 | 5.56f |
| 9 | 5 | HCl (200) | 440 nm irradiation | 43 | <0.30f |
Several reactions were then performed to understand the conditions necessary for BA production to occur from PS. Control experiments (entries 2–4) confirmed that Ph-Acr-Ph, O2 and light were essential for efficient BA generation, and replacing the O2 atmosphere with air (21% O2) resulted in greatly reduced reaction efficacy (Table S1†). It is noteworthy that although the reaction under argon (Table 1, entry 3) did not yield any BA, PS chain cleavage occurred (Fig. S10†), possibly brought about by C–C radical chain cleavage initiated by hydrogen abstraction from the PS backbone by the photoexcited acridinium catalyst (vide infra). This hypothesis is supported by the lack of PS chain cleavage in the dark (entry 4, Fig. S12†), which prevented acridinium excited state formation. A substantial reduction in BA yield resulted without HCl (Table 1, entry 5) and when HCl was replaced with an equivalent quantity of water (Table S1†), indicating the synergistic involvement of Cl− and Ph-Acr-Ph in PS degradation, plausibly through generation of chlorine radicals (vide infra).26 Increasing the HCl loading to 200 mol% afforded higher BA yields despite lowering the Ph-Acr-Ph loading to 5 mol% (entry 6), though larger quantities of HCl (500 mol%) only led to marginally more BA production (entry 7). Notably, 200 mol% HCl alone without Ph-Acr-Ph only afforded BA with 6% yield (Table S1†), underscoring the catalyst's importance even in the presence of large excess of HCl. When HCl was replaced with HBr in the presence of Ph-Acr-Ph, much poorer BA yields resulted (entry 8). While Br− is easier to oxidize than Cl− to generate their respective radicals (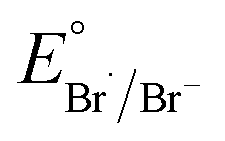 = 1.60 V vs. SCE,
= 1.60 V vs. SCE,  = 2.03 V vs. SCE),27 HAT from PS by Cl˙ is favoured due to the stronger H–Cl bond formed (bond dissociation energy for H–Cl = 103 kcal mol−1; H–Br = 87 kcal mol−1).28,29 The best BA yields were obtained using DCE/MeCN 6:1 (v/v), with MeCN, CHCl3 and different DCE-containing solvent mixtures affording much less BA (Table S1†). Finally, the reaction is tolerant to different excitation wavelengths, with 440 nm irradiation affording similar BA yields (entry 9) as 390 nm.
= 2.03 V vs. SCE),27 HAT from PS by Cl˙ is favoured due to the stronger H–Cl bond formed (bond dissociation energy for H–Cl = 103 kcal mol−1; H–Br = 87 kcal mol−1).28,29 The best BA yields were obtained using DCE/MeCN 6:1 (v/v), with MeCN, CHCl3 and different DCE-containing solvent mixtures affording much less BA (Table S1†). Finally, the reaction is tolerant to different excitation wavelengths, with 440 nm irradiation affording similar BA yields (entry 9) as 390 nm.
Fig. 1A outlines a possible mechanism for photoredox oxidative PS conversion to BA, based on our empirical observations and by analogy to reported mechanisms of acridinium-mediated arene oxidations.22,23 Photoexcitation of the catalyst generates its excited state (Acr+*), which promotes a one-electron oxidation of PS to form a reactive PS+˙ intermediate that can undergo deprotonation to generate a benzylic radical (Cycle 1).30,31 The generated PS+˙ may also oxidise Cl− to Cl˙ (calculated Eox of model PS compound, pentan-3-yl-benzene, > Eox(Cl−)) (Section S5†). In addition, the Cl˙ generated from Acr+*26 can promote HAT from the PS backbone (Cycle 2).18 The common benzylic radical formed from both catalytic cycles can be trapped by O2 to form a peroxo intermediate that can further undergo homolytic O–O cleavage to form an oxo-radical species. β-Scission then forms a primary radical chain end and a phenyl ketone that can further react, propagating chain cleavage to ultimately give BA and formic acid as the terminal oxidized products. Although a [PS⋯H+] adduct from interactions of PS with acids has been proposed to facilitate PS degradation by acting as a photosensitiser for 1O2 generation,19 this is unlikely to play a significant role under our present reaction conditions, as the presence of HCl and HBr alone without acridinium catalysts afforded only poor BA yields at 390 nm irradiation (Tables 1, S1†).
 | ||
| Fig. 1 (A) Proposed mechanism for photoredox catalysed oxidative degradation with acridinium salts; (B) computed photophysical (λabs and fabs) and excited state redox (Ered(Acr+*)) properties of three model acridinium salts, with their empirical BA yields. Computed Eox and Ered values are w.r.t. SHE. # Yields are calculated from an average of two repeat experiments. | ||
Due to the mechanistic complexity of photoredox oxidative PS degradation, a full mechanism-based in silico approach for catalyst screening is extremely demanding computationally.32–34 Thus, as a reasonable compromise for computational workload and accuracy, a descriptor-based approach was adopted.35,36 To perform the catalyst screening logically, photophysical and thermodynamic parameters satisfying essential mechanistic criteria from Fig. 1A were chosen.37 Firstly, the ability of Acr+* to initiate Cycles 1 and 2 must be thermodynamically feasible, i.e. Ered(Acr+*) > Eox(PS) and Eox(Cl−). Secondly, the acridinium salt should possess appropriate photophysical properties allowing access to a high concentration of Acr+* during the reaction. In this regard, the catalyst should have λabs close to the irradiation wavelength (390 nm) and possess high oscillator strength (fabs). Experimentally, the oscillator strength for an electronic transition can be calculated using the area under its corresponding absorption band.38 As the height of the absorption band is simply the molar absorptivity at a given wavelength, the oscillator strength calculated using TD-DFT can be used to obtain the molar absorptivity. In the context of the present study, a higher value for the oscillator strength (or molar absorptivity) would thus imply a larger concentration of acridinium salt being electronically excited upon photostimulation, which should in turn lead to an improvement in the overall BA yield. In order to evaluate the descriptors that could serve as a reasonable predictors of catalyst performance, we computed the Ered(Acr+*), λabs and fabs values of three model acridinium salts (Ph-Acr-Ph, Ph-Acr-Me and Mes-Acr-Me) (details of TD-DFT calculations in Section S5†), and evaluated them against the yields of BA obtained from PS under identical reaction conditions (Fig. 1B). Notably, we verified that TD-DFT calculations were able to provide reasonable estimates of the actual experimental values of Ered(Acr+*) and λabs (Tables S2 and S4† with associated discussions). In all cases, BA was produced, albeit with different yields. This supported the thermodynamic feasibility of both catalytic cycles with calculated Ered(Acr+*) for all catalysts > Eox(PS) and Eox(Cl−). Amongst the descriptors, fabs emerged as the best predictor of catalyst performance, with higher values correlating to better BA yields.
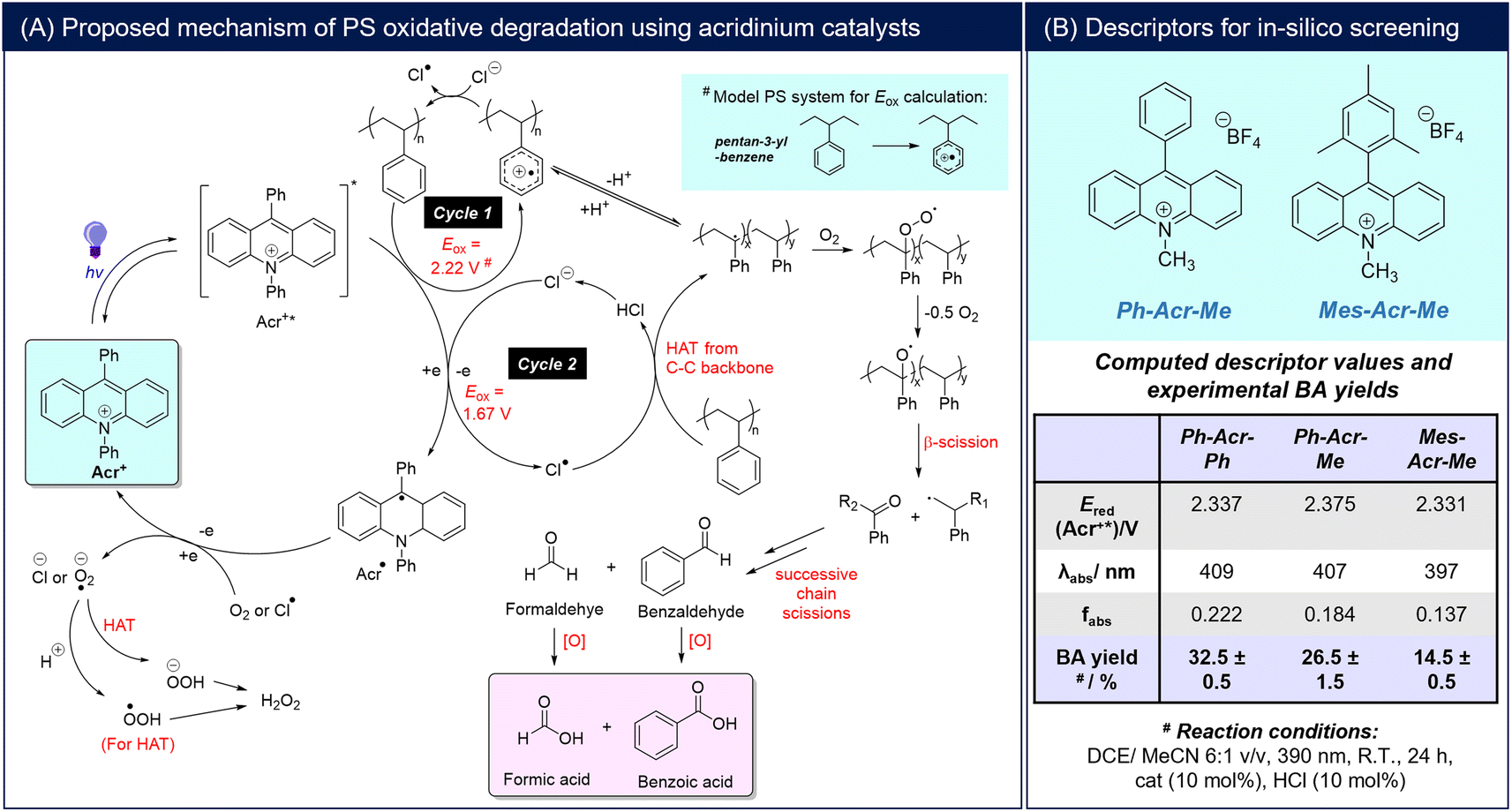
Combinatorial catalyst screening was then performed in silico on a family of acridinium salts bearing different 9-/N-substituents to identify candidates that can possibly outperform Ph-Acr-Ph for PS degradation (Fig. 2A). Despite only varying the 9-/N-substituents, the combinatorial catalyst design space is vast, with >1000 possible substituent candidates for each position from a Reaxys® substructure search. As a proof-of-concept, we selected a group of substituents representing electron-rich and electron-deficient aromatics, alkyl and extended π-systems for in silico screening (Section S5†). Fig. 2B illustrates the effects of each 9-/N-substituent on the acridinium salt's fabs values. Generally, 9-substituents exert a larger influence on the fabs compared to N-substituents, as they contribute to both the locally-excited and charge transfer excited states in the acridinium salts (see Section S5†). Amongst the para-functionalised aryl substituents at the 9-position, fabs decreases as substituents become more electron-deficient (MeOPh > Ph ≈ FPh > CF3Ph), reflected by more positive Hammett substituent constants (Fig. 2A). Notably, this trend does not apply to the N-substituents, with the N-naphthyl unit affording the highest fabs values for each 9-substituent series. In accordance with expectations however, more electron-withdrawing aryl units generally render the activated Acr+* catalyst stronger oxidants, with calculated Ered(Acr+*) values decreasing in the order CF3Ph > FPh > Ph > MeOPh regardless of their location on the N- or 9-positions (Table S5†).
 | ||
| Fig. 2 Acridinium catalyst prediction and experimental validation: (A) general synthetic scheme of acridinium salts, the substituent chemical space for in silico screening and selected Hammett substituent constants (σp);39 (B) scatter plot of acridinium fabs with different substituents at the 9- and N-positions; (C)(i) scatter plot of Ered(Acr+*) and fabs of the screened chemical space, with the region in pink enclosing candidates predicted to have better PS degradation performance than the Ph-Acr-Ph baseline; (ii) BA yields from PS resins and real-life waste using the selected FPh-Acr-Np catalyst, and time course experiments showing the evolution of BA yields and PS molecular weight with increasing reaction duration. Yields are calculated from an average of two repeat experiments (*yields in parentheses consider removal of insoluble additives such as dyes). | ||
Although fabs was determined to be the primary predictor of BA yield, it is insufficient by itself for catalyst selection. The importance of the candidates also satisfying Ered(Acr+*) > Eox(PS) is evident from the MeOPh-Acr-Y family of catalysts. Although they possess the highest fabs values amongst the candidates screened, their calculated Ered(Acr+*) values were unfavorable (Fig. 2C(i)) (see Section S5† for a full discussion).40,41 Indeed, MeOPh-Acr-Ph was synthesized and experimentally verified to not produce any BA from PS degradation using 390 or 440 nm irradiation (Table S1†). With this established, an acridinium candidate with better PS degradation performance than the Ph-Acr-Ph benchmark can be identified in the chemical space bounded by fabs(candidate) > fabs(Ph-Acr-Ph) and Ered(Acr+*) > Eox(PS) (Fig. 2C(i)). The candidate possessing the highest fabs value within this space, FPh-Acr-Np, was identified as the target catalyst (Fig. 2C(ii)) and synthesized (Section S2†). Gratifyingly, it could convert PS into BA with superior yields (of up to 55%) at 5 mol% catalyst loading compared to Ph-Acr-Ph at both low and high HCl loadings, further validating the effectiveness of our descriptor-based screening approach. It is noteworthy that when we lowered the FPh-Acr-Np loading to 1 mol% to evaluate the limits of catalyst loading, BA could still be obtained in 16.5% yield (Fig. 2C(ii)). Compared with existing reported homogeneous photoredox catalysts reported to degrade PS into oxygenated aromatics (Section S6†),18–21,42,43 the catalytic turnover of FPh-Acr-Np was amongst the best, and is especially advantaged by the low catalyst loadings required using our reaction conditions.
Time course experiments were then performed to elucidate the influences of FPh-Acr-Np on PS chain degradation and BA evolution compared with Ph-Acr-Ph. For both catalysts, tracking BA production by 1H NMR analysis revealed that negligible quantities of BA were produced in the first hour (Fig. S50†), but thereafter increased continuously throughout the entire reaction duration (Fig. 2D(ii)). Despite this, a drastic drop in average Mn was observed in the first hour for both reactions catalysed by FPh-Acr-Np and Ph-Acr-Ph. This indicated that the first hour was dominated by propagating oxidative radical chain scission of the PS polymer backbones (Fig. 1A), before formation of benzoic acid, the terminal product of the oxidative degradation, could occur. Significantly, PS deconstruction catalysed by FPh-Acr-Np reduced the average Mn of the polymer at significantly faster rate compared to that catalysed by Ph-Acr-Ph. From the 42.7 kDa PS resin, the Mn of the oxidized oligomers reached 1.26 kDa and 6.90 kDa within the first hour for FPh-Acr-Np and Ph-Acr-Ph, whilst requiring 5 and 9 h to reach the lower limit of GPC molecular weight detection, respectively. This further demonstrated the superiority of FPh-Acr-Np for PS degradation, further validating the effectiveness of our screening approach.
Finally, we demonstrated the generality of FPh-Acr-Np for PS oxidative upcycling using several real-life PS products (Fig. 2C(ii)). All products tested were well-tolerated, generating BA in synthetically-useful yields and demonstrating that FPh-Acr-Np was compatible with common additives such as plasticisers. It is noteworthy that although the dye in the black lid was shown to inhibit degradation,18 we overcame this problem using a solvent system that selectively dissolved the PS polymer but not the black dye, effectively upcycling the polymer into BA.
3. Conclusion
In conclusion, we have demonstrated for the first time that acridinium salts can be potent photoredox catalysts for oxidative degradation of PS to afford industrially-relevant BA at near-ambient temperature and pressure. Through the identification of key thermodynamic and photophysical descriptors, an in silico screening of acridinium analogues enabled the rapid identification of a high-performing FPh-Acr-Np catalyst candidate that could afford superior BA yields compared to all other benchmark acridinium catalysts attempted. The synergistic activity of FPh-Acr-Np and HCl for PS deconstruction allowed low catalyst loadings to be used (<5 mol%), and was also highly-compatible with real-life PS waste. Advantageously, the potential low toxicity of acridinium salts, evident from their many applications in bioimaging,44–46 may be beneficial if trace metal contamination is of concern for the BA's targeted application (especially in biomedical or food and beverage applications). Although only a sample of the acridinium chemical design space was sampled herein, our proof-of-concept study highlights the immense untapped potential of computer-aided catalyst design for low-energy and highly efficient upcycling of plastics and possibly, biomass (e.g. lignin), into essential chemicals.Data availability
All experimental details and data supporting the findings of this study are available within the paper and its ESI.†Author contributions
J. Y. C. L, S. J. A. and M. H. C. conceptualised the research. A. O. and K. L. O. C. performed synthesis and characterisation of all the catalyst. A. O. performed all the experimental screenings, data curation and validation. W. W. L. performed all GPC analyses of product mixtures. Z. C. W. and S. J. A. performed all the computational studies, data curation and validation. J. Y. C. L. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Y. C. Lim. is grateful to the NRF Fellowship (Award number: NRF-NRFF15-2023-0007) and the A*STAR Central Research Fund (UIBR) for generous funding support of this work. M. H. Chua acknowledges funding support by the A*STAR 2020 Career Development Fund (Grant Number: C210112042). This work was supported by the A*STAR Computational Resource Centre through the use of its high performance computing facilities. The authors acknowledge Dr Hazel Lau (A*STAR, IMRE) for assistance with ESI mass spectrometry analyses.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef.
- J. Maul, B. G. Frushour, J. R. Kontoff, H. Eichenauer, K.-H. Ott and C. Schade, Polystyrene and styrene copolymers, in Ullmann's Encyclopedia of Industrial Chemistry, 2007 Search PubMed.
- S. S. Yang, A. M. Brandon, D. F. Xing, J. Yang, J. W. Pang, C. S. Criddle, N. Q. Ren and W. M. Wu, Progresses in Polystyrene Biodegradation and Prospects for Solutions to Plastic Waste Pollution, IOP Conf. Ser.: Earth Environ. Sci., 2018, 150(1), 012005 CrossRef.
- A. Rahimi and J. M. García, Chemical recycling of waste plastics for new materials production, Nat. Rev. Chem, 2017, 1(6), 0046 CrossRef.
- J. Savoldelli, D. Tomback and H. Savoldelli, Breaking down polystyrene through the application of a two-step thermal degradation and bacterial method to produce usable byproducts, Waste Manage., 2017, 60, 123–126 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Critical advances and future opportunities in upcycling commodity polymers, Nature, 2022, 603(7903), 803–814 CrossRef CAS PubMed.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Upcycling to Sustainably Reuse Plastics, Adv. Mater., 2021, 2100843 Search PubMed.
- C. W. S. Yeung, M. H. Periayah, J. Y. Q. Teo, E. T. L. Goh, P. L. Chee, W. W. Loh, X. J. Loh, R. Lakshminarayanan and J. Y. C. Lim, Transforming Polyethylene into Water-Soluble Antifungal Polymers, Macromolecules, 2023, 56(3), 815–823 CrossRef CAS.
- J. Y. Q. Teo, A. Ong, T. T. Y. Tan, X. Li, X. J. Loh and J. Y. C. Lim, Materials from waste plastics for CO2 capture and utilisation, Green Chem., 2022, 24, 6086–6099 RSC.
- M. Y. Tan, L. Goh, D. Safanama, W. W. Loh, N. Ding, S. W. Chien, S. S. Goh, W. Thitsartarn, J. Y. C. Lim and D. W. H. Fam, Upcycling waste poly(ethylene terephthalate) into polymer electrolytes, J. Mater. Chem. A, 2022, 10(46), 24468–24474 RSC.
- Z. Xu, F. Pan, M. Sun, J. Xu, N. E. Munyaneza, Z. L. Croft, G. Cai and G. Liu, Cascade degradation and upcycling of polystyrene waste to high-value chemicals, Proc. Natl. Acad. Sci. U.S.A., 2022, 119(34), e2203346119 CrossRef CAS.
- A. Ong, J. Y. Q. Teo, Z. Feng, T. T. Y. Tan and J. Y. C. Lim, Organocatalytic Aerobic Oxidative Degradation of Polystyrene to Aromatic Acids, ACS Sustainable Chem. Eng., 2023, 11(34), 12514–12522 CrossRef CAS.
- Market Volume of Benzoic Acid Worldwide From 2015 to 2020, With a Forecast for 2021 to 2026, https://www.statista.com/statistics/1245227/benzoic-acid-market-volume-worldwide/, accessed 02 Feb 2023 Search PubMed.
- A. Pifer and A. Sen, Chemical Recycling of Plastics to Useful Organic Compounds by Oxidative Degradation, Angew. Chem., Int. Ed., 1998, 37(23), 3306–3308 CrossRef CAS.
- C. Rabot, Y. Chen, S.-Y. Lin, B. Miller, Y.-M. Chiang, C. E. Oakley, B. R. Oakley, C. C. C. Wang and T. J. Williams, Polystyrene Upcycling into Fungal Natural Products and a Biocontrol Agent, J. Am. Chem. Soc., 2023, 145(9), 5222–5230 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Mixed plastics waste valorization through tandem chemical oxidation and biological funneling, Science, 2022, 378(6616), 207–211 CrossRef CAS PubMed.
- Y. Zhang, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Photoredox-Catalyzed Plastic Waste Conversion: Nonselective Degradation versus Selective Synthesis, ACS Catal., 2023, 13(6), 3575–3590 CrossRef CAS.
- S. Oh and E. E. Stache, Chemical Upcycling of Commercial Polystyrene via Catalyst-Controlled Photooxidation, J. Am. Chem. Soc., 2022, 144(13), 5745–5749 CrossRef CAS.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, Chemical Recycling of Polystyrene to Valuable Chemicals via Selective Acid-Catalyzed Aerobic Oxidation under Visible Light, J. Am. Chem. Soc., 2022, 144(14), 6532–6542 CrossRef CAS.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Integrated strategy for the synthesis of aromatic building blocks via upcycling of real-life plastic wastes, Chem, 2022, 8(9), 2472–2484 CAS.
- R. Cao, M.-Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Catalytic oxidation of polystyrene to aromatic oxygenates over a graphitic carbon nitride catalyst, Nat. Commun., 2022, 13(1), 4809 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116(17), 10075–10166 CrossRef CAS.
- A. Tlili and S. Lakhdar, Acridinium Salts and Cyanoarenes as Powerful Photocatalysts: Opportunities in Organic Synthesis, Angew. Chem., Int. Ed., 2021, 60(36), 19526–19549 CrossRef CAS PubMed.
- B. Zilate, C. Fischer and C. Sparr, Design and application of aminoacridinium organophotoredox catalysts, Chem. Commun., 2020, 56(12), 1767–1775 RSC.
- A. Soyemi and T. Szilvási, Trends in computational molecular catalyst design, Dalton Trans., 2021, 50(30), 10325–10339 RSC.
- K. Ohkubo, K. Mizushima and S. Fukuzumi, Oxygenation and chlorination of aromatic hydrocarbons with hydrochloric acid photosensitized by 9-mesityl-10-methylacridinium under visible light irradiation, Res. Chem. Intermed., 2013, 39(1), 205–220 CrossRef CAS.
- A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, Estimation of Standard Reduction Potentials of Halogen Atoms and Alkyl Halides, J. Phys. Chem. B, 2011, 115(4), 678–684 CrossRef CAS.
- S. Oh and E. E. Stache, Mechanistic Insights Enable Divergent Product Selectivity in Catalyst-Controlled Photooxidative Degradation of Polystyrene, ACS Catal., 2023, 10968–10975 CrossRef CAS.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Light-Promoted Bromine-Radical-Mediated Selective Alkylation and Amination of Unactivated C(sp3)–H Bonds, Chem, 2020, 6(7), 1766–1776 CAS.
- S. Fukuzumi and K. Ohkubo, Organic synthetic transformations using organic dyes as photoredox catalysts, Org. Biomol. Chem., 2014, 12(32), 6059–6071 RSC.
- C. Qin, T. Shen, C. Tang and N. Jiao, FeCl2-Promoted Cleavage of the Unactivated C–C Bond of Alkylarenes and Polystyrene: Direct Synthesis of Arylamines, Angew. Chem., Int. Ed., 2012, 51(28), 6971–6975 CrossRef CAS PubMed.
- G. G. Gerosa, M. O. Marcarino, R. A. Spanevello, A. G. Suárez and A. M. Sarotti, Re-Engineering Organocatalysts for Asymmetric Friedel–Crafts Alkylation of Indoles through Computational Studies, J. Org. Chem., 2020, 85(15), 9969–9978 CrossRef CAS PubMed.
- K. N. Houk and P. H.-Y. Cheong, Computational prediction of small-molecule catalysts, Nature, 2008, 455(7211), 309–313 CrossRef CAS.
- Y. Zou, J. J. Wong and K. N. Houk, Computational Exploration of a Redox-Neutral Organocatalytic Mitsunobu Reaction, J. Am. Chem. Soc., 2020, 142(38), 16403–16408 CrossRef CAS PubMed.
- N. Fey, Lost in chemical space? Maps to support organometallic catalysis, Chem. Cent. J., 2015, 9(1), 38 CrossRef.
- C. Robertson and S. Habershon, Fast screening of homogeneous catalysis mechanisms using graph-driven searches and approximate quantum chemistry, Catal. Sci. Technol., 2019, 9(22), 6357–6369 RSC.
- K. J. Kron, A. Rodriguez-Katakura, R. Elhessen and S. Mallikarjun Sharada, Photoredox Chemistry with Organic Catalysts: Role of Computational Methods, ACS Omega, 2021, 6(49), 33253–33264 CrossRef CAS PubMed.
- S. Grimme, Calculation of the Electronic Spectra of Large Molecules, ed. K. B. Lipkowitz, R. Larter and T. R.Cundari, 2004 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91(2), 165–195 CrossRef CAS.
- G. Jones, D. Yan, S. R. Greenfield, D. J. Gosztola and M. R. Wasielewski, Anilide Linker Group as a Participant in Intramolecular Electron Transfer, J. Phys. Chem. A, 1997, 101, 4939–4942 CrossRef CAS.
- S. A. Jonker, F. Ariese and J. W. Verhoeven, Cation complexation with functionalized 9-arylacridinium ions: possible applications in the development of cation-selective optical probes, Recl. Trav. Chim. Pays-Bas, 1989, 108(3), 109–115 CrossRef CAS.
- Z. Peng, R. Chen and H. Li, Heterogeneous Photocatalytic Oxidative Cleavage of Polystyrene to Aromatics at Room Temperature, ACS Sustainable Chem. Eng., 2023, 11(29), 10688–10697 CrossRef CAS.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, Bridging Plastic Recycling and Organic Catalysis: Photocatalytic Deconstruction of Polystyrene via a C–H Oxidation Pathway, ACS Catal., 2022, 12(14), 8155–8163 CrossRef CAS PubMed.
- M. Wen, X. Wang, T. Wang, Y. Sun, M. Fan, M. Li, J. Zhu, D. Zhang, X. Cui and Y. Shan, Acridinium Benzoates for Ratiometric Fluorescence Imaging, Chem.–Eur. J., 2020, 26(15), 3247–3251 CrossRef CAS.
- A. Scorilas, K. Agiamarnioti and K. Papadopoulos, Novel biotinylated acridinium derivatives: new reagents for fluorescence immunoassays and proteomics, Clin. Chim. Acta, 2005, 357(2), 159–167 CrossRef CAS.
- A. Natrajan, D. Sharpe, J. Costello and Q. Jiang, Enhanced immunoassay sensitivity using chemiluminescent acridinium esters with increased light output, Anal. Biochem., 2010, 406(2), 204–213 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06388g |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |